
 |
 |
 |
 |
 |
Revisão
|
|
| Metabolômica de plantas: métodos e desafios Plant metabolomics: methods and challenges |
|
Alan C. PilonI,II,III,#; Denise M. SelegatoII,IV,#; Richard P. FernandesII; Paula C. P. BuenoI,V; Danielle R. PinhoI; Fausto Carnevale NetoVI; Rafael T. FreireVII; Ian Castro-GamboaII; Vanderlan S. BolzaniII; Norberto P. LopesI,*
I. Núcleo de Apoio à Pesquisa em Produtos Naturais e Sintéticos, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto - SP, Brasil Recebido em 01/11/2019 *e-mail: npelopes@fcfrp.usp.br Metabolomics has played a central role in various areas of plant sciences, offering new perspectives for the advancement of agriculture, drug discovery, chemical ecology and taxonomy. Plant metabolomics (identification and quantification) aims to understand the relationship between biological systems and genetic, pathological and or environmental stimuli in terms of differential expression of the metabolism. Owing to the unique challenges, such studies require multidisciplinary skills involving biology, chemistry, statistics, and computer science for the extraction and complete understanding of information. In this sense, this review summarizes the main procedures that involve the steps of plant metabolomic study (design of experiments, sample preparation, analytical methods and data analysis), providing a comprehensive overview, showing the main challenges and limitations and possible solutions for the different approaches used. INTRODUÇÃO O conhecimento e a compreensão do valor e dos riscos das substâncias encontradas em plantas, sejam elas in natura ou processadas, foi fundamental para a sobrevivência e evolução da humanidade. Se as antigas civilizações já tinham um vasto conhecimento sobre o uso e aplicação de plantas, é nos tempos modernos que tivemos as tecnologias capazes de extrair, separar e identificar as substâncias responsáveis por propriedades medicinais e nutricionais.1-3 Em geral, o produto final dos eventos bioquímicos dos organismos resulta no que denominamos de metabolismo. Em plantas, microorganismos e animais, ele ainda pode ser subdivido em duas categorias. O metabolismo primário, relacionado à síntese e polimerização de monômeros (blocos construtores) em biopolímeros através de reações de redução e condensação por vias enzimáticas altamente especializadas e eficientes (baixa plasticidade enzimática em relação aos substratos),4 e o metabolismo secundário ou especial, que está diretamente envolvido na produção de metabólitos responsáveis pela adaptação e sobrevivência à míriade de interações entre organismos e o seu meio ambiente. Ao contrário dos primários, esses metabólitos são expressos por enzimas com grande plasticidade dos seus sítios.5-7 Ao longo dos últimos dois séculos, diferentes áreas da Bioquímica e Química de Produtos Naturais têm buscado compreender a função específica desses metabólitos (primários e secundários) tanto nos organismos de origem, como também para uso e bem-estar da humanidade. Os avanços tecnológicos das plataformas analíticas bem como a capacidade de processamento dos dados gerados permitiram romper com a visão reducionista sobre a função metabólica para uma visão integrativa sobre o funcionamento dos organismos. Entretanto, os conceitos de reducionismo e o holismo não são excludentes, mas representam a totalidade do funcionamento dos seres vivos. É a partir dessa mudança de paradigma, que as ciências "ômicas" floresceram nas diferentes comunidades científicas.8 A metabolômica foi concebida no final dos anos de 1990 a partir dos trabalhos com genômica funcional de leveduras por Oliver e Ferenci.9 Em 2000, Fiehn e colaboradores do Max-Planck Institute of Plant Physiology publicaram os primeiros trabalhos sobre a aplicação da metabolômica em plantas.10 Ao longo dos anos, várias definições foram elaboradas sobre a metabolômica, considerando tanto o tamanho molecular das substâncias (< 1500 Da), quanto a classe estrutural.1-3,11 Talvez a definição mais difundida seja a ciência que investiga a totalidade da expressão qualitativa e quantitativa do metabolismo primário e secundário dos organismos. Contudo, a metabolômica estabelece que o entendimento dos organismos ocorre através da análise comparativa de perfis metabólicos entre indivíduos e/ou populações sujeitos às diferentes condições genéticas, ambientais ou patológicas. Os estudos metabolômicos eram inicialmente divididos em duas categorias: estudos alvo (do inglês, targeted), nos quais metabólitos ou classes metabólicas são investigados como resposta a um dado estímulo e, os estudos não-alvo (do inglês, untargeted) nos quais se avalia todo o conjunto de substâncias de modo indiscriminado. Atualmente, existem outras subcategorias que visam refletir os diferentes objetivos e especificidades dos estudos metabolômicos, como mostrado na Tabela 1.

A anotação e/ou elucidação e quantificação de "todo" o conjunto de metabólitos presentes em um sistema biológico é uma tarefa complexa em termos analíticos. Atualmente, não existe qualquer técnica analítica que seja capaz de medir todos os metabólitos em um único experimento é necessário o acoplamento de técnicas analíticas ortogonais afim de suprir as deficiências individuais. Em plantas, essa situação é agravada pela diversidade estrutural das substâncias oriundas do metabolismo secundário.14 O Quadro 1 mostra alguns dos desafios analíticos para a realização de estudos metabolômicos em plantas.

