
 |
 |
 |
 |
 |
Artigo
| Electrochemical determination of 2-naphthylamine in perfume samples using boron-doped diamond electrode |
|
Guilherme N. Rodrigues; Weberson P. Silva; Diego P. Rocha; Eduardo M. Richter; Rodrigo A. A. Munoz; Alex D. Batista*
Instituto de Química, Universidade Federal de Uberlândia, 38400-092 Uberlândia - MG, Brasil Recebido em 11/09/2019 *e-mail: alexbatista@ufu.br This work presents a simple electrochemical method for 2-naphthylamine (2NAP) determination in perfume by differential-pulse voltammetry (DPV) employing a boron-doped diamond (BDD) electrode cathodically treated. Simple dilution of sample in supporting electrolyte was performed before electrochemical analysis. A borate buffer solution of pH 10.0 containing 20% (v/v) ethanol provided stable responses of 2NAP without electrode fouling. Modulation time, step potential and modulation amplitude were optimized at 20 ms, 6 mV and 70 mV, which achieved a limit of detection (LOD) of 0.0046 μmol L−1 with a linear range between 0.5 and 90 μmol L−1, and RSD < 2.9%, respectively. Recovery values for spiked samples with 2NAP were between 102 and 104%. The proposed method is feasible to be applied for 2NAP detection and quantification on perfume samples with simple sample preparation. INTRODUCTION Dyes are chemicals present in different manufactured areas, as in the textile, pharmaceutical, food, cosmetics and other industries to give color to different products. Polycyclic aromatic amines (PAA) are intermediary products obtained during several dye synthesis. They have been proved related to the occurrence of bladder cancer.1-4 2-naphthylamine (2NAP) is a PAA largely employed during XX century as an intermediary product in the azo dye synthesis, as antioxidant in rubber productions and for 2-chloronaphthalene synthesis. 2NAP is classified by the International Agency for Research on Cancer as carcinogenic to humans.3 Due to its effects, 2NAP is regulated by different agencies, such as the Occupational Safety and Health Administration from U.S.A. that strictly restricts the exposure to 2NAP.5 Its use is prohibited in the European Union, Switzerland, United Kingdom since 1998, 1938 and 1952, respectively.3,6 In Japan, the production and use of dyes containing 2NAP were prohibited in 1972 and the occurrence of bladder cancer decreased since then.6 The proved carcinogenicity effects on humans of the PAA led to several studies focused on the determination of 2NAP and other PAA in different matrixes. Tomkins and Ho7 proposed the determination of 2NAP and other PAA in fossil fuel samples by GC-MS after derivatization trifluoroacetic anhydride. Grimmer et al.8 and Niu et al.9 performed the determination of 2NAP and other PAA in urine samples from smokers by GC-MS and GC-MS/MS, respectively, Li et al.10 performed the determination of five PAA in samples of river water and wastewater, including 1-naphthylamine and benzidine by hydrophilic interaction liquid chromatography. Few studies are dedicated for determination of 2NAP employing electrochemical techniques, such as voltammetry or potentiometry.1,11-14 Wang11 determined 2NAP and other PAA by potentiometry using a modified carbon paste electrode as the working electrode. Zima et al.14 employed electrochemical detection using a carbon paste electrode based on glassy carbon spherical microparticles with an organic pasting liquid on a HPLC system for determination of 2NAP and different PAA. Zavázalová et al.12 determined 2NAP and other PAA using HPLC with amperometric detection and boron doped diamond electrode as working electrode. Zhu et al.13 determined 2NAP on a carbon electrode modified with 3,4,9,10-perylene tetracarboxylic acid noncovalently bounded to multiwalled carbon nanotubes. Nevertheless, because of the low concentrations of PAA and the matrix interferents in complex samples, some tedious and time-consuming pretreatment procedures, such as derivatization,15 solid phase extraction,16,17 liquid-liquid extraction18 and solid-phase microextraction9 are required, which increase costs and difficulties of the analysis.19 Despite the advantages of the proposed procedures for 2NAP determination, most of them are based on chromatographic techniques due to the complexity of the matrixes as well as time-consuming sample preparation and separation steps. The aim of this work was to develop an electroanalytical procedure for 2NAP determination in perfume samples, a typical cosmetic sample which may contain residues of 2NAP. The strategy for the sensitive detection of 2NAP was based on the differential-pulse voltammetry (DPV) technique employing boron-doped diamond (BDD) electrode.
