
 |
 |
 |
 |
 |
Revisão
|
|
| Há algo novo no reconhecimento molecular aplicado à química medicinal? Is there anything new about the molecular recognition applied to medicinal chemistry? |
|
Harold H. FokoueI; Pedro S. M. PinheiroI; Carlos A. M. FragaI; Carlos M. R. Sant'AnnaII,*
I. Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, 21941-902 Rio de Janeiro - RJ, Brasil Recebido em 07/10/2019 *e-mail: santanna@ufrrj.br In Medicinal Chemistry, the knowledge, the study and the understanding of the intermolecular interactions inherent to the molecular recognition of a ligand by its target bioreceptor are essential. This is because understanding these interactions, which define the molecular complementarity between the two, is the key to analyze critically structure-activity relationship studies, allowing the optimization of the modulation of a target bioreceptor by a ligand. Thus, it is necessary the constant study and analysis of intermolecular interactions related to the ligand-bioreceptor complexes. In general, these interactions are all electrostatic in nature, being traditionally classified into two large groups, the electrostatic forces themselves (such as those present in ion-dipole or ion-ion interactions) and van der Waals forces. Despite being well known, theoretical and experimental studies have revealed an increasing number of interactions mediated by these forces. This work presents and discusses intermolecular interactions considering "classical interactions" and "new interactions" in the context of Medicinal Chemistry, taking into account their current definitions, some theoretical principles of each interaction and addressing examples in which these interactions were important for complementarity between a ligand and a biorreceptor. INTRODUÇÃO Em Química Medicinal, o estudo do reconhecimento molecular tem um papel importante nas diversas estratégias de planejamento e/ou desenvolvimento de novas entidades químicas que modulem um determinado alvo biológico.1-3 O reconhecimento molecular nos sistemas biológicos baseia-se na existência de interações intermoleculares específicas entre duas estruturas moleculares, o chamado ligante e seu biorreceptor alvo, geralmente uma proteína.4 Desde o modelo chave-fechadura introduzido por Emil Fischer em 18941,5 e do modelo do encaixe induzido proposto por Koshland,1,5 estudos continuam sendo feitos para se identificar e compreender essas interações.6 As interações intermoleculares são mediadas por forças bem conhecidas. Embora sejam essencialmente todas de natureza eletrostática, tradicionalmente prefere-se classificá-las em dois grupos, as forças eletrostáticas propriamente ditas (como as presentes em interações íon-dipolo ou íon-íon) e as forças de van der Waals. Estas últimas são forças residuais atrativas ou repulsivas entre átomos que não estão ligados por ligações covalentes ou por forças eletrostáticas entre íons ou entre íons e espécies neutras (IUPAC Gold Book).7,8 Dependendo da natureza das espécies envolvidas, as forças de van der Waals podem ainda ser divididas em forças entre dipolos permanentes (Forças de Keesom),9 forças entre dipolos permanentes e dipolos induzidos (Forças de Debye)7 e forças entre dipolos instantâneos e dipolos induzidos (Forças de dispersão de London).10 Apesar das forças envolvidas serem bem conhecidas, resultados teóricos e experimentais têm revelado um número crescente de interações mediadas por elas, algumas um tanto inesperadas. Nesse trabalho, apresentaremos as que podem ser consideradas como interações clássicas e as consideradas como novas interações, incluindo exemplos de suas participações na Química Medicinal, muitas representadas através de estruturas cristalográficas disponíveis. Estruturas cristalográficas são uma evidência experimental bastante convincente e ilustrativa da presença provável de uma determinada interação em um sistema proteína-ligante. No entanto, lembramos aos leitores que essas estruturas devem ser entendidas como "instantâneos" dentre as várias possibilidades estruturais em equilíbrio, que se interconvertem com uma cinética específica na temperatura da biofase. Em razão da extensão do assunto, aspectos termodinâmicos e cinéticos não fazem parte do escopo do presente texto, ficando a cargo do leitor interessado o aprofundamento destas questões em literatura específica.
LIGAÇÕES DE HIDROGÊNIO De acordo com a IUPAC, ligações de hidrogênio são interações atrativas entre um átomo de hidrogênio de uma molécula ou fragmento molecular X-H, em que X é mais eletronegativo que H, e um átomo ou grupo de átomos na mesma molécula ou em uma molécula diferente.11 Dessa forma, uma ligação de hidrogênio poderia ser representada esquematicamente por X-H∙∙∙Y-Z, em que os três pontos representam a interação, X-H o doador da ligação de hidrogênio e o aceptor poderia ser um átomo ou ânion Y, ou um fragmento ou uma molécula Y-Z. Em todo caso, o aceptor trata-se de uma região rica em elétrons, como por exemplo, um par de elétrons não ligantes de Y ou uma ligação π de Y-Z.11 É importante destacar que quanto maior é a eletronegatividade do átomo ou grupo de átomos representado como X, mais forte é a ligação de hidrogênio, o que explica a definição clássica de que doadores de ligações de hidrogênio são representados por grupos N-H, O-H e F-H. Isso se relaciona diretamente com a intensidade da carga parcial positiva que se forma sobre o átomo H pela ligação com o átomo mais eletronegativo X.12 O compartilhamento simples de átomos de H entre dois átomos eletronegativos representa o tipo mais comum de ligação H, mas atualmente é proposto que existam tipos adicionais de ligações de H mais fracas; por exemplo, interações N-H∙∙∙S e até mesmo C-H∙∙∙π (também chamada ligação de hidrogênio-π), com resíduos de aminoácidos aromáticos atuando como aceitadores.13,14 Esta última interação será tratada mais adiante neste trabalho (Item 7). Um tipo de ligação H que é encontrado em estruturas de proteínas é a ligação de hidrogênio bifurcada. Neste caso, o átomo H com uma carga ligeiramente positiva é deslocalizado entre dois átomos aceptores. Estes são mais frequentemente vistos entre as cadeias laterais de resíduos Ser/Thr com Asp/Glu como aceptores.15 Apesar de sua origem eletrostática, ligações de hidrogênio também apresentam características de transferência de carga entre o doador e o aceptor da ligação, levando à formação de uma ligação com caráter covalente parcial.11 A energia associada a uma ligação de hidrogênio varia de 2 a 10 kcal mol-1, o que pode parecer pouco, mas é importante lembrar que essas interações são flexíveis e dinâmicas, sendo constantemente quebradas e reformadas nas condições fisiológicas.16 Há uma variação bastante grande nas propriedades da ligação de hidrogênio, como foi observado em estruturas das proteínas: as distâncias entre o átomo H do doador (X) e o átomo aceptor (Y) da ligação H ficam tipicamente entre 1,7 e 2,4 Å, e o ângulo X-H∙∙∙Y pode estar entre 130° e 170°.17 Como as átomos de hidrogênio são difíceis de observar diretamente por difração de raios-X, as ligações de hidrogênio geralmente são inferidas quando a distância entre os átomos X e Y é menor do que 3,5 Å e os ângulos no doador/aceptor são maiores do que 90°.13 De uma forma mais específica, é possível destacar alguns exemplos interessantes onde este tipo de interação foi essencial no desenvolvimento de compostos com atividade biológica. Um exemplo são alguns inibidores de proteínas cinases que atuam se ligando no sítio do ATP nessas enzimas. Esses inibidores necessitam realizar interações por ligações de hidrogênio em uma importante região da enzima chamada hinge (dobradiça), como pode ser observado na Figura 1, em que o derivado pirazolo-piridínico (1) realiza ligações de hidrogênio com os resíduos Tyr134, Val135 e Glu97 da proteína cinase GSK3 beta (PDB: 5OY4).18
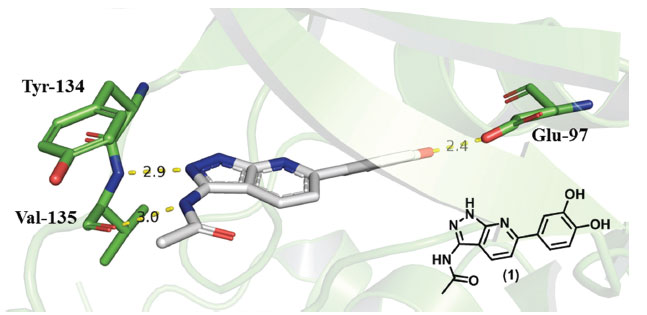 Figura 1. Interações por ligação de hidrogênio (linhas pontilhadas amarelas) observadas entre (1) (átomos de C em branco) e os resíduos de aminoácido Tyr134, Val135 e Glu97 (átomos de C em verde) da proteína cinase GSK3β (PDB: 5OY4).18 Distâncias em Å. Figura gerada com o programa PyMOL.
