
 |
 |
 |
 |
 |
Artigo
|
|
| Influência do impedimento estérico de ligantes ancilares em complexos a base de Ru aplicados em metáteses do álcool e acetato cinamílicos Influence of the steric hindrance of ancillary ligands in Ru-based complexes applied in metathesis of cinnamyl alcohol and cinnamyl acetate |
|
Vanessa B. VieiraI; Eliada A. da SilvaII; Valdemiro P. Carvalho Jr.III; Benedito S. Lima-NetoII,*; José L. S. SáI,*
I. Centro de Ciências da Natureza, Universidade Estadual do Piauí, 64002-150 Teresina - PI, Brasil Recebido em 11/06/2019 *e-mail: benedito@iqsc.usp.br, zeluizquimica@gmail.com Cinnamyl alcohol (AC) and cinnamyl acetate (ACM) were used as substrates in olefin metathesis reactions catalyzed by RuCl2(PCy3)2(=CHPh), first-generation Grubbs catalyst-G1, and RuCl2(PCy3)(H2IMes)(= CHPh), second-generation Grubbs catalyst-G2. The reactions occurred in the same reaction conditions for both substrates, 50 °C, for 24 h, in the proportions Ru:substrate of 1:1 and 1:10 mol. At the end of each experiment, the reaction mixture was evaluated by GC-MS and NMR of 13C{1H}. The results revealed different products when G1 and G2 are applied in the metathesis of AC, 1,5-diphenyl-2-pentene and stilbene, respectively. When ACM is the substrate, no product is noted with G1 and stilbene was the compound obtained with G2 as catalyst. In this study, we have presented a discussion about the electronic and steric influence of the ancillary ligands in the yield and type of product formed in the catalytic process. INTRODUÇÃO A metátese de olefinas vem se tornando uma poderosa ferramenta de síntese para a formação de ligações carbono-carbono de maneira seletiva. Uma das grandes vantagens é possibilitar a obtenção de moléculas insaturadas através da reorganização ou troca dos elementos que formam a dupla ligação, tendo em vista que o preparo delas por outro método seria muito difícil.1,2 Essa reação consiste em uma alternativa atraente devido à grande variedade de materiais de partida e atividade dos catalisadores,3,4 sendo amplamente utilizada em sínteses orgânicas, na química medicinal e na química de polímeros.5,6 Muitos complexos têm sido usados em metátese de olefinas, no entanto, vários metais de transição são inapropriados para serem utilizados como catalisadores nessas reações. Um dos aspectos é afinidade por grupos funcionais ou por moléculas presentes no meio,7-9 assim, é importante o desenvolvimento de catalisadores que reajam preferencialmente com a olefina do substrato.10 Em 1988, o grupo de Grubbs descreveu o desenvolvimento dos primeiros sistemas de catalisadores de rutênio. Os catalisadores de Grubbs são complexos Ru-alquilidenos que apresentam atividade em reações de metátese de olefinas,11 mostrando-se serem bem mais resistentes a interferentes orgânicos do que catalisadores com os complexos de Ti, W, Mo,7,12 despertando mais interesse por se apresentarem melhores que os de Schrock, quanto ao fato de não serem sensíveis ao ar e à umidade, por não serem oxofílicos e manterem sua reatividade na presença de grupo funcionais, reagindo preferencialmente com a olefina do substrato.8,12,13 Tais características produzem alto grau de liberdade e seletividade, permitindo que os compostos à base de rutênio encontrem vasta aplicação como catalisadores. O desenvolvimento de catalisadores de Ru abriu caminhos para transformações de uma ampla gama de substrato nas reações de metátese de olefinas, no entanto, diversos pesquisadores que trabalham com metátese de olefinas estão se voltando para estudo de substituição gradativa de compostos derivados do petróleo por outras fontes de carbono. A preocupação com o uso excessivo de produtos derivados de petróleo tem direcionado diversos ramos da ciência para a busca de materiais alternativos desenvolvidos à base de fontes renováveis.14-17 A utilização de produtos naturais para formulação de produtos aplicáveis industrialmente oferece muitas vantagens, e a principal é a capacidade de renovação. Possui também grande disponibilidade e baixo custo, podendo ser aplicado sobre eles diversas químicas, demonstrando assim, sua versatilidade. Essas substâncias químicas podem ser obtidas em larga escala a partir de plantas cultiváveis, que permite aproveitar a complexidade química gerada por processos bioquímicos naturais na síntese