
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Determinação de docetaxel e metabólitos em plasma por UPLC-MS/MS: desenvolvimento de metodologia analítica e aplicação clínica Determination of docetaxel and metabolites in plasma by UPLC-MS/MS: method development and clinical application |
|
Suziane RaymundoI; Victória Vendramini MüllerI; Mariane TegnerI; Andiara ArtmannI; Natália B. AndriguettiI; Gilberto SchwartsmannII; Rafael LindenI; Marina V. AntunesI,*
I. Instituto de Ciências da Saúde, Universidade Feevale, 93525-075 Novo Hamburgo - RS, Brasil Recebido em 09/08/2019 *e-mail: marinaantunes@feevale.br A highly sensitive ultra-performance liquid chromatography tandem mass spectrometry method for the determination of Docetaxel and metabolites (M1+M3) in human plasma samples was developed and validated. The method employs a liquid-liquid extraction and a reversed phase separation Acquity® C18 column BEH C18 (150 x 2.1 mm, 1.7 µm). Mobile phase was a mixture of formic acid 0.1% (eluent A) and acetonitrile plus 0.1% formic acid (eluent B) in isocratic mode. Total analytical run time was 7 min. The interval of method linearity interval was 50 to 3000 ng mL-1 for docetaxel and 5.0 to 300 ng mL-1, for M1+M3 metabolites. Intra-assay and inter-assay precision was coefficient of variation 2.5-15.7% and 5.9-12.6%, respectively and accuracy was 89.0-114.0%. The method was successfully applied in the measurement of docetaxel plasma levels and area under the curve calculation in 31 cancer patients, demonstrating adequate analytical performance for the clinical application in the therapeutic drug monitoring and pharmacokinetics studies. INTRODUÇÃO O docetaxel (Taxotere®) é um derivado semi-sintético do teixo Europeu Taxus Baccata, sendo utilizado para tratamento de uma variedade de tumores, particularmente tumores de mama, próstata, ovário, pulmão, adenocarcinoma gástrico, câncer gástrico e cabeça e pescoço.1,2 Seu uso foi aprovado em 1996 pelo Food and Drug Administration (FDA) e vem demonstrando benefícios aos pacientes, como aumento da expectativa de vida e redução da progressão do tumor, apresentando atividade antitumoral duas vezes mais potente que o seu precursor placlitaxel.3 A dose clinicamente recomendada do docetaxel (DTX) varia entre 60 a 100 mg/m2 de acordo com o tipo de tumor, sendo atualmente baseada na superfície de área corporal do paciente, administrada por infusão intravenosa 1 hora a cada 3 semanas.1,4-7 Apesar do benefício terapêutico, os efeitos hematológicos como neutropenia, neutropenia febril, anemia e trombocitopenia são fatores limitantes na dose do fármaco, levando a interrupção da quimioterapia e consequentemente redução na taxa de cura.5 O metabolismo do DTX é mediado pelas enzimas hepáticas CYP3A4 e CYP3A5 do citocromo P450, que através de uma reação de oxidação na cadeia lateral de terc-butil-propionato forma os quatro metabólitos M2, M4 e diastereoisômeros M1 e M3, com atividade citotóxica reduzida.3,5,8 A entrada do DTX ao hepatócitos é dada através do transportador de influxo SLCO1B3, e sua eliminação ocorre através de proteínas transportadoras de efluxo, ABCB1 (glicoproteína-P), ABCC2 e a proteína de resistência multi-droga 2 (MRP2).2,9-11 Variações na resposta terapêutica do DTX podem estar relacionadas a polimorfismos nos genes CYP3A4, CYP3A5, ABCC2, ABCB1, SLCO1B3 e CYP1B1, levando à redução do metabolismo do fármaco e à toxicidade.2,8,12,13 Considerando que o DTX possui uma farmacocinética variável e uma gama de efeitos adversos o interesse pelo seu monitoramento terapêutico de fármacos (MTF) tem emergido na oncologia. A área de superfície corporal (ASC) é um parâmetro indicativo da exposição sistêmica ao DTX e tem sido sugerida como um preditor de neutropenia nos pacientes tratados com o fármaco.14-18 No entanto, diferentemente de outros quimioterápicos em que a ASC já está bem estabelecida, para o DTX ainda não há um valor consensual, tendo sido relatado que a melhor resposta terapêutica está associada com valores alvo de ASC entre 2,5 a 3,7 e 4,9 mg.h/L para doses de 50, 75 e 100 mg/m2 respectivamente.18 Ensaios para determinação do DTX têm sido descritos na literatura, sendo que a utilização do LC-MS/MS para determinação de fármacos em fluidos biológicos é especialmente atrativa, devido a sua alta sensibilidade e seletividade.19-21 Em vista disso, o objetivo do presente estudo foi desenvolver e validar um método simples e rápido para a determinação do DTX e metabólitos M1 e M3 em plasma por LC-MS/MS. O método foi aplicado na avaliação da exposição ao DTX em um grupo de pacientes oncológicos no Sul do Brasil, a partir da estimativa de sua ASC em um modelo de amostragem limitado.15 Além disso, a dosagem dos metabólitos permite avaliar as razões de metabolismo para CYP3A, podendo ser utilizada como uma alternativa para a fenotipagem dessa via. Vale ressaltar que este é o primeiro estudo com aplicação clínica no monitoramento dos níveis de DTX e seus metabólitos M1+M3 em uma população brasileira.