INTERAÇÕES ENVOLVENDO CAVIDADES-σ A existência das chamadas cavidades-σ, regiões de baixa densidade eletrônica presentes na superfície de átomos e opostas a ligações σ formadas por eles, foi primeiro associada a halogênios. Essas regiões se tornam maiores e a densidade eletrônica correspondente se torna menor à medida que se desce no grupo 17 da tabela periódica, o que é proposto por alguns autores como estando relacionado com o aumento da polarizabilidade do átomo de halogênio19 (Figura 2). A localização das cavidades-σ, em oposição à ligação σ, indica a participação do orbital anti-ligante σ*. De fato, à medida que se desce no grupo 17 em moléculas com ligações C-X, a energia do orbital σ* diminui, apresentando uma capacidade aceptora de elétrons crescente (Figura 2).
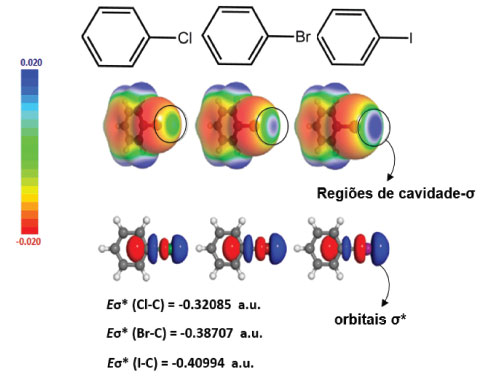 Figura 2. Regiões de cavidade-σ em halogênios, adaptado de Beno e colaboradores.20 Unidade dos mapas de potencial eletrostático em a.u (1 a.u.= 627,50 kcal/mol, 1 a.u.= 2625,5 kJ/mol). Método utilizado para geração dos mapas: B3LYP/6-311G(d).
Ligações de halogênio são interações em que átomos de halogênios interagem favoravelmente através das regiões de cavidade-σ, que apresentam um potencial eletrostático relativamente positivo, com átomos ou grupos capazes de atuar como bases de Lewis.21 De acordo com definição recomendada pela IUPAC, uma ligação de halogênio típica pode ser representada pelos três pontos em R-X∙∙∙Y, em que R-X é o doador da ligação de halogênio, sendo X qualquer halogênio apresentando uma região de cavidade σ e R é um grupo ou átomo ligado covalentemente ao halogênio. Y é o aceptor da ligação de halogênio, sendo uma espécie apresentando ao menos uma região nucleofílica para interagir com o doador da interação.22 Este tipo de interação já vem sendo explorada de forma racional na busca de novos compostos que apresentem atividade biológica. Tomando como exemplo a série de compostos (2) a (5) desenvolvidos para a inibição covalente reversível da catepsina L humana (hCatL),23 é possível perceber pelos dados de CI50 que há uma estabilização da interação conforme se aumenta a massa atômica dos átomos de halogênio. A partir da análise das estruturas cristalográficas dos complexos da enzima com (3) (PDB: 2XU4), (4) (PDB: 2YJC), (5) (PDB: 2YJ2), e (6) (PDB: 2YJ8),23 isso foi relacionado com a capacidade desses compostos realizarem ligações de halogênio com o átomo de oxigênio de um resíduo de glicina (Gly61) (Figura 3).
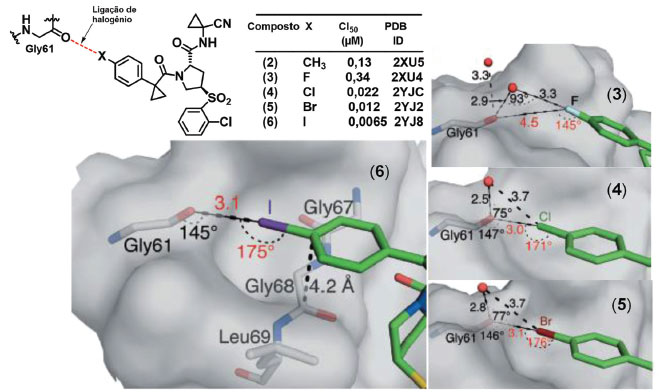 Figura 3. Estruturas químicas dos compostos (3), (4), (5), (6) e modo de interação com hCatL (Adaptado de Hardegger e colaboradores).23 Linhas tracejadas pretas: ligações de hidrogênio, de halogênio e interação amida π. As distâncias e ângulos das ligações de hidrogênio e da interação amida π estão em preto e ligações de halogênios em vermelho. Distâncias em Å. Figura gerada com os programas PyMOL e ChemDraw.
Tesch e colaboradores relataram que o perfil inibidor da enzima glicogênio sintase cinase 3β humana (GSK-3β) do derivado sulfonilidrazônico PIK-75, com alta potência (CI50= 10 nM), é devido ao efeito conformacional da sua estrutura em incomum forma de U dentro do sítio ativo de GSK-3β (PDB: 6GN1), provavelmente estabilizada por uma ligação de halogênio intramolecular entre os substituintes NO2 e Br (Br··· NO2).24 Interações de cavidade-σ também estão atraindo interesse para outros átomos, como alguns calcogênios. Diferentemente dos halogênios, como os elementos do grupo 16 participam de duas ligações σ em moléculas orgânicas, surgem em suas superfícies duas regiões de cavidade-σ. Átomos de enxofre em sistemas aromáticos, por exemplo, apresentam regiões de cavidade-σ em suas laterais (opostas às ligações S-C) (Figura 4), permitindo interações tanto intramoleculares como intermoleculares com bases de Lewis.20,25,26
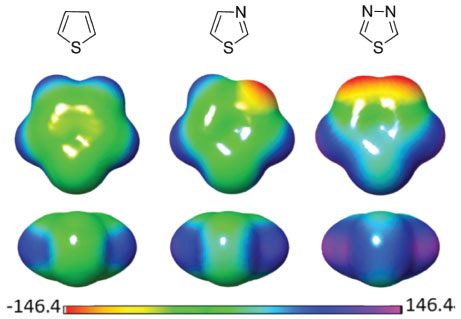 Figura 4. Regiões de cavidade-σ em átomos de enxofre presentes em anéis heterocíclicos de 5 membros. Unidade dos mapas de potencial eletrostático: kJ/mol. Método utilizado para geração dos mapas: M06-2X-D3/6-31++G**. Adaptado de ZHANG e colaboradores.26
Essas interações mediadas por átomos de enxofre foram revisadas mais recentemente por BENO e colaboradores, os quais fizeram uma perspectiva sobre a aplicabilidade dessas interações para o planejamento racional de candidatos a fármacos.20 Nesse trabalho, foi destacado o potencial das interações de cavidade-σ intramoleculares na indução de estabilizações conformacionais, que podem, consequentemente, influenciar no modo de interação de compostos bioativos com os seus respectivos alvos.27,28 Isso pode ser observado claramente para (7) (PDB: 3BX5)29 e (8) (PDB: 3NWW)30 (Figura 5). Esses compostos foram desenvolvidos para a inibição da serina/treonina cinase p38α e, através da análise de seus respectivos modos de interação, foi possível deduzir que estes inibidores equipotentes alcançaram suas conformações bioativas através de interações de cavidade-σ 1,4-O∙∙∙S e 1,4-N∙∙∙S intramoleculares, que levaram à exposição dos átomos de nitrogênio dos anéis tiazólicos presentes em suas estruturas para a realização de interações por ligação de hidrogênio com o resíduo de metionina na posição 109 (Met109) característico da região do hinge dessa cinase.