A maioria dos experimentos em metabolômica de plantas são realizados utilizando da combinação de ferramentas analíticas de separação e detecção, conhecidas como abordagens hifenadas ou acopladas. Dentre as combinações mais comuns estão as técnicas de separação cromatográfica (cromatografia líquida e gasosa) acopladas a detectores de ultravioleta ou espectrometria de massas, como por exemplo a CLAE-UV-DAD, CLAE-EM ou CG-EM.15 A ressonância magnética nuclear (RMN), inicialmente aplicada aos estudos de metaboloma humano, também vem sendo empregada em estudos de plantas, tanto para a elucidação estrutural de moléculas previamente isoladas e purificadas, quanto para o estudo de frações enriquecidas ou mesmo extratos brutos de alta complexidade.16,17 A literatura está repleta de estudos metabolômicos de plantas aplicados à caracterização de amostras18-24 ou substâncias,25 no controle de qualidade,26-29 no melhoramento genético/transgênico de plantações,30-33 no desenvolvimento de medicamentos,34-37 em ecologia-química,38-40 no entendimento de funções bioquímicas14,33,41,42 e na distribuição e evolução taxonômica.43,44 Talvez a tarefa mais desafiadora para um cientista que almeja utilizar a metabolômica seja entender todas as múltiplas e interdisciplinares etapas, assim como as metodologias analíticas mais adequadas considerando as vantagens e limitações do objeto de estudo. Nesse sentido, as comunidades de metabolômica têm-se mobilizado para a criação de protocolos que auxiliem a execução, validação e a troca de informações inter- ou intra-laboratoriais. Iniciativas como COordination of Standards In MetabOlomicS (COSMOS - http://www.cosmos-fp7.eu), Metabolomics Workbench (http://www.metabolomicsworkbench.org), ArMet (Architecture for Metabolomics) e MSI (Metabolomics Standard Initiative) representam alguns desses esforços para a criação de repositórios públicos para análise, monitoramento e disseminação de dados.8 Em geral, essas plataformas exigem informações sobre as diretrizes do planejamento experimental, tais como, coleta, preparação de amostra e extração de metabólitos; dados sobre a(s) plataforma(s) analíticas utilizadas, instrumentos e configurações de análise; os tipos de normalização e escalonamento dos dados pós-análise e os tratamentos quimiométricos e estatísticos utilizados para redução da complexidade dos dados.8 A Figura 1 demonstra, através de um fluxo de informações (etapas), os requerimentos básicos exigidos pelas comunidades científicas para o desenvolvimento de um experimento em metabolômica de plantas. A primeira etapa consiste na aplicação de um planejamento experimental criterioso, considerando a população a ser estudada (viabilidade), os procedimentos de preparação de amostra, seguido pela aquisição dos dados (associação com metadados) seu pré-processamento e finalmente a análise dos dados (estatística e quimiometria) e a interpretação dos resultados.45
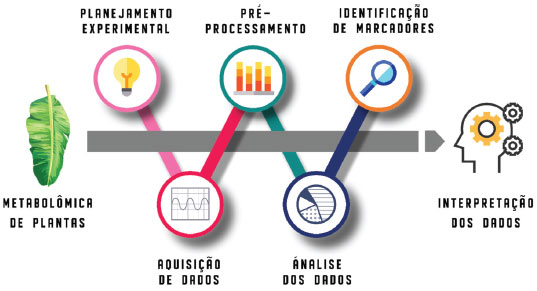 Figura 1. Tópicos que envolvem a aplicação de um estudo metabolômico em plantas
Cada etapa envolve inúmeras escolhas em termos das metodologias e abordagens analíticas disponíveis, que por sua vez, delimitam as etapas subsequentes. Assim, o propósito desta revisão é descrever os principais protocolos, métodos e softwares utilizados em cada uma das etapas (Figura 1) apontando limitações, desafios e possíveis soluções. Delineamos um manuscrito que agrega de princípios básicos a estratégias específicas visando auxiliar no planejamento e na execução experimental em estudos promovidos por diferentes públicos da comunidade científica.
PLANEJAMENTO EXPERIMENTAL Os planejamentos experimentais (PE) constituem o primeiro passo que concerne à experimentação científica.46 Em metabolômica, os PE devem ser elaborados contextualizando todo o conjunto de etapas uma vez que cada escolha interfere nos resultados e suas interpretações.47 A maioria dos planejamentos experimentais aplicados em metabolômica iniciam pela determinação do tamanho amostral, o número de variáveis importantes e seus respectivos níveis.48 Como os dados metabolômicos são tipicamente multidimensionais e correlacionados, a determinação do número de amostras por ferramentas estatísticas pode não ser abrangente. Para contornar esse problema, Nyamundanda e colaboradores desenvolveram um software capaz de determinar o tamanho amostral com base nos objetivos e nas técnicas analíticas, descartando a necessidade de experimentos piloto.49 Estudos similares podem ser encontrados nos trabalhos de Müller et al.,48 Tibshirani,50 Liu & Wang51 e Lin et al.52 Para a investigação sobre como os fatores ambientais ou fatores experimentais, tais como o uso de solventes orgânicos no processo de extração e/ou separação cromatográfica, podem afetar os perfis metabolômicos de plantas, é necessário realizar experimentos de uma forma sistematizada. Os planejamentos experimentais (do inglês, Design of Experiments - DoE) arranjam sistematicamente o conjunto de variáveis dentro de um espaço amostral delimitado, diminuindo o número de experimentos e maximizando a extração de informações, como interações entre variáveis e a distribuição dos efeitos.53 Vejamos um exemplo: estamos interessados em identificar quais dentre as variáveis temperatura, nutrição e estresse hídrico (por exemplo, 25-35 °C, 1-10 mmol N L-1, 100-1000 mL dia-1, respectivamente) afetam a produtividade de frutos e consequentemente a dinâmica do perfil metabólico. Assim, a primeira etapa do PE consiste na escolha das variáveis com maior efeito na produção de frutos. Essa seleção pode ser feita através de dados de literatura incluindo os diferentes níveis. Quando não existe informação sobre o impacto das variáveis na resposta (por exemplo, o efeito do estresse hídrico sobre a frutificação), aplica-se um modelo que possibilite a triagem de variáveis dominantes, isso é, avalia-se um grande número de variáveis. No geral, modelos baseados em planejamentos fatoriais completos ou fracionários são os mais utilizados para esse fim, uma vez que o número de experimentos é relativamente reduzido.54-57 Nos casos em que as variáveis dominantes são conhecidas, é possível aplicar um modelo de otimização das variáveis. Dentre os modelos mais utilizados, o planejamento composto central arranja as variáveis dentro de um espaço amostral com cinco diferentes níveis (5 variações). Suponhamos que, para o nosso estudo da frutificação, temperatura, nutrição e estresse hídrico sejam as três variáveis dominantes e que desejamos aplicar um planejamento fatorial completo de dois níveis para determinar a melhor condição de crescimento de frutos. Inicialmente, as variáveis são codificadas (-1 para o nível inferior e +1 para o nível superior) e arranjadas de modo sistemático para que todos níveis possam ser analisados (Figura 2). O número de experimentos é calculado pela potenciação NV (em que N é o número de níveis e V é o número de variáveis) totalizando oito experimentos (Figura 2). Para esse exemplo, o peso ou o número de frutos poderiam ser considerados como resposta para o modelo.