PARTE EXPERIMENTAL Reagentes e soluções O Docetaxel, Docetaxel-D5 e metabólitos (M1+M3) foram obtidos da Research Chemicals (Toronto, Canada). Ácido fórmico, metanol e acetonitrila em grau HPLC foram adquridos da Merck (Darmstadt, Germany). O acetato de sódio foi obtido da Mallinckrodt Baker (Xalostoc, Mexico). Água purificada foi obtida através de um sistema Elga Purelab Ultra® da Veolia Labwater (High Wycombe, UK). Preparação dos reagentes e soluções Soluções-mãe de DTX e metabólitos (M1 e M3) foram preparadas pela dissolução em metanol afim de se obter a concentração de 1 mg mL-1. A partir das soluções mãe foram preparadas soluções estoque metanólicas nas concentrações de 60; 50; 30; 20; 14; 10; 5,0; 2,0 e 1,0 µg mL-1 para o DTX e 6,0; 5,0; 3,0; 2,0; 1,4; 1,0; 0,5; 0,2 e 0,1 µg mL-1 para a mistura dos metabólitos diastereoisômeros M1+M3. Amostras calibradoras foram preparadas em plasma isento dos analitos diluindo-se as soluções estoque em 1:20, como descrito no item linearidade. Solução de DTX-D5 (padrão interno, PI) foi preparada por dissolução em metanol, na concentração de 1,0 µg mL-1. Equipamentos e condições cromatográficas Foi utilizado um cromatógrafo líquido de alta eficiência Dionex Ultimate 3000 acoplado a espectrômetro de massas sequencial (UPLC-MS/MS). A análise foi realizada em um espectrômetro TSQ Quantum Access MAX triplo quadruplo, controlado por software Xcalibur, proveniente da Thermo Scientific (San Jose, Estados Unidos). A separação cromatográfica foi realizada em coluna Acquity BEH C18 (150 x 2,1 mm; 1,7 µm), proveniente da Waters (Milford, Estados Unidos), mantida a 30 °C durante a análise. A fase móvel utilizada foi composta por mistura de água e acetonitrila com 0,1% de ácido fórmico (45:55, v/v), eluída a uma vazão de 0,2 mL min-1. Nessas condições o tempo total da corrida cromatográfica foi de 7 minutos. A ionização foi realizada com fonte electrospray (EIS) no modo positivo, cujas condições foram definidas após infusão de solução contendo os padrões analíticos na concentração de 1 µg mL-1 em metanol, com voltagem de 4000 V, gás nebulizador nitrogênio com fluxo de 30 Arb, gás auxiliar nitrogênio com fluxo de 10 Arb, gás de colisão argônio, temperatura de vaporizador 370 °C e temperatura de capilar 284 °C. A aquisição foi realizada no modo MRM através das transições m/z 830→247 para quantificação e m/z 830→303 e 549 para qualificação do DTX, m/z 844→300 para quantificação e m/z 844→317 e 549 para qualificação dos metabólitos M1+M3, m/z 835→247 para quantificação e m/z 835 e 554 para qualificação do PI docetaxel-D5. Preparo das amostras As amostras foram processadas em extração líquido-líquido, onde uma alíquota de 500 µL de plasma foi acrescentada de 50 µL do padrão interno (PI) (docetaxel-D5 1 µg/mL) e 3,5 mL do solvente de extração metil-tert-butil-éter (MTBE). Após homogeneização por 10 minutos os extratos foram centrifugados a 3.000 rpm por 10 minutos. Uma alíquota de 3,2 mL da fase orgânica foi transferida para novo tubo e evaporada a 45 ºC em concentrador a vácuo. O extrato seco foi retomado com 100 µL da mistura de fase móvel e acetato de sódio 0,2 mmol L-1 (90:10, v/v). O acetato de sódio foi incluído na fase móvel usada para retomar os extratos formar adutos de sódio e induzir a sua fragmentação. Os extratos foram centrifugados e posteriormente filtrados com membrana de PTFE com poro de 20 µm. Uma alíquota de 25 µL foi injetada em sistema UPLC-MS/MS para a detecção dos adutos de sódio do DTX, metabólitos e PI. Validação do método A validação do método empregou parâmetros essenciais para fornecer a confiabilidade e aceitabilidade da análise preconizados pelo Food and Drug Administration,22 tais