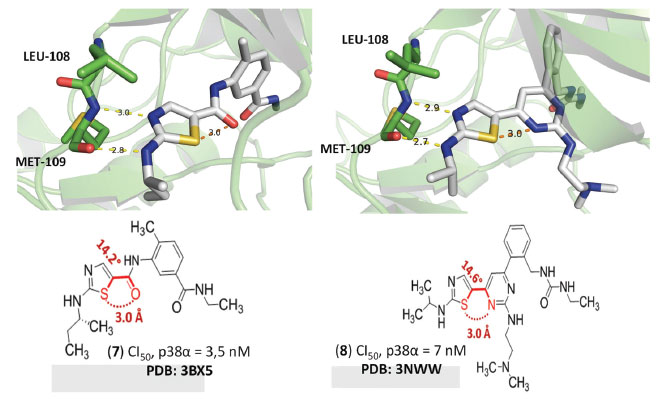 Figura 5. Modos de interação de (7) (PDB: 3BX5) e (8) (PDB: 3NWW) cocristalizados com p38α. Linhas tracejadas amarelas (ligações de hidrogênio) e laranjas (ligações de calcogênio). Distâncias em Å. Figura gerada com os programas PyMOL e ChemDraw.
É interessante destacar que alguns autores consideram a ligação de hidrogênio como um caso especial das interações de cavidade-σ. Uma vez que um átomo de hidrogênio tem apenas um elétron de valência, e que está participando da ligação X-H, pode-se prever que a região externa do hidrogênio tenha um potencial positivo com seu máximo ao longo da extensão da ligação. Devido à ausência de quaisquer outros elétrons de valência no hidrogênio, suas laterais também possuem densidades eletrônicas relativamente baixas e, portanto, o potencial positivo da cavidade-σ se estende mais para trás, em direção ao átomo X.19,24,31,32 INTERAÇÃO CÁTION-π A interação cátion-π é uma interação molecular não covalente entre a face de um sistema π rico em elétrons e um cátion adjacente (Figura 6A). Essa interação é um exemplo de ligação não covalente entre um monopolo (cátion) e um quadrupolo (sistema π). Foi estabelecido que a atração eletrostática entre o sistema π e o cátion é a principal força contribuindo para a interação, mas cálculos demonstraram que a presença de efeitos indutivos também tem um papel importante.33 Essa interação ocorre preferencialmente quando o cátion está localizado sobre o plano do sistema π a uma distância média de 2 a 4 Å.4,33
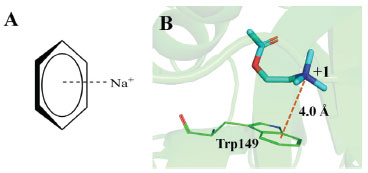 Figura 6. Exemplos de interação cátion-π: A) interação entre um benzeno e um íon Na+ B) Interação da acetilcolina com o resíduo Trp149 da subunidade a do receptor nicotínico.34 Figura gerada com os programas PyMOL e ChemDraw.
Estudos em fase gasosa estabeleceram que a energia da interação cátion-π é considerável; por exemplo, quando o benzeno interage com um íon Li+, é de -38 kcal/mol, e com um íon NH4+, é de -19 kcal mol-1.33 A interação cátion-π permanece energeticamente significativa em meio aquoso e em condições biológicas. Estudos demonstraram que as interações cátion-π podem estabilizar as energias de interação em -2 a -5 kcal mol-1, tornando-as competitivas com ligações de hidrogênio e interações entre pares de íons nas interações ligante-receptor e proteína-proteína. A interação cátion-π é abundante na natureza e estudos de estruturas de proteínas mostraram sua importância no reconhecimento molecular entre proteínas ou ligante-proteína.4,33 Em Química Medicinal, exemplos de interação cátion-π envolvida no reconhecimento molecular ligante-sítio receptor são citados na literatura. A acetilcolina (ACh) se encaixa no sítio catalítico da acetilcolinesterase (AChE) e no sítio de interação dos receptores nicotínicos (nAChR) devido à interação cátion-π entre seu grupo amônio quaternário e um resíduo de triptofano (Figura 6B).34 Interações cátion-π entre o grupo amônio quaternário do ligante (glicina betaína) e dois resíduos de triptofano (Figura 7) mostraram-se fundamentais para afinidade no sítio de ligação da proteína ProX (código PDB: 1R9L), que é envolvida na captação de glicina-betaína e prolina-betaína, em Escherichia coli.35
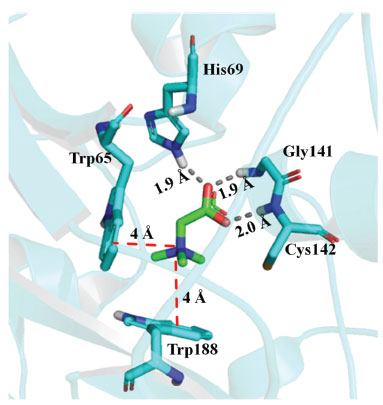 Figura 7. Exemplo de interações cátion-π do amônio quaternário da glicina betaína com os resíduos Trp65 e Trp188 em proteína ProX (código PDB: 1R9L) de Escherichia coli.35 Linhas tracejadas em vermelho: interações cátion-π. Outras interações (ligações de hidrogênio) também estão representadas. Figura gerada com o programa PyMOL.