EXPERIMENTAL Reagents and samples 2-Naphthylamine (2NAP) was purchased from Sigma-Aldrich (Milwaukee, WI, USA), acetic acid and phosphoric acid from Synth (Diadema, Brazil), sodium hydroxide and boric acid from AppliChem Panreac (Barcelona, Spain) and ethanol and sulfuric acid from Vetec (Rio de Janeiro, Brazil). All solutions were prepared with deionized water (Millipore Direct-Q3) with a resistivity not less than 18 MΩ cm. Reagents were of analytical grade and used without further purification. A borate buffer of 0.1 mol L-1 (pH = 10.0) was used as the supporting electrolyte. The Britton-Robinson (BR) buffer solution used in cyclic voltammetric studies was composed of a mixture of 0.04 mol L-1 acetic acid, boric acid and phosphoric acid and its pH was adjusted with sodium hydroxide. Then ethanol was added to the buffer solution in the ratio 80/20 v/v (water/ethanol). 2NAP standard solution was prepared by dissolution in ethanol. Aliquots of perfume samples were diluted in supporting electrolyte before the analysis. Instruments and apparatus Electrochemical measurements were performed employing a μAutolab Type III potentiostat (Metrohm Autolab, Netherlands) controlled by NOVA 1.11 software. As auxiliary and reference electrodes, a platinum wire and mini Ag/AgCl saturated with KCl20 were employed, respectively. The working electrode consisted of a thin film of BDD (~1.2 μm of thickness) with a doping level of around 8000 ppm deposited on a polycrystaline silicon wafer (0.7 x 0.7 cm) with 1 mm of thickness (NeoCoat SA, La Chaux-de-Fonts, Switzerland). Every day, before using, the BDD electrode was pretreated applying +0.01 A for 1000 s in 0.04 mol L-1 Britton-Robinson buffer solution (anodic pretreatment) or -0.01 A for 1000 s in 0.1 mol L-1 H2SO4 solution (cathodic pretreatment) prior to the electrochemical measurements. All results presented in the work were performed without removal of the dissolved oxygen and at room temperature. The experiments were carried using house made electrochemical cell where the working electrode area is defined with a rubber O-ring with 0.4 cm diameter (electrode area = 0.13 cm2) similar to the procedure in previous works.21,22 For comparison, a glassy-carbon electrode (GCE) disk (area = 0.07 cm2, BASi Inc., USA) was used as working electrode.
RESULTS AND DISCUSSION The electrochemical oxidation of 2NAP was previously investigated on BDD electrodes.12 In this work, during electrochemical oxidation of aminobiphenyls using differential pulse voltammetry, the formation of polyamine films on the BDD surface was observed. Consequently, a fast decrease (∼ 80 %) in the oxidation current was noted due to the gradual decrease in the electrode area. According to the authors, these polyamine films can be removed from the electrode surface through immersion in methanol and bubbling with nitrogen for 15 s.23 In addition, as also described in this paper,12 the formation of dimers and polymeric films is more intense in acid than in alkaline medium. Therefore, in order to avoid this periodic cleaning step (immersion in methanol), studies were carried out in alkaline media and with the addition of ethanol (lower toxicity than methanol) to the supporting electrolyte. According to previous works,24,25 the presence of 20% (v/v) of ethanol in the supporting electrolyte greatly reduced the electrode fouling. Based on this information, the electrochemical behavior of 2NAP was evaluated in pH range 8-12 using BR buffer solution containing 20% (v/v) of ethanol. Better current response was observed in BR solution pH 10. This pH value was reevaluated using 0.1 mol L-1 borate buffer (pH = 10.0) with 20% (v/v) ethanol and similar response was obtained. Therefore, this solution (without acetic and phosphoric acids) was selected as the supporting electrolyte in subsequent studies. Cyclic voltammograms of 2NAP using the optimized conditions are presented in Figure 1A using BDD as the working electrode. For comparison, the oxidation of 2NAP was also evaluated using GCE as the working electrode (Figure 1B).
