
 |
 |
 |
 |
 |
Artigo
| Theoretical calculations about the thermal stability and detonation character of nitramino-substituted pyrrole |
|
Butong Li*; Lulin Li; Yu Wang*
School of Chemistry and Materials Science, Guizhou Education University, Guiyang 051008, China Recebido em 27/11/2019 *e-mail: butong.lee@gmail.com, yuwang_gznc@163.com A series of nitramino-derivatives of pyrrole were designed through introducing the nitramino group into pyrrole ring to look for high-energy-density compounds. The molecular stabilities are explored thoroughly based on the calculated heats of formation, bond dissociation energy, and bond order at the B3PW91/6-311+G(d,p) level. The results indicated that the molecules designed in this paper have enough stability not only thermally but also kinetically. To evaluate the potential application as high-energy-density compounds, the detonation velocity and detonation pressure are calculated by using the Kamlet-Jacobs equation. Based on the calculation both of stability and detonation characters, four nitramino-derivatives of pyrrole (D1: 2,3-trinitramino-1H-pyrrole, D2: 2,3,4-trinitramino-1H-pyrrole, D3: 2,3,5-trinitramino-1H- pyrrole and E: 2,3,4,5- tetranitramino- 1H-pyrrole) are screened out as potential high-energy-density molecules for further study. INTRODUCTION For several decades, looking for high-energy-density materials (HEDMs) is ongoing.1-6 In this field, C-H-N-O type compound attract the most attention and many species with high stability and excellent detonation characters are found.7-10 Concluded from these compounds, the improved detonation characters can be derived from the high nitrogen content, large ring tension, more energetic groups, and so on. Based on these finding, the general strategy to design new high-energy-density compound is usually to introduce energy energetic groups into a parent body with large nitrogen content. Pyrrole is belong to nitrogen heterocycle and naturally possess higher nitrogen content than that of alkane. In addition, five hydrogen atoms are located on its five-membered ring and further modification by energetic groups can be achieved. Therefore, it is a good parent body to design new high-energy-density compounds. Pyrrole is found first in 1870, and it has been applied in a lot of fields including medicine,11 natural molecule,12 electroluminescence devices,13 and so on. However, the application in the field of high-energy-density compounds is not entirely explored for the derivative of pyrrole until now, although the interaction between the nitro groups and the pyrrole ring has been proved to stable the derivatives effectively. In addition, the excellent detonation characters have been confirmed in theory.14 It can be concluded that pyrrole is really an acceptable parent body to design new high-energy-density materials. Except nitro groups, nitramino group is another one drawing the attention. Compared to nitro group, nitramino can improve the detonation characters more effectively. Only two nitramino groups can increase the specific weight of tetrazole salt to a surprising 2.177 g cm-3, which is better than that of hexanitro- hexaazaisowurtzitane (CL-20) and almost the highest value already known for H-C-N-O type explosive.15 This implied us the complex structures like CL-20 are not necessary for the next-generation explosive, and the planar molecule accompanied with nitramino groups is the point to increase the specific weight and detonation characters. Therefore, the hydrogen atoms of pyrrole were substituted by nitramino groups successively to design new high-energy-density compounds, and the thermal stability, kinetic stability, and the detonation characters are explored by using the density functional method accompanied with the Kamlet-Jacobs equation.
CALCULATION METHOD All geometries were optimized by using the G03 package at the B3PW91/6-311+G(d,p) level the on a cluster of computers.16 The heats of formation (HOFs) of nitramino-substituted derivatives of pyrrole were evaluated through the isodesmic reaction17 as following: 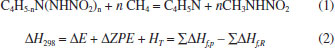 in which ΔH298 is the enthalpy change of the reaction at 298 K; ΔE, ΔZPE, and ΔHT were the change of the total energy, the zero point energy, and the enthalpy change from 0 to 298 K, respectively. ΣΔHƒ,p and ΔHƒ,R are the sums of the heats of formation of the products and the reactants, respectively. As to the reference compounds of CH4, CH3NO2 and C4H5N, the heats of formation are obtained from the NIST web book. The empirical Kamlet-Jacobs equation was developed in 1968 to predict the detonation pressure and detonation velocity of high-energy-density compounds composed of H, C, N, and O atoms,18,19 and the reliability has been proved extensively.20-22 Therefore, it was adopted in this paper and the equations are as following: 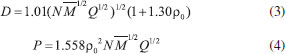 in which D is the detonation velocity (km s-1); P is the detonation pressure (GPa); N is the moles of gas produced per gram of explosive; The strength of chemical bond, which could be evaluated by bond dissociation energy (BDE), can be used to measure the strength of a chemical bond and is fundamental to understanding chemical processes. The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction A - B → A + B, which is defined as the bond dissociation enthalpy of the bond A-B. Therefore, at 0 K, BDE can be represented as eq (5): 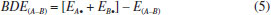 BDE(A-B) is the BDE of the bond A−B, E(A-B), EA• and EB• are the total energies of parent molecule and the corresponding radicals, respectively.