INTERAÇÃO ÂNION-π Interações do tipo de ânion-π são interações entre sistemas aromáticos deficientes em elétrons e ânions, em que o sistema aromático pode estar carregado positivamente ou ser um sistema neutro relativamente pobre em elétrons (como em anéis triazínicos ou sistemas perfluorados), caracterizando-se, assim, como um tipo especial das interações iônicas ou interações íon-dipolo.36 Essas interações podem ocorrer entre ânion e um anel aromático com um ângulo entre 0° a 90° entre a linha que une o ânion e o centro do anel e o plano do anel (Figura 8).36
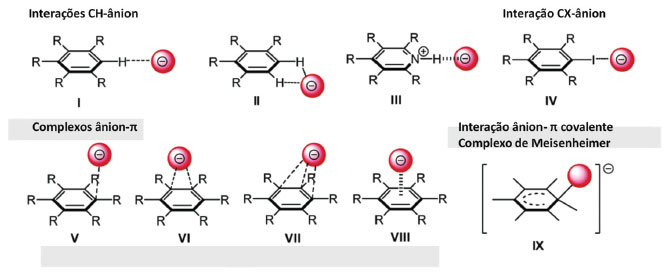 Figura 8. Possíveis modos de interação entre um ânion e um anel aromático.36 Ligações de hidrogênio não-clássicas CH-ânion (I, II, III); ligações de halogênio CX-ânion (IV); complexos ânion-π (V, VI, VII, VIII); Interações ânion-π covalente (IX); o grupo R deve ser retirador de elétrons, como átomos de flúor. (Figura gerada com o programa Chemdraw)
Um ânion pode interagir com o sistema π através de quatro modos diferentes (Figura 8): ligações de hidrogênio CH-ânion (I, II, III); ligações de halogênio CX-ânion (IV); complexos ânion-π (V, VI, VII, VIII), que são interações ânion- π não covalentes, onde o ânion reside acima do plano do anel aromático; e complexos de Meisenheimer, que são interações ânion-π covalentes (IX), formado entre um nucleófilo e um anel contendo grupos retiradores de elétrons. A interação ânion-π raramente ocorre em sistemas biológicos, uma vez que as cadeias laterais de aminoácidos aromáticos ricos em elétrons repelem cargas negativas próximas a suas nuvens π. Egli e Sarkhel afirmam que esses tipos de interação são pouco encontrados na natureza porque fenilalanina, tirosina e triptofano não podem ser positivamente polarizados.37 Uma pesquisa no PDB revelou que os aminoácidos aromáticos interagem com as cadeias laterais de aspartato e glutamato preferencialmente através das interações na borda do anel, com um ângulo entre o plano do anel aromático e o plano do carboxilato de 0°, como nas estruturas I e II da Figura 8.36 Há controvérsias sobre a origem dessa interação. As explicações convencionais das interações atrativas entre ânions e anéis aromáticos substituídos, que dependem da polarização induzida pelo substituinte do sistema π, podem ser falhas. Em vez disso, propôs-se que a interação do tipo ânion-π pode surgir principalmente da interação direta do ânion com os dipolos locais induzidos pelos substituintes, como representado na Figura 9. A interação do ânion com o próprio anel aromático permaneceria energeticamente desfavorável, apesar dos substituintes. Especificamente, as energias de interação para um conjunto de 83 complexos Cl-···C6H6-nXn (n=0-4, 6) puderam ser qualitativamente reproduzidas usando um modelo aditivo simples, no qual os substituintes não são ligados ao anel aromático e os efeitos de polarização π seriam impossíveis.38
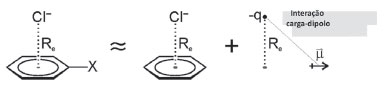 Figure 9. Modelo eletrostático dos efeitos de substituintes em complexos Cl-··· C6H6-nXn (Adaptado de Wheeler e colaboradores).38 As energias de interação de Cl- com benzenos substituídos podem ser simuladas pela adição de uma interação carga eletrostática-dipolo à interação Cl-···C6H6. Os substituintes não afetam de forma significativa uma mudança na interação do ânion com o próprio anel aromático.38 (Figura gerada com o programa Chemdraw)
Essas interações ainda não vêm sendo utilizadas no desenho racional de compostos com atividade biológica, sendo geralmente exploradas em estudos de formação de complexos de estruturas de pequenas moléculas depositadas no Cambridge Crystallographic Data Centre (CCDC)39 ou em estudos teóricos.40
INTERAÇÃO DE EMPILHAMENTO AMIDA-π O empilhamento amida-π é uma interação molecular não covalente entre a face de um sistema π rico em elétrons (polarizado pela presença de heteroátomos, por exemplo) e um grupamento amida (Figura 10). O empilhamento amida-π é um tipo de interação dipolo-dipolo.
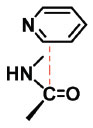 Figura 10. exemplo de interação amida- π.41 Figura gerada com o programa Chemdraw.
A força que resulta do empilhamento amida-π depende da orientação e da distância dos dois dipolos presentes. Estudos computacionais com o sistema N-metil-acetamida/piridina demonstraram que a energia da interação amida-π se torna progressivamente mais favorável à medida que ângulo entre os dipolos é aumentado a partir de 0º, alcançando os valores mais favoráveis entre 180º e 240º; para este mesmo sistema, a distância ótima entre os planos das duas estruturas foi de 3,4 Å.41 Um exemplo onde o empilhamento amida-π entre um anel aromático de uma proteína e um grupo amida foi importante para afinidade do ligante foi descrito no estudo que descreve o modo de interação de inibidores contendo um anel oxazola no sítio S1 do fator Xa (uma serina protease). Dois inibidores do fator Xa (9, 10) contendo um anel oxazola tiveram uma geometria de interação muito semelhante, mas como uma diferença de 11 vezes na afinidade de ligação. Essa diferença é devida à orientação dos momentos dipolares do anel oxazola e da ligação peptídica do esqueleto entre os resíduos Cys191 e Gln192 (Figura 11). Neste caso, a interação amida-π mostrou-se determinante para a potência.42
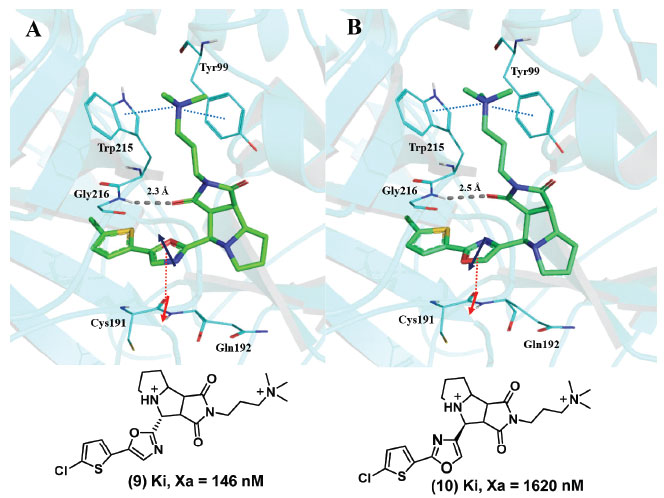 Figura 11. A) Inibidor do fator Xa (9) com o momento dipolar do anel oxazólico invertido em relação ao da ligação peptídica entre Cys191 e Gln192 (PBD: 2Y5G). B) Inibidor do fator Xa (10) com o momento dipolar do anel oxazólico em paralelo com o da ligação peptídica entre Cys191 e Gln192, (PDB: 2Y5H) (linha tracejada azul: interações cátion-π, linha tracejada vermelha: interações amida-π) (adaptado de Salonen et colaboradores)42 Figura gerada com o programa PyMOL.
INTERAÇÕES ORTOGONAIS As interações ortogonais são também um tipo de interação dipolo-dipolo e ocorrem quando dois grupos funcionais dipolares em orientação aproximadamente ortogonal estão próximos (Figura 12). Elas são vistas entre os grupos C-X.C=O, C=O.C=O e também quando dipolos como C≡N, NO2, S=O e espécies nucleofílicas fracas como compostos contendo as funções éter, sulfeto, álcool e imina interagem entre si ou com uma carbonila. Essas interações acontecem quando os dipolos fazem entre si um ângulo entre 70º e 110º com uma distância média de 3 a 4 Å. Dados cristalográficos mostram muitas vezes a perda parcial da planaridade do C da carbonila aceptora em até 5°, devido às interações ortogonais. Estudos mostraram que a interação ortogonal entre C-F.C=O tem uma contribuição de energia livre de ligação em meio apolar de -0,2 a -0,3 kcal/mol, enquanto as interações entre C=O.C=O podem chegar a -0,7 kcal/mol.43,44,45
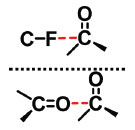 Figura 12. exemplo de interação ortogonais.43 Figura gerada com o programa Chemdraw.
