
 |
 |
 |
 |
 |
Artigo
| Electrochemical incineration of short-chain carboxylic acids with Nb-supported boron doped diamond anode: supporting electrolyte effect into the electrogenerated oxidant species (hydroxyl radicals, hydrogen peroxide and persulfate) |
|
Jorge Leandro Aquino de QueirozI; Dayanne Chianca de MouraI; Elaine Cristina M. de Moura SantosI; Bernardo A. Frontana-UribeII,III; Carlos A. Martínez-HuitleI,*
I. Universidade Federal do Rio Grande do Norte, Instituto de Química, 59072-970 Lagoa Nova - RN, Brasil II. Centro Conjunto de Investigación en Química Sustentable UAEMéx-UNAM, Carretera Toluca-Ixtlahuaca Km. 14.5, 50200, Toluca, México III. Instituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, 04510, México City, México Recebido em 24/05/2019 *e-mail: carlosmh@quimica.ufrn.br This work describes the electrochemical oxidation study of three short chain carboxylic acids (acetic, oxalic and formic 0.18 mol L-1) using different electrolysis conditions (electrolyte composition and current density) in an electrochemical reactor fitted with niobiumsupported/BDD electrode (Nb/BBD) as anode and titanium plate as cathode. The experiments were focused to elucidate if the oxidation of these carboxylic acids involved a direct or a mediated oxidation pathway, as well as their electrochemical incineration efficiency using this electrochemical reactor. The amount of oxidizing species (hydroxyl radicals, hydrogen peroxide, and persulfate) produced at Nb/BDD electrode was spectroscopically determined. The electrochemical kinetics study in H2SO4 and HClO4 solutions, as well as the chemical oxygen demand and total organic carbon analysis for each one of the carboxylic acids studied in both media, gave us information to propose a degradation mechanism for the electrochemical degradation of the carboxylic acids in each electrolytic media. The degradation in H2SO4 is promoted by the persulfate production, whereas in HClO4 by •OH. INTRODUCTION In the last years, the research of new efficient and cost-effective methods for the decontamination of waters has led to develop of several advanced oxidation processes (AOPs). Among the AOPs, electrochemical methods like direct electrochemical oxidation1,2 (DEO, the organic compounds interact with the anode surface) and indirect electrochemical oxidation3-5 (IEO, oxidation occurs via the reaction with electro-generated oxidant species) have received a lot of attention, due to their advantageous characteristics.1,2 Depending on the electrolysis conditions, different oxidant species can be electrochemically produced. For example using boron doped diamond electrode (BDDE) •OH can be efficiently formed during water discharge (Eq. 1),1,5 but others oxidants can be generated, depending on the supporting electrolyte composition. In this way, hydrogen peroxide (H2O2) can be produced when dissolved molecular oxygen (O2) is reduced1,6 or by the oxidation of water (Eq. 2) or combination of hydroxyl radicals (Eq. 3).1,7 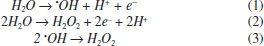 Persulfates are formed by combination of sulfate radicals, produced by direct electron transfer (Eq. 4) or reaction of sulfate anion with hydroxyl radical (Eq. 5 to 7).8 Sulfate radicals also can favor the organic compounds degradation as shown in Eq. 8.9 Both species acts as strong oxidants for removing efficiently the organic matter in solution. When the solutions contain sulfates, persulfates (S2O82-) play an important role during the electrochemical degradation of organic pollutants (Eq. 9).2,5 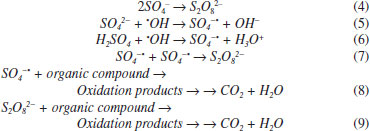 Peroxide, persulphate and hydroxyl radical can all be considered green oxidants because the products of their electronic transfer are harmless and can be part of a catalytic cycle. Thanks to these characteristics, its use is very attractive in the treatment of contaminated effluents.10 The electrogeneration of these oxidants by using silicon-supported/BDD electrode (Si/BBD) as anode has been confirmed.5,9 Then, in consideration of the fact that, the electrode contains an electrocatalytic conductive diamond layer, it was of our interest to study the effect of other material support for the BDD on the oxidants produced by the electrolysis. In this way, the change from Si/BDD to Nb/BDD should not involve dramatic changes in the results if the main degradation reaction pathways follow homogeneous reactions generated by the electrogenated oxidants, on the other hand if direct electrolysis occurs the results should be very different. Nevertheless, both anodes have been successfully used to degrade organic compounds (e.g.: pharmaceuticals, dyes, petroleum, pesticides and herbicides,1-5,11-15 no attempts have been reported yet regarding the use of different electrolytes to promote the electrogeneration of different oxidants at Nb/BDD anode. Thus, this investigation describes the comparative study of two supporting electrolytes (H2SO4 and HClO4) to determine its role during the electrochemical oxidation of short-chain carboxylic acids acetic acid (AA), oxalic acid (OA) and formic acid (FA) at Nb/BDD anode under different current densities. This strategy let to understand the formation and role of the strong oxidant species electrogenerated in the experimental conditions. The short-chain carboxylic acids were chosen as model compounds, because they have been identified as intermediates or final products of in various AOPs, since they are more difficult to oxidize than the initial contaminants.4,16 This characteristic make of them interesting compounds to determine the limitation of the AOPs as well as useful to the determination of reactions mechanisms.18-20