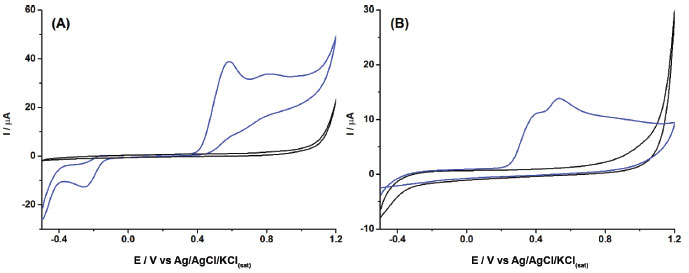 Figure 1. Cyclic voltammograms obtained before (black line) and after (blue line) the addition of 1 mmol L-1 of 2NAP in 0.1 mol L-1 borate buffer (pH = 10.0) with 20% (v/v) ethanol: (A) BDD electrode (area = 0.13 cm2); (B) GCE (area = 0.07 cm2). Scan rate: 50 mV s-1; step potential: 5 mV
As can be observed in Figure 1, the current density detected with the BDD electrode (40/0.13 = 285 μA cm2) is greater than that detected with the GCE (15/0.07 = 214 μA cm2). The best analytical response combined with the greatest resistance to fouling made the BDD electrode the best choice for the determination of 2NAP. The mechanism of the electrochemical oxidation was previously reported and the first electrochemical process (+ 0.58 V) involves one electron transfer resulting in the formation of a cation radical while the second one (+ 0.80 V) is a result of another electron transfer with the formation of the respective hydroxylamine derivative.13 The precision and antifouling properties of the BDD surface were evaluated after cathodic and anodic treatments. Consecutive measurements of a solution containing 2NAP were carried out in the presence of sample matrix (perfume samples). This experiment revealed that the cathodic-treated BDD surface presented higher stability for 2NAP detection (RSD < 5% for n=5) while the response of 2NAP on the anodic-treated BDD surface decreased quickly. Next, the parameters of differential-pulse voltammetry were studied. The modulation amplitude was varied from 10 to 100 mV and a constant increase in current was verified with a plateau at 70 mV, which was selected for further measurements. The modulation time was varied from 10 to 50 ms and the current response decreased continuously with time. The value of 20 ms was selected due to the obtaining of DPV recordings with low standard deviation (<1%, n=3) in comparison with those obtained with 10 ms (5%). The step potential was varied from 1 to 10 mV and a dramatic increase in current was verified from 1 to 3 mV, followed by a plateau. The value of 6 mV was selected due to the low standard deviation (<1%) for repetitive DPV recordings (n=3). Table 1 summarized the ranges of DPV parameters and selected values.
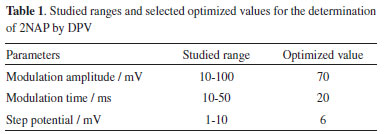
Figure 2A presents the DPV recordings for increasing amounts of 2NAP from 0.5 to 105 μmol L-1 under conditions listed in Table 1. A linear response was verified from 0.5 to 90 μmol L-1 with a correlation coefficient of 0.999 (the analytical curve is shown in Figure 2B). Using this data, the limit of detection (LOD) was calculated as 0.0046 μmol L−1 (LOD = 3 x δ/S in which δ is the standard deviation of the blank for n=10 and S the slope of the analytical curve, according to IUPAC guideline). However, this value is much lower than the first concentration of the linear calibration curve. The LOD was also calculated experimentally by the measuring the response of increasing concentrations of 2NAP and the first detectable signal occurred for 0.010 μmol L−1, so this LOD value can be considered a more realistic one.