RESULTS AND DISCUSSION Heats of formation and molecular stabilities Heats of formation is a parameter to evaluate the stability and energy content of high-energy-density compounds. In general, greater is the heat of formation, more energy content and less stability a molecule has. It can be calculated in theory by using the isodesmic reaction, and the reliability are proved by many former research. So, the method was used in this paper and the final results are listed in Table 1. The optimized structures are shown in Figure 1.
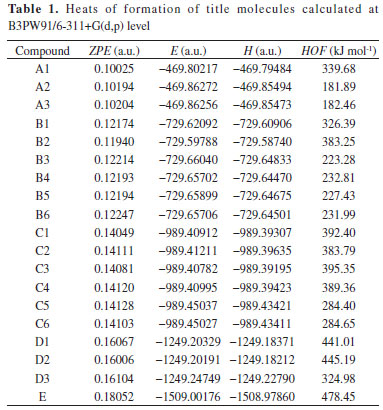
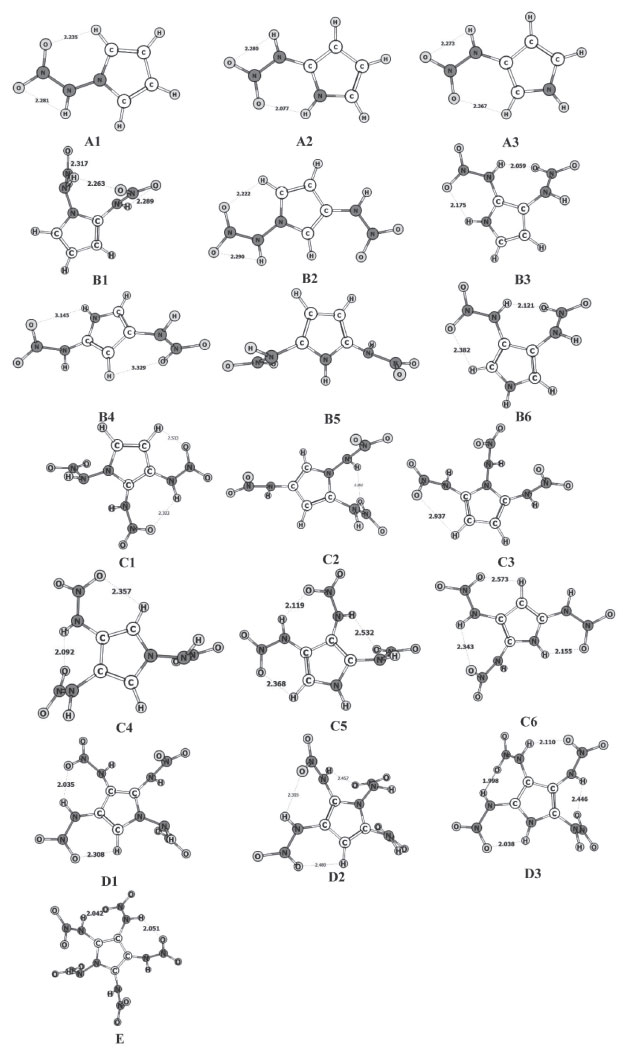 Figure 1. Structures of title molecules optimized at the B3PW91/6-311+G(d,p) level
All of the heats of formation are all positive values, which is the representative feature of high-energy-density compound and indicate the energetic nature of title molecules. The linear regression is performed between the substituent number and the HOFs, but no evident linear correlation are found for the low R2 value. This situation may be attributed to the hydrogen bonding effect. However, a rising tendency still can be confirmed, that is the HOF is increased from 181.89 kJ mol-1 of mono-substituted derivatives to 478.45 kJ mol-1 of penta-substituted derivatives. Whatever, the positive and large heats of formation of title molecules are suited to the basic requirements of high-energy-density compounds, and the high detonation characters can be expected. Bond dissociation energy and bond orders The kinetic stability is very important for high-energy-density compounds, which is described generally by the bond dissociation energy in theory. In detonation reaction, the weakest bond can initiate the entire reaction and named as the "trigger bond". Therefore, the kinetic stability of compounds can be tested by the bonding strength of trigger bond. Here, the wiberg bond orders were calculated to sort the trigger bond out by the least value, and all of them are located at the N−N bonds of nitramino group. The calculations of bond dissociation energy were performed only for the trigger bonds to save time. The final results are listed in Table 2.