EXPERIMENTAL Materials The short-chain carboxylic acids (AA, OA and FA) were purchased from J. T. Baker and used without further purification. Distilled water was used in all solutions and experiments. The synthetic solutions (FA 0.18 mol L-1, AA 0.18 mol L-1 and OA 0.18 mol L-1) were prepared using 0.25 mol L-1 HClO4 and H2SO4 as supporting electrolytes. Electrochemical measurements Linear polarization analyses were performed at room temperature (25 °C) with a Metrohm AUTOLAB potentiostat model PGSTAT302N. The three-electrode cell was constituted by a Ag/AgCl (KCl 3 mol L-1) as reference electrode, Pt wire as the counter electrode and Nb/BDD (Metakem, Germany) as working electrode with an exposed geometric area of 1 cm2. Quasi-steady polarization curves were carried out at a scan rate of 50 mV s-1 and with a 2.44 mV step potential, by using the aforementioned BDD working electrode in 0.25 mol L-1 HClO4 and H2SO4 as supporting electrolytes. Large scale electrolysis system Bulk EO were carried out in a single compartment undivided cell (batch mode) containing 0.5 L of solution under constant stirring and galvanostatic conditions with a MINIPA MPL-3305M power supply. Nb/BDD film (Metakem, Germany) was used as anode, while titanium was employed as cathode. The electrodes were plates with a geometric area of about 18 cm2 and were placed parallel each other with an inter-electrode gap of 1.2 cm. The experiments were performed at 25 °C (thermostatic bath), using a current density (j) range of 30, 60, 90 and 120 mA cm-2 during 240 min. Analytical methods Chemical oxygen demand (COD) and total organic carbon (TOC): The short-chain carboxylic acids concentration elimination was monitored by COD which was determined by pre-COD dosage tubes and these were then heated in a Hanna Instruments HI 839800 thermo reactor for 2 h at 150 °C. Shimadzu TOC-V CPH was used to quantify TOC in samples. Hydroxyl radicals determination: Hydroxyl radicals were detected by the bleaching of N,N-dimethyl-4-nitrosoaniline (RNO). The original yellow solution was decolorized through reaction between RNO and •OH forming a colorless adduct.21 The bleaching of the 2 ×10-5 mol L-1 RNO solution in 0.25 mol L-1 H2SO4 and in 0.25 mol L-1 HClO4 was followed by using the spectrophotometer previously described reading the absorbance at 350 nm. Hydrogen peroxide determination: The spectrophotometric methodology using ammonium metavanadate proposed by Nogueira et al.22 was used for detection and quantification of H2O2 formed in solution. Also, some parameters described by Oliveira et al.23 were used. Persulfates determination: The spectrophotometric methodology proposed by Liang et al.24 was used for persulfate quantification. Efficiency parameters Energy consumption: Energetic requirements of the each experiment were estimated (in kWh m−3) means of the expression:4 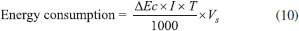 where t is the time of electrolysis (h), Ec (V) and I (A) are the average cell voltage and the electrolysis current, respectively; and Vs is the sample volume (m3). Total current efficiency (TCE): Percentage of TCE for electrochemical oxidations of the carboxylic was estimated by using the initial and final COD values, following relationship:4 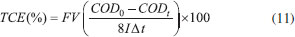 where I is the current (A), F the Faraday constant (96,487 C mol−1), V is the electrolyte volume (dm3), 8 is the oxygen equivalent mass (g eq.−1) and t is the electrolysis time, allowing for a global determination of the overall efficiency of the process. Mineralization current efficiency (MCE): This parameter was calculated using the relationship established by Brillas et al.3,4 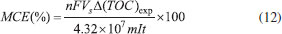 were n is the number of electrons consumed in the oxidation of the compound (2 for formic and oxalic and 8 for acetic acid), F is the Faraday constant (=96487 C mol-1), Vs is the solution volume (L), Δ(TOC)exp is the experimental TOC decay (mg L-1), 4.32 x 107 is a conversion factor (=3600 s h-1 12000 mg of C mol-1), m is the number of carbon atoms in the molecule (1 for FA and 2 for AA and OA), I is the applied current (A) and t is the time (h). The reactions of oxidation of formic, acetic and oxalic acids, respectively, are shown in Eq. 13 - 15:25 
RESULTS AND DISCUSSION Supporting electrolyte effect into the electrogenerated of oxidants species (hydroxyl radicals, hydrogen peroxide and persulfate) UV-visible measurements were used for detecting hydroxyl radicals formed at Nb/BDD electrode during water anodic discharge. The indirect technique for the detection of low concentrations of hydroxyl radicals is carried out with a spin trapping (RNO) compound in order to produce a more stable radical (spin adduct). The used compound for this task is particularly advantageous and selective, because under these conditions of concentration and pH it is electrochemically inactive at positive potentials, and the addition reaction occurs at a very high rate constant (1.3×1010 L mol−1s−1).20,21 Figure 1 shows the adsorption spectrum of aqueous RNO solution (2×10-5 mol L-1) during galvanostatic electrolysis with Nb/BDD anode by applying 60 mA cm-2, in 0.25 mol L-1 H2SO4 (Figure 1a) and HClO4 (Figure 1b) as supporting electrolytes.