Um exemplo de interação C-F.C=O e C=O.C=O é mostrado na estrutura cristalográfica da cinase Abl humana cocristalizado com o nilotinibe (11), um inibidor de tirosina cinase de segunda geração (código PDB: 3CS9). São observadas uma interação C-F.C=O e uma C=O.C=O com distâncias de 3,1 e 3,5 Å, respectivamente, que são essenciais para a afinidade com o sítio de interação (Figura 13).45
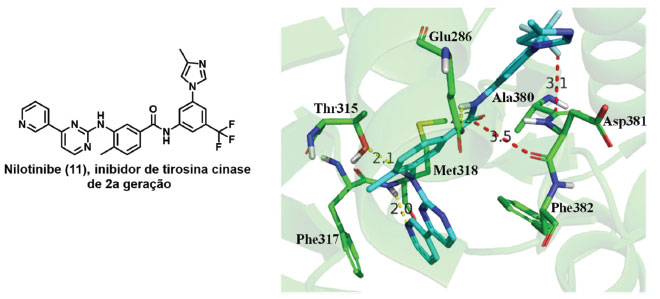 Figura 13. Exemplo de interações ortogonais: Interação C-F···C=O e C=O···C=O do nilotinibe (11) na estrutura cristalográfica de código PDB 3CS9.45 Linhas tracejadas em amarelo: ligações de hidrogênios; em vermelho: ligações ortogonais. Distâncias em Å. Figura gerada com os programas PyMOL e ChemDraw.
INTERAÇÃO ALQUILA-ARILA A interação alquila-arila é considerada por alguns autores como um tipo de ligação de hidrogênio com anel aromático não clássica (CH-π), que ocorre quando uma ligação C-H de um grupo alquila é polarizada na presença de um anel com sistema π (Figura 14).46 É uma interação do tipo dipolo-dipolo induzido fraca, mas pode ter um papel significativo na conformação e na estabilidade de estruturas de complexos, como na formação de complexos ligante-proteína e proteína-proteína. Geralmente o grupo C-H está perpendicular ao sistema π e aponta para seu centro a uma distância de 3,4 a 4,4 Å.46 A energia da interação CH-π, depende do tipo de fragmento molecular ligado ao CH doador, sendo favorecida por grupos retiradores de elétrons, e do sistema π receptor, apresentando valores que variam entre -1 a -5 kcal/mol.46,47
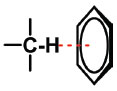 Figura 14. exemplo de interação alquila-arila.
O total de energia de estabilização de cerca de 0,5-1,0 kcal mol -1 por interação é suficiente para tornar essa interação um contribuinte potencialmente importante para a estabilidade geral das proteínas, que em muitos casos não é mais do que algumas kcal/mol.47 Brandl e colaboradores descreveram que essas interações são chave em reconhecimentos que incluem a formação de complexos de proteínas com ligantes ou cofatores, como o grupo heme, piridoxal-5-fosfato, nucleotídeos, carboidratos e peptídeos ligados.46,47 Shimohigashi e colaboradores mostraram que as interações CH-π são importantes no desenho e no reconhecimento de alguns inibidores de serina proteases. 48 As interações alquila-arila entre o ligante e a proteina são importantes para estabilidade da conformação do composto α-ceto-oxazólico (12). Na estrutura cristalográfica da enzima hidrolase de amida de ácido graxo (código 3K83 no PDB), observaram-se interações CH-π entre os resíduos Val491 e Thr488 com grupos fenila do inibidor (12) (Figura 15),49 além de interações de empilhamento T (ver item a seguir) com os resíduos Phe192 e Phe381.
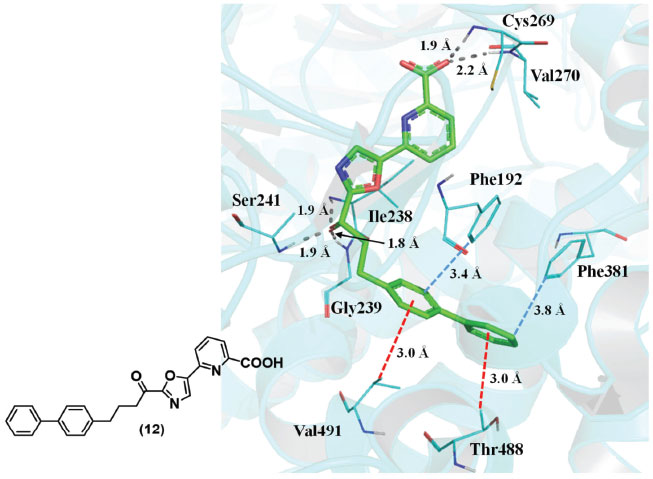 Figura 15. Exemplos de interações CH-π entre o sistema π do inibidor (12) e CH da proteína (código 3K83 no PDB).49 Linhas tracejadas em cinza: ligações de hidrogênio; em vermelho: interações CH-π; em azul: empilhamento T. Figura gerada com o programa PyMOL.
EMPILHAMENTO π O empilhamento π é uma interação de tipo dipolo-dipolo (ou, mais exatamente, quadrupolo-quadrupolo), entre anéis aromáticos e/ou hetero-aromáticos, devido à polarização das nuvens eletrônicas ou, em alguns casos, à atração eletrostática entre os elétrons π do anel aromático com carga parcial positiva σ em um átomo contido em sua estrutura.4,36 No empilhamento π, as nuvens π polarizáveis podem estar exatamente paralelas uma em relação à outra, com os átomos de cada anel envolvido localizados uns sobre os outros (empilhamento sanduíche), ou podem estar deslocadas, quando os átomos de um anel estão deslocados em relação aos do outro (empilhamento deslocado), tanto em distância quanto em ângulo, que é o modo de interação mais comum e energeticamente mais favorável (Figura 16). O ângulo entre os planos dos anéis pode chegar até 90º (empilhamento T) com uma distância de cerca de 3 a 4 Å (que poderia ser descrita também como uma interação CH (aromático) -π) (Figura 16). A energia de interação quando os sistemas π estão deslocados em paralelo ou em forma de T é de aproximadamente -2,50 kcal/mol. Quando os sistemas estão empilhados a energia de interação é menos favorável, -1,48 kcal/mol; contudo, se a polaridade é invertida, por exemplo a partir da fluoração do anel de benzeno, a interação empilhada π-π é favorecida.4,36,50
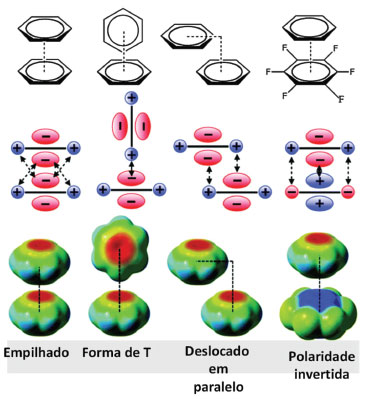 Figura 16. Exemplos de empilhamento π e respectivas orientações (adaptado de Matthews e colaboradores).50
Essas interações são importantes contribuintes para a estabilidade de estruturas de proteínas e para a formação de complexos proteína-ligante, estando também presentes nas estruturas de ácidos nucleicos, como é evidente a partir da análise do empilhamento dos pares de bases nitrogenadas na dupla hélice do DNA e do RNA. Mesmo sendo fracas, as interações de empilhamento π foram reconhecidas como sendo importantes no enovelamento e na estabilidade das proteínas. Elas podem não só determinar a estrutura, mas também modular as propriedades físicas dos resíduos nos sítios ativos da enzima.4,36,51,52,53 A literatura fornece vários exemplos ilustrativos: Ibarra e colaboradores mostraram que as interações de empilhamento estão associadas à diminuição do valor de pKa do resíduo Tyr9 que está localizado nas proximidades do resíduo Phe10 no sítio ativo da glutationa S-transferase, enzima que catalisa a conjugação da glutationa a substratos eletrofílicos;54 Quiocho e colaboradores mostraram que as interações de empilhamento têm um papel essencial no reconhecimento do mRNA-cap por proteínas;55 Boehr e colaboradores mostraram que as interações de empilhamento π desempenham um papel proeminente no reconhecimento da subunidade heterocíclica da adenina por cinases.56 Interações π participam no reconhecimento molecular do ligante (13) pela proteína β-cetoacil-ACP sintase, como ilustrado através do complexo co-cristalizado com código PDB 2VBA (Figura 17A).57 Pode ser observado um empilhamento π deslocado entre o grupo tiazol do ligante e o grupo fenila de Phe392. Podem ser observados também empilhamentos T entre os grupos fenila do ligante e do resíduo Phe201 e entre o anel tiazola e os grupos fenila de Phe201 e de Phe229. Além disso, pode ser observada uma interação amida-π entre a fenila de (13) e a ligação peptídica entre Ala162/Cys163 (Figura 17A).
