
 |
 |
 |
 |
 |
Artigo
|
|
| Interações competitivas de complexos de rutênio contendo dimetilsulfóxido e ligantes N-heterocíclicos com albumina de soro humano Competitive interactions of ruthenium complexes containing dimethylsulfoxide and N-heterocyclic ligands with human serum albumin |
|
Guilherme Luiz B. Neto; Eduardo Alexandre M. Baptista; Gabriel Hideki S. Becca; Helena S. Nakatani; Vagner R. de Souza*
Departamento de Química, Universidade Estadual de Maringá, 87020-900 Maringá - PR, Brasil Recebido em 25/07/2019 *e-mail: vrsouza2@uem.br Two ruthenium complexes of the type [RuCl(dmso)(L)2]Cl {L = 4,4'-dimethyl-2,2'-bipyridine (dmbpy); 4,4'-dinonyl-2,2'-bipyridine (dnbpy)} have been synthesized and characterized by elemental analysis, 1H NMR, FTIR, electronic spectra, and molar conductivity. The IR spectral studies revealed that the DMSO molecule is S-bound ( νSO = 1100 cm-1; 1079 cm-1). For complexes, MLCT bands were observed around 390 nm and 440 nm. The measurements of conductivity revealed the presence of 1:1 electrolyte. Interactions of ruthenium complexes with human serum albumin were examined by fluorescence spectroscopy. The results revealed a combined quenching mechanism of HSA fluorescence by [RuCl(dmso)(dmbpy)2]Cl, with binding constant of 5.82 ± 0.08 x 105 mol-1 L (297 K), 5.29 ± 0.06 x 105 mol-1 L (303 K), and 4.68 ± 0.06 x 105 mol-1 L (313 K), whereas the [RuCl(dmso)(dnbpy)2]Cl caused static quenching predominantly, with binding constant of 9.87 ± 0.05 x 105 mol-1 L (297 K), 3.41 ± 0.04 x 105 mol-1 L (303 K), and 0.89 ± 0.05 x 105 mol-1 L (313 K). The binding process occurred spontaneously and was mainly driven by enthalpy, as evidenced by thermodynamic parameters. Site marker competitive experiment showed that ruthenium complexes bind to the warfarin binding site in subdomain IIA of HSA. INTRODUÇÃO A síntese de um composto com potencial ação farmacológica é a primeira etapa de um longo processo laboratorial, como mostram os guias para a condução de estudos não clínicos de segurança necessários ao desenvolvimento de medicamentos elaborados pela Agência Nacional de Vigilância Sanitária - ANVISA.1 Nos ensaios pré-clínicos é necessário descobrir como direcionar o fármaco para um local específico do organismo e como as barreiras naturais do organismo podem interagir com ele.2 Por isso, na elaboração de agentes terapêuticos é importante estudar a interação desses derivados com biomoléculas, pois as biomoléculas presentes no plasma sanguíneo têm a capacidade de se ligarem aos fármacos provocando sua inativação.3,4 Das biomoléculas plasmáticas, albumina e globulinas são encarregadas pelo armazenamento e transporte de íons, metabólitos e nutrientes, como glicose, gorduras, vitaminas e aminoácidos pela corrente sanguínea até os tecidos do corpo humano.5 Em organismos saudáveis, a concentração de albumina no sangue está entre 3,5 e 4,5 g/100 mL, que corresponde a aproximadamente 60% das proteínas plasmáticas,6 enquanto os teores de globulinas são 4% de globulina-α1, 8% de globulina-α2, 12% de globulina-β e 16% de globulina-γ. Por ser a proteína mais abundante no sangue e corresponsável pela difusão de fármacos para os diferentes tipos de tecidos do corpo humano, a interação de novos compostos com albumina é uma demanda da área medicinal.7-10 A albumina utilizada nos ensaios pré-clínicos é obtida a partir do fracionamento industrial do plasma humano coletado por doação de sangue.11 Os processos de obtenção e fracionamento da albumina do plasma sanguíneo elevam o preço da proteína comercial 12 e, por isso, a maioria dos ensaios in vitro troca a proteína humana (HSA) pela albumina do soro bovino (BSA).13 A substituição da proteína humana pela bovina é possível devido às semelhanças estruturais dessas biomoléculas.14 As informações disponíveis no banco de dados de proteínas (RCSB Protein Data Bank)15 mostram que as proteínas humana e bovina apresentam algumas diferenças com relação às suas sequências de aminoácidos, mas conservam alto grau de similaridade estrutural. Considerando que o acesso à informação estrutural das diferentes proteínas é gratuito e de fácil obtenção no banco de dados de proteínas, optamos por não reproduzi-la integralmente nesse trabalho, mas destacar alguns aspectos da albumina humana. A albumina humana é formada por uma sequência de 585 aminoácidos, perfazendo uma cadeia polipeptídica simples com massa molar próxima a 66000 g mol-1, sustentada por α-hélices e unida por dezessete pontes dissulfeto.16 As ligações do tipo dissulfeto, -S-S, aliadas às ligações de hidrogênio -NH·····O=C e interações iônicas do tipo -NH3+·····-OCOR entre os fragmentos de aminoácidos são responsáveis pela manutenção da estrutura terciária da proteína e pelo aparecimento de cavidades com diferentes graus de hidrofobicidade, as quais determinam a seletividade proteica e o armazenamento de fármacos.17-19 As observações realizadas por Sudlow e colaboradores na década de 1970 envolvendo mudanças conformacionais da estrutura proteica induzidas pela inclusão de compostos orgânicos, como warfarina, ibuprofeno, flurbiprofeno, ácido esteárico, fenilbutazona e oxifenilbutazona, permitiram determinar microrregiões responsáveis pelo armazenamento de compostos exógenos nos diferentes tipos de albuminas.20 Essas regiões ficaram conhecidas como sítios I e II de Sudlow.21 Estudos de difração de raios X detalharam os diferentes microambientes proteicos, que resultaram nas subdivisões IA, IB, IIA, IIB, IIIA e IIIB desses sítios, como pode ser observado nas ilustrações disponíveis gratuitamente no banco de dados de proteínas,15 cuja representação simplificada está apresentada na Figura 1.
