
 |
 |
 |
 |
 |
Artigo
|
|
| Determinação de multirresíduos de agrotóxicos em méis produzidos na região do Triângulo Mineiro por UHPLC-MS/MS Determination of pesticide multiresidues in honey produced in the Triângulo Mineiro region of Brazil by UHPLC-MS/MS |
|
Renata A. G. Silva; Anizio M. Faria*
Instituto de Ciências Exatas e Naturais do Pontal, Universidade Federal de Uberlândia, 38304-402 Ituiutaba - MG, Brasil Recebido em 19/09/2019 *e-mail: anizio@ufu.br Excessive use of pesticides can lead to the accumulation of residues in different parts of plants, endangering various species and contaminating foods. Honey can be used as an indicator of agricultural contamination and pre-mature bee mortality by pesticides. However, to quantify pesticide residues in honey, high sensitivity analytical methodologies are required. In this work, a new method was proposed for the determination of seven pesticides, widely used in Triângulo Mineiro, in honey samples based on QuEChERS method and quantification by UHPLC-MS/MS. The method was optimized and validated according to the protocol recommended by the European Community. The method presented LOQ in the range of 0.6-4.0 μg kg-1, lower than the MRLs established for the pesticides studied. Also, the methodology showed high accuracy, with recoveries ranging from 89-107%, and good accuracy, RSD <14%. Honey samples obtained in the Triângulo Mineiro region and subjected to the determination of pesticide residues were analyzed by the analytical methodology, indicating the presence of methomyl and imidacloprid residues in all samples. These results indicate that beehives close to the grain and sugarcane crops are being affected by the application of pesticides in the Triângulo Mineiro Region, Brazil. INTRODUÇÃO O agronegócio é um setor estratégico para economia brasileira, sendo responsável, em média, por mais de 20% do produto interno bruto e mais de 40% das exportações de mercadorias, nos últimos anos.1 A região do Triângulo Mineiro possui participação significativa neste setor, estando entre os maiores produtores de cana-de-açúcar e de grãos do país. No entanto, o agronegócio brasileiro é fundamentado em um modelo de produção que é dependente do uso intensivo de agrotóxicos.2 O Brasil é o terceiro maior mercado mundial de agrotóxicos, ficando atrás apenas de Japão e Coréia, tanto em termos de terras cultiváveis quanto em termos de quantidade por alimento produzido.3,4 Embora os agrotóxicos tenham a finalidade de otimizar a produção agrícola, o uso excessivo e desregrado representa um risco potencial para a saúde humana e para o meio ambiente, uma vez que alguns princípios ativos possuem alta toxicidade, alta persistência e baixas taxas de degradação, podendo permanecer nos alimentos ou nos solos, serem lixiviados para as águas subterrâneas ou superficiais, e até mesmo se concentrar em diferentes partes das plantas com sua contínua aplicação.5-7 A expansão da área de cultivo, a permanência de resíduos de agrotóxicos nas plantas e a pulverização foliar de agrotóxicos têm sido responsáveis pela contaminação e, até mesmo, a mortalidade de diversas espécies de animais.3,7,8 Em especial, o desaparecimento de abelhas polinizadoras devido ao uso intenso de agrotóxicos tem despertado uma enorme preocupação mundial.3,9-11 As abelhas, que desempenham um papel fundamental de polinização, ao visitar culturas que fazem uso de agrotóxicos, retém no corpo ou no sistema respiratório os resíduos desses agentes químicos, causando sua morte pré-matura e contaminando os produtos apícolas.12 Logo, o mel, como principal produto apícola, pode ser usado como um indicador para avaliação do impacto ambiental causado por agrotóxicos em uma determinada região agricultável, inclusive sobre as colmeias de abelhas.12 No entanto, para detectar e até mesmo quantificar os resíduos de agrotóxicos no mel é necessário o uso de metodologias analíticas de alta detectabilidade, uma vez que a presença desses compostos nas amostras podem estar em níveis de μg kg-1 (ppb, partes por bilhão). Dentre as técnicas analíticas disponíveis, a cromatografia líquida de ultra-alta eficiência (UHPLC, ultra high-performance liquid chromatography) combinada com a espectrometria de massas em série (MS, mass spectrometry) apresenta o maior nível de detectabilidade para um grande número de moléculas, bem como a possibilidade de confirmação de suas identidades, sendo considerada a ferramenta ideal para determinação de contaminantes em alimentos.13-15 Outro fator fundamental para a obtenção de resultados confiáveis nas análises de resíduos de contaminantes em amostras alimentícias é a preparação da amostra. A presença de pequenas quantidades dos resíduos de agrotóxicos em meio a um concentrado de substâncias constituintes da matriz alimentícia e potenciais interferentes da análise torna a preparação da amostra a etapa crítica de todo o