como: seletividade, linearidade, sensibilidade, precisão, exatidão, estabilidade, efeito matriz e rendimento da extração. Seletividade Foram avaliadas amostras de plasma de 6 indivíduos que não eram usuários do fármaco e processadas conforme descrito no item "preparação das amostras" para verificar a presença de picos cromatográficos que possam interferir na detecção do analito e padrão interno (PI). Linearidade A linearidade dos modelos de calibração foi avaliada em 9 níveis, em sextuplicata para cada nível. Soluções calibradoras foram preparadas diluindo-se 20 vezes as soluções de trabalho DTX e M1+M3 para se obter concentrações em plasma de: 50; 100; 250; 500; 700; 1.000; 1.500; 2.500; e 3.000 ng mL-1 para DTX e 5; 10; 25; 50; 70; 100; 150; 250; e 300 ng mL-1 para M1+M3. As 6 réplicas de cada calibrador foram extraídas conforme item "preparação das amostras". Curvas de calibração foram construídas por cálculos da razão entre a área dos picos dos analitos e a área do padrão interno (y), e relacionando essas razões com concentração nominal das amostras de calibração (x). A homocedasticidade dos dados de calibração foi avaliada com o teste F com nível de confiança de 95%. As curvas foram ajustadas através de regressão linear empregando-se diversos fatores ponderais (1/x, 1/x0,5, 1/x2, 1/y, 1/y0,5, 1/y2). Os modelos de calibração foram avaliados pelos coeficientes de correlação (r) e pelo erro percentual cumulativo (Σ%ER), de acordo com Almeida et al.23 Sensibilidade A sensibilidade foi determinada através do ponto mais baixo da curva de calibração (50 e 5 ng mL-1 para DTX e M1+M3, respectivamente). Esse experimento foi incluído na avaliação da precisão e exatidão do método (limite inferior de quantificação, LIQ) e foi avaliado em triplicata durante três dias. O coeficiente de variação (CV%) máximo aceitável intra ensaio e inter ensaio foi de 20%, e a exatidão entre 80% e 120%. Ensaios de precisão e exatidão A precisão e exatidão do método foram avaliadas com amostras controle contendo DTX e M1+M3 nas concentrações 75 e 7,5 ng mL-1 (controle de qualidade baixo - CQB); 90 e 75ng mL-1 (controle de qualidade médio - CQM), 2.700 e 250 ng mL-1 (controle de qualidade alto - CQA), respectivamente. As amostras foram processadas em triplicata e repetidas durante 5 dias diferentes. A precisão intra ensaio (variação entre as análises de um mesmo dia) e inter ensaio (variação entre os dias analisados) foram calculadas pela análise de variância (ANOVA), enquanto que a exatidão foi calculada como percentagem média obtida do valor teórico adicionado na amostra. O coeficiente de variação (CV%) aceito para a precisão foi de até 15%, e a exatidão entre 85 e 115%. Estabilidade A avaliação da estabilidade foi realizada através da análise de amostras de plasma controle de qualidade (CQB e CQA) em sextuplicata, os quais foram processados conforme descrito anteriormente. Os extratos obtidos de cada controle foram reunidos e injetados sob condições de uma corrida analítica com intervalos de 1 h, durante 12 h. A estabilidade foi avaliada através da diferença percentual entre o valor das áreas dos analitos obtidas no tempo inicial e as obtidas nos demais tempos, sendo considerada aceitável se todos os valores encontrados estivessem na faixa de 90 a 110% da razão das áreas entre os analitos e o PI obtida no tempo inicial. A estabilidade em ciclos de congelamento e descongelamento foi avaliada em amostras controle (CQB e CQA), as quais foram preparadas em triplicata para cada controle e analisadas no dia da preparação das amostras. Após três ciclos de congelamento e descongelamento as amostras foram reanalisadas em triplicata. Esse teste foi avaliado através