industrial de produtos da indústria química.18 Várias famílias de produtos naturais contêm ligações duplas carbono-carbono, as quais são particularmente adequadas para transformações catalíticas.19 Uma classe de compostos orgânicos naturais interessantes são os fenilpropanóides, nos quais podemos destacar os grupos cinâmicos derivados da canela, que podem ser obtidos através das folhas e da casca interna das árvores do gênero Cinnamomum, uma planta perene tropical que, além do seu uso em culinária, pode ser utilizada em medicamentos e em cosméticos,20 apresentando constituintes químicos como o álcool e o acetato cinamílicos.21 Esses produtos são utilizados como fragrâncias de perfumes, cosméticos, produtos de higiene pessoal. Essas moléculas são obtidas naturalmente das plantas, podendo ser extraídas e purificadas. No entanto, são encontradas em pequenas quantidades, e sua demanda industrial é geralmente preenchida por síntese químicas para obter melhores rendimentos.22 O presente estudo propõe realizar reações de autometátese dos grupos cinâmicos: álcool cinamílico e acetato cinamílico, pois são idênticos estruturalmente com diferentes grupos funcionais, obtendo-se assim dímeros desses compostos, bem como avaliar a atividade e seletividade dos catalisadores de Grubbs de 1ª e 2ª geração. Os rendimentos e as análises de CG-EM e RMN 13C {1H} realizadas são apresentados nesse estudo. É importante ressaltar que esse estudo visa contribuir na atuação dos catalisadores de Grubbs com moléculas simples e de baixo custo, além do entendimento da influência de diferentes grupos funcionais aos catalisadores.
PARTE EXPERIMENTAL Reações de autometátese Para as reações de metátese, em um balão de fundo redondo (100 mL) foram adicionados 10 mL de clorofórmio, previamente degaseificados por saturação com gás N2, por um período de 30 min. Em seguida, adicionou-se o substrato AC no balão degaseificado com solvente. Logo após, adicionaram-se à mistura 10 mg do catalisador. Utilizaram-se as relações Ru: substrato nas proporções de 1:10, 1:1 mol. As reações aconteceram em sistema de refluxo sob agitação constante, com temperatura controlada de 50 ºC, pelo período de 24 h. Após o tempo transcorrido, a reação foi finalizada com a adição de 3 µL de etilvinil éter. Uma alíquota desta mistura foi retirada para a análise por Cromatografia Gasosa acoplada a Espectrometria de Massas (CG-EM) e Ressonância Magnética Nuclear (RMN) 13C {1H}. Todo o procedimento descrito anteriormente também foi realizado com o substrato acetato cinamílico (ACM). Os catalisadores utilizados nas reações são os catalisadores de 1º e 2º geração de Grubbs, que foram usados separadamente para cada experimento realizado, utilizando as duas proporções estudadas com cada catalisador, na presença do AC ou ACM como substrato. Na Figura 1 estão dispostos os produtos de metátese dos grupos cinâmicos esperados e baseados no mecanismo da reação de autometátese.
 Figura 1. Ilustração do mecanismo de autometátese para o AC e ACM
Caracterizações Análise de cromatografia gasosa acoplada ao espectômetro de massas (CG-EM) As análises de CG-EM dos dímeros sintetizados foram conduzidas usando um aparelho GCMS-QP2010, fabricado pela Shimadzu, em uma coluna RTX®-5MS (30 m x 0.25 mm x 0.25 µm). A temperatura iniciou-se em 80 ºC e foi aumentando até 100 °C. Foram realizadas duas rampas de aquecimento: a primeira, a temperatura 10 ºC mantida por 1 min; a segunda, a temperatura aumentava 4 ºC/min durante 10 min, até chegar a 250 ºC. A análise ocorria por 40 min, com fluxo de coluna total de 1,0 mL min-1 e a razão split de 1,0. O espectrômetro de massas é acoplado à cromatografia gasosa. As moléculas fragmentadas são detectadas pelo equipamento Shimadzu GCMS-QP2010, entrando na fonte de ionização por impacto eletrônico com energia cinética de 70 eV. O equipamento utilizado é pertencente à Universidade Estadual do Piauí (UESPI). Ressonância magnética nuclear (RMN) As análises de RMN foram realizadas utilizando equipamento Bruker DRX-400 ou Bruker DRX-500, obtendo espectros de RMN de 13C {1H} e 1H. As amostras foram preparadas em atmosfera inerte, utilizando clorofórmio deuterado (CDCl3) como solvente. Esse equipamento é pertencente ao Instituto de São Carlos (IQSC-USP), e se encontra na central de Análises Químicas Instrumentais (CAQI).