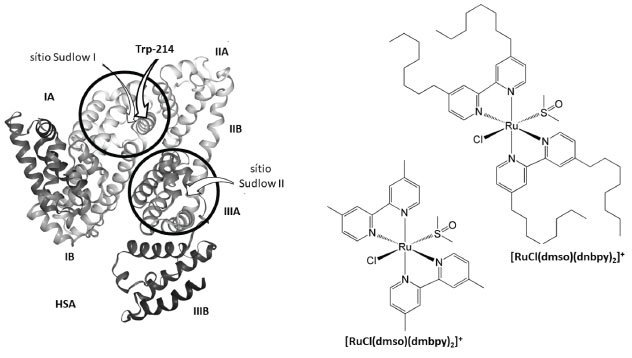 Figura 1. Representação simplificada da estrutura terciária da albumina do soro humano (HSA), destacando os sítios de Sudlow e as suas subdivisões, e representação estrutural dos compostos de rutênio
A existência de diferentes ambientes para a ligação de fármacos na estrutura proteica permite que a albumina armazene concomitantemente dois ou mais medicamentos e funcione como um reservatório multidrogas temporário no plasma sanguíneo retardando a chegada dos princípios ativos aos sítios alvos.22 De acordo com a literatura,23 muitos fármacos ocupam o mesmo microambiente proteico, por exemplo: subdomínio IB: azapropazona e indometacina; subdomínio IIA: ácido 3-carboxi-4-metil-5-propil-2-furanopropanóico, ácido 3,5-diiodosalicílico, iodipamida, oxifenbutazona, fenilbutazona e warfarina; subdomínio IIIA: tiroxina, diflunisal, diazepam, propofol, sulfato de indoxila e halotano. Dessa forma, a absorção e a biodisponibilidade de um fármaco em relação aos demais ocorrem via mecanismo de competição pelo sítio de ligação da albumina.24 Nesse contexto, a distribuição, ação e eliminação dos fármacos no organismo humano dependem da força de ligação desses derivados com a cadeia polipeptídica da biomolécula, que pode ser inferida a partir dos valores de constante de ligação.25 O método utilizado para calcular esse parâmetro está baseado no modelo de supressão da fluorescência intrínseca das proteínas.26 O equacionamento desse modelo, fundamentado na colisão de moléculas no estado excitado com a espécie supressora (Q) elaborado por Stern-Volmer,27 foi difundido por Lakowicz28 e resultou na equação abaixo: 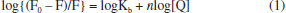 em que F0 = intensidade de fluorescência na ausência do supressor, F = intensidade de fluorescência na presença do supressor, Kb é a constante de ligação, n é o número de sítios proteicos ocupados e [Q] é a concentração da espécie supressora. Estudos de supressão de fluorescência de proteínas com derivados metálicos com potencial ação farmacológica29 têm contribuído para determinar a força de interação entre as espécies exógenas e a proteína. Nesse conjunto de compostos, complexos contendo ligantes de natureza lábil aliado à presença de grupos periféricos com características hidrofóbicas formam adutos mais estáveis com o arcabouço proteico, o que minimiza a sua biotransformação e eliminação. Contudo, o grau de distribuição desses derivados nos tecidos corporais depende do conteúdo de lipídios no tecido, do coeficiente de partição lipídio-água, da permeabilidade capilar, do débito cardíaco, da perda de função hepática, da perda da massa muscular, entre outros fatores externos associados ao envelhecimento, obesidade e gravidez.30 Assim, a ação alostérica de complexos metálicos31 e a natureza hidrofóbica de ligantes N-heterocíclicos nos motivaram a estudar a interação dos compostos [RuCl(dmso)(dmbpy)2]Cl e [RuCl(dmso)(dnbpy)2]Cl, sendo dmbpy = 4,4'-dimetil-2,2'-bipiridina e dnbpy = 4,4'-dinonil-2,2'-bipiridina, com a albumina do soro humano (HSA), Figura 1. A presença de grupos periféricos hidrofóbicos na esfera de coordenação do centro metálico afeta o tempo de retenção do composto no ambiente proteico e, consequentemente, altera os processos de absorção, distribuição e metabolismo do composto na corrente sanguínea, visto que a interação do composto com os receptores intracelulares envolve a transposição da membrana plasmática composta principalmente por lipoproteínas.32 O processo de inserção dos compostos no arcabouço proteico foi monitorado pela supressão de fluorescência da albumina do soro humano. Também avaliamos a afinidade dos complexos por diferentes sítios de ligação da HSA frente às moléculas bioativas ibuprofeno e warfarina, visto que a compreensão do mecanismo de substituição de espécies na estrutura proteica permite entender e controlar a ação terapêutica dos compostos no organismo humano.
PARTE EXPERIMENTAL Todos os reagentes utilizados neste trabalho foram de grau analítico e empregados sem purificação prévia: RuCl3.nH2O (Sigma-Aldrich), 4,4'-dimetil-2,2'-bipiridina (Sigma-Aldrich), 4,4'-dinonil-2,2'-bipiridina (Sigma-Aldrich), DMSO (Sigma-Aldrich), Albumina do soro humano (HSA, sólido liofilizado, ≥ 96%, Sigma-Aldrich), ibuprofeno (Sigma-Aldrich) e warfarina (Sigma-Aldrich). O precursor [RuCl2(dmso)4] foi sintetizado conforme metodologia descrita na literatura.33 A análise por espectrometria de massas foi realizada por infusão direta em um espectrômetro de massas Quadrupolo -Time of Flight (Bruker, Impact II), utilizando água como solvente para o complexo [RuCl(dmso)(dmbpy)2]Cl e acetonitrila para o complexo [RuCl(dmso)(dnbpy)2]Cl. Espectros de RMN de 1H foram obtidos à temperatura ambiente em um espectrômetro Varian Inova operando a 300 MHz, utilizando acetona deuterada para o complexo [RuCl(dmso)(dmbpy)2]Cl e água deuterada para o complexo [RuCl(dmso)(d-bpy)2]Cl. Espectros vibracionais na região do infravermelho (4000 a 400 cm-1) foram obtidos para amostras em pastilha de KBr, utilizando espectrofotômetro FTIR modelo 8300 Shimadzu. Espectros eletrônicos foram obtidos em espectrofotômetro de varredura rápida Hewlett-Packard modelo 8453, acoplado a um microcomputador. Foram utilizadas soluções com concentrações na faixa de 1 x 10-6 a 1 x 10-3 mol L-1 (solvente acetonitrila). Medidas de condutância molar foram realizadas em um condutivímetro de bancada modelo Simpla EC150 utilizando banho termostatizador. O complexo [RuCl(dmso)(dmbpy)2]Cl foi dissolvido em água destilada, enquanto que o [RuCl(dmso)(dnbpy)2]Cl foi dissolvido em acetonitrila. Os teores de C, H, N foram determinados no equipamento Perkin Elmer CHN-2000. Para análise de supressão de fluorescência, cada complexo foi dissolvido em 25 mL de solução tampão TRIS-HCl pH 7,4 (solução estoque), preparado conforme descrito na literatura,34 e o valor foi monitorado utilizando pHmetro digital modelo PHM 240 Meter Lab. A força iônica do meio foi ajustada com solução de KCl 0,10 mol L-1. A concentração de complexo na solução estoque foi de 1 x 10-3 mol L-1. Para o complexo [RuCl(dmso)(dnbpy)2]Cl foi adicionado 1 mL de solvente DMSO à solução tampão devido à solubilidade do composto em água, perfazendo 4% de DMSO na solução inicial. Síntese dos complexos de rutênio [RuCl(dmso)(dmbpy)2]Cl: 0,50 g (1 x 10-3 mol) de [RuCl2(dmso)4] foram adicionados em 100 mL de etanol contendo 0,40 g (2,2 x 10-3 mol) de 4,4'-dimetil-2,2'-bipiridina (dmbpy). A mistura reacional foi mantida sob agitação e refluxo por 90 minutos. Após esse período, a solução etanólica de coloração amarela foi mantida a temperatura ambiente até o dia seguinte (aproximadamente 14 h), que resultou na formação de um sólido amarelo, que foi filtrado e lavado com etanol gelado (rendimento: 70%). O resultado de análise elementar é coerente com a fórmula RuCl2(C12H12N2)2(CH3)2SO - experimental (calculado): C: 50,42 (50,44); H: 4,95 (4,90); N: 9,10 (9,12)%. Condutividade molar (em água): 135 ohm-1 cm2 mol-1. FTIR: 1618 cm-1 (νC=C); 1595 cm-1 (νC=N); 1100 cm-1 (νS-O); 680 cm-1 (νC-S-C). UV/Vis: λ = 310 nm (ε = 4,4 x 104 L mol-1 cm-1); λ = 390 nm (ε = 5,7 x 103 L mol-1 cm-1); λ = 440 nm (ε = 4,8 x 103 L mol-1 cm-1). O mesmo procedimento foi utilizado para a obtenção do complexo [RuCl(dmso)(dnbpy)2]Cl utilizando, nesse caso, 0,90 g (2,2 x 10-3 mol) do ligante 4,4'-dinonil-2,2'-bipiridina (dnbpy). Rendimento: 50%. Análise elementar {RuCl2(C28H44N2)2(CH3)2SO} - experimental (calculado): C: 65,18 (65,21); H: 8,94 (8,89); N: 5,20 (5,25) %. Condutividade molar (em acetonitrila): 108 ohm-1 cm2 mol-1. FTIR: 1620 cm-1 (νC=C); 1590 cm-1 (νC=N); 1079 cm-1 (νS-O); 685 cm-1 (νC-S-C). UV/Vis: λ = 300 nm (ε = 3,1 x 104 L mol-1 cm-1); λ = 385 nm (ε = 2,5 x 103 L mol-1 cm-1); λ = 430 nm (ε = 1,9 x 103 L mol-1 cm-1). A obtenção dos complexos foi confirmada por Ressonância Magnética Nuclear de 1H, Espectrometria de massas e Espectroscopia vibracional de absorção na região do infravermelho, cujos espectros estão apresentados na Figuras 1S, 2S e 3S. Supressão de fluorescência Os espectros foram obtidos em um espectrofluorímetro da Varian, modelo Cary Eclipse, com compartimento de amostra com banho termostatizado, com largura das fendas de excitação e de emissão igual a 5 nm. As amostras foram excitadas em 280 nm e as leituras de emissão de fluorescência foram realizadas na faixa de 300 a 550 nm. Os espectros de emissão foram obtidos nas temperaturas de 24, 30 e 37 ºC. Para a obtenção dos espectros, utilizou-se 3 mL de solução tampão de HSA 5 x 10-6 mol L-1 e fez-se adições sucessivas de 15 µL da solução estoque do complexo [RuCl(dmso)(dmbpy)2]Cl (e 5 µL do complexo [RuCl(dmso)(dnbpy)2]Cl). Considerando a absorção dos complexos de rutênio na região analisada (Figura 4S), os valores de fluorescência foram corrigidos utilizando a equação:28 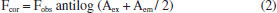 em que Fcor e Fobs são os valores de fluorescência corrigido e observado, respectivamente, e Aex e Aem são os valores de absorbância nos comprimentos de onda 280 e 350 nm, respectivamente. Influência das sondas warfarina e ibuprofeno O mesmo equipamento e condições experimentais descritos anteriormente foram utilizados para avaliar a influência das moléculas de warfarina e ibuprofeno no processo de interação da proteína com complexos de rutênio. Para a obtenção dos espectros, fizeram-se adições sucessivas da solução do complexo de rutênio em 3 mL de solução de HSA (5 x 10-6 mol L-1) contendo as respectivas sondas warfarina ou ibuprofeno.