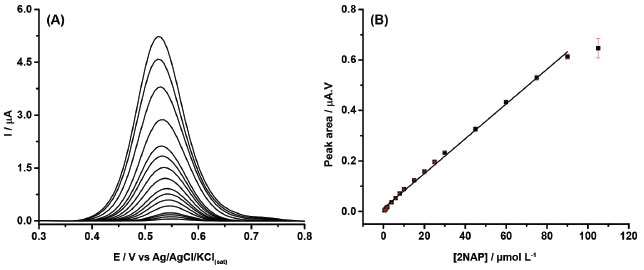 Figure 2. (A) Baseline-corrected DPV recordings obtained with increasing concentrations of 2NAP (0.5 - 105 μmol L-1) and (B) respective analytical curve (r = 0.999). Modulation amplitude: 70 mV; modulation time: 20 ms; step potential: 6 mV. Supporting electrolyte: 0.1 mol L-1 borate buffer (pH = 10.0) with 20% (v/v) ethanol
The precision of the cathodic-treated BDD electrode for 2NAP sensing was estimated through a repeatability study. Figure 3 shows the DPV recordings for consecutive measurements (n=10) of 2NAP at three concentration levels (0.5, 2.0 and 5.0 μmol L−1). The relative standard deviation values were lower than 2.9% for all tested concentrations, which indicates the cathodic-treated BDD electrode under selected conditions of DPV and electrolyte provides stable and precise responses to 2NAP.
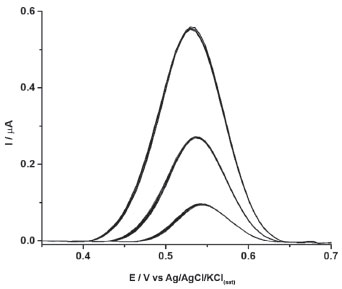 Figure 3. Intra-day repeatability study obtained by DPV from successive measurements (n = 10) of solutions containing 0.5, 2.0 and 5.0 μmol L-1 of 2NAP. Other experimental conditions as indicated in Figure 2
After demonstrating the feasibility of the proposed sensor for 2NAP determination, two perfume samples were analyzed after 100-fold dilution in supporting electrolyte. Both samples did not present a response in DPV under selected conditions (< LOD). Therefore, both samples were spiked with 40 μmol L-1, which after dilution (100-fold) would generate solutions with amounts of 2 μmol L-1. Figure 4A and 4C shows the DPV recordings of both perfume samples before and after spiked with 2NAP, and three additions of 2NAP standard solutions. The standard addition method was used to quantify the 2NAP concentration in both spiked perfume samples.
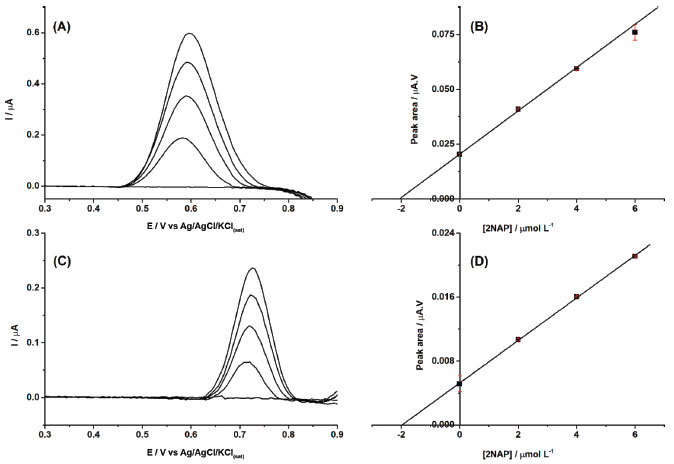 Figure 4. Baseline-corrected DPV recordings obtained for two perfume samples (A and C) before and after spiked with 40 μmol L-1 (100-fold dilution in supporting electrolyte) and addition of increasing concentrations of 2NAP (2 μmol L-1). (B and D) Respective analytical curves using the standard addition method (r = 0.998 and 0.99, respectively). Other experimental conditions as indicated in Figure 2
The results in Figure 4 show that a cathodic-treated BDD electrode in a supporting electrolyte of 0.1 mol L-1 borate buffer (pH = 10.0) with 20% (v/v) ethanol can be used for the quantification of 2NAP residues in perfume samples, with high precision and linearity, and with a low LOD value. The sample matrix did not show any electrochemical signal in the potential window, so no electroactive interferences were verified in the analysis of these cosmetic samples. On the other hand, the results of Figure 4 also show that the slopes (0.00989 and 0.00266) of the calibration curves of both samples are very different (matrix effect). However, using the standard addition method, the matrix effect was canceled, and recovery values were between 102 and 104% (n=3). Probably, the matrix effect also caused the small shift of the peak potential of 2NAP. Although no interferences were observed in both analyzed samples, a selectivity study was performed. Figure 5 shows DPV recordings obtained before (black line) and after (blue line) the addition of 2.0 μmol L-1 of 2NAP. After this, two components commonly added to perfumes such as ethylene glycol (A; red line) and glycerol (B; red line) were added to the solutions. As can be observed, small variations were observed in DPV recordings for solutions containing 2.0 μmol L-1 of 2NAP after addition of the interference compounds.