RESULTADOS E DISCUSSÃO Para a investigação da atividade dos catalisadores, com destaque para as propriedades estéricas e eletrônicas dos ligantes ancilares, reagiram-se diferentes proporções de AC, à 50 ºC por 24 h, catalisadas por G1 e G2. Os resultados foram analisados por CG-EM (Figura 2) e resumidos na Tabela 1.
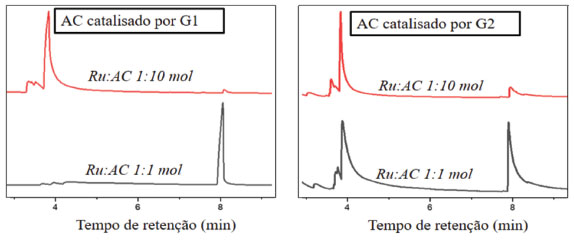 Figura 2. Cromatograma dos produtos das reações de metátese de AC catalisadas por G1 e G2. Condições reacionais: razão Ru:substrato de 1:1 ou 1:10 mol, 50 ºC por 24 h

O pico em menor tempo de retenção de cada cromatograma é referente ao substrato, e o segundo pico é relativo ao produto formado. Na Tabela 1 estão demonstrados os resultados obtidos nas análises de CG-EM para AC. Os rendimentos foram aferidos por integração dos sinais do substrato e do produto na mistura. Primeiramente se integrou o sinal do substrato e o rendimento do produto foi medido em relação à área do substrato. Os valores de rendimentos foram aferidos pela integração dos picos observados nos cromatogramas. Quando o catalisador foi G1 e o substrato AC, em ambas as proporções estudadas, o único produto observado foi o 1,5-difenil-2-penteno. Com Ru:AC de 1:10 mol, a conversão do substrato foi de cerca de 5%; na proporção de 1:1 mol, a formação de produto alcançou cerca de 76%. Essa tendência também acontece com G2 (cerca de 18% com Ru:AC de 1:10; cerca de 40% com Ru:AC de 1:1 mol), no entanto, o único produto obtido com G2 como catalisador foi o estilbeno (Tabela 1). Os diferentes rendimentos de produtos de metátese, tanto com G1 e G2 como catalisadores, variando-se as proporções de 1:1 para 1:10 mol, indicam um caráter associativo nas reações. Num mecanismo de caráter associativo, a principal variável de sistema aplicados em sínteses é a quantidade de substrato, como na proporção Ru:AC de 1:1 mol, sendo que nessa proporção foram obtidos maiores valores de formação de produtos com os diferentes catalisadores. No contrário, espera-se para mecanismos dissociativos que a variação da quantidade de substrato não afete o rendimento da reação. O maior rendimento observado em todos os experimentos de metátese de AC foi cerca de 76% com G1, na proporção de 1:1 mol, e o produto formado foi o 1,5-difenil-2-penteno, indicando que o menor impedimento estérico deste catalisador proporcionou maior reatividade. Diminuindo-se a quantidade de substrato inicial do sistema, a influência eletrônica do G2 foi mais pronunciada. Comparando-se os valores de 5,5%, para G1, e 18,2%, para G2, nota-se que o deslocamento eletrônico sinérgico-trans NHC®Ru®substrato auxiliou a saturação dos orbitais π* da olefina, vencendo o impedimento estérico na proporção de 1:10 mol. Muitos trabalhos, bem como a teoria de metátese de olefinas, apontam diferentes resultados baseados nas diferentes contribuições eletrônicas dos catalisadores. Ressonância magnética 13C {1H} de AC com G1 e G2 como catalisadores Os resultados dos ensaios de metátese com G1 e G2 como catalisadores também foram analisados por RMN de 13C{1H}. Assim como as análises por CG-EM, não foram observados indícios de outros produtos formados além do estilbeno e do 1,5-difenil-2-penteno. O principal objetivo dessas análises foi de confirmar a formação destes produtos e descartar a formação dos diálcoois a partir da troca dos grupos da olefina (ver Figura 1).23-25 Devido aos baixos rendimentos de produtos formados a partir das reações com AC com G1 e G2 na proporção Ru:AC de 1:10 mol, não foi possível observar picos nos espectros de RMN de 1,5-difenil- 2-penteno e estilbeno. Por outro lado, as análises dos produtos da reação do AC com G1 e G2 na proporção de 1:1 mol comprovaram os produtos formados (Figura 3b).