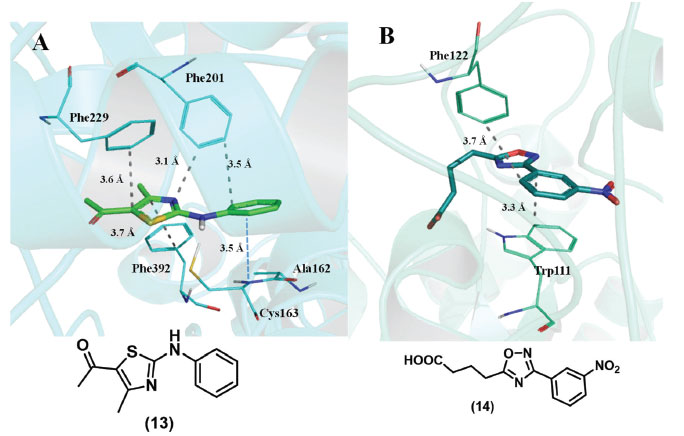 Figura 17. Exemplos de interação π-π: A) Inibidor (13) co-cristalizado com a enzima β-cetoacil-ACP sintase (PDB: 2VBA). B) Inibidor (14) co-cristalizado com a enzima aldose redutase (PBD: 2IKG). Linhas tracejadas em cinza: empilhamento π; em azul: interação amida-π. Figura gerada com o programa PyMOL.
Como outro exemplo, foram observadas interações de empilhamentos π e T do grupo fenila do inibidor de aldose redutase (14) com os grupos fenila de Trp111 e Phe122, que contribuem para sua estabilidade no sítio de catalítico da enzima (Figura 17B) (PBD: 2IKG).58
INTERAÇÕES HIDROFÓBICAS As interações hidrofóbicas são interações importantes, mas em geral mal compreendidas em Química Medicinal. De acordo com a IUPAC, as interações hidrofóbicas podem ser definidas como a tendência dos hidrocarbonetos (ou de grupos lipofílicos em solutos, semelhantes a hidrocarbonetos) em formar agregados intermoleculares em meio aquoso, e as interações intramoleculares análogas. O nome surge da atribuição do fenômeno da aparente repulsão entre água e hidrocarbonetos. No entanto, o fenômeno deve ser atribuído ao efeito dos grupos semelhantes a hidrocarbonetos na interação água-água.7 O termo alternativo "ligação hidrofóbica", por introduzir ainda menos clareza na descrição do fenômeno, deve ser evitado.8 Quando estruturas ditas hidrofóbicas são inseridas em água, as moléculas de água se juntam em forma de "gaiola" em torno das superfícies. Quando estas estruturas hidrofóbicas se agregam, a área superficial do agregado é menor do que a soma das áreas combinadas de cada superfície isolada e, portanto, moléculas de água são liberadas para o interior do solvente. Isso maximiza a quantidade de moléculas de água livres e, portanto, a entropia.4 Além disso, essas moléculas reinseridas no interior do solvente passam a fazer mais ligações de hidrogênio do que fariam estando localizadas na superfície das estruturas. Assim, a interação hidrofóbica resulta principalmente do aumento da entropia das moléculas de água e das novas ligações de hidrogênio que surgem quando as moléculas de água que envolviam as moléculas hidrofóbicas entram em contato com outras, tendo, portanto, componentes entrópico e entálpico favoráveis.4 Mesmo que a complexação do ligante hidrofóbico no interior da proteína resulte em custos associados à perda parcial de entropias translacional, rotacional e conformacional do ligante, o ganho entrópico das moléculas do solvente deslocadas do sítio é superior a estes. Um bom exemplo do efeito do deslocamento de moléculas de água na atividade inibitória foi mostrado no trabalho de Ritschel e colaboradores,59 onde as interações hidrofóbicas foram um dos fatores que contribuíram para aumentar em cerca de 20 vezes a afinidade do composto (18) no sítio de ligação da t-RNA-guanina transglicosilase (TGT) em relação ao (15) devido ao efeito do deslocamento concomitante de moléculas de água pela introdução dos grupos metila e CH2CH2NH-cicloexila (Figura 18).
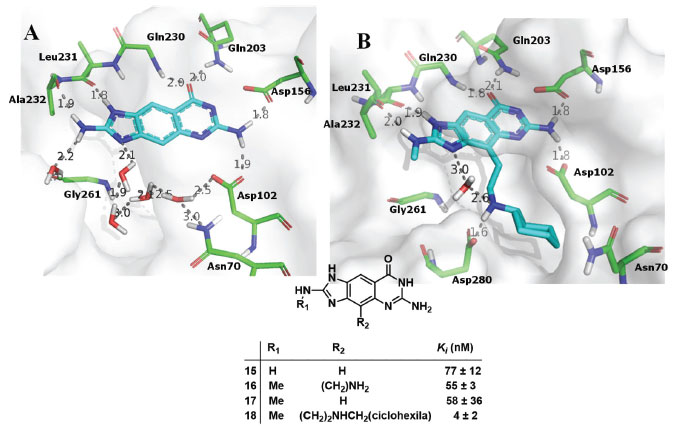 Figura 18. Exemplos de interações hidrofóbicas: A) perfil de interação do composto (15) no sítio de ligação da TGT (PDB: 2Z7K). B) Perfil de interação do composto (18) no sítio de ligação da TGT (PDB: 3EOS), cujo deslocamento de moléculas de água vistas em A aumenta a afinidade.59 Tracejado em cinza: ligações de hidrogênio. Figura gerada com o programa PyMOL.