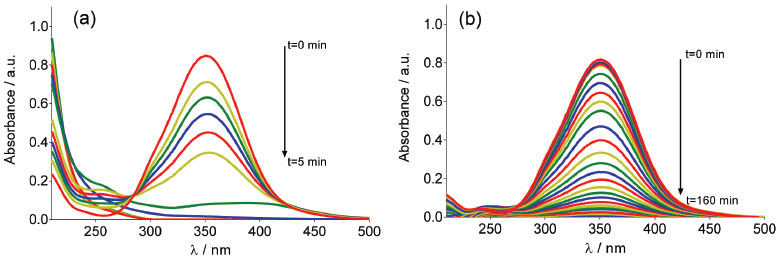 Figure 1. Adsorption spectra of aqueous RNO solution (2×10-5 mol L-1) obtained at different electrolysis-time intervals under galvanostatic conditions, in (a) 0.25 mol L-1 H2SO4 and (b) 0.25 mol L-1 HClO4 as supporting electrolytes, at Nb/BDD anode by applying 60 mA cm-2 at 25 °C
A decrease in absorbance at 350 nm during electrolysis is observed in both medium. Although the results showed that, there was accumulation of •OH radicals at the Nb/BDD anode surface, when H2SO4 was used as supporting electrolyte RNO absorbance decreases faster (5 min), whereas with HClO4 medium requires more almost 3 hrs. This effect indicates that, when electrolysis is carried out in H2SO4, the adduct formed by reaction between RNO and •OH radicals is formed faster and can be in first instance be rationalized as a major production of •OH species. However, as was mentioned in the introduction, in a solution containing with sulfates the electrogeneration of persulfate ions is feasible. Taking into consideration this fact, the persulfate formation is attained in parallel, promoting the degradation of RNO under these experimental conditions, and consequently, accelerating the disappearance of RNO absorption peak. In fact, the UV-vis spectrum showed the existence of intermediates that absorb irradiation approximately at 190 and 250 nm. Meanwhile, at HClO4, the results clearly demonstrated that, only •OH radicals are produced at BDD surface, which react with RNO to form adduct species, avoiding their degradation. In fact, no absorption bands were achieved at wavelengths below 280 nm. Similar behavior was achieved at all j values, indicating that significant concentrations of •OH radicals are electrochemically generated in HClO4, but their concentration depends on the j used (Figure 2). In fact, higher j provoked a more rapid disappearance of the RNO adsorption, confirming that in these conditions the production of •OH radicals was favored. These results are in agreement with the proposed model by Comninellis in which the BDD electrocatalytic material is considered a nonactive anode due to the significant production of •OH radicals, physically adsorbed, that favor the complete combustion of organic pollutants.1,5,7,21
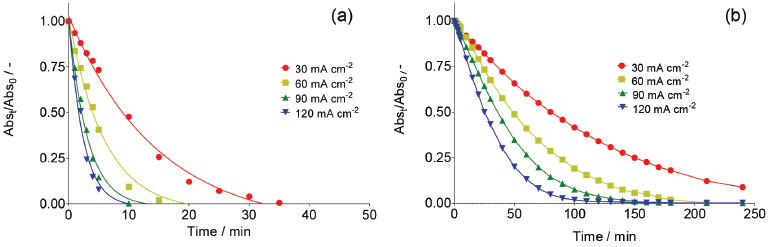 Figure 2. Normalized absorbance decay of RNO solution as consequence of hydroxyl radicals electrogeneration at Nb/BDD anode by applying 30, 60, 90 and 120 mA cm-2 during electrolysis in (a) 0.25 mol L-1 H2SO4 and (b) 0.25 mol L-1 HClO4 at 25 °C
The formation of other oxidants than •OH at the BDD surface during the electrolysis is feasible, for example H2O2 and S2O82- (Eq. 1-9).25,26 For this reason, its quantification is necessary to have a better picture of the oxidizing agents' role during carboxylic acids degradation. Figure 3 shows the evolution of H2O2 concentration as a function of electrolysis time for BDD electrodes by applying different j in H2SO4 (Figure 3a) and HClO4 (Figure 3b) at 25°C. This figure shows clearly that the production of H2O2 is significantly higher at HClO4 medium than that formed at H2SO4. It also increases when an increase on the j is attained at HClO4.