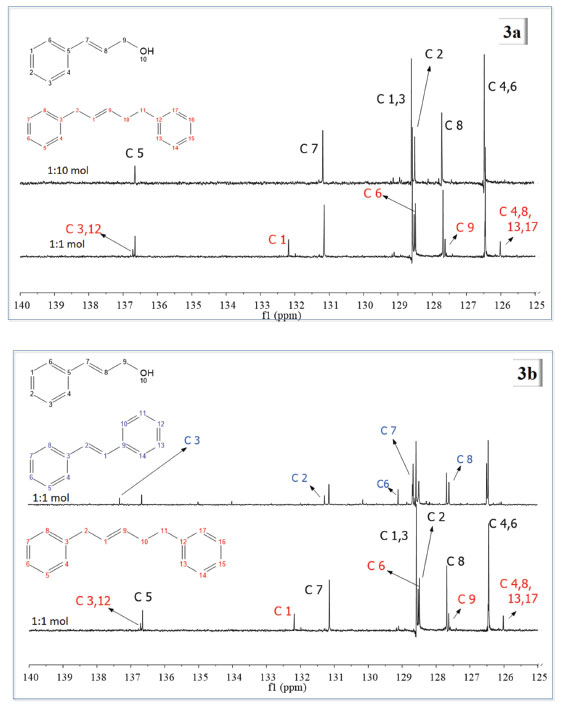 Figura 3. (a) Espectros de RMN de 13C {1H} para os produtos das reações de metátese de AC catalisada por G1, com Ru:AC de 1:10 e 1:1 mol. (b) Espectros de RMN de 13C {1H} para os produtos das reações de metátese do AC com G2 (espectro com numeração em azul) e G1
Na Figura 3a compararam-se os espectros resultantes das reações de metátese do AC com G1 como catalisador, nas duas proporções estudadas. Os traços de 1,5-difenil-2-penteno obtidos nos estudos com Ru:AC de 1:10 mol não foram observados, portanto, apenas o substrato que se encontrava em maior concentração foi notado. Os espectros foram analisados e os picos dos produtos atribuídos e comparados com os do substrato. Os carbonos C4,6 e C1,3 (figura com numeração em preto) encontram-se em d típicos de carbono aromático, 126,5 e 128,5 ppm, respectivamente. Devido ao efeito indutivo do C9, o d para C8 foi observado em campo mais alto que C7, que aparece em um d típico para olefinas. C5 é o carbono mais eletronicamente desblindado, com pico relativo em cerca de 136,5 ppm devido ao efeito anisotrópico gerado pelo anel aromático e pela ligação C7=C8. Para o produto 1,5-difenil-2-penteno (molécula com numeração em vermelho), destacam-se os picos dos C3 e 12 que são os mais desblindados da molécula. C9 aparece como o carbono olefínico em campo mais alto, e os carbonos C4, C8, C13 e C17 são os carbonos aromáticos mais blindados, com pico relativo em 126 ppm. Na Figura 3b, estão apresentados os espectros do produto da metátese do AC com G2 como catalisador (figura com numeração em azul) e G1 (figura com numeração em vermelho e preto), ambos em ensaios com proporção Ru:AC de 1:1 mol. Nos dois casos, além dos produtos de metátese, também foram observados picos referentes ao substrato. Com G2 como catalisador, o pico referente ao C3 do estilbeno é o mais desblindado, com δ em cerca de 137,5 ppm. Nesse espectro não se observa o sinal referente aos carbonos C3 e C12 (molécula com numeração em vermelho) do 1,5-difenil-2-penteno, indício de que esse composto não foi formado. Assim como C1, C4, C8, C13 e C17 da molécula de 1,5-difenil-2-penteno. Para a molécula de