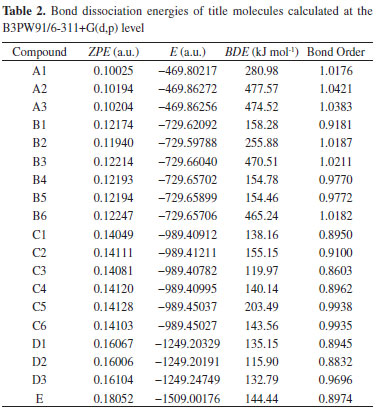
From Table 2, it is found the bond dissociation energies of title molecules are quite large, from 477.57 kJ mol-1 of A2 to 115.90 kJ mol‑1 of D1. Compared to the bond dissociation energy (145.62 kJ mol-1) of RDX, the bond dissociation energy of title molecules are pretty large enough to stable the molecules kinetically. The attempt to correlate the bond order with the bond dissociation energies was also performed, but the consistent relationship can not be found for all of the data. For instance, the bond order (1.01760) of A1 is less than that (1.01870) of B2, but the bond dissociation energy (280.98 kJ mol-1) of former is greater than that (255.88 kJ mol-1) of later. It is well known the bond order can not be detected in experiment, so the bond dissociation energy is a more reliable parameter to predict the molecular stability. Detonation characters The detonation characters are determinate characters for high-energy-density molecules. In general, the detonation pressure and detonation velocity can be calculated and used to evaluate these characters by using the Kamlet-Jacobs equation in theory. The final data are listed in Table 3 with the reference data of RDX and HMX.
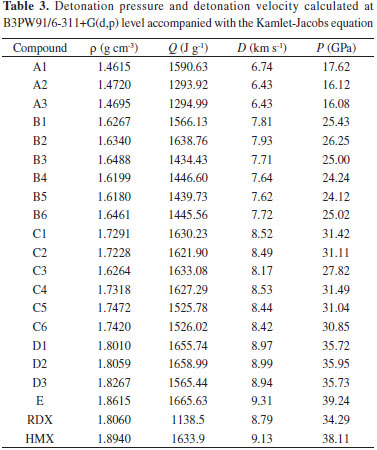
From Table 3, the excellent detonation pressure and detonation velocity can be found for the tetra- and penta-substituted derivatives, which entirely exceed the 8.79 km s-1 and 34.29GPa of RDX. More abstractive is the specific weight. For a long time, a specific weight above 2.0 g cm-3 is regarded as the fundamental condition for next generation high-energy-density compounds. However, the largest specific weight is 1.8615 g cm-3 for penta-substituted pyrrole accompanied with the detonation pressure of 39.24GPa and detonation velocity of 9.31km/s, which are all better than that of HMX. Obviously, large specific weight is not always the determinate factor to improve the detonation characters and the nitramino is an effective group to design high-energy-density compounds with low specific weight.
CONCLUSION Based on our calculation, all of the heat of formation are positive for title molecules, which satisfy the basic requirement of high-energy-density compounds. Furthermore, the large values of heats of formation implied the energetic nature of title molecules. All of derivatives have large bond dissociation energies better than that of RDX and HMX. So, excellent kinetic stability are confirmed for title molecules. The detonation pressure and detonation velocity are confirmed for all of the derivatives, in which the tetra- and penta-substituted derivatives of pyrrole have better parameters than those of RDX and HMX and are screened out as the potential high-energy-density compounds. In addition, it is found high specific weight is not the most important integrant to design high-energy-density compounds and the nitramino group can improve the detonation characters with a low specific weight.