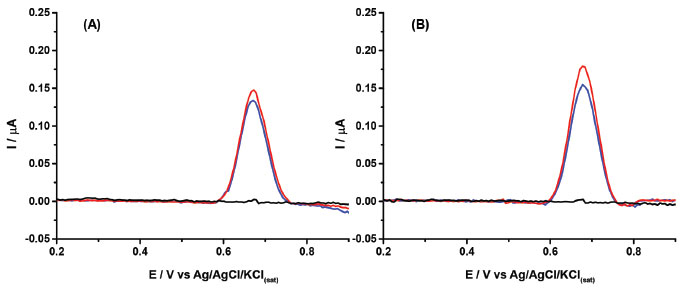 Figure 5. Baseline corrected DPV recordings obtained before (black line) and after (blue line) the addition of 2.0 μmol L-1 of 2NAP in 0.1 mol L-1 borate buffer (pH = 10.0) and, finally, after addition 8.94 mmol L-1 of ethylene glycol (A; red line) or 6.84 mmol L-1 of glycerol (B; red line). Other experimental conditions as indicated in Figure 2
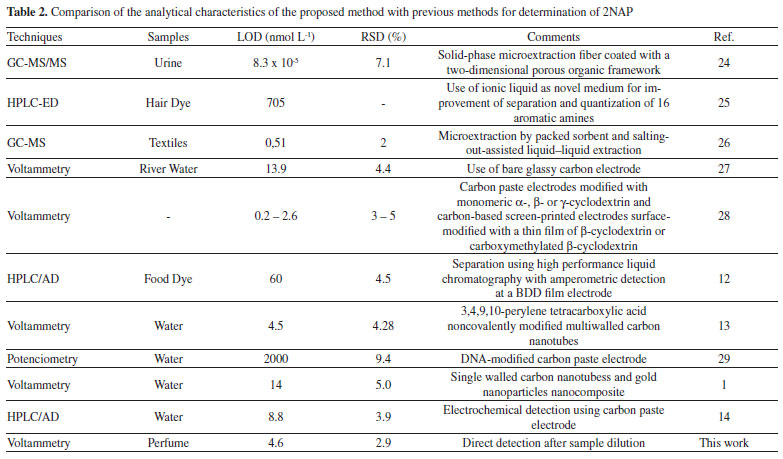
Table 2 compares the analytical characteristics of the proposed method with previous methods for determination of 2NAP. The proposed procedure presented one of the lowest LOD among the electroanalytic techniques, without any electrode modification and a minimal sample pretreatment (dilution). Despite separation techniques achieved lower LOD, they use very sophisticated detector allied with sample pretreatment techniques with high enrichment factor, which resulted in very low LOD. The proposed procedure also achieved low RSD, highlighting its precision, even for a complex matrix, such as perfumes.
CONCLUSIONS We demonstrated that a cathodic-treated BDD electrode can be applied for 2NAP determination in perfume samples. The presence of 20% (v/v) ethanol in the supporting electrolyte reduced electrode fouling as well as the selection of a buffer solution at higher pH values (pH = 10). A highly stable and precise response of the BDD electrode was verified from 0.5 to 90 μmol L-1 of 2NAP with a LOD value of 0.0046 μmol L−1. Such analytical characteristics enabled the determination of residues of 2NAP in perfume samples, with adequate recovery values. Therefore, the proposed electrochemical method can be extended to other cosmetic samples that do not contain electroactive species in their composition.
ACKNOWLEDGMENTS The authors are grateful to FAPEMIG (APQ-02272-18), CNPq (465389/2014-7-INCTBio, 425114/2018-0 and 307271/2017-0) and CAPES (financial code 001).