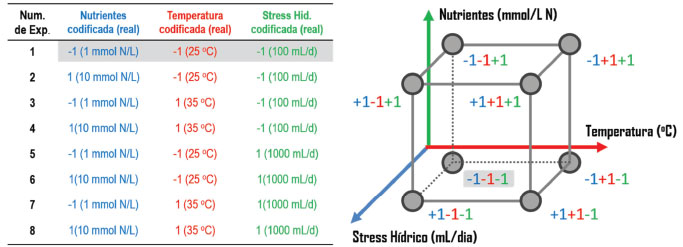 Figura 2. Planejamento fatorial 23 aplicado ao estudo de otimização dos fatores de crescimento de frutos. A tabela corresponde ao número de experimentos 23 (8 experimentos) e o respectivo arranjo sistemático das variáveis (codificado e real). Foram considerados dois níveis, sendo os inferiores codificados com os valores de -1 (Nutrientes: 1 mmol N L-1; Temperatura: 25 oC e Stress hídrico: 100 mL dia-1) e os superiores com +1 (Nutrientes: 10 mmol N L-1; Temperatura: 35 °C e Stress hídrico: 1000 mL dia-1). O gráfico representa a disposição de cada experimento (conjunto de variáveis) dentro do espaço amostral. O experimento em destaque na tabela é representado no gráfico. Mais informações sobre a forma e a distribuição de planejamentos em estudos metabolômicos podem ser encontradas em Pereira Filho (2015)58
Uma vez realizados os experimentos, a resposta do sistema em estudo (peso/número de frutos) pode ser correlacionada com dados cromatográficos, espectrométricos ou espectroscópicos, possibilitando determinar os sinais (metabólitos) associados, bem como modelar as respostas dentro do espaço amostral para encontrar a condição ideal de frutificação (Figura 2). A partir das variáveis e de seus níveis, o planejamento permite a construção de mapas e superfícies de contorno apontando as condições ideais para cada variável. Para o estudo da frutificação, seria possível, inclusive, avaliar temperatura, nutrientes e quantidade de água baseado na relação com possíveis metabólitos associados para produção de frutos nos diferentes experimentos. Até esse momento, estamos considerando apenas os modelos que utilizam de variáveis independentes, isso é, a variação da temperatura não afeta a quantidade de nutrientes fornecidos para o nosso estudo. No entanto, existem casos em que o interesse seja a análise e otimização de variáveis dependentes, como é o caso da escolha da melhor proporção de misturas de solventes para o processo de extração, ou a escolha do melhor e mais adequado gradiente de fase móvel em um sistema cromatográfico (por exemplo, a proporção de MeOH e ACN que compõe a fase orgânica). Para esses casos, recomenda-se modelos baseados em simplex-centroide ou estrela.58 A aplicação desses modelos no uso de diferentes solventes orgânicos no processo de extração e na otimização da separação cromatográfica em estudos de metabolômica de plantas foi explorado por Pilon et al.,59 Bueno et al.,60 Zhang et al.,61 Ramandi et al.,62 Iyadivam et al.,63 Rai et al.64 e Asati et al.65 De forma geral, os planejamentos consistem em uma primeira etapa de seleção das variáveis, por exemplo aquelas que mais influenciam no processo extrativo ou no método de análise a ser empregado, seguido de uma etapa de otimização destas variáveis ou fatores. Atualmente, existem diversas ferramentas capazes de organizar os planejamentos experimentais, realizar os cálculos e avaliar a relevância estatística de um estudo. Dentre elas estão os pacotes em Matlab66 da plataforma R (AlgDesign, Conf.design: e Crossdes), assim como os softwares, Minitab67 e Piruette.68 É possível realizar os cálculos em plataformas gratuitas como o Octave, (https://www.gnu.org/software/octave), assim como diretamente no pacote Office Excel. Pereira Filho apresenta um tutorial de aplicação e análise de amostras utilizando de planejamentos experimentais, bem como disponibiliza uma sequência de vídeos explicativos.69,70
AMOSTRAGEM Procedimento de coleta e inibição enzimática O processo de amostragem pode levar, em determinados casos, a resultados tendenciosos, sendo um dos principais fatores de erro em metabolômica de plantas. A contaminação, conversão ou degradação dos compostos pode gerar diferenças no decorrer de segundos, o que torna fundamental os cuidados com a coleta e manuseio de amostra.71 No ato da coleta, alguns fatores devem ser considerados sobre as condições físicas, químicas, biológicas e geográficas do conjunto amostral. Se o material vegetal foi cultivado e coletado em casas de vegetação instaladas em institutos, universidades, herbários ou reservas, deve-se levar em conta que mesmo sob condições controladas, pequenas mudanças na intensidade de luz, irrigação, temperatura, gradientes de dióxido de carbono e efeitos sazonais podem alterar o perfil metabólico de plantas geneticamente idênticas. Para evitar esses resultados, denominados "efeitos de borda", as plantas devem ser periodicamente redistribuídas, de forma aleatória, entre o centro e as bordas da casa de vegetação.72,73 Em coletas de campo, muito comuns quando o estudo contempla variáveis agronômicas ou ecológicas, as condições ambientais são dificilmente controladas. Assim, deve-se demarcar os indivíduos através de sistemas de posicionamento global (GPS). Ferramentas online como Google Maps ou equipamentos específicos oferecem esse tipo de serviço com alto grau de precisão. Isso garante o monitoramento e o controle de experimentos conduzidos a longo prazo e planejados para a realização de coletas sazonais, semestrais ou anuais.74 Em estudos de campo não se conhece, a priori, o tipo do solo, a incidência de herbivoria e o estado ontogenético dos indivíduos. Isso, invariavelmente, aumenta a complexidade para a interpretação dos resultados referente a um dado estímulo genético ou ambiental. Assim, locais que ofereçam informações adicionais como índices pluviométricos, características do solo, altimetria, irradiação ultravioleta, vento predominante, variação diária de temperatura, entre outros fatores, são de grande importância e devem ser preferencialmente escolhidos. A padronização no ato de coleta é um dos alicerces da metabolômica de plantas e requer a consideração de fatores como o tipo de tecido a ser utilizado (folhas, frutos, flores, caule, raiz), os períodos sazonais, os horários de coleta (ciclo circadiano) e os estágios ontogenéticos (crescimento e reprodução).75,76 Em casos em que as coletas são realizadas em diferentes localidades, recomenda-se a padronização de acordo com o estágio de desenvolvimento da planta. Isso pode ser realizado através de referências bibliográficas ou através de bases de dados dedicadas a ontologia, como a Plantontology (www.plantontology.org). É importante também definir a posição do órgão ou parte da planta que foi coletada, e informações acerca da exposição solar (se está localizado no dossel ou sob a sombra).77,78 Uma vez coletado o material vegetal, a atividade enzimática deve ser inibida imediatamente para garantir a integridade metabólica.79 Em pequenos órgãos, tecidos dissecados ou até pequenas plantas inteiras, utiliza-se de choques térmicos para a desnaturação de proteínas. O uso de condições ácidas (pH < 2) ou de solventes quentes não fornece garantias de integridade metabólica, como hidrólises e/ou clivagens glicosídicas, gerando artefatos e possíveis equívocos na interpretação dos resultados.80-82 A imersão em nitrogênio líquido permite o resfriamento imediato a -180 °C, em metanol resfriado a -20 °C, ou em gelo seco (dióxido de carbono solidificado) a -78 °C, garantindo a inibição enzimática e auxiliando a etapa de lise celular.72 Secagem e homogeneização A presença de água em matrizes vegetais afeta negativamente várias etapas dos estudos metabolômicos, pois modifica a eficiência do processo de extração por solventes orgânicos (alterando a proporção de solventes previamente estabelecida), prejudica