processo de análise.16,17 Diversos métodos de preparação têm sido explorados para a extração seletiva e eficiente de contaminantes orgânicos em alimentos, porém o método QuEChERS (Quick, Easy, Cheap, Rugged, Effective, and safe) tem se apresentado como o mais adequado para diversas matrizes.16,18-22 O método QuEChERS foi originalmente desenvolvido para a extração de resíduos de agrotóxicos em frutas e vegetais e modificado para atender às mais diversas características das diferentes matrizes de alimentos.23 Diante desse contexto, o objetivo deste trabalho foi desenvolver um método analítico de alta detectabilidade para a determinação de resíduos de agrotóxicos em méis produzidos na região do Triângulo Mineiro, empregando o método QuEChERS e a técnica UHPLC-MS/MS. Os agrotóxicos selecionados para o estudo são aqueles mais comercializados e mais empregados nas culturas de cana-de-açúcar, soja, sorgo, citrus e milho na Região do Triângulo Mineiro. De acordo com o levantamento os principais agrotóxicos empregados nas diferentes culturas da Região do Triângulo Mineiro são o metomil, o imidaclopride, o acetamipride, o acefato, o diflubenzurom, o tiametoxam e o carbendazim, cujas propriedades químicas e toxicológicas são apresentadas na Tabela 1S e Figura 1S do material suplementar. O método foi validado de acordo com o documento de orientação da Comunidade Europeia sobre o controle de qualidade analítico e procedimentos de validação de metodologias para determinação de resíduos de agrotóxicos em alimentos, documento SANTE/11813/2017.24
PARTE EXPERIMENTAL Materiais Todos os solventes empregados no trabalho foram de grau LC-MS e todos os reagentes foram de grau analítico. Água ultrapura foi produzida por um ultrapurificador Millipore Milli-Q 8. Os solventes e água ultrapura foram filtrados individualmente em membranas de poli(tetrafluoretileno) de 0,22 μm de poros da Millipore. Padrões analíticos de alta pureza, > 99%, dos princípios ativos acefato, acetamipride, carbendazim, diflubenzuron, imidaclopride, metomil e tiametoxam foram adquiridos da Dr. Ehrenstofer (Augsburg, Alemanha) e Sigma Aldrich (St. Louis, MO, EUA). Soluções padrão estoque de 100 mg L-1 da mistura dos agrotóxicos foram preparadas em acetonitrila e metanol. Soluções padrão de trabalho em diferentes concentrações foram preparadas a partir da diluição da solução estoque para realização dos estudos de otimização e validação do método. Extração de resíduos de agrotóxicos de amostras de mel empregando QuEChERS Para a otimização do método de extração QuEChERS dos agrotóxicos, amostras de mel isentas de agrotóxicos foram adquiridas no comércio local de Uberaba, MG. Para confirmação da ausência de resíduos de agrotóxicos no mel, o branco das amostras foi analisado por UHPLC-MS/MS nas condições de análise do método, não sendo detectado quaisquer sinais dos compostos em estudo. Para a extração dos resíduos de agrotóxicos do mel foi empregado o método QuEChERS, no qual amostras de 5,000 g de mel foram pesadas em tubos de centrífuga de polipropileno. Em seguida, foram adicionados 5,00 mL de água ultrapura e agitado em vórtex por 1 min. 4,00 mL de acetonitrila foram então adicionados à solução e novamente agitada em vórtex por 1 min. Logo após, 6,000 g de MgSO4 anidro e 1,5000 g de CH3COONa foram adicionados ao tubo e a solução foi agitada em vórtex por 3 min. O tubo foi centrifugado a 3000 rpm por 10 min e o extrato orgânico foi recuperado, sendo submetido a uma etapa de clean-up para a eliminação de potenciais interferentes na análise cromatográfica e minimização do efeito de matriz. Na etapa de clean-up, três sorventes foram avaliados no intuito de eliminar os interferentes sem perdas dos analitos: amina primária secundária (PSA, primary secundary amine), sílica e uma mistura de MgSO4 e C18. Alíquotas de 1 mL da fase orgânica do processo de extração foram transferidas para tubos de centrífuga de polipropileno contendo os 50 mg dos sorventes em estudo. O tubo foi agitado em vórtex por 30 s e então a solução foi filtrada em membranas de PVDF de 0,2 μm de diâmetro de poros. 100 μL do extrato limpo foram transferidos para vials e diluídos com 900 μL de água ultrapura. A solução do vial foi agitada manualmente e 2 μL desta foram injetados no sistema UHPLC-MS/MS. Validação do método analítico O método proposto foi validado de acordo com o documento SANTE/11813/2017 proposto pela Comunidade Europeia que estabelece procedimentos para validação de metodologias de análise e determinação de resíduos de agrotóxicos em alimentos. As amostras de mel, isenta de resíduos de agrotóxicos, foram usadas como testemunhas e fortificadas com soluções padrão dos agrotóxicos em concentrações conhecidas para os estudos de validação. Curvas analíticas para os sete agrotóxicos foram construídas pelo método do padrão externo e por calibração na matriz, fortificando a matriz e submetendo-a ao