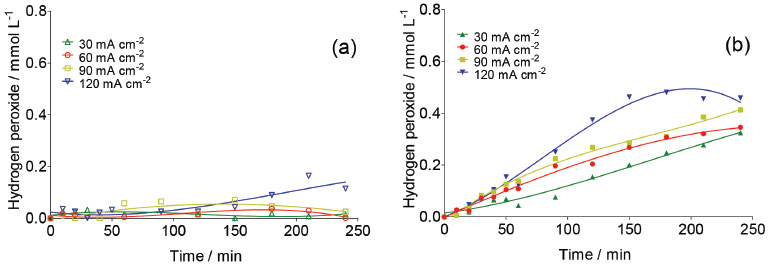 Figure 3. Concentration of electrogenerated H2O2 at Nb/BDD surface in (a) 0.25 mol L-1 H2SO4 and (b) 0.25 mol L-1 HClO4 by applying 30, 60, 90 and 120 mA cm-2 at 25 °C
This behavior is can be attributed to the electrochemical formation of free hydroxyl radicals at BDD electrode surface because, the higher concentration •OH radicals the higher the recombination between them to form H2O2 (Eq. 3). However, it is also expected that after its formation, hydrogen peroxide in solution can be decomposed through reaction at the anode surface (Eq. 16) or cathode surface (Eq. 17); therefore its concentration reaches a plateau. 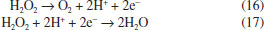 Concentrations of H2O2 are lower at Nb/BDD than those reported in the literature for Si/BDD,7 suggesting that the electrochemical activity for hydrogen peroxide formation is minor. In H2SO4 medium, the main reactions at water discharge should be the formation of persulfate (Eqs. 4 and 7) via oxidation of sulfate ions in solution7,27 and the oxygen evolution (Eq. 18). In contrast, using HClO4 the main reaction at water discharge should be oxygen evolution (Eq. with concomitant production of •OH radicals, due to water oxidation (Equations 1 and 15 respectively). 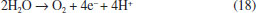 To evaluate the amount of persulfate formed at water oxidation in H2SO4, electrolyzes at different current density were performed. Concentration of these species was determined in the anolyte during electrolyzes by ISCO method.24 Figure 4 shows the influence of current density on the S2O82− concentration. As can be observed, an increase on the S2O82- concentration is achieved when j increases from 30 to 90 mA cm-2. However, when higher j is applied, a decrease on the persulfate concentration is attained. This behavior is due to the faster oxygen evolution at higher j, decreasing the electrosynthesis of persulfate.
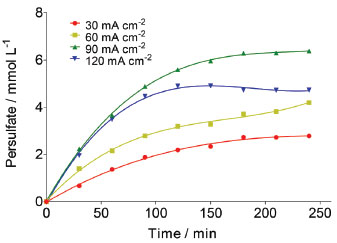 Figure 4. Electrogenerated S2O82− at BDD anode by applying 30, 60, 90 and 120 mA cm-2 during electrolysis in 0.25 mol L-1 H2SO4 at 25 °C
In order to gain insight about these important reactions in both supporting electrolytes, potentiodynamic measurements of the water discharge were carried out. Figure 5 shows linear polarization curves and the corresponding Tafel plots of Nb/BDD electrode obtained in HClO4 and H2SO4 at a scan rate of 50 mV s−1. Current-potential curves are almost identical, although the reactions that take place in HClO4 and H2SO4 media are different. The value of Tafel slope is almost the same for both media (ca. 0.81 V decade-1), being 3.5-folds higher than Si/BDD.7 Therefore, silicon supported BDD is oxidizes faster water and the difference probably is due to the different semimetal/semiconductor properties between both diamond films.