RESULTADOS E DISCUSSÃO A primeira evidência da coordenação dos ligantes N-heterocíclicos ao centro metálico foi o surgimento de bandas de absorção em 440 nm (ε = 4,8 x 103 mol-1 L cm-1), 390 nm (ε = 5,7 x 103 mol-1 L cm-1) e 310 nm (ε = 4,4 x 104 mol-1 L cm-1) para o complexo contendo o ligante 4,4'-dimetil-2,2'-bipiridina, e bandas em 430 nm (ε = 1,9 x 103 mol-1 L cm-1), 385 nm (ε = 2,5 x 103 mol-1 L cm-1) e 300 nm (ε = 3,1 x 104 mol-1 L cm-1) para o complexo contendo o ligante 4,4'-dinonil-2,2'-bipiridina. As bandas na região de 385 a 440 nm foram atribuídas às transições de transferência de carga do íon rutênio(II) para o ligante N-heterocíclico (MLCT).35 A banda na região de 300 nm foi atribuída à transição interna do ligante N-heterocíclico. A ausência dessas bandas no espectro eletrônico do complexo precursor {[RuCl2(dmnso)4] λ = 308 nm (ε = 218 mol-1 L cm-1), λ = 355 nm (ε = 435 mol-1 L cm-1), ambas atribuídas às transições d-d}36 é uma evidência direta da formação de novos complexos de rutênio. As composições das esferas de coordenação foram determinadas pelas análises dos espectros de ressonância magnética nuclear de hidrogênio (Figura 1S), espectros de massas (Figura 2S) e espectros vibracionais (Figura 3S). No espectro de ressonância do derivado [RuCl(dmso)(dmbpy)2]Cl, observou-se três sinais de ressonância na região de campo baixo do espectro com valores de descolamentos químicos iguais a δ = 9,4 ppm, δ = 8,3 ppm e δ = 7,5 ppm atribuídos, respectivamente, aos hidrogênios H6, H5 e H3 dos anéis piridínicos do ligante dmbpy. Os prótons dos grupos CH3 do ligante N-heterocíclico foram atribuídos ao sinal de ressonância com valor de deslocamento químico igual a 2,9 ppm. Na região de campo alto do espectro de ressonância foram observados dois sinais singleto com valor de deslocamento químico igual a 3,2 e 3,4 ppm atribuídos aos prótons do ligante DMSO coordenado ao centro metálico. Considerando o padrão dos sinais de ressonância e os valores de integração desses sinais, pode-se afirmar que duas moléculas de 4,4'-dimetil-2,2'-bipiridina estão ligadas ao centro metálico em posição trans entre si e perfazem o plano equatorial do complexo. Para o complexo de rutênio contendo 4,4'-dinonil-2,2'-bipiridina, [RuCl(dmso)(dnbpy)2]Cl, observou-se um padrão simples de sinais de ressonância com os seguintes valores de deslocamentos químicos para os hidrogênios aromáticos: H6 = 9,6 ppm, H5 = 8,4 ppm e H3 = 7,3 ppm. Esses valores são similares ao apresentado para o sistema contendo o ligante 4,4'-dimetil-2,2'-bipiridina, porém com as multiplicidades dos hidrogênios diferentes, que podem ser justificadas pela diferença do ambiente magnético gerada pelas conformações dos grupos dinonil [-CH2(CH2)7CH3] sobre os anéis piridínicos, que favorecem acoplamentos espaciais dos hidrogênios, gerando os desdobramentos dos sinais. Considerando o padrão dos sinais de ressonância e os valores de integração desses sinais, pode-se concluir que duas moléculas de 4,4'-dinonil-2,2'-bipiridina estão ligadas ao centro metálico em posição trans entre si, perfazendo o plano equatorial do complexo. Os sinais relativos aos hidrogênios alifáticos do fragmento dinonil foram observados na região de 1,5 a 3 ppm do espectro. A presença da molécula de dimetilsulfóxido na esfera de coordenação do íon metálico pode levar à formação de isômeros de ligação, pois o DMSO pode se coordenar ao centro metálico via átomo de enxofre (S-dmso) ou via átomo de oxigênio (O-dmso).37-39 Esses isômeros podem ser distinguidos por espectroscopia vibracional na região do infravermelho.40 De acordo com a literatura,40 o estiramento da ligação S=O (νSO) da molécula DMSO ligada ao metal através do átomo de enxofre é observada na região de 1080 a 1150 cm-1, e para o DMSO coordenado ao metal via átomo de oxigênio, o νSO = 890 a 950 cm-1. Nos espectros vibracionais dos complexos, observou-se uma banda de intensidade média em 1100 cm-1 para o derivado [RuCl(dmso)(dmbpy)2]Cl, e em 1079 cm-1 para o derivado [RuCl(dmso)(dbpy)2]Cl, atribuída ao estiramento assimétrico da ligação S=O do ligante dimetilsulfóxido coordenado ao centro metálico via átomo de enxofre. As principais frequências observadas nos espectros vibracionais dos complexos com as atribuições prováveis estão apresentadas na Tabela 1S. O modo de coordenação do ligante ambidentado DMSO está relacionado com efeitos eletrônicos e estéricos.40 O íon Ru2+, por apresentar carga iônica pequena e raio iônico grande, comporta-se como uma espécie mole (ou eletronicamente polarizável, segundo conceito de Pearson) e, portanto, deve preferir coordenar-se ao átomo de enxofre da molécula de DMSO, que apresenta densidade eletrônica maior e mais polarizável (base mole de Pearson), em vez de ligar-se ao átomo de oxigênio (um átomo com caráter mais duro).41 A presença de ligantes N-heterocíclicos de caráter π-receptor em posição trans entre si na esfera de coordenação induz uma competição pelos elétrons π do íon metálico, o que favorece a manutenção de um ligante com características σ-doadora e π-doadora, como o íon cloreto. Nesse caso, o íon cloreto, um ligante de campo fraco e de característica π-doadora, induz a coordenação do dimetilsulfóxido ao centro metálico pelo átomo de enxofre (ligante de campo forte e de característica π-receptora), o que leva ao aparecimento de um efeito cooperativo nas ligações axiais do complexo do tipo Cl-Ru-S(dmso).37,39 A composição da esfera de coordenação dos complexos foi corroborada pela análise dos espectros de massas dos compostos, pois permitiu obter a razão massa-carga (m/z) dos íons complexos e, com a expansão dos picos de interesse, foi possível observar o padrão da distribuição isotópica do centro metálico. No espectro de massas do complexo [RuCl(dmso)(dmbpy)2]Cl foi observado o pico em 583,0699 atribuído ao íon complexo sem o contra-íon cloreto, [RuCl(dmso)(dmbpy)2]+ m/zcalculado = 583,133. Para o complexo [RuCl(dmso)(dnbpy)2]Cl foi observado o pico em 1031,5847 atribuído ao íon complexo sem o contra-íon cloreto, [RuCl(dmso)(dnbpy)2]+ m/zcalculado =1031,973. Para estabelecer a natureza iônica dos complexos e, consequentemente, definir o tipo de eletrólito formado na reação do precursor [RuCl2(dmso)4] com o ligante dmbpy (ou dnbpy), medidas de condutividade foram realizadas. Os valores de condutividade molar para soluções dos complexos (1,2 x 10-3 mol L-1) indicaram a estequiometria 1:1 de eletrólitos, o que é coerente com a dissociação dos complexos nas espécies iônicas [RuCl(dmso)L2]+ e Cl- em solução (Λm = 135 ohm-1 cm2 mol-1 para [RuCl(dmso)(dmbpy)2]Cl em água; Λm = 108 ohm-1 cm2 mol-1 para [RuCl(dmso)(dnbpy)2]Cl em acetonitrila). Supressão de fluorescência Do conjunto de aminoácidos que compõem a estrutura primária da albumina, os aminoácidos aromáticos triptofano, tirosina e fenilalanina são os responsáveis pela emissão de fluorescência da cadeia peptídica, quando a biomolécula é excitada na região de 280 nm. Fenilalanina e tirosina apresentam rendimentos quânticos pequenos quando excitados nessa região, devido ao processo de ionização e efeitos de polarização induzida no microambiente ao redor desses aminoácidos.16,42 Dessa forma, dos fluoróforos naturais da proteína, a emissão do triptofano, que ocorre em 350 nm, é predominante.16,43 A adição dos complexos de rutênio diminuiu a intensidade dessa banda, como mostrado na Figura 2.