processo de extração. Os parâmetros do método analítico avaliados no estudo de validação foram a seletividade, linearidade, limite de quantificação, exatidão, precisão e efeito de matriz para os sete agrotóxicos estudados. As curvas analíticas foram preparadas na faixa de concentração dos agrotóxicos de 0,5 a 4,5 μg L-1 e cada concentração analisada em triplicata. O limite de quantificação (LOQ, limit of quantification) foi obtido a partir da injeção de soluções de concentrações decrescentes dos agrotóxicos no UHPLC-MS/MS, sendo o LOQ do método definido para a menor concentração do agrotóxico, cuja a extração apresentou recuperação na faixa de 70-120% e um RSD (relative standard deviation) inferior a 20%, conforme preconizado no documento SANTE.24 A exatidão do método foi avaliada a partir de ensaios de recuperação, nos quais amostras de mel foram fortificadas em níveis de 0,5; 2,5 e 4,5 μg L-1 com as soluções padrão dos agrotóxicos e submetidas ao método QuEChERS. A precisão do método foi avaliada quanto à repetibilidade (todas as condições idênticas) e à reprodutibilidade dentro do laboratório (extrações em três dias consecutivos) a partir da realização de extrações QuEChERS de amostras de mel fortificadas com 0,5; 2,5 e 4,5 μg L-1 de soluções padrão dos agrotóxicos. Os ensaios foram realizados em seis replicatas. O grau de precisão foi expresso pelo desvio padrão relativo (RSD) destas determinações. Análises cromatográficas As análises cromatográficas foram realizadas em um sistema de cromatografia de ultra-alta eficiência (UHPLC) acoplado a um espectrômetro de massas em série, empregando uma fonte de ionização electrospray (ESI, electrospray ionization source) e analisador de massas triplo quadrupolo (QqQ) da Waters modelo Xevo TQ-s Micro (Mildford, MA, EUA). O software Mass Lynx foi utilizado para a aquisição e processamento dos cromatogramas e espectros de massas. A separação dos compostos foi realizada em uma coluna analítica HSS T3 da Waters, com 100 mm × 2,1 mm d.i. e com partículas de 1,8 μm. A fase móvel foi composta de (A) 0,1% de solução aquosa de ácido acético e (B) acetonitrila contendo 0,1% de ácido acético eluída sob gradiente inicial de 10% de B, mantido por 0,25 min, aumentado para 98% de B até 7,75 min (0,75 min) e retornado para 10% de B em 10 min. Cada composto gerou essencialmente um único íon [M+H]+ no sistema UHPLC-MS/MS. Os dados do monitoramento de reações múltiplas (MRM, multiple reaction monitoring) foram adquiridos e processados no modo positivo. Os parâmetros da fonte de ionização otimizados foram a temperatura da fonte de 150 °C; voltagem do capilar em 1 kV para todos compostos; vazão e temperatura do gás nebulizador (N2) de 900 L h-1 e 450 °C, respectivamente. O gás de colisão foi o argônio com pressão de 2,5 mTorr. Análise de amostras de mel da região do Triângulo Mineiro Sete amostras de mel, sem qualquer tipo de tratamento, adquiridas de apiários localizados nas cidades de Uberlândia (amostra A), Ituiutaba (amostra B), Capinópolis (amostra C), Patrocínio (amostra D), Nova Ponte (amostra E) e Uberaba (amostras F e G) da região do Triângulo Mineiro foram submetidas ao método analítico validado neste trabalho para a determinação de resíduos de acefato, acetamipride, carbendazim, diflubenzuron, imidaclopride, metomil e tiametoxam, empregando o método QuEChERS e análise por UHPLC-MS/MS. As amostras foram identificadas por letras para preservar as marcas e os fabricantes. As amostras foram armazenadas em frascos de vidro a 4 °C em refrigerador até o momento da análise. A cidade de origem, no entanto, foi empregada para a identificação do potencial foco de contaminação pelos agrotóxicos avaliados.
RESULTADOS E DISCUSSÃO Otimização dos parâmetros do triplo quadrupolo O UHPLC-MS/MS é uma técnica analítica de alta detectabilidade capaz de quantificar resíduos de agrotóxicos em concentrações inferiores a 1 μg L-1. No entanto, para que a obtenção dos sinais sejam reprodutíveis e confiáveis próximos aos limites de quantificação dos compostos o sistema MS/MS requer o ajuste fino dos parâmetros no analisador de massas. Desta forma, soluções dos analitos preparadas em solvente foram infundidas individualmente no detector triplo quadrupolo em combinação com fase móvel proveniente do UHPLC, composta por acetonitrila:H2O (50:50, v/v) acidificada com 0,1% de ácido acético, para otimizar os parâmetros no analisador de massa triplo quadrupolo com fonte de ionização electrospray (ESI). Os analitos apresentaram melhor sinal com a fonte de ionização operando no modo positivo, voltagem do capilar 1 kV, temperatura e fluxo do gás de dessolvatação (N2) 450 °C e 900 L h-1, respectivamente. A Tabela 1 apresenta os parâmetros otimizados para a detecção dos compostos estudados no sistema de detecção espectrométrica de massas que proporcionaram a melhor resposta em termos de detectabilidade e seletividade.