estilbeno, C1 e C2 aparecem com picos no mesmo deslocamento químico, típico de olefinas. Foram propostos esquemas reacionais de AC com os dois catalisadores de Grubbs para um melhor entendimento das reações. Na Figura 4 está apresentado um esquema reacional que demonstra as diferentes etapas para formação do 1,5-difenil-2-penteno. É importante destacar que há uma etapa anterior a esse processo, na qual parte do catalisador G1 catalisa a isomerização do AC. O mecanismo de isomerização já é bem aceito na literatura, iniciando-se pela decomposição do catalisador, gerando uma espécie inativa para metátese e ativa para isomerização de olefinas.26-29
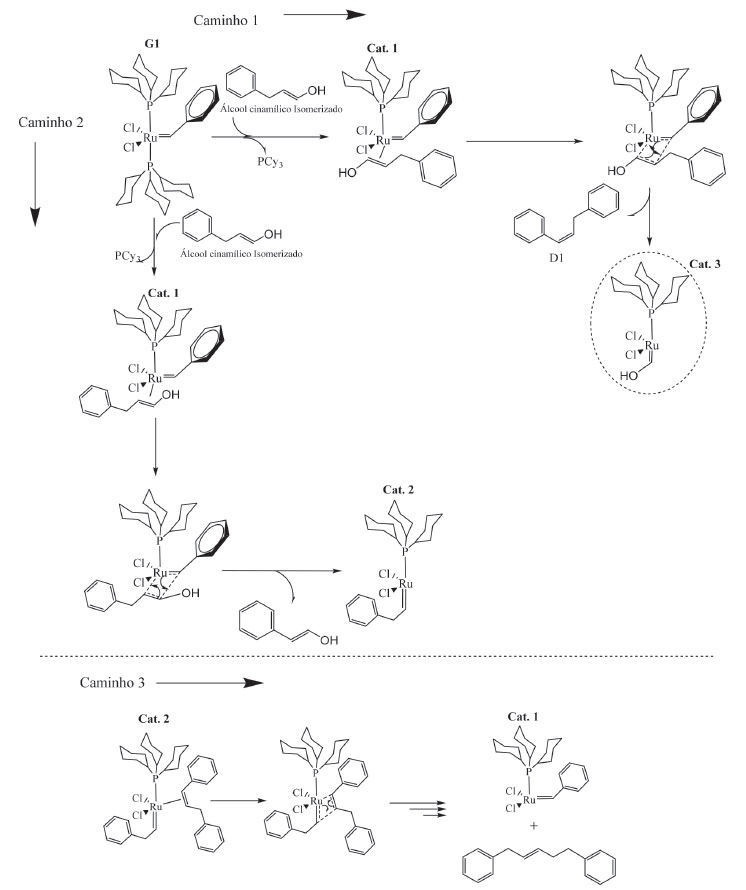 Figura 4. Esquema reacional da metátese de olefinas do AC isomerizado, catalisada por G1
Seguindo-se o caminho 1, Figura 4, há a descoordenação de uma PCy3 e formação da espécie ativa "cat.1", simultaneamente coordenada ao substrato. Há a formação do primeiro intermediário metalociclobutano, troca entre os grupos da olefina e do carbeno, formando o primeiro produto dimérico (D1) e a espécie "cat.3". "cat.3" não parece ser ativo para metátese de olefinas, nas análises por RMN de 13C ou CG-EM nenhum possível produto formado a partir do cruzamento do carbeno Ru=CH-OH foi observado. Seguindo pelo caminho 2, que considera a aproximação do AC de forma contrária à apresentada no caminho 1, ou seja, com os grupos fenis do carbeno e do AC trans-posicionados, há formação de uma nova espécie ativa "cat. 2" e o álcool 2-feniletenol. A formação da espécie ativa "cat.2", em adição ao primeiro dímero formado no caminho 1, foi crucial para o entendimento da síntese do 1,5-difenil-2-penteno; composto notado nas análises por RMN de 13C e CG-EM. A reação entre a espécie ativa "cat. 2" e a primeira espécie dimérica leva à formação do 1,5-difenil-2-penteno. Apesar de neste trabalho não ter sido realizada nenhuma investigação mais profunda a respeito da isomerização do AC, a autometátese desta molécula não isomerizada levaria a dois possíveis produtos, o estilbeno, que não foi observado, ou ao próprio AC (Figura 5). Portanto, discorre-se acerca do mecanismo a partir do AC isomerizado.