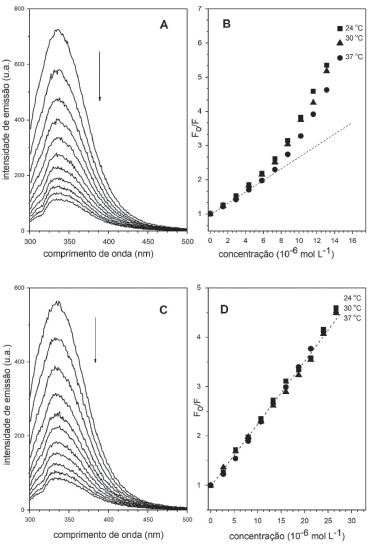 Figura 2. (A) Espectros de supressão de fluorescência da HSA na presença do complexo de [RuCl(dmso)(dmbpy)2]Cl, concentração = 0 a 14,6 x 10-6 mol L-1. (B) Gráfico de Stern-Volmer sistema HSA-[RuCl(dmso)(dmbpy)2] Cl. (C) Espectros de supressão de fluorescência da HSA na presença do complexo de [RuCl(dmso)(dnbpy)2]Cl, concentração = 0 a 26,7 x 10-6 mol L-1. (D) Gráfico de Stern-Volmer sistema HSA-[RuCl(dmso)(dnbpy)2]Cl. Solução tampão pH = 7,4; λexc. = 280 nm; temperatura 37 °C; CHSA = 5 x 10-6 mol L-1
A supressão da fluorescência intrínseca da biomolécula indica o contato entre o fluoróforo e o agente supressor. Assim, é possível inferir a partir dessa supressão que o complexo de rutênio atinge a microrregião em que está alojado o resíduo de triptofano que, nesse caso, está contida no sítio I de Sudlow, na interface dos subdomínios IIA e IIB da cadeia proteica.42 A supressão de fluorescência observada pode ter sido induzida por diferentes processos como reações no estado excitado, rearranjos moleculares, transferência de energia, formação de complexo no estado fundamental e colisões moleculares.28 Para compostos de rutênio,44 a supressão de fluorescência da albumina é consequência da interação do centro metálico com o resíduo de triptofano via histidina e envolvendo o processo de transferência de energia. A característica dessa região é a presença de seis α-hélices conectadas por uma estrutura do tipo laço formada por resíduos de aminoácidos que ocupam as posições 148 a 154 da estrutura polipeptídica. A conformação é mantida através de forças hidrofóbicas envolvendo cadeias laterais de aminoácidos neutros apolares (Leu-219, Phe-223, Leu-238, Leu-260, Ile-264, Ile-290 e Ala-291) e de interações eletrostáticas entre fragmentos laterais alcalinos dos aminoácidos His-242, Arg-257, Lys-195, Lys-199, Arg-218 e Arg-222 com moléculas do solvente. É nesse ambiente, onde forças hidrofóbica e eletrostática coexistem e são corresponsáveis pela conformação local da estrutura proteica, que está localizado o principal grupo emissor de fluorescência da HSA (Trp-214), o qual é utilizado como sonda no monitoramento da interação de espécies exógenas com a proteína.16 Mecanismo de supressão de fluorescência Na maioria dos processos, a supressão de fluorescência pode ser interpretada a partir de dois mecanismos relativamente simples, denominados de mecanismo dinâmico e mecanismo estático.28 Esses mecanismos podem ser entendidos em termos das colisões entre as moléculas de albumina e espécies exógenas que levam à supressão de fluorescência da proteína, e podem ser distinguidos pela dependência que os valores das constantes de supressão têm com a temperatura e viscosidade.28 O mecanismo dinâmico está baseado na colisão do fluoróforo (HSA, Trp-214) no estado excitado com a espécie supressora (complexo de rutênio). Nesse caso, o aumento da temperatura faz aumentar as energias cinéticas das moléculas e faz diminuir a viscosidade do meio reacional e, por consequência, as moléculas do supressor movem-se com mais velocidade para o interior proteico aumentando a frequência de colisões e ocasionando o aumento do valor da constante de supressão (Ksv).28 No mecanismo estático, a etapa determinante é estabelecida pela formação de complexos não-fluorescentes entre as espécies no estado fundamental. Nesse caso, a elevação de temperatura faz diminuir a estabilidade do complexo não-fluorescente (HSA-complexo metálico), visto que as moléculas se movem com mais velocidade, colidem com mais frequência e mais energia. À medida que a temperatura aumenta, os valores de constantes de supressão diminuem.28 O método utilizado para atribuir o mecanismo de supressão baseado no modelo colisional foi elaborado por Stern-Volmer,26,28 cuja abordagem matemática está apresentada na equação abaixo: 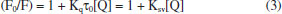 em que F0 é a intensidade de fluorescência da proteína pura, F é a intensidade de fluorescência da proteína na presença do supressor, Ksv é a constante de supressão de Stern-Volmer, a qual é o resultado da multiplicação da constante de velocidade bimolecular de supressão (Kq) e o tempo de vida médio da proteína na ausência do complexo de rutênio (τ0 ~10-8 s), e [Q] é a concentração do supressor (no caso, complexos de rutênio). Essa equação tem a forma da equação geral para uma reta, onde Ksv é a inclinação e 1 é a intercessão de y (F0/F) com a reta. Assim, para um mecanismo de supressão colisional dinâmico ou estático, um gráfico da razão da intensidade de fluorescência da proteína (F0/F) versus a concentração do complexo de rutênio [Q] fornece uma reta, cuja inclinação é a constante de supressão (Ksv). Um mecanismo que não é dinâmico ou estático não produzirá uma linha reta. A partir da equação 3, obteve-se os gráficos de (F0/F) em função da concentração dos complexos de rutênio, como representado na Figura 2. Nesse caso, observamos um perfil não-linear para o sistema HSA-[RuCl(dmso)(dmbpy)2]Cl, e um perfil linear para o sistema contendo o complexo [RuCl(dmso)(dnbpy)2]Cl. O desvio positivo da linearidade observado para o sistema HSA-[RuCl(dmso)(dmbpy)2]Cl (Figura 2B) indica que o mecanismo de supressão de fluorescência da albumina induzida pelo derivado metálico não é exclusivamente dinâmico ou estático, mas sim um mecanismo híbrido ou intermediário (dinâmico + estático),45 no qual deve-se considerar a esfera de ação do complexo metálico. Nesse caso, a equação de Stern-Volmer é modificada e a constante de supressão é dada pela combinação das constantes dos dois mecanismos, conforme equação abaixo:28 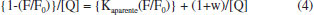 em que