MODELAGEM DE INTERAÇÕES Antes de concluir, dada a crescente utilização de métodos de modelagem molecular para a avaliação dos modos de interação entre ligantes e seus bioalvos, algumas observações devem ser acrescentadas quanto à maneira como essas interações podem ser modeladas por esses métodos. As interações aqui apresentadas são em grande parte mediadas por forças eletrostáticas que surgem das distribuições de cargas nas espécies envolvidas na interação, as quais podem ser obtidas por meio de métodos baseados na Mecânica Quântica, quer seja através do uso de orbitais moleculares, quer seja através da densidade eletrônica. Esses métodos são, em princípio, capazes de uma descrição completa de todas as interações aqui apresentadas. No entanto, devido à complexidade na resolução das equações fundamentais envolvidas, aproximações devem ser introduzidas na resolução das mesmas por razões práticas, as quais acabam por influenciar a extensão com que as interações são descritas.60 A introdução de correlação eletrônica nos métodos pós-Hatree-Fock, por exemplo, leva a uma melhoria considerável na descrição das forças de dispersão.60 Uma proposta em uma direção diferente, o uso dos métodos de orbital molecular semi-empíricos com correções específicas para melhorar a representação de forças de dispersão, ligações de hidrogênio e ligações de halogênio, também mostrou-se bem-sucedida.61,62 Uma descrição mais simples das interações pode ser conseguida com o uso das funções empíricas dos métodos de campo de força. Nestes métodos não-quânticos, cada interação pode ser descrita pela inclusão de uma função específica com parâmetros adequados ajustados a partir de dados empíricos, como uma função para a ligação de hidrogênio, por exemplo. Interações identificadas mais recentemente, como as ligações de halogênio, só estão presentes em campos mais atuais e o leitor interessado em modelar sistemas contendo estas interações deve verificar cuidadosamente essa presença. Alguns métodos foram modificados especificamente para reproduzir essa interação, usando átomos "falsos" (um ponto extra positivamente carregado) na posição em que ficaria a região de baixa densidade eletrônica sobre o átomo do halogênio.63,64 Liu e colaboradores apresentaram um método baseado no conhecimento aplicável a estudos de docagem molecular, usando estruturas cristalográficas depositadas no PDB para gerar potenciais de pares, o qual se mostrou eficaz para representar a ligação de halogênio.65 Um protocolo de simulação de dinâmica molecular foi desenvolvido para modelar a ligação de halogênio em complexos proteína-ligante pela inclusão de um ponto extra carregado para representar a distribuição anisotrópica de carga no átomo de halogênio.66
CONCLUSÕES Embora essencialmente todas se originem de forças eletrostáticas, que podem ser descritas classicamente através da aproximação da atração entre cargas e dipolos (ou quadrupolos, etc), com algumas correções quânticas em alguns casos, a classificação das interações em tipos característicos tem a vantagem de destacar os fragmentos moleculares capazes de estarem envolvidos em cada interação determinada. Com isso, novos ligantes podem ser planejados pela introdução de fragmentos moleculares visando a construção de interações específicas entre eles e seus alvos biológicos, uma estratégia de grande utilidade para o projetista de moléculas bioativas, especialmente nos casos em que a estrutura do alvo é conhecida. Cada adição bem-sucedida de uma interação contribui para a energia de interação, que finalmente se reflete no aumento da afinidade do ligante por um determinado alvo biológico. Neste aspecto, métodos de modelagem molecular, especialmente os métodos quânticos, mas também os métodos clássicos com adaptações específicas para a descrição de novas interações, podem ser ferramentas de auxílio para o desenvolvimento de novos ligantes. Neste trabalho buscamos destacar algumas das interações exploradas classicamente em Química Medicinal, como as ligações de hidrogênio e as interações hidrofóbicas, mas também apresentar ao leitor novas interações que vêm sendo descritas na literatura e exploradas com sucesso no planejamento de novos ligantes bioativos.
AGRADECIMENTOS O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Código de Financiamento 001 (P.S.M.P.), do Conselho Nacional de Pesquisa - CNPq (309229/2018-9) (C.M.R.S.), da Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro - Faperj (E-26/202.146/2015) (H.H.F.) e do INCT-INOFAR (BR573.564/2008-6 e E-26/170.020/2008)) (H.H.F. e C.M.R.S.).
REFERÊNCIAS 1. Verli, H.; Barreiro, E. J.; Quim. Nova 2005, 28, 95. 2. Montanari, C. A.; Bolzani, V.; D. S. Quim. Nova 2001, 24, 105. 3. Campbell, S. F.; Quim. Nova 1991, 14, 196. 4. Bissantz, C.; Kuhn, B.; Stahl, M. J. Med. Chem. 2010, 53, 5061. 5. Koshland, D. E.; Angew. Chemie Int. Ed. English 1995, 33, 2375. 6. Brooijmans, N.; Kuntz, I. D.; Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335. 7. Muller, P.; (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 1077. 8. Nič, M.; Jirát, J.; Kosata, B.; Jenkins, A.; McNaught, A.; organizadores IUPAC Compendium of Chemical Terminology IUPAC: Research Triagle Park, NC, 2009. 9. Keesom, W. H.; KNAW Proc. 1915, 18I, 636. 10. London, F.; Trans. Faraday Soc. 1937, 33, 8b. 11. Arunan, E.; Desiraju, G. R.; Klein, R. A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D. C.; Crabtree, R. H.; Dannenberg, J. J.; Hobza, P.; Kjaergaard, H. G.; Legon, A. C.; Mennucci, B.; Nesbitt, D. J.; Pure Appl. Chem. 2011, 83, 1637. 12. Oliveira, B. G.; Araújo, R. C. M. U.; Quim. Nova 2012, 35, 2002. 13. Hubbard, R. E.; Haider, M. K.; Hydrogen bonds in proteins: Role and strength. In Encyclopedia of Life Science; John Wiley & Sons Ltd.: Chichester, UK, 2010. 14. Kumar, M.; Balaji, P. V.; J. Mol. Model. 2014, 20, 2136. 15. Torshin, I. Y.; Weber, I. T.; Harrison, R.; W.; Prot. Eng. 2002, 15, 359. 16. Martin, T. W.; Derewenda, S.; Nat. Struct. Biol. 1999, 6, 403. 17. Baker, E. N.; Hubbard, R. E.; Prog. Biophys. Mol. Biol. 1984, 44, 97. 18. Henley, Z. A.; Bax, B. D.; Inglesby, L. M.; Champigny, A.; Gaines, S.; Faulder, P.; Le, J.; Thomas, D. A.; Washio, Y.; Baldwin, I. R.; ACS Med. Chem. Lett. 2017, 8, 1093. 