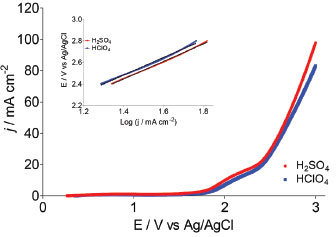 Figure 5. Polarization curves and Tafel analysis for Nb/BDD electrode in 0.25 mol L-1 H2SO4 and 0.25 mol L-1 HClO4 at 50 mV s-1 at 25°C
Taken into consideration the data obtained in potentiodynamic measurements can be concluded that even if the main reaction products are different (O2 and •OH radicals in HClO4 and O2 and S2O82− in H2SO4); the same electrochemical reaction is generating them in both electrolytes. Then, this indicates that the first step during water discharge on Nb/BDD electrode is the O2 evolution (Eq. 16 or 18) with simultaneous generation of hydroxyl radicals (Eq. 1). After, and depending on the supporting electrolyte, the electrogenerated hydroxyl radicals can react to produce persulfate in case of H2SO4 electrolyte (Eq. 5 and 6), or can generate H2O2 by the reaction of hydroxyl radicals in case of HClO4 electrolyte (Eq. 3). In both cases, recalcitrant organic matter degradation follows indirect electrochemical oxidation pathways that include oxidation with •OH and the other electrogenerated strong oxidants. It is important to remark that, in the case of H2SO4 medium, the co-existence of direct oxidation mechanism cannot be feasible due to the significant production of persulfate and •OH radicals at diamond surface. COD decay In order to determine the efficiency of the EO on very difficult to oxidize (recalcitrant) short-chain carboxylic acids (AA, OA and FA) and to understand the participation of the strong oxidants produced at Nb/BDD surface in both supporting electrolytes, bulk electrolyzes were carried out. COD and TOC were monitored during 240 min of electrolysis at different j values. Figure 6 shows the COD decays for the studied carboxylic acids as a function of time at the Nb/BDD anode. The results demonstrate that COD removal is strongly influenced by the j as well as the carboxylic acid added to the electrolyte. In the case of AA in H2SO4 media, no significant COD removals were achieved at 30 and 60 mA cm−2 (26% of COD removal was achieved in each case). At 90 mA cm−2, 50% of COD was attained; but an increase on the j (120 mA cm-2) promotes a decrease on the COD removal achieving up to 25%. This behavior suggests that the BDD anode favors a set of electrochemical reactions, involving most probably the oxygen evolution and persulfate formation as was concluded in the previous section (Figure 4).7,19,20,28-30 Indirect oxidation of AA via •OH and persulfate could be attained by applying 30, 60 and 90 mA cm-2, however, the production of some intermediates more difficult to oxidize (small amounts of FA and traces of OA),30,31 limit an higher COD removal. Conversely, with 120 mA cm-2 oxygen evolution was favored, reducing the production of persulfates (see, Figure 4), as well as hydroxyl radicals and consequently, limiting an efficient degradation of AA and its intermediates. Comparing these results with the behavior observed at HClO4, very similar elimination profiles of AA were reached (Figure 6), but slight differences (in terms of efficiency) were observed at lower j, where degradation rate was slightly faster when sulfate medium is used. This experiment indicates that AA can be oxidized by the strong oxidants generated in both media, but the production of persulfates in sulfate medium, oxidizes faster in the bulk the short-chain carboxylic acid.
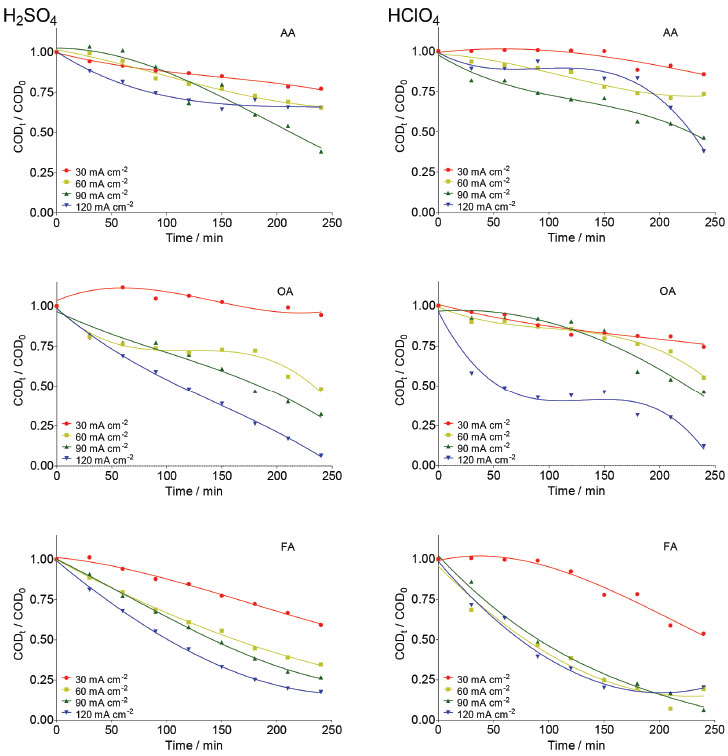 Figure 6. Effect of j on the normalized COD decay as a function of electrolysis time during the EO of AA, OA and FA at Nb/BDD anode surface in 0.25 mol L-1 H2SO4 (left) and 0.25 mol L-1 HClO4 (right) applying 30, 60, 90 and 120 mA cm-2