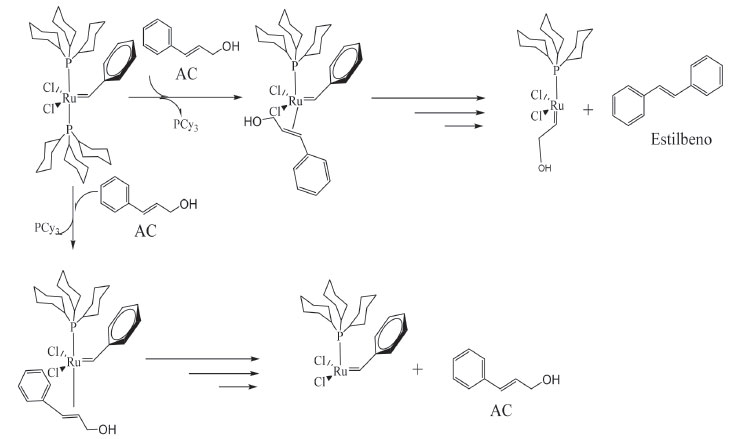 Figura 5. Esquema reacional para a reação de autometátese do AC, catalisada por G1
Ainda na proporção de 1:1 mol de Ru;AC, agora com G2 como catalisador, nota-se a formação de cerca de 40% de produto, mas de estilbeno, não 1,5-difenil-2-penteno, como observado para G1. Acredita-se que o impedimento estérico do ligante ancilar NHC de G2 (ver Figura 1) promoveu a limitação espacial tal que, mesmo com grande quantidade de catalisador, não permitiu a formação do 1,5-difenil-2-penteno. Principalmente na etapa análoga ao caminho 3 da Figura 4.3, no qual uma espécie dimérica deve coordenar-se ao centro metálico. Além disso, a formação do estilbeno como único produto da metátese de AC com G2 como catalisador aponta que a molécula de AC não foi previamente isomerizada, com uma reação direta entre G2 e AC, formando estilbeno em uma única etapa (Figura 1S). Autometátese do ACM Reações catalisadas por G1 e G2 Para a avaliação da atividade dos catalisadores, reagiram-se diferentes proporções de ACM, a 50 ºC por 24 h, catalisadas por G1 e G2. Os resultados foram obtidos por CG-EM (Figura 6) e resumidos na Tabela 2.
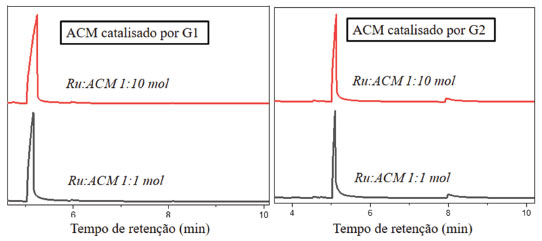 Figura 6. Cromatograma dos produtos das reações de metátese de ACM catalisadas por G1 e G2. Condições reacionais-razão Ru:substrato de 1:1 ou 1:10 mol, 50 ºC por 24 h

Da mesma forma dos cromatogramas para os resultados da metátese de AC, os gráficos de ACM carregam as mesmas características, sendo que os gráficos do lado esquerdo são referentes às análises do ACM com G1, já os do lado direito representa as análises do ACM com G2. A Tabela 2 representa os resultados de rendimento para as reações de metátese com ACM, com os diferentes catalisadores de Grubbs. Nas reações com o ACM, pôde-se observar menores valores de conversão de substrato em relação aos experimentos com AC (Tabela 1), obtendo o estilbeno como produto nas reações tanto com G1 como G2. Nos experimentos realizados para ACM, não houve formação de produto de metátese com G1 como catalisador, tanto com Ru:ACM de 1:10 quanto 1:1 mol. Já utilizando G2 como catalisador, esse apresentou atividade, porém, menor que a observada nos ensaios com AC, os rendimentos foram cerca de 15 e 18%, com Ru:ACM de 1:10 e 1:1 mol, respectivamente, enquanto para AC foram 18 e 40%, respectivamente. Os ensaios de metátese também foram analisados por RMN, no entanto, da mesma forma que as análises de CG-EM, não foram observados picos nos espectros de RMN devido aos baixos rendimentos de produto. De fato, os dois grupos funcionais oxigenados diferentes das moléculas de AC e ACM devem gerar diferentes reatividades aos respectivos sistemas, muito observado em diferentes literaturas de metátese de olefinas.30,31 Esse fato corrobora com os resultados encontrados para os diferentes substratos deste trabalho. De acordo com os baixos rendimentos de produtos formados de ACM, foi proposta a realização de esquemas reacionais para um melhor entendimento das reações de ACM com os diferentes catalisadores de Grubbs. Na Figura 7 está apresentado um esquema reacional que demonstra a reação de autometátese do ACM com G1.