da diferença percentual entre a média das concentrações obtidas no dia da preparação das amostras controle e as médias obtidas no ciclo, sendo considerado aceitável se todos os valores encontrados estiverem na faixa de 90 a 110% das concentrações obtidas nas amostras preparadas antes do ciclo. Efeito matriz e rendimento da extração Para avaliação do efeito matriz e rendimento da extração foram preparadas três séries de amostras controle de qualidade (CQB, CQM e CQA) e extraídas conforme segue: (A) Soluções contendo DTX e M1+M3 e padrão interno (DTX-5) foram adicionadas a fase móvel e injetadas diretamente no UPLC-MS/MS de forma que a concentração final seja equivalente a 100% da extração. (B) Amostras de cinco fontes diferentes foram extraídas, e re-extraídas com PI, DTX e M1+M3 em fase móvel. (C) Amostras de plasma de cinco fontes diferentes enriquecidas com os controles de qualidade foram extraídas em quintuplicada. O efeito matriz (ME) em ionização foi estimado com as percentagens de redução ou aumento de DTX e M1+M3 e a área do padrão interno na re-extração das amostras enriquecidas com DTX, M1+M3 e PI (B), comparando com a injeção direta das soluções (A), usando o cálculo ME= [100%-B/A%]. O rendimento da extração (RE) foi calculado comparando a área do analito com a área do padrão interno antes da extração (C) e após a extração (B), usando a formula RE= C/B%. Aplicação do método O método desenvolvido foi aplicado nas dosagens de 31 amostras de plasma obtidas de pacientes em tratamento com o DTX no Hospital de Clínicas de Porto Alegre (HCPA). Os pacientes incluídos no estudo possuem câncer de próstata, mama ou pulmão, idade superior a 18 anos e somente foram aceitos após terem recebido esclarecimentos em relação ao estudo e assinarem o Termo de Consentimento Livre e Esclarecido (TCLE). O estudo foi aprovado pelo Comitê de Ética em Pesquisa do Hospital de Clínicas de Porto Alegre sob o parecer 1.055.723. Para calcular a exposição ao DTX através da estimativa da ASC, foi empregado um modelo de amostragem limitada descrito e validado por Engels et al.24 e adaptado por Kraff et al.25 Dessa forma, duas amostras de 8 mL de sangue venoso em tubo contendo EDTA foram colhidas dos participantes, como segue: a primeira no dia da administração do DTX, 5 minutos antes do final da infusão; e a segunda 60 ± 10 minutos após a infusão com o DTX. Após as coletas, as amostras de plasma foram centrifugadas e armazenadas em freezer -80 °C até as análises. Para a realização do cálculo da ASC em modelo bayesiano, foi utilizada uma planilha Excel® disponibilizada pelo Departamento de Farmácia Clínica da Universidade Bonn (Bonn, Alemanha), na qual foram preenchidos os dados de dose utilizada, horários de inicio e término da infusão, horários da primeira e segunda coletas, bem como, as concentrações obtidas nas duas amostras coletadas.25
RESULTADOS E DISCUSSÃO Cromatografia e preparação da amostra O UPLC-MS/MS mostrou-se apropriado para determinação do DTX e M1+M3 em amostras de plasma. Foi utilizada uma coluna de fase reversa octadecil e fase móvel com eluição isocrática, que permitiu uma separação eficiente dos analitos dentro de uma corrida de 7 minutos, com tempos de retenção de 5,5 para DTX e PI e 2,8 minutos para M1+M3. O UPLC-MS/MS é um método que apresenta intrinsecamente alta seletividade, possibilitando a aquisição dos cromatogramas através de três transições do íon molecular para seus fragmentos (1 de quantificação e 2 de confirmação). Dessa forma, durante as análises de seletividade não foram identificados picos interferentes que possam impactar na detecção dos analitos. Na Figura 1 são apresentados cromatogramas adquiridos no método.