ACKNOWLEDGMENTS This project was supported by the Natural Science Foundation of Guizhou Province (Nos. [2018]5778-09 and J20181122) and the Natural Science Foundation of Guizhou Education University (No. 14BS017 and 2019ZD001)
REFERENCES 1. Li, B.; Zhou, M.; Peng, J.; Li, L.; Guo, Y.; J. Mol. Model. 2019, 25, 23. 2. Cheng, L. P.; Xu, Y. Q.; Wang, G. L.; He, H. H.; J. Mol. Model. 2012, 18, 1927. 3. Janning, J. D.; Ball, D. W.; J. Mol. Model. 2010, 16, 857. 4. Ghule, V. D.; Sarangapani, R.; Jadhav, P. M.; Tewari, S. P.; J. Mol. Model. 2011, 17, 1507. 5. Kumar, A. S.; Ghule, V. D.; Subrahmanyam, S.; Sahoo, A. K.; Chemistry 2013, 19, 509. 6. Bittererová, M.; Brinck, T.; Östmark, H.; J. Phys. Chem. A 2000, 104, 11999. 7. Kolb, J. R.; Rizzo, H.; Propellants, Explos., Pyrotech. 1979, 4, 10. 8. Li, B.; Chi, W.; Li, L.; Chinese J. Struct. Chem. 2016, 35, 1306. 9. Li, B.; Li, L.; Chen, S.; J. Mol. Model. 2019, 25, 298. 10. Simpson, R.; Urtiew, P.; Ornellas, D.; Moody, G.; Scribner, K.; Hoffman, D.; Propellants, Explos., Pyrotech. 1997, 22, 249−255. 11. Richards, J. J.; Reed, C. S.; Melander, C.; Bioorg. Med. Chem. Lett. 2008, 18, 4325. 12. Domagala, A.; Jarosz, T.; Lapkowski, M.; Eur. J. Med. Chem. 2015, 100, 176. 13. Ono, N.; Tsukamura, C.; Nomura, Y.; Hironaga, H.; Murashima, T.; Ogawa, T.; Adv. Mater. 1997, 9, 149. 14. Li, B.-T.; Li, L.-L.; Li, X.; Mol. Simul. 2019, 45, 1459. 15. Fischer, D.; Klapotke, T. M.; Stierstorfer, J.; Angew. Chem., Int. Ed. Engl. 2015, 54, 10299. 16. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian, Inc., Pittsburgh PA 2003, Gaussian 03, Revision B.01. 17. Hehre, W. J.; Ditchfield, R.; Radom, L.; Pople, J. A.; J. Am. Chem. Soc. 1970, 92, 4796. 18. Kamlet, M. J.; Ablard, J. E.; J. Chem. Phys. 1968, 48, 36. 19. Kamlet, M. J.; Jacobs, S. J.; J. Chem. Phys. 1968, 48, 23. 20. Liu, T.; Jia, J.; Li, B.; Gao, K.; Chin. J. Struct. Chem. 2019, 38, 688. 21. Rice, B. M.; Hare, J. J.; J. Phys. Chem. A 2002, 106, 1770. 22. Chi-Durán, I.; Enríquez, J.; Manquián, C.; Fritz, R. A.; Vega, A.; Serafini, D.; Herrera, F.; Singh, D. P.; ACS Omega 2019, 4, 14398. |
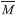 is the mean molecular weight of the gaseous detonation products; Q is the heats of detonation (cal g-1); ρ0 is the theory specific weight (g cm-3) and calculated from the molar weight (M) divided by the average value molar volume (V), which was defined as the volume of 0.001 electrons Bohr-3 electron density envelope and obtained from the arithmetic average value of 1000 singe point molar volumes by using Monte Carlo integration.
is the mean molecular weight of the gaseous detonation products; Q is the heats of detonation (cal g-1); ρ0 is the theory specific weight (g cm-3) and calculated from the molar weight (M) divided by the average value molar volume (V), which was defined as the volume of 0.001 electrons Bohr-3 electron density envelope and obtained from the arithmetic average value of 1000 singe point molar volumes by using Monte Carlo integration.On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access