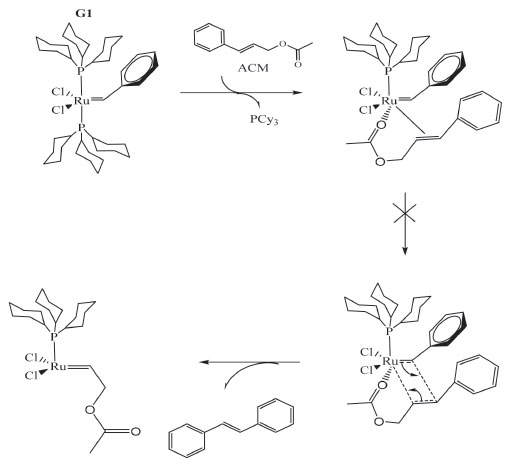 Figura 7. Esquema reacional para a reação de autometátese do ACM, catalisada por G1
Acredita-se que houve coordenação do oxigênio C=O do grupo carboxílico ao metal simultaneamente à coordenação da olefina, o que levaria a um sistema estabilizado, com uma etapa a mais para formação do produto. É um entendimento da comunidade científica que a formação de um sistema quelato pode dificultar a metátese de olefinas, porque o sistema perde mobilidade, fica rígido.8 Esse efeito, associado ao pouco impedimento estérico de G1 gerado pela PCy3, aumentou a estabilidade do coordenação adicional do C=O, impedindo assim a continuação da etapa de formação do intermediário metalociclobutano e posterior formação do produto esperado-estilbeno (Figura 2S). Da mesma forma como explicado anteriormente, acredita-se na coordenação do oxigênio C=O de ACM ao metal simultaneamente à da dupla olefínica do substrato, formando um sistema quelato. No entanto diferente de G1, que o ligante ancilar PCy3 aumentou a estabilidade da coordenação do C=O, a presença do grupo NHC de G2 diminuiu a dupla coordenação, somado ao efeito sinérgico trans, proporcionaram a continuidade da etapa da reação de metátese, levando à formação do intermediário metalociclobutano, e posterior desenvolvimento do estilbeno, produto a partir de ACM. É fato que o oxigênio do grupo C-OH do AC possui elétrons livres capazes de se coordenarem ao centro metálico, análogo ao seu similar ACM. No entanto, para um complexo a base de Ru (II), 6 elétrons no subnível d externo, a retrodoação Ru®O=C torna a ligação com ACM mais forte. Nós realizamos experimentos adicionais com o trans-β-Metilestireno, molécula similar ao AC e ACM sem nenhum grupo funcional oxigenado para avaliar a teoria da dupla coordenação de ACM. O rendimento com G2 como catalisador alcançou mais de 90% de estilbeno formado, corroborando com a proposta que a formação de um sistema quelato com ACM diminuiu, ou impediu no caso de G1, a metátese do ACM. Esse resultado foi obtido quando em ensaios a 50 ºC por 24 h, com a razão Ru: trans-β-Metilestireno de 1:10 mol. A proposta de fragmentação para os produtos de metátese, baseados nos respectivos espectros de massa, está apresentada no Material Suplementar.32,33
CONCLUSÃO G1 e G2 foram avaliados como catalisadores na metátese de olefinas do AC e ACM, por 24 h a 50 ºC, tendo como a principal variável de sistema a proporção Ru:substrato; de 1:10 e 1:1 mol. Os resultados indicaram que o impedimento estérico dos ligantes ancilares PCy3 e NHC conduziram à formação de dois diferentes produtos, 1,5-difenil-2-penteno para G1 e estilbeno para G2. O maior ângulo de cone do NHC impediu a continuidade das sucessivas reações de metátese, levando à formação do estilbeno, um dímero menor que o 1,5-difenil-2-penteno. Na proporção Ru:AC de 1:1 mol, G1 apresentou maior rendimento que G2, base para a proposta de um mecanismo de caráter associativo. Já na proporção de 1:10 mol, o efeito eletrônico sinérgico NHC®Ru®AC foi preponderante em relação ao efeito estérico. Nas reações com ACM, pôde-se concluir que houve a formação de dupla coordenação do substrato com o metal, pela ligação olefínica e pelo oxigênio do grupo C=O, dificultando de maneira geral a metátese. E, corroborando com os dados de AC, o maior impedimento estérico de G2 dificultou a dupla coordenação, levando assim à maior atividade desse catalisador em relação a G1 na metátese de ACM.
MATERIAL SUPLEMENTAR Os esquemas reacionais para a metátese do AC e ACM com G2 como catalisador, espectros de massa e sequências de fragmentação do estilbeno e 1,5-difenil-2-penteno estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) (Código de Financiamento 001), do CNPq, da FAPEPI e da FAPESP: Proc. FAPESP 2017/06329-59 (Benedito S. Lima-Neto) e FAPESP 2018/06340-1 (Valdemiro P. Carvalho Jr.)