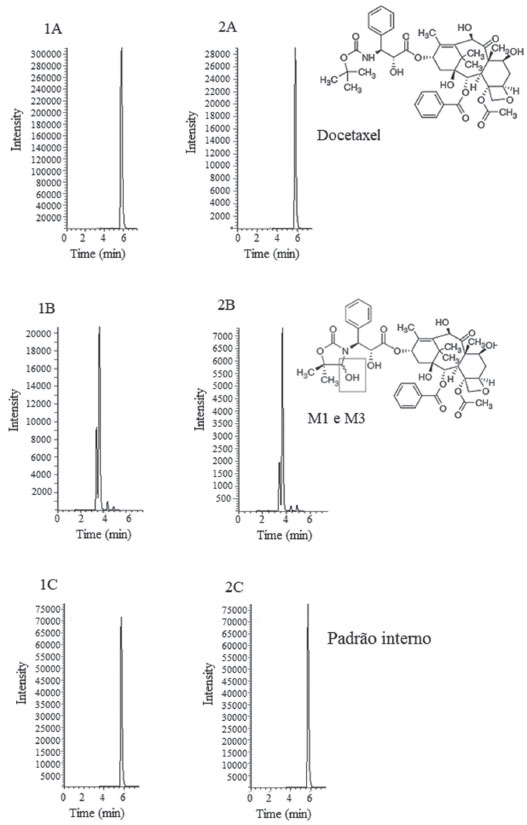 Figura 1. Cromatogramas obtidos nas análises de plasma. (A) Amostra de paciente coleta 1 1A: Docetaxel 1.125,6 ng mL-1, 1B: M1+M3 25,8 ng mL-1, 1C: Padrão interno; (B) Amostra de paciente coleta 2 2A: Docetaxel 85,1 ng mL-, 2B: M1+M3 23,9 ng mL-1, 2C: Padrão interno; Transições: DTX (m/z 830 → 247; 830 → 303; 830 → 549), Padrão interno (m/z 835 → 554; 835 → 427; 835 → 427), M1/M3 (m/z 844 → 300; 844 → 317;844 → 549)
A técnica utilizada para o preparo das amostras foi relativamente simples, com extração líquido-líquido, seguida de evaporação da fase orgânica. O acetato de sódio adicionado a amostra permitiu a formação dos adutos de sódio e uma ionização mais eficiente dos analitos com alcance de uma maior sensibilidade no ensaio. Validação do método O método foi linear nos intervalos de 50 a 3000 ng mL-1 para o DTX e 5 a 300 ng mL-1 para os metabólitos. Todas as curvas de calibrações incluíram este amplo intervalo, devido a grande diferença nas concentrações encontradas entre as coletas 1 e 2 realizadas neste estudo. Os dados da curva de calibração possuem heterocedasticidade significativa, com Fexp = 326,72 e 25,43 (Ftab (9. 5. 0,95) = 3,48) para os DTX e metabólitos, respectivamente. Diferentes fatores ponderais foram avaliados para a seleção modelo de regressão a ser empregado. Dentre os modelos avaliados, a regressão utilizando o fator ponderal 1/x apresentou menor ΣResíduos, com valores de -3,41 × 10-13 e -2,7 × 10-13 e menor Σ%ER, com valores de 1,47 × 10-13 e 1,86 × 10-13 e com coeficientes de correlação de 0,999 (DTX) e 0,9929 (M1+M3). Os resultados de precisão e exatidão foram adequados, conforme a Tabela 1. A precisão intra-ensaio apresentou CV de 2,5 a 10,1% e a precisão inter-ensaio CV entre 5,9 e 12,6%, demonstrando adequada repetibilidade do método. A exatidão foi estimada entre 89 e 114%, também dentro dos critérios de aceitação para métodos bioanalítcos preconizados pelo Food and Drug Administration.22
O efeito matriz foi avaliado para verificar a ocorrência de supressão de íons ou efeito de aumento causado por co-eluição de moléculas originarias da matriz da amostra com os compostos de interesse. Esta avaliação demonstrou um efeito de ionização entre -12 e +15%, indicando que a co-eluição com os componentes da matriz parecem ter efeito mínimo com os compostos de interesse (Tabela 1). As amostras foram estáveis durante o período de 12 horas, com variação máxima de 6,1% para o DTX e 5,5% para M1+M3, permitindo o processamento de grandes lotes analíticos. A avaliação da estabilidade após três ciclos de congelamento e descongelamento atenderam os critérios de aceitação com variações entre 98,2 e 109,1% para o DTX e 99,4 e 110,3% M1+M3, respectivamente (Tabela 2).