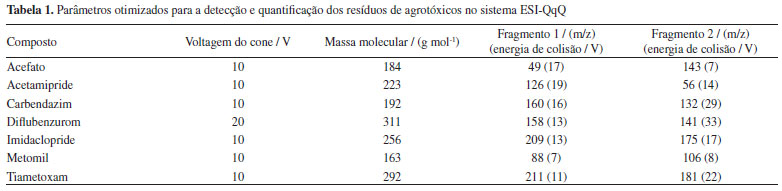
Extração de resíduos de agrotóxicos de amostras de mel empregando QuEChERS A extração dos resíduos de agrotóxicos em mel foi realizada pelo método QuEChERS, por este se apresentar como a melhor alternativa de preparação de amostras de alimentos quando se objetiva a extração de resíduos de agrotóxicos. Para aplicação na extração de acefato, acetamipride, carbendazim, diflubenzurom, imidaclopride, metomil e tiametoxam de amostras de mel, o método QuEChERS foi otimizado quanto ao processo de clean-up dispersivo, uma vez que em análise preliminar a recuperação dos agrotóxicos no método original, empregando o PSA, não foi satisfatória para todos os compostos. Três adsorventes foram avaliados para a remoção eficiente de potenciais interferentes da matriz sem a perda dos compostos em estudo. Os sorventes avaliados foram a amina primária secundária (PSA), a sílica pura (SiO2), e uma mistura de sulfato de magnésio e octadecilsilano (C18). Amostras de mel fortificadas com 4,5 μg L-1 da solução padrão de agrotóxicos foram submetidas à extração QuEChERS, seguida da etapa de clean-up dispersivo com os diferentes adsorventes e as taxas de recuperação dos agrotóxicos são apresentados na Figura 1.
 Figura 1. Efeito do adsorvente na etapa de clean-up dispersivo do método QuEChERS para a recuperação dos resíduos de agrotóxicos de amostras de mel empregando UHPLC-MS/MS
Dos adsorventes avaliados, o que apresentou as melhores taxas de recuperação para todos os agrotóxicos, com menor dispersão dos resultados foi a mistura MgSO4:C18 (3:1, m/m), Figura 1. Os extratos da amostra obtidos com o uso dessa mistura de adsorventes também se apresentaram mais límpidos. Isso ocorreu, provavelmente pelo MgSO4 remover a água residual do extrato, diminuindo, portanto, sua polaridade e impedindo a precipitação de co-extrativos polares provenientes da matriz do mel. O sorvente C18, por sua vez, remove com boa eficiência os interferentes apolares, tais como graxas e lipídios característicos do mel.25 Considerando que os coeficientes de partição octanol/água (logP) para os agrotóxicos em estudo são menores que 2, exceto para o diflubenzurom, que o logP é 3,88,26 os compostos são, de uma forma geral, substâncias mais polares e a presença do C18 na mistura confere menor risco à perda dos analitos nesta etapa. A SiO2 não foi eficiente para a remoção de componentes da matriz do mel presentes no extrato, resultando em uma taxa de recuperação acima de 100% para o acefato, carbendazim e metomil. O sorvente PSA também proporcionou extratos límpidos, porém, para o acefato, dilfubenzurom e imidaclopride a taxa de recuperação foi abaixo de 70%, indicando que houve a retenção dos agrotóxicos no adsorvente no processo de clean-up. O extrato incolor se deve às ligações de hidrogênio formadas entre o PSA e os pigmentos do mel, removendo-os do extrato. No entanto, provavelmente o PSA também estabeleceu ligações de hidrogênio com algumas moléculas dos agrotóxicos, retendo-as e reduzindo as suas taxas de recuperação.15 A mistura de sulfato de magnésio:C18 (3:1, m/m) foi então empregada no método para determinação de resíduos de agrotóxicos em mel como o material adsorvente na etapa de limpeza dos extratos obtidos pelo método QuEChERS. Validação do método O método proposto para a determinação de resíduos dos setes agrotóxicos em amostras de mel, empregando o método QuEChERS e análise por UHPLC-MS/MS, foi submetida ao estudo de validação de acordo com o protocolo proposto pela Comunidade Europeia - documento SANTE/11813/2017.24 O protocolo europeu é o mais empregado e reconhecido dentre aqueles que fazem referência à validação de metodologias analíticas para determinação de resíduos de agrotóxicos em alimentos, por apresentar procedimentos e critérios bem estabelecidos para aceitação dos parâmetros analíticos de uma metodologia. Os parâmetros validados no método analítico proposto neste trabalho foram: linearidade, seletividade/especificidade, limite de quantificação, exatidão e precisão. A incerteza do método analítico foi determinada empregando metodologia Top-Down, a qual utiliza um determinado nível de informação obtida em estudo de validação que corresponde a todo o processo de medição para resultar em uma incerteza global do ensaio, conforme descrito no guia Eurachem.27 As fontes de incertezas consideradas foram o preparo de soluções, o ajuste do modelo linear para as curvas analíticas, a reprodutibilidade dentro do laboratório (precisão intermediária) e as taxas de recuperação dos agrotóxicos. De acordo com o documento SANTE/11813/2017, os íons precursores e de transições (quantificação e identificação) devem apresentar relação sinal/ruído (S/R) superior a 3, e a razão das transições de quantificação/confirmação nas amostras e nos padrões não devem diferir de ± 30%. Portanto, duas transições foram selecionadas para cada composto, conforme apresentadas na Tabela 1, obedecendo os critérios definidos pelo protocolo. A Figura 2 apresenta os cromatogramas de íons totais (TIC) obtidos de uma amostra isenta dos agrotóxicos e os cromatogramas de uma amostra fortificada com todos os agrotóxicos com concentração de 2,5 μg L-1. A inexistência de picos cromatográficos no tempo de retenção dos compostos estudados no cromatograma da amostra de mel, isenta dos agrotóxicos, evidencia a ausência de interferências e a seletividade/especificidade do método analítico.