o armazenamento de amostras e influencia as subsequentes análises instrumentais.82,83 Sinais de água (1H RMN) em extratos brutos são um exemplo dos efeitos de distorção e de sobreposição aos sinais de interesse.72 Dentre os procedimentos para a remoção de água destacam-se o uso de estufa com ar circulante a 40 °C, secagens a temperatura ambiente e a vácuo, e através de processos de sublimação, como é o caso da liofilização.84 A liofilização é a técnica mais utilizada para amostras sensíveis à altas temperaturas e para inibição de hidrolases e fosfatases.72,82 Contudo, existe uma preocupação quanto a perda de metabólitos devido a adsorção irreversível em paredes celulares e membranas durante o processo de congelamento.84 O processo de liofilização, realizado em duas etapas, é iniciado com o congelamento dos tecidos ou fluídos em tubos ou frascos, e em seguida submetidos ao processo de sublimação (a pressão reduzida e baixas temperaturas, aproximadamente ≤ -40 °C). Amostras liofilizadas devem ser armazenadas à -80 °C, em frascos vedados para se evitar a reabsorção de água. A amostra deve ser triturada ou moída para reduzir o tamanho dos tecidos. A padronização desse processo (controle granulométrico) é fundamental para estudos metabolômicos uma vez que garante tanto a homogeneidade amostral na extração, quanto a proporcionalidade na área de contato entre a matriz e o solvente extrator. O uso de almofariz e pistilo85 é recorrente em metabolômica, apesar de não ser recomendada para estudos com um grande número de amostras, por ser laboriosa e demandar uma etapa adicional de tamisagem. Moinhos de bolas, criogênicos e homogeneizadores verticais vem sendo empregados com sucesso.72,82 Em geral, eles promovem uma rápida trituração do material mantendo uniformidade do grânulo e contam com acessórios capazes de realizar múltiplas moagens em microescala (como, por exemplo, a moagem simultânea de 10 tubos de 1,5 mL ou 2,0 mL).82,86 Idealmente, o processo de coleta, moagem e secagem deveriam ser imediatamente sucedidos pela extração e análise. Quando isso não é possível recomenda-se o armazenamento das amostras em freezer a -80 ºC. O condicionamento das amostras a temperaturas de -20 ºC e 4 ºC é utilizado em alguns casos, porém, não é recomendado devido a possibilidade de algumas reações bioquímicas e atividades enzimáticas ocorrerem, até mesmo em temperaturas inferiores a -20 ºC, especialmente em amostras com a presença de sais ou solventes orgânicos.87-89 Processo de extração A etapa de extração determina o intervalo ou cobertura química, considerando aspectos de polaridade e solubilidade dos metabólitos a serem detectados pelas técnicas analíticas. É importante que essa etapa seja realizada de modo simples, rápido e em poucas etapas, uma vez que a metabolômica exige um consideravel número amostras e, portanto, de experimentos. Diferentes técnicas de extração têm sido discutidas a fim de produzir perfis metabólicos representativos, como por exemplo a extração com solventes em procedimentos de maceração, por arraste a vapor, fluído supercrítico, líquidos iônicos e extrações por solvente sob alta temperatura e pressão.90,91 As extrações baseadas em solventes orgânicos ainda constituem a melhor escolha em estudos metabolômicos devido a eficiência e compatibilidade com as principais plataformas analíticas, como CLAE-EM, CG-EM e RMN. Entretanto, novos métodos de extração seletiva, com baixa toxicidade e/ou custo, têm sido propostos, como o uso de NADES (Natural Deep Eutectic Solvents).92-94 A seleção do método extrativo pode ser realizada de duas maneiras: em análise direcionada de determinado(s) metabólito(s) alvo(s), o que consiste em uma abordagem aplicada em Profiling Analysis e Target Analysis, ou em busca indiscriminada de perfis metabólicos, como Fingerprint Analysis e o Metabolomics. Geralmente, a busca inespecífica exige ensaios, preferencialmente concebidos através de planejamentos experimentais, para a avaliação da capacidade de extração dos solventes assim como a proporção entre solvente e o material vegetal. O trabalho de Pilon et al.,59 por exemplo, explora através de planejamentos experimentais o efeito de quatorze solventes orgânicos sobre o rendimento e o número de substâncias no processo de extração em folhas de Jatropha assim como avalia as propriedades físico-químicas dos solventes associadas aos resultados de extração. O tipo de extração afeta diretamente80,95-97 a escolha das plataformas instrumentais, assim como as ferramentas de identificação e os tratamentos de dados. Embora as otimizações dos parâmetros de extração sejam importantes, é fundamental estar consciente de que o desenvolvimento de métodos específicos pode, em certos casos, impedir a comparação com outros estudos ou mesmo tornar incompatível o uso de plataformas analíticas, ou influenciar as etapas de identificação de compostos, como por exemplo, o uso de bases de dados. A seleção do solvente deve considerar, também, a toxicidade, o poder de solubilização, a seletividade, a taxa de dissolução, a reatividade química e o pH, sendo o solvente ideal aquele que apresentar a menor toxicidade com o maior poder de solubilização.82,98 A mistura metanol-água tem sido amplamente utilizada em estudos metabolômicos devido a sua menor seletividade, sendo capaz de extrair uma ampla gama de metabólitos tais como açúcares, ácidos orgânicos, alcaloides, compostos fenólicos, dentre outros.82 Se possível, a introdução de extratos brutos (devidamente diluídos) diretamente no equipamento analítico evita etapas tais como a evaporação e redissolução de amostra. Para a RMN, existe um menor número de limitações na escolha do solvente extrator, embora solventes de média polaridade sejam frequentemente escolhidos, a exemplo das misturas de metanol:água. Para CG-EM, a escolha do solvente é limitada às características dos compostos voláteis. Nesse caso, para a análise de metabólitos polares, etapas adicionais de derivatização são imprescindíveis. Para as misturas aquosas, o monitoramento do pH é fundamental para se evitar a formação de artefatos durante o processo de extração. Maltese e colaboradores99 demonstraram os efeitos da água e outros solventes nesse processo. Além disso, o pH está diretamente associado aos resultados de análise, podendo afetar os deslocamentos químicos de hidrogênios lábeis em RMN, tempos de retenção em CLAE e a ionização de compostos em espectrometria de massas, principalmente na ionização por eletrospray (ESI-EM). O uso de tampões possibilita minimizar esses efeitos na RMN, embora para CLAE-EM cuidados adicionais devem ser tomados para se evitar a precipitação de sais metálicos na fonte de ionização. Sistemas bifásicos compostos por clorofórmio:metanol:água na proporção de 2:1:1 (v/v) têm se mostrado eficiente tanto na extração de compostos polares quanto apolares. O trabalho proposto por Lisec e colaboradores100 é um dos mais aplicados em metabolômica de plantas utilizando de CG-EM (Figura 3). Resumidamente, em 100 mg de material fresco é adicionado uma mistura clorofórmio:metanol:água (2:1:1), seguido pela separação da fase polar (contendo açúcares, álcoois, ácidos orgânicos, aminoácidos, aminas e um conjunto complexo de compostos oriundos do metabolismo secundário), da fase lipofílica (rica em fosfolípides, triglicerídeos, ácidos graxos, terpenos e outras classes de menor polaridade do metabolismo secundário).