REFERÊNCIAS 1. Hoveyda, A. H.; Zhugralin, A. R.; Nature 2007, 450, 243. 2. Buffon, R.; Chemkeys 2009, 1, 5. 3. Schmidt, B.; Krehl, S. Em Olefin Metathesis: Theory and Practice; Grela, K., ed.; John Wiley & Sons: Hoboken, 2014. 4. Fogg, D. E; Santos, E. M.; Coord. Chem. Ver. 2004, 248, 2365. 5. Rybak, A.; Fokou, P. A.; Meier, M. A. R.; Eur. J. Lipid Sci. Technol. 2008, 110, 797. 6. Sá, J. L. S.; Lima-Neto, B. S.; J. Mol. Catal. A: Chem. 2009, 304, 187. 7. Frenzel, U.; Nuyken, O.; J. Polym. Sci. Pol. Chem. 2002, 40, 17. 8. Grubbs, R. H.; Tetrahedron 2004, 60, 7117. 9. Bielawski, C. W.; Grubbs, R. H.; Prog. Polym. Sci. 2007, 32, 29. 10. Matos, J. M. E; Batista, N. C.; Carvalho, R. M.; Santana, S. A. A.; Puzzi, P. N.; Sanches, M.; Lima-Neto, B. S.; Quim. Nova 2007, 30, 431. 11. Nguyen, S. T.; Ziller, W.; Grubbs, R. H.; J. Am. Chem. Soc. 1993, 115, 9858. 12. Deraedt, C.; D'Halluin, C. M; Astruc, D.; Eur. J. Inorg. Chem. 2013, 95, 4881. 13. Burdett, K. A; Harris, L. D.; Margl, P.; Maughon, B. R.; Mokhtar-Zadeh, T.; Saucier, P. C.; Wasserman, E.P.; Organometallics 2004, 23, 2027. 14. Biermann, U.; Bornscheuer, U.; Meier, M. A. R.; Metzger, J. O.; Schäfer, H. J.; Angew. Chem., Int. Ed. 2011, 18, 17. 15. Islam, M. R.; Beg, M. D. H.; Jamari, S. S.; J. Appl. Polym. Sci. 2014, 131, 40787. 16. Malacea, R.; Dixneuf, P. H. Em Great Challenges in Synthesis, Catalysis and Nanotechonology; Dragutan, V., eds.; Springer: New York, 2010, p. 185. 17. Renewable Bioressources: Scope and Modification for Non-food Applications, Verhé, R. G., eds.; Wiley: Hoboken, 2004. p. 208. 18. Swift, K. A. D.; Top. Catal. 2004, 27, 143. 19. Borré, E.; Dinh, T. H.; Caijo, F.; Crévisy, C.; Mauduit, M.; Synthesis 2011, 2125. 20. Nogueira, R. C.; De Cerqueira, H. F.; Soares, M. B. P.; Expert Opin. Ther. Pat. 2010, 2, 20. 21. Ranasinghe, P.; Pigera, S.; Premakumara, G. A.; Galappaththy, P.; Constantine, G. R.; Katulanda, P.; BMC Complem. Altern. Med. 2013, 13, 275. 22. Zucca, P.; Littarru, M.; Rescigno, A.; Sanjust, E.; Biosci. Biotech. Biochem. 2009, 5, 73. 23. Jean-Louis Hérisson, P.; Chauvin, Y.; Makromol. Chem. 1971, 1, 141. 24. Blackwell, H. E.; O'Leary, J. D.; Chatterjee, A. K.; Washenfelder, R. A.; Bussmann, D. A.; Grubbs, R. H.; J. Am. Chem. Soc. 2000, 122, 59. 25. Chauvin, Y.; Angew. Chem., Int. Ed. 2006, 45, 3740. 26. Hong, S. H.; J. Am. Chem. Soc. 2007, 25, 129. 27. Alexander, K. A; Emily, A.; Paulhus, E. A; Lazarus, G. M. L; Nicholas, E.; Leadbeater, N. E.; J. Org. Chem. 2015, 74, 812. 28. Grandner, J. M., Shao, H.; Grubbs, R. H.; Liu, P.; Houk, K. N.; J. Org. Chem. 2017, 19, 10595. 29. Dos Santos, E. N.; Granato, A.V.; Santos, A. G.; ChemSusChem 2017, 10, 749. 30. Dinger, M. B.; Mol, J. C.; Organometallics 2003, 5, 22. 31. Marvey, B. B.; Segakweng, C. K.; Vosloo, M. H. C.; Int. J. Mol. Sci. 2008, 9, 615. 32. Güsten, H; Klasinc, L; Marsel, J.; Milivojevic, D. A.; Org. Mass Spectrom. 1972, 6, 175. 33. Güsten, H; Klasinc, L; Kramer, V.; Marsel, J.; Org. Mass Spectrom. 1974, 8, 323. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access