A extração líquido-líquido com metil-tert-butil-éter (MTBE) mostrou alta recuperação dos analitos com média de 90% para DTX e 91% para os metabólitos. O método apresentou sensibilidade satisfatória, sendo que o limite inferior de quantificação foi menor do que o nível mais baixo encontrado em amostras clínicas. Cabe destacar ainda, que em estudos anteriores20,21,26 os metabólitos formados a partir do metabolismo do DTX, não foram quantificados como no presente estudo. O desenvolvimento do método analítico descrito aqui, permitiu a quantificação dos metabólitos, mesmo que as concentrações presentes nas amostras clínicas sejam baixas. Entretanto, em contraste com Rosing et al.,27 em que a detecção por LC-UV foi um fator limitante, a detecção por espectro de massas utilizada neste estudo permitiu adequada sensibilidade para detecção de M1+M3. No estudo de Guitton et al.,28 apesar da quantificação dos metabólitos M1+M3, o limite superior de quantificação do método de 1000 ng mL-1 não abrange a maioria das amostras clínicas obtidas antes do final da infusão, sendo necessária etapas de diluição das amostras, além disso a corrida cromatográfica possui tempo reduzido. Aplicação do método O método desenvolvido foi aplicado em 31 amostras de pacientes em tratamento quimioterápico com o DTX para câncer de mama (n=8), da próstata (n=12), pulmonar (n=5) e esofágico/gástrico (n=7), sarcomatoide (n=1) e osteossarcoma quadril (n=1). Todos os pacientes receberam uma dose padrão do quimioterápico de 75 mg m-2 por infusão intravenosa durante 1 hora. O DTX foi administrado em regime de monoterapia ou associado a ácido zoledrônico, leuprorrelina, carboplatina e trastuzumab, cisplatina, prednisona e ciclofosfamida. As concentrações do DTX plasmáticas medidas 5 minutos (± 5 minutos) antes do final da infusão estiveram no intervalo de 779,0 a 3842,2 ng mL-1, com média de 2116,9 ng mL-1 (Shapiro-Wilk = 0,9808 p =0,82). As concentrações do fármaco mensuradas 60 minutos (± 10 minutos) após o término da infusão foram significativamente menores do que as inicialmente medidas, apresentando concentrações entre 49,78 e 394,8 ng mL-1, com mediana de 97,15 ng mL-1 (Shapiro-Wilk = 0,769 p <0,001). Isso ocorre em virtude do rápido tempo de meia-vida do fármaco (4, 36 minutos e 11,1 horas para as fases α, β e γ, respectivamente).29 A estimativa da ASC dos pacientes seguiu um modelo Bayesiano, seguindo amostragem limitada proposto por Engels et al.15 e adaptado por Kraff.25 Os valores de ASC ficaram dentro do intervalo de 2,4 e 4,9 mg h L-1, com mediana de 3,0 mg h L-1 (P25-75 de 2,72‑3,27 mg h L-1) (Shapiro-Wilk = 0,8875; p <0,01). O coeficiente de variação percentual (CV%) dos valores de ASC foi de 18,1%, apresentado variabilidade interpaciente menor que a encontrada em estudos anteriores (23 a 56%), realizados com pacientes europeus, asiáticos e americanos.15,18,21,30,31 Entretanto, até o momento, ainda não há relatos em relação a farmacocinética do DTX na população brasileira, dessa forma é importante considerar diferenças étnicas, diferenças no metabolismo, transporte e excreção do fármaco. Dentre os pacientes avaliados 4 (12,9%) apresentaram ASC acima do alvo terapêutico previamente proposto. Conforme descrito por Ma et al.,18 uma ASC de 2,5 a 3,7 mg h L-1 seria adequada para efeitos clínicos benéficos durante a quimioterapia, com redução nas taxas de toxicidade grave em 24% dos pacientes asiáticos avaliados. Em contrapartida, em nosso estudo 3 (9,7%) dos pacientes obtiveram a ASC marginalmente inferior ao limite proposto para faixa terapêutica, com valor de ASC de 2,4 mg h L-1. Entretanto, cabe ressaltar que apesar dos valores de ASC não estarem bem definidos, de acordo com a faixa terapêutica previamente estabelecida 22,6% dos pacientes avaliados no estudo seriam candidatos ao MTF. Adicionalmente ao monitoramento do DTX, foi realizada a dosagem dos metabólitos M1+M3, cujos níveis plasmáticos antes do término da infusão foram de 8,13 a 93,87 ng mL-1 e após a infusão de 5,01 a 50,47 ng mL-1. As razões metabólicas [(DTX)]/[(M1+M3)] apresentaram-se altamente variáveis entre os pacientes, sendo de 8,30 a 312,51 durante a infusão e de 1,43 a 14,74 em coleta realizada 1 h após o final da infusão. A dosagem dos metabólitos e estabelecimento das razões metabólicas pode ser uma promissora estratégia para a fenotipagem da CYP3A4/5, devendo ser validada em amostras clínicas em um grupo maior de pacientes, de forma a estabelecer valores de corte populacionais, podendo ser um marcador adicional em estudos farmacocinéticos do DTX. O método proposto apresenta excelente custo benefício na avaliação da terapia com o DTX. O 'turnaround time" estimado para a determinação da ASC foi inferior a um dia, com custo estimado na ordem de poucas dezenas de reais. Esse tempo permite a otimização da terapia do DTX em condições clínicas, uma vez que a quimioterapia é administrada a cada três semanas.