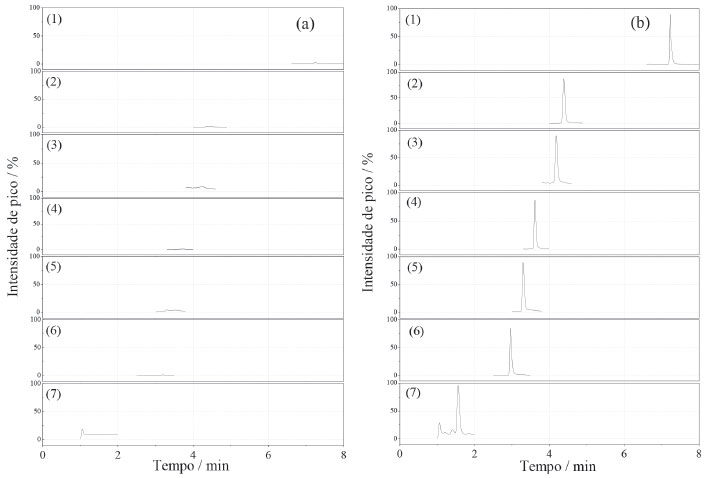 Figura 2. Cromatogramas de íons totais para os extratos das amostras de mel (a) sem a fortificação e (b) com a fortificação de soluções padrão a 2,5 μg L-1 dos agrotóxicos (1) diflubenzurom, (2) acetamipride, (3) imidaclopride, (4) tiametoxam, (5) metomil, (6) carbendazim e 7 (acefato), obtidos por UHPLC-MS/ MS, de acordo com as condições apresentadas na seção experimental
As curvas analíticas foram construídas pelo método do padrão externo (em acetonitrila:água, 10:90 v/v) e pela fortificação na matriz do mel. A faixa de concentrações empregadas na construção das curvas analíticas para os agrotóxicos variou de 0,07 a 4,50 μg L-1, sendo essa a faixa de trabalho (range) definida neste estudo. A partir das curvas analíticas foram determinadas também a linearidade do método proposto, expressa pelo coeficiente de correlação linear (r) entre as concentrações e as respectivas áreas de pico dos compostos, e a sensibilidade analítica, expressa pelo coeficiente angular da equação da reta. Os parâmetros analíticos do método obtidos das curvas analíticas são apresentados na Tabela 2. O método apresentou maior sensibilidade analítica para o carbendazim e menor para o imidaclopride, ainda assim apresentando limites de quantificação da ordem de 0,07 μg L-1. As curvas analíticas apresentaram excelentes ajustes ao modelo linear, > 99,1%, com coeficientes de regressão superiores a 0,991, estando em acordo com o recomendado pelos principais protocolos de validação.24,28-30
Os limites de quantificação (LOQ) para os agrotóxicos pelo método proposto foram obtidos pela menor concentração extraída dos compostos que apresentou taxa de recuperação na faixa de 70-120% com RSD menor que 20%. Os valores de LOQ foram de 0,07 μg L-1 para o extrato de carbendazim, que corresponde a 0,6 μg kg-1 de resíduos de carbendazim no mel; 1,3 μg kg-1 (0,15 μg L-1) para o acetamipride e o tiametoxam e 4,0 μg kg-1 (0,50 μg L-1) para o diflubenzurom, acefato, imidaclopride e metomil. Esses valores são inferiores aos estabelecidos como limite máximo de resíduos (LMR) para os agrotóxicos em mel pela Comunidade Europeia, que estabelece limites mais rigorosos para o consumo do mel.31 Os valores de LOQ para os resíduos de agrotóxicos obtidos neste método são inferiores também a outras metodologias propostas para determinação de resíduos de agrotóxicos em mel, empregando diferentes métodos de preparação de amostras e técnicas analíticas na literatura.32-35 A Tabela 2S do material suplementar apresenta uma comparação entre os parâmetros analíticos do método proposto neste trabalho com outros métodos desenvolvidos para determinação de resíduos de agrotóxicos em mel. Essa alta detectabilidade do método está associada à técnica de UHPLC-MS/MS, que apresenta alta detectabilidade no modo MRM. A partir das curvas analíticas construídas a partir das soluções dos agrotóxicos no extrato da amostra de mel e em solvente avaliou-se o efeito de matriz no método, o qual deve ser considerado e estimado em análises quantitativas por cromatografia líquida com detector por espectrometria de massas com fonte de ionização electrospray. Os componentes da matriz podem melhorar ou suprimir o sinal dos analitos e o efeito pode variar de amostra para amostra. No caso da fonte de ionização ESI os componentes da matriz podem afetar a eficiência de ionização dos analitos. Para reduzir os efeitos da matriz foi importante empregar uma preparação de amostra e uma separação cromatográfica eficientes, além disso foi realizada a diluição do extrato, 1:10, antes da injeção cromatográfica para a reduzir os efeitos de matriz. Desta forma, os efeitos de matriz foram negativos para acetamipride, metomil e tiametoxam, provavelmente devido à competição por ionização entre os analitos e outros componentes da amostra que não foram eliminados na preparação de amostra, enquanto os agrotóxicos acefato, carbendazim, diflubenzurom e imidaclopride foram acometidos por efeitos de matriz positivos, Figura 3. Nesse caso, provavelmente os componentes do mel, não eliminados no processo de extração, favoreceram a ionização dos analitos, gerando um sinal analítico maior que o esperado para os compostos. Portanto, a matriz exerce influência nas análises cromatográficas, sendo necessária a construção das curvas analíticas em extratos da matriz de mel, para minimizar os efeitos da mesma.