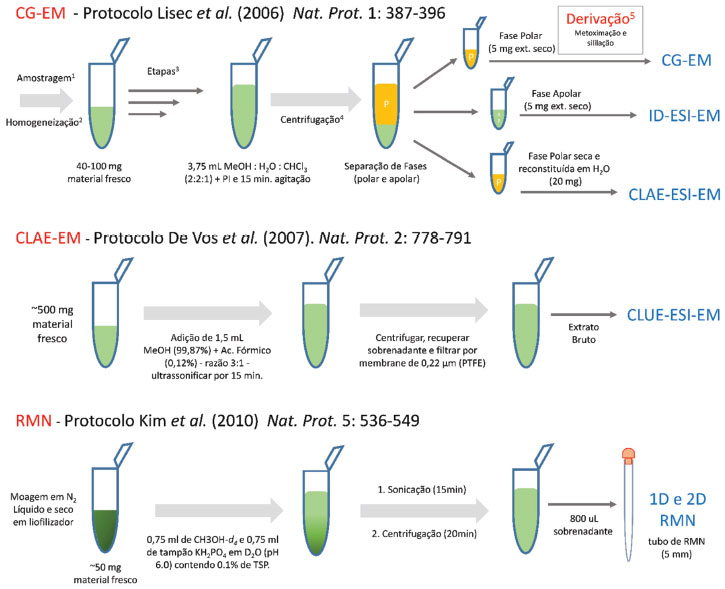 Figura 3. Protocolos padrão de preparação de amostra mais utilizados em metabolômica por CLAE-EM, CG-EM e RMN. 1amostragem, pesagem e inibição enzimática; 2uso de moinho de bolas por 2 minutos a 20 Hz, 3consiste das etapas de C a G no protocolo de Lisec e colaboradores (2006); 4centrifugação por 15 minutos a 2000 x g
A CLAE-EM é o método preferencial para análise de perfis metabólicos semi-polares, tais como glucosinolatos, saponinas, alcaloides, poliaminas, ácidos fenólicos e flavonoides.101 De Vos e colaboradores102 propuseram um protocolo de extração partindo de 500 mg de material vegetal fresco com adição de 1,5 mL de metanol acidificado (99,875% metanol e 0,125% de ácido fórmico), Figura 3. Esse protocolo estabelece uma relação de 3:1 entre a razão do volume de solvente e o material fresco e resulta em uma concentração final de 75% de metanol e 0.1% de ácido fórmico, assumindo que o material vegetal apresenta um conteúdo de 95% de água. É interessante destacar que o metanol pode ser substituído pela a mesma proporção em acetona. O extrato é diretamente analisado por CLAE-EM em fase reversa, embora ele possa ser concentrado usando de rotaevaporadores ou speed vacuum. Esse método de extração foi eficiente na análise de plantas incluindo Arabidopsis, tomate, batata e morango, dentre outros.102-105 Por fim, o protocolo desenvolvido por Kim e colaboradores78 explora a análise de metabólitos secundários, como os compostos fenólicos, e primários, como açúcares e aminoácidos, utilizando 1H RMN. Esse método contempla a extração do material vegetal com uso de solvente deuterado (MeOH-d4) em tampão fosfato de sódio pH 6,0, Figura 3. Equipamentos para melhorar a taxa de difusão e adsorção de solventes na matriz vegetal têm aumentado consideravelmente a eficiência de extração.106 A extração assitida por ultrassom tem se mostrado eficiente e eficaz devido a simplicidade, rapidez e baixa limitação em relação aos tipos de solventes utilizados. Ao contrário da extração assistida por micro-ondas (dependente de solventes sensíveis à energia da radiação em micro-ondas), o ultrassom não leva a problemas de superaquecimento de amostra, evitando explosões e degradação de compostos voláteis. Em geral, o tempo de extração e o uso de variações da temperatura levam a um maior número de compostos extraídos; no entanto, eles aumentam as chances de reações e aceleram o processo de degradação.82
PRÉ-TRATAMENTO DE AMOSTRAS E AQUISIÇÃO DE DADOS Pré-tratamento das amostras Uma vez que os extratos são obtidos, a próxima fase envolve a seleção do método analítico. Nessa etapa, é fundamental a escolha de uma técnica reprodutível, simples e rápida, permitindo a detecção de analitos em diferentes concentrações. Tanto em cromatografia líquida quanto em cromatografia gasosa, a remoção de interferentes é realizada por cartuchos de extração em fase sólida (SPE - solid-phase extraction). A seleção e preparação dos cartuchos baseia-se em princípios cromatográficos, como solubilidade e adsorção, e podem ser otimizados também no fracionamento seletivo das amostras. A ampla variedade de adsorventes em sílica gel ou resinas sintéticas modificadas (fase reversa: C2, C8, C18, fenil; fase normal: ciano, NH2, diol; troca iônica: sax, scx e wcx; adsortivas: alumina e florisil) permitem a separação de grupos específicos de metabólitos. Partições do tipo líquido-líquido são utilizadas para a separação de substâncias e a obtenção de frações enriquecidas. O uso sequencial de solventes com o aumento de polaridade concentra as diferentes classes metabólicas. Já o aumento da volatilidade de açúcares e outros compostos polares para a análise em CG-EM pode ser realizado por reações de derivatização.107 Muitos protocolos foram desenvolvidos nas últimas décadas,107-110 principalmente reações de acilação, oximação, sililação e esterificação. Tipicamente, as derivatizações ocorrem em duas etapas: a metoximação, na qual a metoxiamina reduz a ciclização de açúcares e protege os grupos carbonila. Em seguida, ocorre a sililação, na qual derivados de trimetilsilano substituem hidrogênios ácidos por grupos silanóis, reduzindo o ponto de ebulição do analitos. Dentre os agentes sililantes mais comuns estão o bis-(trimeltilsilil)trifluoroacetamida (BSTFA) e o N-metil-N-(trimetilsilil) trifluoroacetamida (MSTFA).109,111 Na sililação, os efeitos de BSTFA e MSTFA são similares, embora os derivados de MSTFA possuam um menor ponto de ebulição e, consequentemente, um menor tempo de retenção comparado ao BSTFA. Isso é importante no momento da escolha das bases de dados para correspondência espectral e por tempo de retenção.112 A remoção de interferentes dos solventes ou de partículas insolúveis é outro fator fundamental para a qualidade dos resultados. Independente da técnica de separação utilizada, recomenda-se filtrar todos os reagentes e extratos. Para CLAE, as membranas mais utilizadas são de PTFE hidrofílica de 0,45 e 0,22 µm, para solvente e amostra, respectivamente. Em RMN, a presença de interferentes é ainda mais preocupante, uma vez que a maioria dos estudos por essa técnica não utiliza de uma etapa prévia de separação e os sinais dos contaminantes podem interferir na interpretação dos compostos relevantes. A remoção de partículas suspensas (material insolúvel) é essencial para a homogeneidade de campo, garantindo qualidade do shimming e, consequentemente, resolução dos deslocamentos químicos. Essas partículas são comumente removidas por centrifugação a 13000 rpm ou por filtração (PTFE). Dentre os interferentes mais comuns, destacam-se os açúcares livres (multipletos entre 3,0 - 5,0 ppm), os ácidos graxos (1,0 - 1,3 ppm) e os ftalatos (7,0 - 8,0 ppm). Instrumentação analítica Devido à falta de uma técnica capaz de