CONCLUSÃO Foi desenvolvido e validado um método para determinação simultânea de DTX e seus metabólitos M1+M3 em plasma por UPLC-MS/MS. A preparação das amostras mostrou-se simples e eficiente, através de uma extração líquido-líquido. O método demonstrou desempenho analítico adequado para aplicações clínicas, sendo o primeiro estudo realizado em uma população brasileira em pacientes durante quimioterapia com o DTX. Os parâmetros de validação mostraram que o método oferece desempenho analítico satisfatório, e desta forma pode ser utilizado como ferramenta para o MTF do DTX. Cabe destacar, ainda, que a partir da estimativa da ASC foi possível identificar os potenciais candidatos para o monitoramento terapêutico do DTX.
AGRADECIMENTOS Os autores agradecem à Universidade Feevale, ao Hospital de Clínicas de Porto Alegre, à CAPES e à Fapergs (processo 16/0191-0) pelo apoio financeiro.
REFERÊNCIAS 1. Montero, A.; Fossella, F.; Hortobagyi, G.; Valero, V.; Lancet Oncol. 2005, 6, 229. 2. Baker, S. D.; Verweij, J.; Cusatis, G. A.; Van Schaik, R. H.; Marsh, S.; Orwick, S. J.; Franke, R. M.; Hu, S.; Schuetz, E. G.; Lamba, V.; Messersmith, W. A.; Wolff, A. C.; Carducci, M. A.; Sparreboom, A.; Clin. Pharmacol. Ther. 2009, 85, 155. 3. Joerger, M.; Cancer Chemother. Pharmacol. 2016, 77, 221. 4. Tetzlaff, E. D.; Cheng, J. D.; Ajani, J. A.; Ther. Clin. Risk Manage. 2008, 4, 999. 5. Awada, Z.; Haider, S.; Tfayli, A.; Bazarbachi, A.; El-Saghir, N. S.; Salem, Z.; Shamseddine, A.; Taher, A.; Zgheib, N. K.; OMICS 2013, 17, 353. 6. Pereira, L. C.; Nogueira, T. A.; Barbosa, L. A. de O.; Calil-Elias, S.; Castilho, S. R. de.; Braz. J. Pharm. Sci. 2015, 51, 551. 7. Reddy, L. H.; Bazile, D.; Adv. Drug Deliv. Rev. 2014, 71, 34. 8. Bosch, T. M.; Huitema, A. D. R.; Doodeman, V. D.; Jansen, R.; Witteveen, E.; Smit, W. M.; Jansen, R. L.; Van Herpen, C. M.; Soesan, M.; Beijnen, J. H.; Schellens, J. H. M.; Clin. Cancer Res. 2006, 12, 5786. 9. Kiyotani, K.; Mushiroda, T.; Kubo, M.; Zembutsu, H.; Sugiyama, Y.; Nakamura, Y.; Cancer Sci. 2008, 99, 967. 10. van Waterschoot, R. A. B.; Lagas, J. S.; Wagenaar, E.; Rosing, H.; Beijnen, J. H.; Schinkel, A. H.; Int. J. Cancer 2010, 127, 2959. 11. de Weger, V. A.; Beijnen, J. H.; Schellens, J. H. M.; Anti-Cancer Drugs 2014, 25, 488. 12. Paci, A.; Veal, G.; Bardin, C.; Levêque, D.; Widmer, N.; Beijnen, J.; Astier, A.; Chatelut, E.; Eur. J. Cancer 2014, 50, 2010. 13. Krens, S. D.; McLeod, H. L.; Hertz, D. L.; Pharmacogenomics 2013, 14, 555. 