Kaparente é a constante de supressão resultante da combinação da parte estática (Ks) e dinâmica (Kd) do mecanismo híbrido, e (1-w) é a fração da proteína que está sob a influência da esfera de ação do supressor. Assim, para um mecanismo de supressão híbrido, um gráfico de {1-(F/F0)}/[Q] versus a razão de fluorescência (F/F0) fornece uma reta, cuja inclinação é a constante de supressão aparente (Kaparente). A fração da proteína que está sob a influência da esfera de ação do supressor é obtida a partir do coeficiente linear da reta {(1+w)/[Q]} e é dependente da concentração do supressor. Usando a equação 4 nos dados obtidos para o sistema HSA-[RuCl(dmso)(dmbpy)2]Cl, tem-se os gráficos mostrados na Figura 3, que apresentaram perfis lineares e, a partir do tratamento matemático adequado, obteve-se os valores de constante de supressão (Kaparente). Nesse caso, verifica-se que à medida que a temperatura aumenta, os valores de constantes de supressão tendem a diminuir {1,31 ± 0,02 x 104 mol-1 L (24 oC); 1,14 ± 0,03 x 104 mol-1 L (30 oC); 0,95 ± 0,04 x 104 mol-1 L (37 oC)}, o que permite inferir que a etapa predominante do processo de supressão de fluorescência da HSA pelo complexo [RuCl(dmso)(dmbpy)2]Cl é a formação de aduto não fluorescente entre as espécies no estado fundamental. Os valores para a esfera de ação do complexo metálico no ambiente proteico variaram de 0,4 a 0,9 (ou 40 a 90%) dependendo da concentração do supressor.
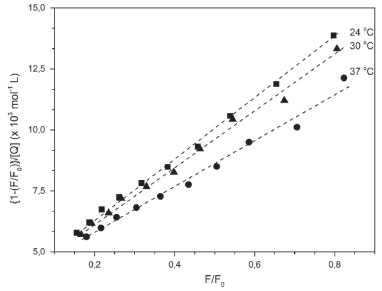 Figura 3. Gráfico de {1-(F/F0)}/[Q] versus a razão de fluorescência da HSA na presença do complexo de [RuCl(dmso)(dmbpy)2]Cl
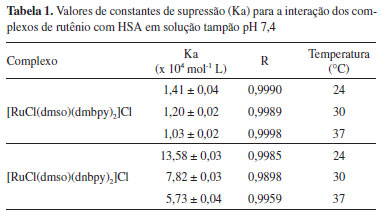
Para o sistema HSA-[RuCl(dmso)(dnbpy)2]Cl, como mencionado anteriormente, tem-se os gráficos com perfis lineares (Figura 2D) e, a partir do tratamento matemático adequado, obteve-se os valores de constante de supressão {Ksv = 13,6 ± 0,2 x 104 mol-1 L (24 oC); 13,2 ± 0,3 x 104 mol-1 L (30 oC); 12,5 ± 0,3 x 104 mol-1 L (37 oC)}. Nesse caso, verifica-se também que à medida que a temperatura aumenta, os valores de constante de supressão tendem a diminuir, o que é coerente com a formação de um aduto proteína - complexo de rutênio. Em todos os casos, a constante de supressão de fluorescência da albumina tende a diminuir com o aumento da temperatura, porém, a presença de grupos periféricos com maior caráter hidrofóbico aumenta a magnitude dessa constante, refletindo a influência das forças de van der Waals na formação desses adutos não fluorescentes no estado fundamental.46 Como nos dois sistemas o mecanismo que justifica o processo de supressão é baseado na ligação dos complexos de rutênio ao microambiente proteico onde está localizado o principal grupo emissor de fluorescência da HSA (Trp-214), gerando espécies não fluorescentes no estado fundamental, é possível inferir a acessibilidade dos complexos ao arcabouço proteico e, por consequência, determinar a constante de supressão da fração acessível. A equação de Stern-Volmer,28 que descreve a acessibilidade de supressores ao fluoróforo, leva em conta a concentração do agente supressor e a razão entre as intensidades de fluorescência da proteína pura e na presença do supressor, expressas pela equação: 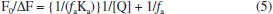 em que ΔF = F0-F, ƒa é a fração acessível do supressor à proteína, e Ka é a constante de supressão da fração acessível. Os gráficos de Stern-Volmer, parametrizados em termos de F0/ΔF e 1/[Q], apresentaram perfis lineares para ambos os complexos (Figura 4). Com base nas equações das retas, calculamos os valores de constante de supressão das frações acessíveis dos complexos de rutênio à proteína (Tabela 1). Observando os dados de supressão da interação dos complexos com a HSA, nota-se a diminuição dos valores de Ka em função do aumento da temperatura, característico do mecanismo de supressão estático. A interação do complexo de rutênio com a HSA resultou em um valor médio do parâmetro ƒa de 0,9, definindo que aproximadamente 90% da fluorescência inicial da albumina está acessível ao derivado metálico.
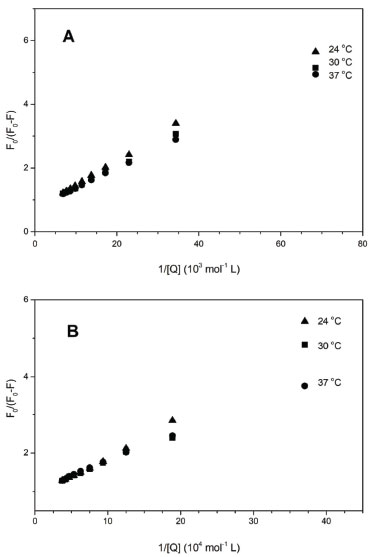 Figura 4. Gráficos de Stern-Volmer parametrizados em termos de F0/ΔF e 1/ [Q] para os sistemas HSA-[RuCl(dmso)(dmbpy)2]Cl (A) e HSA-[RuCl(dmso) (dnbpy)2]Cl (B)
Determinação da constante de ligação (Kb) e número de sítios de ligação (n) A interação dos complexos de rutênio com a HSA pode ser escrita genericamente da seguinte forma:  Nela, destaca-se a presença do íon complexo e da proteína em equilíbrio dinâmico com o aduto proteína-complexo formado em solução. A formação desse aduto apresenta uma constante de ligação (Kb), que é influenciada pela temperatura, pela solubilidade das espécies em água ou lipídeos e pela natureza das interações químicas (atração eletrostática, ligação covalente ou forças de van der Waals) do complexo com a estrutura proteica, e pode ser utilizada para avaliar a afinidade do composto pela proteína. Valor elevado de constante de ligação (Kb > 105) indica que a espécie exógena liga fortemente à proteína e, por consequência, diminui sua ação farmacológica, pois há menor quantidade da espécie exógena livre para atravessar a membrana celular e interagir com os receptores biológicos.47 O valor da constante de ligação pode ser calculado usando a equação modificada de Stern-Volmer apresentada na parte introdutória desse artigo (eq. 1), que utiliza a variação de fluorescência do sistema em função da concentração da espécie supressora, em termos de cologaritmo. Os gráficos parametrizados em termos de log{(F0-F)/F} e log[Q] estão apresentados na Figura 5 e os valores das constantes de ligação são mostrados na Tabela 2.