analisar todo o conteúdo do metaboloma, diversos métodos analíticos vêm sendo empregados complementarmente, com o objetivo de se minimizar as deficiências individuais das ferramentas analíticas, e assim aumentar o nível de separação, detecção, estabilidade, resolução, sensibilidade, velocidade e a amplitude do intervalo dinâmico de detecção.113 Dentre as técnicas mais comuns, destacam-se a cromatografia gasosa e líquida acoplada a espectrometria de massas (CG-EM ou CLAE-EM, respectivamente), a injeção direta em espectrometria de massas (ID-EM) e a ressonância magnética nuclear (RMN). Porém, outras técnicas como a eletroforese capilar acoplada a espectrometria de massas (EC-EM),114,115 cromatografia em camada delgada de alta eficiência (HPTLC - High performance thin-layer chromatography)116-118 e a cromatografia líquida com detecção por arranjos de diodos (CLAE-DAD)119,120 também são utilizadas na análise de extratos de plantas, com aplicações metabolômicas significativas. A Tabela 2 mostra algumas das plataformas analíticas em uso nos estudos metabolômicos de plantas.
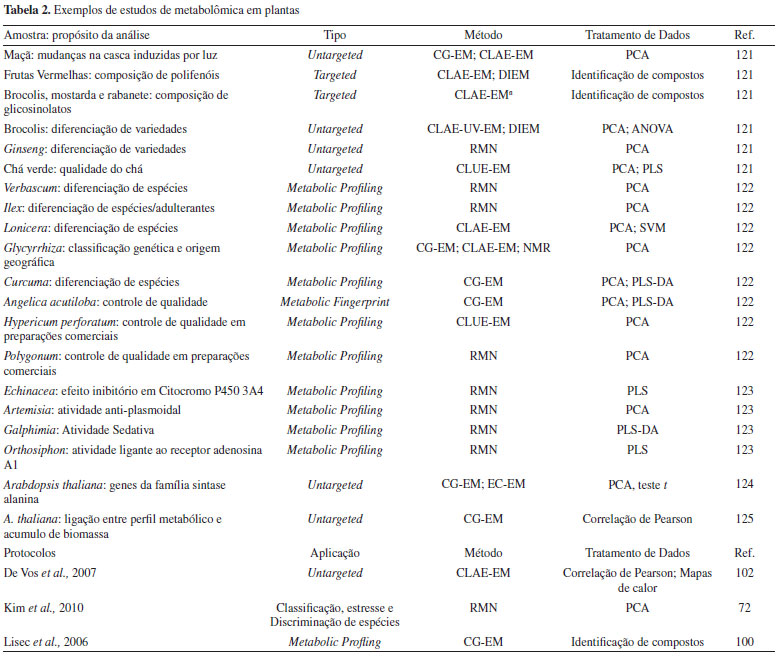
Cromatografia em fase gasosa acoplada a espectrometria de massas (CG-EM) A cromatografia em fase gasosa acoplada à espectrometria de massas foi a primeira técnica analítica utilizada para detecção e quantificação de compostos voláteis e semi-voláteis (< 650 Da)126,127 em estudos metabolômicos. Ácidos orgânicos, álcoois, aminoácidos, ácidos graxos, esteróis, catecolaminas e outros produtos naturais, fármacos e toxinas podem ser analisados por CG-EM, mas necessitam de uma etapa de derivatização, como já mencionado no tópico de pré-tratamento de amostras.128 A maioria dos trabalhos utilizando CG-EM em metabolômica de plantas utilizam de fonte de ionização por elétrons (para fins de padronização da fragmentação) e poucos têm feito uso de analisadores de massas de alta resolução. Os principais protocolos de GC-EM utilizam de colunas capilares de sílica fundida composta por 5% de grupos fenil ou 100% de polidimetilsiloxano junto com uma pequena coluna de guarda para aumentar a sensibilidade.128,129 Uma das vantagens do uso da CG-EM consiste no processo de identificação molecular baseada na combinação de informações ortogonais, isso é, o uso de tempos ou índices de retenção de compostos combinados às informações espectrais de massas obtidos sob uma energia padronizada de ionização (usualmente 70 eV).128,129 Essa combinação não é só importante por ser altamente sensível e reprodutível, mas principalmente por possibilitar o desenvolvimento de bibliotecas de compostos (padrões) para identificação estrutural de picos cromatográficos. A biblioteca NIST14 (Mass Spectral Library collection of the U.S. National Institute of Standards and Technology) é composta por mais de 240 mil estruturas moleculares. A GMD, ou Golm Metabolome Database, mantida pelo Max-Planck Institute of Plant Physiology é uma base de dados dedicada à experimentos de metabolic pro?ling de plantas com mais de 1400 substâncias de referência obtidos in-house. A principal vantagem dessa base é ser de acesso aberto, integrada à outras bases de dados, e desenvolvida para experimentos do metabolismo vegetal.130 As bibliotecas em CLAE-EM/EM são significantemente menores. A mesma NIST14 conta com apenas 8171 compostos em sua biblioteca CLAE-EM/EM enquanto a Metlin131 apresenta aproximadamente 12 mil compostos, sendo que ambas não possuem valores de tempo de retenção.128 Os dados espectrais obtidos por cromatografia gasosa acoplada a espectrometria de massas podem ser processados em ferramentas e softwares livres como o Automated Mass Spectral Deconvolution Software (AMDIS). Esse software pode processar várias extensões de arquivos, abrangendo toda a gama de marcas comerciais de cromatógrafos, como Bruker, Agilent, Thermo, Shimadzu, etc. AMDIS também possui algoritmos específicos de deconvolução espectral a partir de picos cromatográficos e funções que permitem a comparação dos espectros deconvoluídos com bibliotecas de espectros puros.132 Vale ressaltar que a CG-EM é capaz de analisar compostos com massa até aproximadamente 650 Da e que a metabolômica visa contemplar substâncias com até 1500 Da. Além disso, a CG-EM possui grandes dificuldades para a análise de fosfatos (Bis, Di e Tri, NADH ou ATP), enquanto os monofosfatos podem ainda ser observados via derivatização com trimetilsilano. Outra desvantagem do uso da CG-EM em estudos metabolômicos está na análise de aminas biogênicas, principalmente em estudos do metabolismo primário para a avaliação de aminoácidos. Essas funções podem sofrer efeitos de matrizes e afetar a altura e os valores absolutos das intensidades de pico devido as diferentes formas de derivatização dos grupos amino. Para esses casos, é importante avaliar as condições de limpeza do injetor, liner, seringa e os primeiros 15 cm da coluna.128,130 Cromatografia líquida acoplada a espectrometria de massas (CLAE-EM) A Cromatografia Líquida de Alta Eficiência (CLAE) é a técnica analítica de separação mais recente para aquisição de perfis cromatográficos, podendo ser acoplada a diferentes analisadores e detectores.133 O processo de separação em CLAE procede através dos diferentes estados de equilíbrio entre analito e as fases móvel (solventes orgânicos e aquosos) e estacionária (coluna cromatográfica).134 Em metabolômica, essa técnica se tornou especialmente importante a partir do início dos anos 90 após o seu acoplamento a fontes de ionização em pressão atmosférica (APCI - atmospheric-pressure chemical ionization, APPI - APCI - atmospheric-pressure photo ionization e ESI - electrospray ionization). Dentre os métodos de ionização em pressão atmosférica "brandos", a ESI é o método de escolha para a maioria dos estudos