14. Yamamoto, N.; Tamura, T.; Murakami, H.; Shimoyama, T.; Nokihara, H.; Ueda, Y.; Sekine, I.; Kunitoh, H.; Ohe, Y.; Kodama, T.; Shimizu, M.; Nishio, K.; Ishizuka, N.; Saijo, N.; J. Clin. Oncol. 2005, 23, 1061. 15. Engels, F. K.; Loos, W. J.; Van Der Bol, J. M.; De Bruijn, P.; Mathijssen, R. H. J.; Verweij, J.; Mathot, R. A. A.; Clin. Cancer Res. 2011, 17, 353. 16. Ozawa, K.; Minami, H.; Sato, H.; Cancer Chemother. Pharmacol. 2008, 62, 551. 17. Björkhem-Bergman, L.; Bäckström, T.; Nylén, H.; Rönquist-Nii, Y.; Bredberg, E.; Andersson, T. B.; Bertilsson, L.; Diczfalusy, U.; Drug Metab. Dispos. 2013, 41, 1488. 18. Ma, Y.; Lin, Y.; Li, Y.; Zhao, H.; Zhang, Y.; Sheng, J.; Yang, Y.; Huang, Y.; Zhang, L.; Annals of Oncology 2015, 26 (suppl 9), ix128.3-ix128. 19. Ciccolini, J.; Catalin, J.; Blachon, M. F.; Durand, A.; J. Chromatogr. B 2001, 759, 299. 20. Wang, L. Z.; Goh, B. C.; Grigg, M. E.; Lee, S. C.; Khoo, Y. M.; Lee, H. S.; Rapid Commun. Mass Spectrom. 2003, 17, 1548. 21. Yamaguchi, H.; Fujikawa, A.; Ito, H.; Tanaka, N.; Furugen, A.; Miyamori, K.; Takahashi, N.; Ogura, J.; Kobayashi, M.; Yamada, T.; Mano, N.; Iseki, K.; J. Chromatogr. B 2012, 893-894, 157. 22. U. S. Food and Drug Administration; Guidance for Industry, Bioanalitical Method Validation, disponível em https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry, acessada em Fevereiro 2020. 23. Almeida, A.; Castel-Branco, M.; Falcão, A.; J. Chromatogr. B 2002, 774, 215 24. De Graan, A. J. M.; Elens, L.; Sprowl, J. A.; Sparreboom, A.; Friberg, L. E.; Van Der Holt, B.; De Raaf, P. J.; De Bruijn, P.; Engels, F. K.; Eskens, F. A. L. M.; Wiemer, E. A. C.; Verweij, J.; Mathijssen, R. H. J.; Van Schaik, R. H. N.; Clin. Cancer Res. 2013, 19, 3316. 25. Stefanie Theresa Kraff. Pharmakokinetische Dosisindividualisierung von Fluorouracil und Taxanen, 2015. 26. Sheu, M. T.; Wu, C. Y.; Su, C. Y.; Ho, H. O.; Sci. Rep. 2017, 7, 14609. 27. Rosing, H.; Lustig, V.; van Warmerdam, L. J.; Huizing, M. T.; ten Bokkel Huinink, W. W.; Schellens, J. H.; Rodenhuis, S.; Bult, A.; Beijnen, J. H.; Cancer Chemother. Pharmacol. 2000, 45, 213. 28. Guitton, J.; Cohen, S.; Tranchand, B.; Vignal, B.; Droz, J. P.; Guillaumont, M.; Manchon, M.; Freyer, G.; Rapid Commun. Mass Spectrom. 2005, 19, 2419 29. Kenmotsu, H.; Tanigawara, Y.; Cancer Sci. 2015, 106, 497. 30. Veyrat-Follet, C.; Bruno, R.; Olivares, R.; Rhodes, G. R.; Chaikin, P.; Clin. Pharmacol. Ther. 2000, 68, 677. 31. Yamamoto, N.; Tamura, T.; Kamiya, Y.; Sekine, I.; Kunitoh, H.; Saijo, N.; J. Clin. Oncol. 2000, 18, 2301. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access