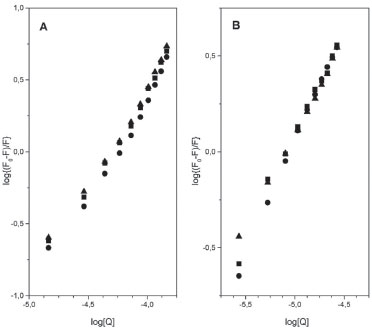 Figura 5. Gráficos parametrizados em termos de log{(F0-F)/F} e log[Q] para os sistemas HSA-[RuCl(dmso)(dmbpy)2]Cl (A) e HSA-[RuCl(dmso) (dnbpy)2]Cl (B)
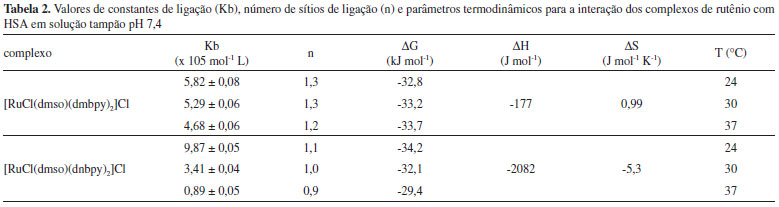
Nesse caso, a constante de ligação dos complexos à HSA é da ordem de 105 mol L-1, indicando que a formação do aduto complexo-HSA é favorável e que os derivados metálicos apresentam elevada afinidade pelos sítios de ligação da proteína. Entre vários sítios de ligação existentes na estrutura proteica, a interação dos complexos de rutênio com a HSA resultou na ocupação de apenas um sítio de ligação, como mostrado na Tabela 2. Essa informação é importante, pois em vários tratamentos de doenças como hipertensão, insuficiência cardíaca e na quimioterapia, é necessária a administração de dois ou mais medicamentos ao mesmo tempo. Nessas situações, é necessário saber o número de microambientes proteicos disponíveis para as interações, visto que a HSA atua como um reservatório temporário de multidrogas no plasma sanguíneo retardando a chegada dos princípios ativos aos receptores. A estabilidade dos adutos formados também deve ser analisada sob o ponto de vista termodinâmico, uma vez que a estabilidade desses sistemas está diretamente relacionada com a interação química dos complexos metálicos com a molécula de HSA. Parâmetros termodinâmicos Na concepção de Ross e Subramanian,48 a partir da constante de ligação é possível determinar os parâmetros termodinâmicos da interação dos complexos metálicos com a HSA e correlacioná-los com os diferentes tipos de forças intermoleculares envolvidos na inclusão do composto na cavidade proteica. Para a determinação dos valores da variação da entalpia (ΔH) e da entropia (ΔS) foi aplicada a Equação de Van't Hoff (eq. 6):28 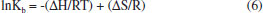 em que ΔH = variação de entalpia, ΔS = variação de entropia, R = 8,314 J mol-1 K-1 e T é a temperatura em Kelvin. Para a variação da energia livre de Gibbs (ΔG) foi utilizada a relação ΔG = - RT lnKb, onde Kb é a constante de ligação, R = 8,314 J mol-1 K-1 e T é a temperatura em Kelvin. Os parâmetros termodinâmicos obtidos a partir do tratamento matemático adequados dos gráficos mostrados na Figura 6 estão na Tabela 2.
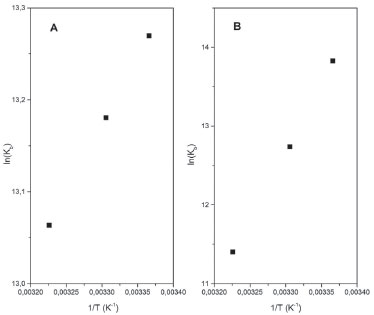 Figura 6. Gráficos de van't Hoff para determinação de parâmetros termodinâmicos dos sistemas HSA/[RuCl(dmso)(dmbpy)2]Cl (A) e HSA/[RuCl(dmso) (dnbpy)2]Cl (B)
A interação do complexo [RuCl(dmso)(dmbpy)2]Cl com a HSA é espontânea, conforme mostram os valores de ΔG < 0, e controlada essencialmente pela entalpia (ΔH < 0) com pouca contribuição do parâmetro entrópico. Considerando as correlações observadas por Ross e Subramanian,48 é possível inferir que a inserção e manutenção do complexo de rutênio na estrutura proteica é determinada por interações eletrostáticas do tipo íon-dipolo. O complexo induz uma alteração estrutural da proteína e expõe fragmentos polares dos aminoácidos, em especial, dos resíduos de lisina (Lys-195, Lys-199) e arginina (Arg-218, Arg-222, Arg-257), presentes na interface dos sítios de Sudlow. Essa recomposição da estrutura do ambiente proteico em torno do complexo metálicos maximiza as interações íon-dipolo no aduto, visto que em pH 7,4 os resíduos estão carregados (pIlisina = 9,74; pIarginina = 10,8)49 justificando, assim, o predomínio das interações eletrostáticas nesse sistema. A presença do ligante torna a contribuição entálpica maior, decorrente de um caráter covalente maior da ligação entre o complexo [RuCl(dmso)(dnbpy)2]Cl e as sequências de aminoácidos presentes na estrutura proteica. Assim, considerando as correlações observadas na literatura,48 é possível inferir que a inserção do complexo na HSA é dirigida por forças de van der Waals. Influência das sondas warfarina e ibuprofeno na interação dos complexos de rutênio com HSA A molécula de albumina tem dois sítios de ligação para a maioria dos compostos exógenos classificados como sítios I e II de Sudlow. O sítio I é maior, mais flexível e menos estereosseletivo e sua conformação é mantida predominantemente por forças hidrofóbicas.16 O sítio II é menor, mais seletivo e sua estrutura é mantida pela combinação de forças hidrofóbicas, ligações de hidrogênio e atração eletrostática.16 Os dados termodinâmicos mostraram que os complexos de rutênio têm afinidades diferentes pela estrutura da HSA: o complexo [RuCl(dmso)(dmbpy)2]Cl encontra-se em um ambiente proteico mais polarizado, ao passo que o [RuCl(dmso)(dnbpy)2]Cl prefere uma região mais hidrofóbica. Para complementar a caracterização desses sistemas, monitoramos o comportamento dos complexos em soluções contendo HSA e moléculas marcadores (ou sondas) de sítios proteicos. Nesse caso, utilizamos o composto warfarina como molécula marcadora do sítio I de Sudlow, especificamente no subdomínio IIA, e o composto ibuprofeno como molécula marcadora do subdomínio IIIA do sítio II de Sudlow.50 A interação dos complexos de rutênio com a HSA contendo warfarina e ibuprofeno pode ser escrita genericamente de duas maneiras,51 em que Y representa as sondas: 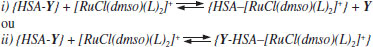 Na primeira representação (i), o complexo de rutênio compete com a sonda pelo sítio de ligação da proteína. Essa competição implica na ruptura da ligação sonda-HSA e, consequentemente, na diminuição do valor da constante de ligação (Kb) do complexo pelo sítio de ligação da albumina, pois altera a porcentagem de complexo no ambiente proteico. Na segunda representação (ii) não há competição entre complexo e sonda pelo sítio de ligação. Nesse caso, o complexo de rutênio desloca a sonda para outro sítio de ligação, configurando um mecanismo associativo complexo-HSA-sonda e isso leva a um aumento da constante de ligação do sistema. A Figura 7 ilustra a variação de fluorescência da HSA contendo warfarina e ibuprofeno na presença do complexo [RuCl(dmso)(dmbpy)2]Cl. A presença de ibuprofeno aumenta a intensidade de fluorescência da albumina, ao passo que a presença de warfarina diminui a intensidade de fluorescência e desloca o máximo de emissão da HSA (λemissão ~350 nm para HSA pura; λemissão ~390 nm para HSA+warfarina). A adição de complexo de rutênio diminui a intensidade da banda, em ambos os sistemas.