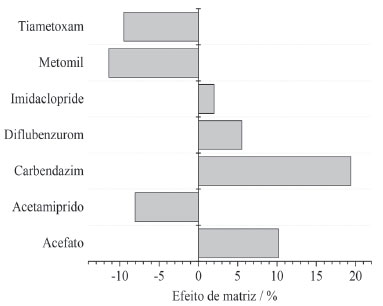 Figura 3. Efeitos de matriz (%) na quantificação dos agrotóxicos por UHPLC-MS/MS, determinados a partir de soluções padrão dos agrotóxicos preparadas em solvente e nos extratos de mel
A exatidão e precisão do método foram avaliadas em três diferentes níveis de fortificação (0,5 2,5 e 4,5 μg L-1) das amostras de mel com soluções padrão dos agrotóxicos e submetidas às extrações, em seis replicatas, pelo método QuEChERS e análise dos extratos por UHPLC-MS/MS em um mesmo dia, para avaliar a repetibilidade, e também em três dias distintos, para avaliar a reprodutibilidade dentro do laboratório (precisão intermediária), como preconizado no documento SANTE/11813/2017. A exatidão do método foi expressa pela taxa de recuperação alcançada para os agrotóxicos, sendo obtida uma faixa de recuperações de 89,3 a 106,6% (Figura 4). O protocolo de validação europeu estabelece como critério de aceitação da exatidão do método recuperações na faixa de 70 a 120%, considerando a complexidade das matrizes alimentícias e a presença de resíduos em níveis traço. Dessa forma, o método proposto neste trabalho pode ser considerado de boa exatidão para a determinação de resíduos de agrotóxicos em amostras de mel.
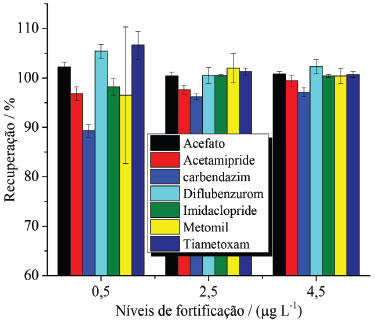 Figura 4. Porcentagens de recuperação dos resíduos de agrotóxicos de amostras de mel empregando a extração pelo método QuEChERS e a quantificação por UHPLC-MS/MS. N = 6
A precisão do método foi avaliada a partir dos parâmetros de repetibilidade e reprodutibilidade dentro do laboratório de acordo com o documento SANTE/11813/2017. A precisão é expressa pelo desvio padrão relativo (RSD) dos resultados de recuperação para seis réplicas das amostras fortificadas em três níveis de concentração e submetidas às condições idênticas de processamento (repetibilidade) e variando uma das condições do método (reprodutibilidade dentro do laboratório), que neste estudo foi o dia de realização das extrações. Os resultados obtidos para esta avaliação estão apresentados na Tabela 3, na qual se observa que todos os valores de RSD foram inferiores a 13,8%. De acordo com o protocolo de validação europeu, para que uma metodologia analítica possa ser considerada precisa é necessário que a dispersão dos resultados, medida pelos valores de RSD no estudo da repetibilidade e reprodutibilidade, dentro do laboratório, sejam inferiores a 20%. Logo, o método proposto para a determinação de resíduos dos sete agrotóxicos, empregados em larga escala em diferentes culturas na região do Triângulo Mineiro, em amostras de mel pode ser considerado de boa precisão.