metabolômicos, permitindo a formação de espécies protonadas, desprotonadas, adutos e em casos particulares, íons moleculares a partir da remoção de elétrons.135 A ESI possui uma maior extensão de ionização e detecção de compostos polares e moléculas com maior peso molecular quando comparado a APCI e APPI.136-138 No entanto, um problema comum em estudos metabolômicos é a busca pela molécula protonada ou desprotonada. É importante destacar que o balanço entre as espécies formadas pode ser modificado, mas é, antes de mais nada, uma propriedade físico-química do analito. Determinadas estruturas sequer formam o íon protonado, mesmo em condições de pH < 2.139 Com o desenvolvimento dos analisadores do tipo time of flight (ToF), Orbitrap e FT-ICR, foi possível obter espectros em alta resolução e a determinação de fórmulas elementares a partir dos valores de massa/carga dos íons detectados, auxiliando o processo de identificação e quantificação de metabólitos - fundamentais para qualquer estudo metabolômico. Além do poder de resolução, muitas configurações instrumentais permitem a fragmentação de metabólitos através de câmaras de colisão, como a dissociação induzida por colisão (CID). A Tabela 3 resume a performance de analisadores de massas utilizados em estudos metabolômicos.136

Diferentes tipos de coluna (dimensões e química da fase ligada) podem ser utilizados. Para análises exploratórias, as colunas de fase reversa C8 e C18 são preferencialmente utilizadas devido ao melhor poder de resolução para os compostos de média polaridade em matrizes biológicas. A CLAE-EM é adequada para análise de compostos semi-polares (50-1500 Da) sem a necessidade de qualquer tipo de derivatização. Em plantas, a CLAE-EM é utilizada para análise de diversos grupos de metabólitos secundários, como alcaloides, saponinas, ácidos fenólicos, fenilpropanoides, flavonoides, glicosinolatos, poliamidas, terpenos, esteróis e derivados.133 É importante destacar que o número de metabólitos primários disponíveis comercialmente é expressivamente maior que produtos do metabolismo secundário. Consequentemente, a elucidação estrutural por uso de padrões internos é inviável e limitaria o potencial da CLAE-EM na pesquisa de plantas. Nesse sentido, uma série de softwares e serviços online oferecem ferramentas de extração de pico (métodos de deconvolução), alinhamento, remoção de efeitos indesejados (efeitos de matriz) e principalmente, ferramentas para visualização e tratamento de dados, como é o caso de Mzmine2,140 XCMS141 e o GNPS.140-143 Para o desenvolvimento e a otimização do método cromatográfico, diversas variáveis podem ser avaliadas, incluindo o modo cromatográfico (normal, reverso, troca iônica, HILIC, etc.), o tipo de coluna (largura, comprimento, diâmetro do poro, dimensão interna, composição da fase estacionária, etc.), o detector (UV-VIS, CAD, FLU, EM, EM/EM, RMN), parâmetros físico-químicos (temperatura, pressão, vazão do da fase móvel) e a composição da fase móvel (seletividade, classe de solvente, proporção da mistura e presença de modificadores). Estes parâmetros são comumente testados em análise exploratória isocrática ou em gradiente, permitindo a otimização através da variação singular das variáveis. Na análise sistemática, planejamentos fatoriais são aplicados, como descrito no tópico planejamento experimental, objetivando a visualização das interações entre as variáveis testadas. Eletroforese capilar acoplada a espectrometria de massas (EC-EM) A EC, apesar de ser uma técnica menos difundida nos laboratórios de química de produtos naturais, também exerce um papel importante em estudos metabolômicos. A EC-EM se caracteriza pela separação de compostos com o uso de campos eletromagnéticos em fase líquida condutiva resultando em um fluxo eletro-osmótico.133,134,136 A EC utiliza colunas capilares de sílica e sua eficiência de separação é comparável aos sistemas de CLUE (cromatografia líquida de ultra eficiência) e CG. A grande vantagem da EC-EM é a possibilidade de análise de ampla faixa de metabólitos de íons inorgânicos (ainda pouco explorado em metabolômica) até grandes proteínas. Como a separação da EC se baseia na relação massa/carga, o poder de resolução e separação é muito alto, alcançando a distinção de isômeros e diastereoisômeros.134,136 A EC é uma técnica rápida, de baixo custo, que requer pequeno volume de amostra e pouca ou nenhuma preparação de amostra. Embora possua uma série de vantagens, existem algumas limitações da EC no acoplamento a fontes de ionização à pressão atmosférica. A adição de solventes adicionais e tampões de baixa volatilidade, podem contaminar e aumentar a supressão de ionização dos analitos no acoplamento com ESI.134,136 Dessorção/ionização a laser auxiliada por matriz (MALDI-EM) e Infusão direta a espectrometria de massas (ID) A dessorção/ionização a laser auxiliada por matriz137 (MALDI) é uma técnica que vem ganhando espaço no campo da metabolômica 131,138 principalmente pela alta velocidade de análise144,145 e pela possibilidade de gerar "imagens" de íons em tecidos biológicos,133,146 incialmente em animais e, mais recentemente, em plantas.147,148 Devido à possibilidade de focalização, potência laser e a boa resolução do MALDI (íons), os dados gerados podem ser correlacionados a diferentes escalas de cores e, através da sobreposição com fotografias, é possível gerar mapas e imagens espectrais. De fato, existem outras técnicas de imagem por massas tal como espectrometria de massas por íons secundários (SIMS) e a ionização de dessorção por eletronebulização (DESI). Enquanto a SIMS possui uma resolução numa escala de µm, o MALDI e DESI possuem resoluções piores.133 Embora seja uma técnica de alta sensibilidade, alta velocidade de varredura e uma menor influência de contaminantes, o MALDI apresenta