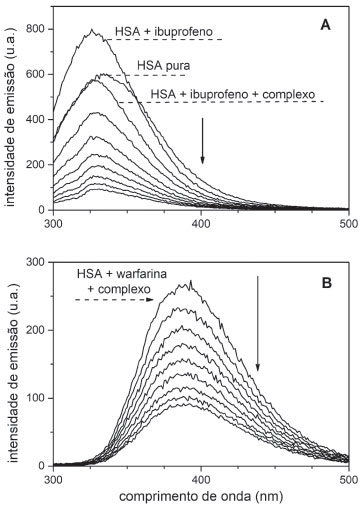 Figura 7. Espectros de supressão de fluorescência da HSA contendo Ibuprofeno (A), e Warfarina (B), na presença do complexo [RuCl(dmso)(dmbpy)2] Cl, concentração = 0 a 14,6 x 10-6 mol L-1. Solução tampão pH = 7,4; λexc. = 280 nm; CHSA = 5 x 10-6 mol L-1. Cibuprofeno = Cwarfarina = 5 x 10-6 mol L-1
Os valores de constante de ligação obtidos a partir da equação modificada de Stern-Volmer apresentada na parte introdutória desse artigo (equação 1) estão apresentados na Tabela 3.

Observando os valores de constante de ligação da interação dos complexos de rutênio com a HSA contendo os marcadores de sítios proteicos, nota-se a variação significativa desse parâmetro na presença de warfarina e pouca influência do ibuprofeno e, portanto, evidencia que os complexos de rutênio estão conectados à HSA no sítio I de Sudlow, especificamente no subdomínio IIA. O fato da constante de ligação do complexo [RuCl(dmso)(dmbpy)2]Cl diminuir na presença de warfarina indica que o complexo compete com a sonda pelo sítio de ligação. No caso do complexo [RuCl(dmso)(dnbpy)2]Cl, o elevado valor da constante de ligação indica um processo associativo, no qual o derivado metálico liga-se fortemente à albumina e desloca a sonda para outro microambiente proteico.
CONCLUSÃO O processo de inserção dos compostos no arcabouço proteico foi monitorado pela supressão de fluorescência da albumina do soro humano. Variando a metodologia, observou-se que os parâmetros associados ao processo de supressão de fluorescência da albumina pelos complexos metálicos seguiram o mesmo padrão, mostrando a predominância do mecanismo estático, característico da ligação do supressor à proteína no estado fundamental. Das evidências da influência das sondas warfarina e ibuprofeno na interação dos complexos de rutênio com a HSA, somadas aos resultados termodinâmicos, pode-se concluir que os complexos de rutênio estão alojados no subdomínio IIA do sítio I de Sudlow e, devido às características dessa região, o complexo [RuCl(dmso)(dnbpy)2]Cl é mantido nesse microambiente através de forças de van der Waals envolvendo cadeias laterais de aminoácidos neutros apolares, ao passo que o complexo [RuCl(dmso)(dmbpy)2]Cl é mantido por interações eletrostáticas envolvendo fragmentos laterais alcalinos dos aminoácidos da estrutura proteica.
MATERIAL SUPLEMENTAR Estão disponíveis em http://quimicanova.sbq.org.br, com acesso livre, espectros de ressonância magnética nuclear de hidrogênio (Figura 1S), espectros de massas (Figura 2S), espectros vibracionais (Figura 3S e Tabela 1S) e espectros eletrônicos (Figura 4S) dos complexos de rutênio sintetizados nesse trabalho.
AGRADECIMENTOS Os autores agradecem à Fundação Araucária/Paraná pelo suporte financeiro na execução do projeto do grupo e ao Programa de Educação Tutorial - PET pelas bolsas concedidas aos alunos.
REFERÊNCIAS 1. http://www.portal.anvisa.gov.br, acessada em Fevereiro 2020. 2. Rosenbaum, S. E.; Basic Pharmacokinetics and Pharmacodynamics: An Integrated Textbook and Computer Simulations, 2nd ed., Wiley: New York, 2016. 3. Li, X.; Jasti, B. R.; Design of Controlled Release Drug Delivery Systems, McGraw-Hill: New York, 2006. 4. Tekade, R.; Drug Delivery Systems, Academic Press: Cambridge, 2019. 5. Jain, A.; Jain, A.; Gulbake, A.; Shilpi, S.; Hurkat, P.; Jain, S. K.; Crit. Rev. Ther. Drug Carrier Syst. 2013, 30, 293. 6. Cohen, D.; Human Serum Albumin: Structure, Binding and Activity, Nova Science Pub Inc.: New York, 2019. 7. Sabbioni, G.; Turesky, R. J.; Chem. Res. Toxicol. 2017, 30, 332. 8. Chackalamannil, S.; Rotella, D.; Ward, S.; Comprehensive Medicinal Chemistry III, 3rd ed., Elsevier: Cambridge, 2017. 9. Hatley, R. J. D.; Macdonald, S. J. F.; Slack, R. J.; Le, J.; Ludbrook, S. B.; Lukey, P. T.; Angew. Chem. 2018, 57, 3298. 10. Levina, A.; Crans, D. C.; Lay, P. A.; Coord. Chem. Rev. 2017, 352, 473. 11. More, J.; Bulmer, M.; Production of Plasma Proteins for Therapeutic Use, Robinson, A. S. ed.; Wiley: New York, 2013, cap. 12. 12. https://www.sigmaaldrich.com, acessada em Fevereiro 2020. 13. Resolução n° 115 - Diretrizes para o uso da albumina, disponível em http://bvsms.saude.gov.br/bvs/saudelegis/anvisa/2004/rdc0115_10_05_2004.html, acessada em Fevereiro 2020. 14. Naveenraj, S.; Anandan, S.; J. Photochem. Photobiol., C 2013, 14, 53. 15. http://www.rcsb.org/pdb/home/home.do, acessada em Fevereiro 2020. 16. Peters Jr., T.; All About Albumin; Biochemistry, Genetics and Medical Applications, 1st ed., Academic Press: San Diego, 1995. 17. Maciazek-Jurczyk, M.; Szkudlarek, A.; Chudzik, M.; Pozycka, J.; Sulkowska, A.; Spectrochim. Acta, A 2018, 188, 675. 18. Chandel, T. I.; Zaman, M.; Khan, M. V.; Ali, M.; Rabbani, G.; Ishtikhar, M.; Khan, R. H.; Int. J. Biol. Macromol. 2018, 106, 1115. 19. Zhang, Y.; Sun, T.; Jiang, C.; Acta Pharm. Sin. B 2018, 8, 34. 20. Sudlow, G.; Birkett, D. J.; Wade, D. W.; Molec. Pharmacol. 1976, 12, 1052. 21. Kragh-Hansen, U.; Pharmacol. Rev. 1981, 33, 17; Peters, T. J.; Adv. Protein Chem. 1985, 37, 161. 22. Zsila, F.; Mol. Pharm. 2013, 10, 1668; Qi, J.; Zhang, Y.; Gou, Y.; Lee, P.; Wang, J.; Chen, S.; Zhou, Z.; Wu, X.; Yang, F.; Liang, H.; Mol. Pharm. 2016, 13, 3098; Bruckmueller, H.; Martin, P.; Kählerm M.; Haenisch, S.; Ostrowski, M.; Drozdzik, M.; Siegmund, W.; Cascorbi, I.; Oswald, S.; Mol. Pharm. 