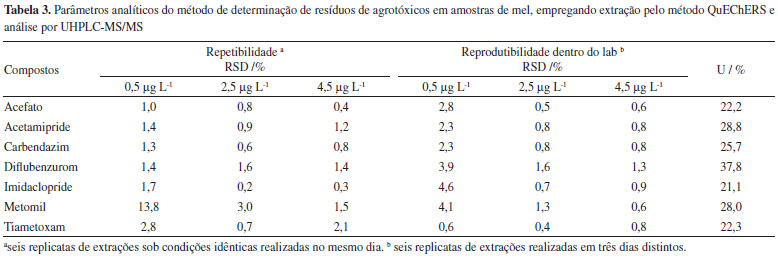
A incerteza de medição para o método analítico proposto para a determinação de resíduos de agrotóxicos em mel foi calculada conforme a metodologia top-down descrita no guia Eurachem.27 O preparo das soluções; o ajuste do modelo linear para as curvas analíticas, obtido pela regressão linear; a precisão intermediária e os ensaios de recuperação foram as principais fontes de incerteza associadas ao método.36,37 Os valores de incerteza expandida (U), expressos em percentagem para cada um dos agrotóxicos estudados, estão apresentados na Tabela 3. Como pode-se observar, a incerteza expandida apresentou valores abaixo de 50%, sendo os valores de incerteza na faixa de 21,1% a 37,8%. Esses resultados satisfazem os critérios de aceitação para metodologias analíticas que visam a determinação de resíduos de agrotóxicos em alimentos estabelecidos no documento SANTE/11813/2017. Dessa forma, o método proposto para determinação dos resíduos de acefato, acetamipride, carbendazim, diflubenzurom, imidaclopride, metomil e tiametoxam em amostras de mel foi validado seguindo os procedimentos sugeridos pelo protocolo europeu, satisfazendo todos os critérios de aceitação. No Material Suplementar é apresentado um resumo dos resultados obtidos para o estudo de validação do método proposto neste trabalho (Tabela 3S). Determinação de resíduos de agrotóxicos em amostras de mel da região do Triângulo Mineiro Sete amostras de mel oriundas de 6 cidades da região do Triangulo Mineiro foram analisadas seguindo o método proposto. A Tabela 4 apresenta os resultados das análises das amostras de mel. Observa-se que em todas as amostras foram detectados os agrotóxicos metomil e imidaclopride. Uma das amostras, identificada como amostra F, obtida de colmeia localizada na cidade de Uberaba, apresentou, além do imidaclopride e metomil, resíduos de carbendazim e de tiametoxam. Os compostos acefato, acetamipride e diflubenzurom não foram detectados em nenhuma das amostras analisadas.
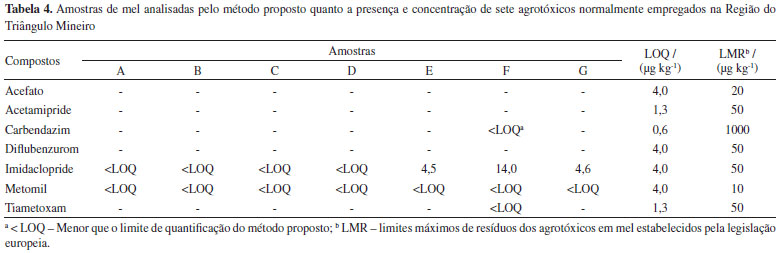
O imidaclopride e o metomil são inseticidas comumente empregados nas culturas de milho e soja que estão espalhadas por toda região do Triângulo Mineiro. Portanto, a presença desses dois resíduos em todas as amostras de mel analisadas, mesmo estando abaixo do LMR, é um forte indicativo do uso demasiado dos agrotóxicos nestes cultivos ao longo de toda a região do Triângulo Mineiro. Os outros dois resíduos detectados em uma das amostras analisadas são comumente empregados nas lavouras soja e de cana-de-açúcar, como fungicida (carbendazim) e inseticida (tiametoxam), respectivamente. Essa amostra é oriunda da cidade de Uberaba, indicando o elevado uso destes agrotóxicos nessa região e a consequente contaminação de colmeias de abelhas.
CONCLUSÃO Neste trabalho foi desenvolvido um método para a determinação de resíduos de agrotóxicos em amostras de mel, empregando o método de extração QuEChERS e a UHPLC-MS/MS. Um dos objetivos no desenvolvimento era a busca por uma metodologia de alta detectabilidade, capaz de detectar e quantificar baixos teores de resíduos de agrotóxicos em amostras de mel, que fornecesse resultados com alta exatidão e precisão para possibilitar a avaliação da contaminação deste produto apícola. De acordo com os resultados de validação e considerando os critérios de aceitação estabelecidos no documento SANTE/11813/2017 da comunidade europeia, o método desenvolvido apresenta todas essas características, alcançando os objetivos propostos. Aplicando o método em amostras de mel adquiridas da região do Triângulo Mineiro foi possível identificar a presença dos compostos metomil, carbendazim, imidaclopride e tiametoxam. Desta forma, podemos concluir que as regiões das quais as amostras de mel foram extraídas estão sob incidência dos agrotóxicos empregados nas culturas de grãos e cana-de-açúcar e que os produtos apícolas obtidos das colmeias nessas regiões estão sob risco real de contaminação por esses agrotóxicos.
MATERIAL SUPLEMENTAR Informações adicionais sobre os agrotóxicos e o método proposto neste trabalho estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem à Financiadora de Estudos e Projetos [processos 01.11.0135.00 e 01.13.0371.00] pelo suporte financeiro, à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) pelo apoio financeiro ao Programa de pós-graduação em Química e à Ourofino Agrociência S.A. pelo apoio e toda a infraestrutura laboratorial e de instrumentação disponibilizada para a realização do trabalho.