During Nb/BDD electrolysis for OA elimination (Figure 6) in H2SO4, no significant COD removal was observed at 30 mA cm−2 (5%). However, when an increase on the j is attained, the elimination of OA from solution was gradually increased, achieving 50%, 70% and 98% of COD removals after 240 min by applying 60, 90 and 120 mA cm−2, respectively. This result indicates that this carboxylic acid requires high concentration of strong oxidant (•OH and H2O2 in HClO4 or •OH and S2O82− in H2SO4) to be degraded. Conversely, at HClO4, lower COD decays were achieved at 30, 60 and 90 mA cm−2, while an 87% of COD was achieved at 120 mA cm−2. In the former behavior, it is clearly that a synergic effect due to the participation of strong oxidants (•OH and persulfate) promotes a quick elimination of OA (Eq. 19). Meanwhile, the oxidation process, in HClO4, competes with the side reaction of oxygen evolution (Eq. 18) and H2O2 production (considered a weaker oxidant), avoiding a significant elimination of OA, in the proximity of the electrode surface and/or in the bulk of the electrolyte.19,25,29 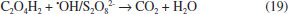 Figure 6 also shows the COD removal of FA in solution by applying 30, 60, 90 and 120 mA cm−2 at Nb/BDD anode in H2SO4. Results clearly showed that, COD removal increases when an increase on the j is attained. At both acidic medium, no enough concentration of oxidants is formed at 30 mA cm-2. Meanwhile, when j increases, the production of •OH/H2O2 radicals in HClO4 and •OH/S2O82- in H2SO4 are favored, increasing significantly the mineralization of FA. Generally speaking, the behavior observed at H2SO4 is similarly achieved at HClO4 (see, Figure 6) because of the small chemical structure of FA, favoring mainly the formation of CO2 (Eq. 20) with any strong oxidant formed at the electrode: 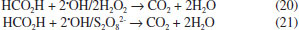 From these experiments, it can be proposed that when the electrochemical incineration is mediated solely by •OH specie, degradation has a close behavior to a heterogeneous electrochemical reaction, due to the physisorbed •OH radicals onto the electrode. In the other hand, in the presence of sulfates the degradation reaction occurs both homogeneously with S2O82- anion and heterogeneously by means of the physisorbed •OH radicals, boostering the organic compounds elimination. From Eq. 11, total current efficiency (TCE) values were estimated (Figure 7) for the electrochemical incineration of AA, OA and FA using Nb/BDD in H2SO4 after 240 min, and these were compared with the values obtained using HClO4. According to the results, the electrical energy furnished for the electrochemical incineration of AA is more efficiently employed than the other experiments. In fact, good TCE values were obtained for the elimination of AA when 30, 60 and 90 mA cm-2 were applied in H2SO4. This indicates that, the elimination is gradually performed via the participation of •OH and persulfate32 in a catalytic way increasing degradation by homogeneous reactions. Over 90 mA cm-2, the TCE decreased notably (less than 50%), indicating that oxygen evolution is the main electrochemical reaction. For this carboxylic acid and HClO4 medium, again higher TCE values were obtained, but only with 60 and 90 mA cm-2 indicating that in this range of j, the •OH are produced efficiently. As observed in H2SO4 media, higher values of j favored oxygen evolution and lower did not generate enough •OH radicals or H2O2 is formed which do not oxidizes efficiently AA. In the case of OA, it shown to be a more recalcitrant acid than AA and TCE values close to 40% were the maximal values obtained in HClO4 media using the highest current density values. Conversely, in H2SO4 media, more than 50% is achieved by applying 90 and 120 mA cm-2 indicating the participation of persulfate improves the elimination. It is well known that one of the most recalcitrant carboxylic acids during organic matter degradation is oxalic acid and is generally detected in the electroincineration using BDD electrodes,5 for this reason, it was not efficiently eliminated. For FA, TCE values increased again in H2SO4 to values close to 90% at low current densities (60 mA cm-2) and decreases as result of oxygen evolution when current density increases; FA had a similar behavior to AA in HClO4.