2017, 14, 2245; Fan, W.; Yung, B.; Huang, P.; Chen, X.; Chem. Rev. 2017, 117, 13556; Gao, M.; Tang, B. Z.; ACS Sens. 2017, 2, 1382; Srinivasarao, M.; Low, P. S.; Chem. Rev. 2017, 117, 12133; Browning, R. J.; Reardon, P. J. T.; Parhizkar, M.; Pedley, R. B.; Edirisinghe, M.; Knowles, J. C.; Stride, E.; ACS Nano, 2017, 11, 8560. 23. Ghuman, J.; Zunszain, P. A.; Petitpas, I.; Bhattacharya, A. A.; Otagiri, M.; Curry, S.; J. Mol. Biol. 2005, 353, 38; Zsila, F.; Mol. Pharm. 2013, 10, 1668. Yang, F.; Zhang, Y.; Liang, H.; Int. J. Mol. Sci. 2014, 15, 3580; Tailor, A.; Waddington, J. C.; Meng, X.; Park, B. K.; Chem. Res. Toxicol. 2016, 29, 1912; Sawada, T.; Serizawa, T.; Advances in Bioinspired and Biomedical Materials, ACS Symposium Series, 2017, vol. 1252, cap. 3. 24. Storr, T.; Ligand Design in Medicinal Inorganic Chemistry, 1st ed., Wiley: New York, 2014. 25. Dasgupta, A. Em Clinical Challenges in Therapeutic Drug Monitoring; Clarke, W., Dasgupta, A., eds.; Elsevier: Cambridge, 2016. 26. Eftink, M. R.; Ross, J. B. A.; Luck, L. A.; Rousslang, K. W.; Subramaniam, V.; Stell D. G.; Gafni, A. Em Topics in Fluorescence Spectroscopy: Protein Fluorescence; Lakowicz, J. R., ed.; Kluwer Academic Publishers: New York, 2000. 27. Stern, O.; Volmer, M.; Physik. Z. 1919, 20, 183. 28. Lakowicz, J. R.; Principles of Fluorescence Spectroscopy, 3a ed., Springer Science, 2006. 29. Kerns, E. H.; Di, L.; Drug-like properties: concepts, structure, design and methods; Elsevier: Cambridge, 2008. Timerbaev, A. R.; Hartinger, C. G.; Aleksenko, S. S.; Keppler, B. K.; Chem. Rev. 2006, 106, 2224. Medici, S.; Peana, M.; Nurchi, V. M.; Zoroddu, M. A.; J. Med. Chem. 2019, 62, 5923. Alexander, J. L.; Thompson, Z.; Cowan, J. A.; ACS Chem. Biol. 2018, 13, 844. Deshpande, A. R.; Pochapsky, T. C.; Ringe, D.; Chem. Rev. 2017, 117, 10474. 30. Blaney, J.; Davis, A. M. Em Structure based design for medicinal chemists, in The Handbook of Medicinal Chemistry: Principles and Practice; Davis, A., Ward, S. E., eds.; RSC: London, 2014. 31. Meier-Menches, S. M.; Gerner, C.; Berger, W.; Hartinger, C. G.; Keppler, B. K.; Chem. Soc. Rev. 2018, 47, 909; Kenny, R. G.; Marmion, C. J.; Chem. Rev. 2019, 119, 1058; Wang, X.; Wang, X.; Jin, S.; Muhammad, N.; Guo, Z.; Chem. Rev. 2019, 119, 1138; Markwalter, C. F.; Kantor, A. G.; Moore, C. P.; Richardson, K. A.; Wright, D. W.; Chem. Rev. 2019, 119, 1456. 32. Selassie, C. D.; Li, R.-L.; Poe, M.; Hansch, C.; J. Med. Chem. 1991, 34, 46; Seelig, J.; Biochim. Biophys. Acta 2004, 1666, 40; Bertrand, B.; Fernandez-Cestau, J.; Ângulo, J.; Cominetti, M. M. D.; Waller, Z; A. E.; Searcey, M.; O'Connell, A. A.; Bochmann, M.; Inorg. Chem. 2017, 56, 5728; Elwell, C. E.; Gagnon, N. L.; Neisen, B. D.; Dhar, D.; Spaeth, A. D.; Yee, G. M.; Tolman, W. B.; Chem. Rev. 2017, 117, 2059; Chen, L.-J.; Yang, H.-B.; Acc. Chem. Res. 2018, 51, 2699. 33. Alessio, E.; Mestroni, G.; Nardin, G.; Attia, W. M.; Calligaris, M.; Sava, G.; Zorzet, S.; Inorg. Chem. 1988, 27, 4099. 34. Morita, T.; Assumpção, R. M. V.; Manual de Soluções, Reagentes e Solventes, 2ª ed., Blucher Editora: São Paulo, 2007. 35. Toyama, M.; Inoue, K.-I.; Iwamatsu, S.; Nagao, N.; Bull. Chem. Soc. Jpn. 2006, 79, 1525. 36. Silva, D. O.; Toma, H. E.; Quim. Nova 1993, 16, 39. 37. Bratsos, I.; Alessio, E.; Eur. J. Inorg. Chem. 2018, 2996. 38. Toyama, M.; Iwamatsu, S.; Inoue, K.-I.; Nagao, N.; Bull. Chem. Soc. Jpn. 2010, 83, 1518. 39. Coe, B. J.; Meyer, T. J.; White, P. S.; Inorg. Chem. 1993, 32, 4012. 40. Calligaris, M.; Coord. Chem. Rev. 2004, 248, 351. 41. Huheey, J. E.; Keiter, E. A.; Keiter, R. L.; Inorganic chemistry: principles of structure and reactivity, 4th Ed., HarperCollins College Publishers; New York, 1993. 42. Cuya Guizado, T. R.; J. Mol. Model. 2014, 20, 2450; Al-Harthi, S.; Lachowicz, J. I.; Nowakowski, M. E.; Jaremko, M.; Jaremko, L.; J. Inorg. Biochem. 2019, 198, 110716. 43. Amiri, M.; Jankeje, K.; Albani, J. R.; J. Fluoresc. 2010, 20, 651. 44. Trynda-Lemiesz, L.; Karaezyn, A.; Keppler, B. K.; Koztowski, H.; J. Inorg. Biochem. 2000, 78, 341. 45. Kohlmann, T.; Naumann, R.; Kerzig, C.; Goez, M.; Phys. Chem. Chem. Phys. 2017, 19, 8735; Kohlmann, T.; Goez, M.; Phys. Chem. Chem. Phys. 2019, 21, 10075. 46. Balakrishnan, G.; Rajendran, T.; Murugan, K. S.; Ganesan, M.; Sivasubramanian, V. K.; Rajagopal, S.; J. Lumin. 2018, 205, 51; Zimmermann, L.; Kempf, J.; Briée, F.; Swain, J.; Mingeot-Leclercq, M.-P.; Décout, J.-L.; Eur. J. Med. Chem. 2018, 157, 1512; Bhand, S.; Lande, D. N.; Pereira, E.; Gejji, S. P.; Weyhermüller, T.; Chakravarty, D.; Puranik, V. G.; Salunke-Gawali, S.; Polyhedron 2019, 164, 96. 47. Dasgupta, A. In Clinical Challenges in Therapeutic Drug Monitoring, 2016, ch. 4; Rabbani, G.; Lee, E. J.; Ahmad, K.; Baig, M. H.; Choi, I.; Mol. Pharm. 2018, 15, 1445. 48. Ross, P. D.; Subramanian, S.; Biochemistry 1981, 20, 3096. 49. Kessel, A.; Ben-Tal, N.; Introduction to Proteins: structure, function, and motion, 2th ed., Chapman and Hall/CRC: London, 2018. 50. Chaves, O. A.; Echerria, A.; Esteves-Souza, A.; Maciel, M. A. M.; Netto-Ferreira, J. C.; J. Braz. Chem. Soc. 2018, 29, 1786; Chaves, O. A.; Tavares, M. T.; Cunha, M. R.; Parise-Filho, R.; Sant'Anna, C. M. R.; Netto-Ferreira, J. C.; Biomolecules 2018, 8, 78. 51. Guo, Q.; Liu, M.; Zhao, Y.; Wu, Y.; Liu, J.; Cai, C.; Shi, Y.; Han, J.; Spectrochim. Acta, A 2019, 222, 117213. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access