REFERENCES 1. George, S.; Lee, H. K.; Anal. Methods 2010, 2, 326. 2. Boyland, E.; Dukes, C. E.; Grover, P. L.; Br. J. Cancer 1963, XVII, 79. 3. IARC; IARC Monogr. 2012, 100F, 83. 4. Fuller, T. W.; Acharya, A. P.; Meyyappan, T.; Yu, M.; Bhaskar, G.; Little, S. R.; Tarin, T. V; Sci. Rep. 2018, 8, 507. 5. Thirteen OSHA-Regulated Carcinogens, available at https://www.cdc.gov/niosh/npg/nengapdxb.html, accessed at February 2020. 6. Olfert, S. M.; Felknor, S. A.; Delclos, G. L.; South. Med. J. 2006, 99, 1256. 7. Tomkins, B. A.; Ho, C. H.; Anal. Chem. 1982, 54, 91. 8. Grimmer, G.; Dettbarn, G.; Seidel, A.; Jacob, J.; Sci. Total Environ. 2000, 247, 81. 9. Niu, J.; Zhao, X.; Jin, Y.; Yang, G.; Li, Z.; Wang, J.; Zhao, R.; Li, Z.; J. Chromatogr. A 2018, 1555, 37. 10. Li, R.; Zhang, Y.; Lee, C. C.; Lu, R.; Huang, Y.; J. Chromatogr. A 2010, 1217, 1799. 11. Wang, J.; Anal. Chem. 1996, 68, 4365. 12. Zavázalová, J.; Dejmková, H.; Barek, J.; Pecková, K.; Electroanalysis 2013, 25, 253. 13. Zhu, G.; Yi, Y.; Han, Z.; Liu, J.; Gai, Z.; Anal. Lett. 2014, 47, 2370. 14. Zima, J.; Dejmkova, H.; Barek, J.; Electroanalysis 2007, 19, 185. 15. Weiss, T.; Angerer, J.; J. Chromatogr. B 2002, 778, 179. 16. Yu, J.; Wang, S.; Zhao, G.; Wang, B.; Ding, L.; Zhang, X.; Xie, J.; Xie, F.; J. Chromatogr. B 2014, 958, 130. 17. Xie, F.; Yu, J.; Wang, S.; Zhao, G.; Xia, Q.; Zhang, X.; Zhang, S.; Talanta 2013, 115, 435. 18. Feng, W.; Zhu, Y.; Ouyang, G.-F.; Chin. J. Anal. Chem. 2015, 43, 957. 19. Jain, A.; Reddy-Noone, K.; Pillai, A. K. K. V; Verma, K. K.; Anal. Chim. Acta 2013, 801, 48. 20. Pedrotti, J. J.; Angnes, L.; Gutz, I. G. R.; Electroanalysis 1996, 8, 673. 21. Silva, L. A. J.; da Silva, W. P.; Giuliani, J. G.; Canobre, S. C.; Garcia, C. D.; Munoz, R. A. A.; Richter, E. M.; Talanta 2016, 165, 33. 22. Silva, W. P.; Silva, L. A. J.; França, C. H.; Sousa, R. M. F.; Muñoz, R. A. A.; Richter, E. M.; Electroanalysis 2017, 29, 1860. 23. Barek, J.; Jandová, K.; Pecková, K.; Zima, J.; Talanta 2007, 74, 421. 24. Niu, J.; Zhao, X.; Jin, Y.; Yang, G.; Li, Z.; Wang, J.; Zhao, R.; Li, Z.; J. Chromatogr. A 2018, 1555, 37. 25. Lizier, T. M.; Zanoni, M. V. B.; Molecules 2012, 17, 7961. 26. Sánchez, M. D. N.; Santos, P. M.; Sappó, C. P.; Pavón, J. L. P.; Cordero, B. M.; Talanta 2014, 119, 375. 27. Zavazalova, J.; Ghica, M. E.; Schwarzova-Peckova, K.; Barek, J.; Brett, C. M. A.; Electroanalysis 2015, 27, 1556. 28. Ferancová, A.; Korgová, E.; Labudaa, J.; Zima, J.; Barek, J.; Electroanalysis 2002, 14, 1668. 29. Wang, J.; Rivas, G.; Luo, D.; Cai, X.; Valera, F. S.; Dontha, N.; Anal. Chem. 1996, 68, 4365. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access