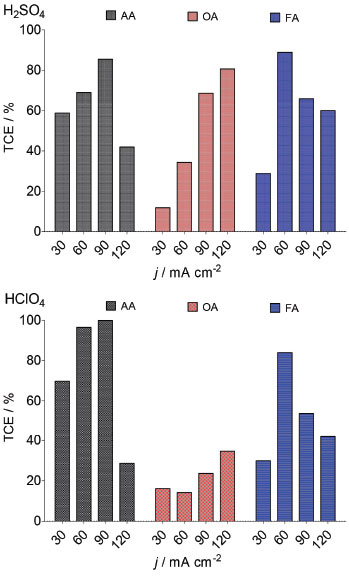 Figure 7. TCE values as a function of applied j during EO of AA, OA and FA in 0.25 mol L-1 H2SO4 and 0.25 mol L-1 HClO4 by using Nb/BDD anode estimated at 240 min of electrolysis
Mineralization of the short chain carboxylic acids The mineralization current efficiency (MCE) was determined using equation 12, which takes into account the change on TOC during the electrolysis. Table 1 shows TOC removals in both supporting electrolytes studied. In the case of FA in both H2SO4 and HClO4, it was at low j values almost entirely mineralized reaching similar MCE values. At the higher j values, the process was limited by mass transport at the end of the process showing a decrease in the MCE. This confirmed that secondary reactions (such as oxygen evolution) occurs in concomitance as was discussed previously.25,33-35
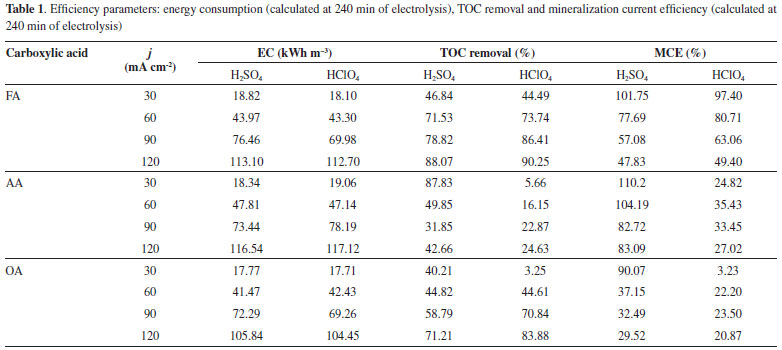
For AA, MCE was significantly high in H2SO4 than that achieved in HClO4, but it was achieved at lower j. This indicates that the mineralization of organic matter (from AA molecule) to CO2 and water, is accomplished in slower, suggesting that it is converted to other simpler compounds that limit the EO process or/and this reaction is in competition with oxygen evolution.24,33,34 In the case of OA, lower mineralization is achieved at 30 and 60 mA cm-2 at both supporting electrolytes. However, an increase on the j promotes an increase on the mineralization level29 after 240 min of electrolysis (Table 1). This result confirms the COD removal efficiencies achieved in H2SO4 and HClO4, but in the case of sulfuric acid, the participation of strong oxidants in reaction cage (•OH and S2O82-) is attained. However, it is evident that, the mineralization of OA into CO2 and H2O is not complete favored due to the complexity of the OA structure.19-21 Efficiency parameters In order to evaluate the efficiency of EO, we compare the results obtained by the energy consumption (EC), at the end of each one of the electrochemical treatments. Table 1 also shows the EC required for removing AA, OA and FA using Nb/BDD anode. As can be observed, during the electrolysis of synthetic wastewater solutions, EC is directly proportional to the j applied, then, an increase on j spent higher electrical energies. Also, EC is independent of the carboxylic acid degraded because similar values were achieved. However, when EC is compared with the % COD removed at each one of the carboxylic acids, it is evident that higher removal efficiencies were achieved for FA oxidation with lower EC. Meanwhile, similar behavior is observed between AA and OA, in terms of COD removals at the end of the electrolysis, but the increase on the energy requirements is directly proportional for OA elimination. Conversely, a significant variation of EC is achieved at AA oxidation with different COD removals.
CONCLUSIONS The supporting electrolyte has important role on the oxidative process pathway due to the strong oxidants produced at Nb/BDD surface. In fact, in the case of HClO4, higher concentrations of hydroxyl radicals and hydrogen peroxide were detected, whereas persulfate production and hydroxyl radicals are the main reactions when H2SO4 is used in solution as electrolyte. The formation of persulfate is performed due to the direct oxidation of sulfate on diamond electrode surface, but the reaction between hydroxyl radicals and sulfate ion is also feasible. This reaction can be conveniently for electrochemical incineration used, treating a synthetic solution containing carboxylic acids, separately. The experiments with sulfuric acid demonstrated a faster removal of organic matter, in terms of COD and TOC, which can be attributed to the simultaneous action of persulfates and hydroxyl radicals. Comparing the results using H2SO4 and HClO4, similar behaviors were achieved in terms of electrochemical degradation for FA, and AA, nevertheless, OA showed to be a more recalcitrant organic compound.
ACKNOWLEDGEMENTS Financial support from National Council for Scientific and Technological Development (CNPq - 465571/2014-0; CNPq - 446846/2014-7 and CNPq - 401519/2014-7) and FAPESP (2014/50945-4) are gratefully acknowledged. Carlos A. Martínez-Huitle acknowledges the funding provided by the Alexander von Humboldt Foundation (Germany) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brazil) as a Humboldt fellowship for Experienced Researcher (88881.136108/2017-01) at the Johannes Gutenberg-Universität Mainz, Germany. B. A. Frontana-Uribe thanks funding provided by PAPIIT-UNAM Project 208919 and CONACYT-MEXICO Project A1-5-18230.