REFERÊNCIAS 1. Barros, G. S. C.; Castro, N. R.; Revista de Economia e Agronegócio 2017, 15, 156. 2. Ferreira, M. J. M.; Viana Júnior, M. M.; Pontes, A. G. V.; Rigotto, R. M.; Gadelha, D.; Ciência & Saúde Coletiva 2016, 21, 743. 3. Santos, C. F.; Otesbelgue, A.; Blochtein, B.; Plos One 2018, 13, 1. 4. http://desafio2050.org/arquivos/pdf/Eficiencia_no_uso_da_Terra_e_Insumos-Caio_Carbonari.pdf, acessada em Fevereiro 2020. 5. Hladik, M. L.; Vandever, M.; Smalling, K. L.; Sci. Total Environ. 2016, 542, 469. 6. Long, E. Y.; Krupke, C. H.; Nat. Commun. 2016, 7, article number 11629. 7. Carvalho, F. P.; Food and Energy Security 2017, 6, 48. 8. van Meter, R. J.; Adelizzi, R.; Glinski, D. A.; Henderson, W. M.; Environ. Toxicol. Chem. 2019, 38, 1052. 9. Sánchez-Bayo, F.; Goulson, D.; Pennacchio, F.; Nazzi, F.; Goka, K.; Desneux, N.; Environ. Int. 2016, 89-90, 7. 10. Schmolke, A.; Kearns, B.; O'Neill, B.; Apidologie 2018, 49, 637. 11. Gierer, F.; Vaughan, S.; Slater, M.; Thompson, H. M.; Elmore, J. S.; Girling, R. D.; Environ. Pollut. 2019, 249, 236. 12. Rissato, R. N.; Galhiane, M. S.; Knoll, F. R. N.; Andrade, R. M. B.; Almeida, M. V.; Quim. Nova 2006, 29, 950. 13. Beltrán, E.; Ibánez, M.; Sancho, J. V.; Cortés, M. A.; Yusà, V.; Hernández, F.; Food Chem. 2011, 126, 737. 14. Frenich, A. G.; Romero-González, R.; Aguilera-Luiz, M. M.; TrAC, Trends Anal. Chem. 2014, 63, 158. 15. Tette, P. A. S.; Oliveira, F. A. S.; Pereira, E. N. C.; Silva, G.; Glória, M. B. A.; Fernandes, C.; Food Chem. 2016, 211, 130. 16. Rejczak, T.; Tuzimski, T.; Open Chem. 2015, 13, 980. 17. Turner, N. W.; Subrahmanyam, S.; Piletsky, S. A.; Anal. Chim. Acta 2009, 632, 168. 18. Prestes, O. D.; Friggi, C. A.; Adaime, M. B.; Zanella, R.; Quim. Nova 2009, 32, 1620. 19. Prestes, O. D.; Adaime, M. B.; Zanella, R.; Sci. Chromatogr. 2011, 3, 51. 20. Lehotay, S. J.; Mass Spectrometry in Food Safety 2011, 747, 65. 21. González-Curbelo, M. Á.; Socas-Rodríguez, B.; Herrera-Herrera, A. V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M. Á.; TrAC, Trends Anal. Chem. 2015, 71, 169. 22. Rejczak, T.; Tuzimski, T.; Food Chem. 2017, 217, 225. 23. Anastassiades, M.; Lehotay, S. J.; Stajnbaher, D.; Schenck, F. J.; J. AOAC Int. 2003, 86, 412. 24. SANTE, Documento SANTE/11813/2017, Analytical quality control and method validation procedures for pesticide residues analysis in food and feed, 2017, disponível em: http://www.eurlpesticides.eu, acessada em Fevereiro 2020. 25. Cabrera, L. C.; Martins, M. L.; Primel, E. G.; Prestes, O. D.; Adaime, M. B.; Zanella, R.; Sci. Chromatogr. 2012, 4, 227. 26. https://pubchem.ncbi.nlm.nih.gov/compound/37123#section=MS-MS, acessada em Fevereiro 2020. 27. https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf, acessada em Fevereiro 2020. 28. Brasil, Agência Nacional de Vigilância Sanitária, Resolução RDC nº 166, de 24 de julho de 2017: Guia para validação de métodos analíticos. 29. Guidance for Industry - Q2B Validation of Analytical Procedures: Methodology, European Agency for the Evaluation of Medical Products (ICH): Londres, 1996. 30. United States Food and Drug Administration (US-FDA); Guidance for Industry, Analytical Procedures and Methods Validation for Drugs and Biologics - Julho, 2015. 31. European Commission, EU pesticides database, disponível em: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=pesticide.residue.selection&language=EN, acessada em Fevereiro 2020. 32. Barganska, Z.; Slebioda, M.; Namiesnik, J.; Food Control 2013, 31, 196. 33. Shamsipur, M.; Yazdanfar, N.; Ghambariam, M.; Food Chem. 2016, 204, 289. 34. Jovanov, P.; Guzsvány, V.; Lazic, S.; Franko, M.; Sakac, M.; Saric, L.; Kos, J.; J. Food Compos. Anal. 2015, 40, 106. 35. Barganska, Z.; Lambropoulou, D.; Namiesnik, J.; CRC Crit. Rev. Anal. Chem. 2018, 48, 447. 36. Carneiro, R. P.; Oliveira, F. A. S.; Madureira, F. D.; Silva, G.; Souza, W. R.; Lopes, R. P.; Food Control 2013, 33, 413. 37. Madureira, F. D.; Oliveira, F. A. S.; Souza, W. R.; Pontelo, A. P.; Oliveira, M. L.; Silva, G.; Food Addit. Contam., Part A 2012, 29, 665. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access