19. Wilcken, R.; Zimmermann, M. O.; Lange, A.; Joerger, A. C.; Boeckler, F. M. Biology.; J. Med. Chem. 2013, 56, 1363. 20. Beno, B. R.; Yeung, K.-S.; Bartberger, M. D.; Pennington, L. D.; Meanwell, N. A.; J. Med. Chem. 2015, 58, 4383. 21. Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G.; Chem. Rev. 2016, 116, 2478. 22. Desiraju, G. R.; Ho, P. S.; Kloo, L.; Legon, A. C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K.; Pure Appl. Chem. 2013, 85, 1711. 23. Hardegger, L. A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; Plancher, J.-M.; Hartmann, G.; Banner, D. W.; Haap, W.; Diederich, F.; Angew. Chemie Int. Ed. 2011, 50, 314. 24. Tesch, R.; Becker, C.; Müller, M. P.; Beck, M. E.; Quambusch, L.; Getlik, M.; Lategahn, J.; Uhlenbrock, N.; Costa, F. N.; Polêto, M. D.; Pinheiro, P. de S. M.; Rodrigues, D. A.; Sant'Anna, C. M. R.; Ferreira, F. F.; Verli, H.; Fraga, C. A. M.; Rauh, D.; Angew. Chemie Int. Ed. 2018, 57, 9970. 25. Murray, J. S.; Lane, P.; Politzer, P.; Int. J. Quantum Chem. 2008, 108, 2770. 26. Zhang, X.; Gong, Z.; Li, J.; Lu, T.; J. Chem. Inf. Model. 2015, 55, 2138. 27. Jackson, N. E.; Savoie, B. M.; Kohlstedt, K. L.; Olvera de la Cruz, M.; Schatz, G. C.; Chen, L. X.; Ratner, M. A.; J. Am. Chem. Soc. 2013, 135, 10475. 28. Pennington, L. D.; Bartberger, M. D.; Croghan, M. D.; Andrews, K. L.; Ashton, K. S.; Bourbeau, M. P.; Chen, J.; Chmait, S.; Cupples, R.; Fotsch, C.; Helmering, J.; Hong, F.-T.; Hungate, R. W.; Jordan, S. R.; Kong, K.; Liu, L.; Michelsen, K.; Moyer, C.; Nishimura, N.; Norman, M. H.; Reichelt, A.; Siegmund, A. C.; Sivits, G.; Tadesse, S.; Tegley, C. M.; Van, G.; Yang, K. C.; Yao, G.; Zhang, J.; Lloyd, D. J.; Hale, C.; St. Jean, D. J.; J. Med. Chem. 2015, 58, 9663. 29. Hynes, J.; Wu, H.; Pitt, S.; Shen, D. R.; Zhang, R.; Schieven, G. L.; Gillooly, K. M.; Shuster, D. J.; Taylor, T. L.; Yang, X.; McIntyre, K. W.; McKinnon, M.; Zhang, H.; Marathe, P. H.; Doweyko, A. M.; Kish, K.; Kiefer, S. E.; Sack, J. S.; Newitt, J. A.; Barrish, J. C.; Dodd, J.; Leftheris, K.; Bioorg. Med. Chem. Lett. 2008, 18, 1762. 30. Lin, S.; Wrobleski, S. T.; Hynes, J.; Pitt, S.; Zhang, R.; Fan, Y.; Doweyko, A. M.; Kish, K. F.; Sack, J. S.; Malley, M. F.; Kiefer, S. E.; Newitt, J. A.; McKinnon, M.; Trzaskos, J.; Barrish, J. C.; Dodd, J. H.; Schieven, G. L.; Leftheris, K.; Bioorg. Med. Chem. Lett. 2010, 20, 5864. 31. Politzer P.; Murray J. S.; Clark, T.; top. curr. chem. 2015, 358, 19. 32. Pinheiro, P. de S. M.; Rodrigues, D. A.; Alves, M. A.; Tinoco, L. W.; Ferreira, G. B.; de Sant'Anna, C. M. R.; Fraga, C. A. M.; New J. Chem. 2018, 42, 497. 33. Dougherty, D.; A. Acc. Chem. Res. 2013, 46, 885. 34. Zhong, W.; Gallivan, J. P.; Zhang, Y.; Li, L.; Lester, H. A.; Dougherty, D. A.; Proc. Natl. Acad. Sci. 1998, 95, 12088. 35. Schiefner, A.; Breed, J.; Bösser, L.; Kneip, S.; Gade, J.; Holtmann, G.; Diederichs, K.; Welte, W.; Bremer, E.; J. Biol. Chem. 2004, 279, 5588. 36. Salonen, L. M.; Ellermann, M.; Diederich, F.; Angew. Chemie Int. Ed. 2011, 50, 4808. 37. Egli, M.; Sarkhel, S.; Acc. Chem. Res. 2007, 40, 197. 38. Wheeler, S. E.; Houk, K. N.; J. Phys. Chem. A 2010, 114, 8658. 39. Wang, D.-X.; Wang, M.-X.; J. Am. Chem. Soc. 2013, 135, 892. 40. Hay, B. P.; Bryantsev, V. S.; Chem. Commun. 2008, 2417. 41. Harder, M.; Kuhn, B.; Diederich, F.; ChemMedChem 2013, 8, 397. 42. Salonen, L. M.; Holland, M. C.; Kaib, P. S. J.; Haap, W.; Benz, J.; Mary, J.-L.; Kuster, O.; Schweizer, W. B.; Banner, D. W.; Diederich, F.; Chem. - A Eur. J. 2012, 18, 213. 43. Paulini, R.; Müller, K.; Diederich, F.; Angew. Chemie Int. Ed. 2005, 44, 1788. 44. Persch, E.; Dumele, O.; Diederich, F.; Angew. Chemie Int. Ed. 2015, 54, 3290. 45. Weisberg, E.; Manley, P. W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S. W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; Kung, A. L.; Mestan, J.; Daley, G. Q.; Callahan, L.; Catley, L.; Cavazza, C.; Mohammed, A.; Neuberg, D.; Wright, R. D.; Gilliland, D. G.; Griffin, J. D.; Cancer Cell 2005, 7, 129. 46. Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M. S.; Chakrabarti, P.; Phys. Chem. Chem. Phys. 2014, 16, 12648. 47. Brandl, M.; Weiss, M. S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R.; J. Mol. Biol. 2001, 307, 357. 48. Shimohigashi, Y., Nose, T., Yamauchi, Y. & Maeda, I.; Biopolymers, 1999, 51, 9 49. Oakley, A. J.; Barrett, S.; Peat, T. S.; Newman, J.; Streltsov, V. A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J. L.; J. Med. Chem. 2010, 53, 6421. 50. Matthews, R. P.; Welton, T.; Hunt, P. A.; Phys. Chem. Chem. Phys. 2014, 16, 3238. 51. Meyer, E. A.; Castellano, R. K.; Diederich, F.; Angew. Chemie Int. Ed. 2003, 42, 1210. 52. Chelli, R.; Gervasio, F. L.; Procacci, P.; Schettino, V.; J. Am. Chem. Soc. 2002, 124, 6133. 53. Bhattacharyya, R.; Samanta, U.; Chakrabarti, P.; Protein Eng. Des. Sel. 2002, 15, 91. 54. Ibarra, C.; Nieslanik, B. S.; Atkins, W. M.; Biochemistry 2001, 40, 10614. 55. Quiocho, F. A.; Hu, G.; Gershon, P. D.; Curr. Opin. Struct. Biol. 2000, 10, 78. 56. Boehr, D. D.; Farley, A. R.; LaRonde, F. J.; Murdock, T. R.; Wright, G. D.; Cox, J. R.; Biochemistry 2005, 44, 12445. 57. Pappenberger, G.; Schulz-Gasch, T.; Kusznir, E.; Müller, F.; Hennig, M.; Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 1208. 58. McDonough, M. A.; Anderson, J. W.; Silvaggi, N. R.; Pratt, R. .; Knox, J. R.; Kelly, J. A.; J. Mol. Biol. 2002, 322, 111. 59. Ritschel, T.; Kohler, P. C.; Neudert, G.; Heine, A.; Diederich, F.; Klebe, G.; ChemMedChem 2009, 4, 2012. 60. Riley, K. E.; Pitonak, M.; Jurecka, P.; Hobza, P.; Chem. Rev. 2010, 110, 5023. 61. Rezác, J., Hobza, P.; J. Chem. Theory Comput. 2011, 8, 141. 62. Rezác, J., Hobza,; Chem. Phys. Lett. 2011, 506, 286. 63. Kolar, M., Hobza, P., Bronowska, A. K.; Chem. Commun. 2013, 49, 981. 64. Ibrahim, M. A. A.; J. Comput. Chem. 2011, 32, 2564. 65. Liu, Y.; Xu, Z.; Yang, Z.; Chen, K.; Zhu, W.; J. Mol. Model. 2013, 19, 5015. 66. Celis-Barros, C.; Saavedra-Rivas, L.; Salgado, J.; Cassels, B.; Zapata-Torres, G.; J. Comput. Aided Mol. Des. 2015, 29, 37. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access