REFERENCES 1. Martínez-Huitle, C. A.; Panizza, M.; Curr. Opin. Electrochem. 2018, 11, 62. 2. Martínez-Huitle, C. A.; Rodrigo, M. A.; Sirés, I.; Scialdone, O.; Chem. Rev. 2015, 115, 13362. 3. Brillas, E.; Sirés, I.; Oturan, M. A.; Chem. Rev. 2009, 109, 6570. 4. Brillas, E.; Martínez-Huitle, C. A.; Appl. Catal., B 2015, 166-167, 603. 5. Panizza, M.; Cerisola, G.; Chem. Rev. 2009, 109, 6541. 6. Espinoza-Montero, P. J.; Vasquez-Medrano, R.; Ibanez, J. G.; Frontana-Uribe, B. A.; J. Electrochem. Soc. 2013, 160, G3171. 7. Michaud, P. A.; Panizza, M.; Ouattara, L.; Diaco, T.; Foti, G.; Comninellis, C.; J. Appl. Electrochem. 2003, 33, 151. 8. Davis, J.; Baygents, J. C.; Farrell, J.; Electrochim. Acta 2014, 150, 68. 9. Garcia-Segura, S.; Dos Santos, E.V.; Martínez-Huitle, C.A.; Electrochem. Commun. 2015, 59, 52. 10. Brito, C. N.; De Araújo, D. M.; Martínez-Huitle, C. A.; Rodrigo, M. A.; Electrochem. Commun. 2015, 55, 34. 11. Cotillas, S.; Clematis, D.; Cañizares, P.; Carpanese, M. P.; Rodrigo, M. A.; Panizza, M.; Chemosphere 2018, 199, 445. 12. Clematis, D.; Abidi, J.; Cerisola, G.; Panizza, M.; ChemElectroChem, 2019, 6, 1794. 13. Klidi, N.; Clematis, D.; Carpanese, M. P.; Gadri, A.; Ammar, S.; Panizza, M.; Sep. Purif. Technol. 2019, 208, 178. 14. Tasca, A. L.; Puccini, M.; Clematis, D.; Panizza, M.; Environ. Pollut., 2019, 251, 285. 15. Dos Santos, E. V.; Sáez, C.; Martínez-Huitle, C. A.; Cañizares, P.; Rodrigo, M. A.; Electrochem. Commun. 2015, 55, 26. 16. Cañizares, P.; García-Gómez, J.; Lobato, J.; Rodrigo, M. A.; Ind. Eng. Chem. Res. 2003, 42, 956. 17. Martínez-Huitle, C. A.; Ferro, S.; De Battisti, A.; J. Appl. Electrochem. 2005, 35, 1087. 18. Cañizares, P.; Paz, R.; Sáez, C.; Rodrigo, M.A.; Electrochim. Acta 2008, 53, 2144. 19. Ferro, S.; Martínez-Huitle, C. A.; De Battisti, A.; J. Appl. Electrochem. 2010, 40, 1779. 20. Fierro, S.; Abe, K.; Comninellis, C.; Einaga, Y.; J. Electrochem. Soc. 2011, 158, F183. 21. Comninellis, C.; Electrochim. Acta 1994, 39, 1857. 22. Nogueira, R. F. P.; Oliveira, M. C.; Paterlini, W. C.; Talanta 2005, 66, 86. 23. Oliveira, M. C.; Nogueira, R. F. P.; Neto, J. A. G.; Jardim, W. F.; Rohwedder, J. J. R.; Quim. Nova 2001, 24, 188. 24. Liang, C.; Huang, C.-F.; Mohanty, N.; Kurakalva, R. M.; Chemosphere 2008, 73, 1540. 25. Gandini, D.; Mahé, E.; Michaud, P. A.; Haenni, W.; Perret, A.; Comninellis, C.; J. Appl. Electrochem. 2000, 30, 1345. 26. Panizza, M.; Cerisola, G.; Appl. Catal., B 2007, 75, 95. 27. Michaud, P. A.; Mahé, E.; Haenni, W.; Perret, A.; Comninellis, C.; Electrochem. Solid-State Lett. 2000, 3, 77. 28. Kapałka, A.; Fóti; G.; Comninellis, C.; J. Appl. Electrochem. 2008, 38, 7. 29. Martínez-Huitle, C. A.; Ferro, S.; De Battisti, A.; Electrochim. Acta 2004, 49, 4027. 30. Panizza, M.; Cerisola, G.; J. Electroanal. Chem. 2010, 638, 28. 31. Kapałka, A.; Lanova, B.; Baltruschat, H.; Fóti, G.; Comninellis, C.; J. Electrochem. Soc. 2008, 155, E96. 32. Rodrigo, M. A.; Cañizares, P.; Sánchez-Carretero, A.; Sáez, C.; Catal. Today 2010, 151, 173. 33. Kapałka, A.; Lanova, B.; Baltruschat, H.; Fóti, G.; Comninellis, C.; Electrochem. Commun. 2008, 10, 1215. 34. Weiss, E.; Groenen-Serrano, K.; Savall, A.; Comninellis, C.; J. Appl. Electrochem. 2007, 37, 41. 35. Labiadh, L.; Barbucci, A.; Cerisola, G.; Gadri, A.; Ammar; S.; Panizza, M.; J. Solid State Electrochem. 2015, 19, 3177. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access