alguns contrapontos como a necessidade de otimização dos parâmetros instrumentais para cada tipo de metabólito, isso é, configuração da intensidade, frequência e foco do laser, número de tiros aplicados por área, composição da matriz, etc. A infusão direta (ID) consiste na técnica de injeção de uma amostra diretamente na fonte de ionização. Em metabolômica, essa técnica é utilizada em fontes de ionização a pressão atmosférica (ionização branda) uma vez que causa uma menor taxa de fragmentação dos íons, resultando em espectros menos complexos. Contudo, essa é a abordagem menos utilizada uma vez que se baseia nos valores de massa/carga, sem dados ortogonais (índice de retenção), que permitiria a detecção de isômeros que possuem tempos de retenção distintos, além dos efeitos de supressão de sinal e de matriz.133 Outras considerações sobre o uso da espectrometria de massas em metabolômica Independente da técnica de separação que precede a análise por espectrometria de massas (CLAE, EC ou CG), recomenda-se a adição de uma solução calibrante em cada análise visando ao ajuste da exatidão de massas/cargas dos íons detectados em intervalo de peso molecular.134,136 Esse processo é particularmente importante para analisadores de massas de alta resolução como Orbitrap, FT-ICR e ToF. Os compostos FC-43 (perfluorotributilamina) e ácido trifluoracético sodiado (Na-TFA), estão entre os calibrantes mais utilizados em metabolômica. Em alguns casos, um conjunto separado de injetores pode realizar a adição contínua de calibrante nas amostras para correção automática dos desvios de massas/cargas (lock mass). Para análises quantitativas em espectrômetro de alta resolução, além do calibrante, deve-se garantir um tuning adequado do espectrômetro, bem como adição de padrão interno de concentração conhecida, permitindo cálculo preciso da área relativa e, consequentemente, da concentração metabólica dos sinais em uma amostra. Diversos softwares estão disponíveis para o pré-tratamento de amostras para análise quantitativa149 como MS-Dial,150 MarkerView (Sciex, EUA), Compound Discoverer (ThermoFisher Scientific, EUA), Data Analysis (Bruker, Alemanha), XCMS,141 MetaboAnalyst151 e MZmine 2,140 sendo os protocolos de aquisição de validação disponíveis na literatura.152-154 Ressonância magnética nuclear A ressonância magnética nuclear (RMN) é uma técnica qualitativa e quantitativa altamente reprodutível e não seletiva, ou seja, não depende das características químicas dos compostos observados, como polaridade e acidez (pKa).155 Em metabolômica, experimentos de RMN uni- e bidimensionais vem sendo extensamente empregados devido a sua alta reprodutibilidade e fácil preparo de amostras, fornecendo informações a respeito da estrutura de diferentes compostos, incluindo metabólitos inéditos, isômeros ou substâncias de difícil ionização ou derivatização para análise por EM.155 Nas últimas décadas, a RMN tem sido aplicada majoritariamente a estudos metabolômicos ao possibilitar a análise simultânea de diversos grupos de metabólitos secundários (flavonoides, alcaloides, terpenoides, etc) e primários (açúcares, ácidos orgânicos e aminoácidos), assim como a comparação direta da concentração dos compostos sem a necessidade de se elaborar curvas de calibração, o que requer o uso de substâncias de referência de alto grau de pureza, muitas vezes raras e de difícil obtenção. Em RMN, o cálculo de quantificação necessita da presença de um sinal de composto de concentração conhecida em todas as amostras analisadas, sendo que esse sinal pode ser obtido por (a) adição de padrão interno; (b) adição artificial de sinal após aquisição por método ERETIC (Electronic REference To access In vivo Concentrations);156 (c) adição artificial de sinal após aquisição por QUANTAS (QUANTification by Artificial Signal);157 ou adição de sinal residual de solvente protonado. No caso da adição de um padrão interno, esse deve ser cuidadosamente selecionado, devendo ser estável no solvente deuterado, não ser reativo ou possuir sinais residuais de água, higroscópio, volátil, e apresentar sinais junto dos sinais das amostras. Esses pré-requisitos são imprescindíveis para garantir a seletividade, precisão e acurácia do método, tornando a quantificação reprodutível e confiável.158 Durante a aquisição de dados para análise quantitativa, diversos parâmetros experimentais devem ser otimizados, incluindo a calibração do pulso, homogeneidade do campo, tunning, número de scans (NS) e tempo de relaxamento T1, sendo o último estimado por sequência de pulso inversão-recuperação. Todas as etapas para otimização foram detalhadamente descritas por Giraudeau e colaboradores e devem ser realizadas em réplicas para garantir uma resposta significativa.159 Diversas revisões relatam os processos de validação e podem ser encontradas nos trabalhos de Pauli et al. (2012, 2014),160,161 Gödecke et al.,162 Maniara et al.163 e Malz & Jancke.164 A aplicação de RMN possui algumas limitações, sendo a maior delas a baixa sensibilidade e a dificuldade de acoplamento com técnicas de separação. Entretanto, vários avanços em instrumentação, hardwares e softwares tem aumentado consideravelmente a sensibilidade da RMN. A potência de magnetos, aumentando a população da resultante magnética de spins, o advento das criossondas, que diminuem o ruído instrumental aumentando a sensibilidade em até 16 vezes e a miniaturização da amostragem com tubos de até 1,6 mm, tem aumentado consideravelmente a sensibilidade de experimentos, chegando a ordem de microgramas. As amostras de RMN para metabolômica são preparadas em solventes deuterados, como metanol-d4, DMSO-d6, D2O e CDCl3, centrifugadas e transferidas a tubos de 5 mm de diâmetro. Tubos de 1-3 mm também estão disponíveis, sendo utilizados para espectrômetros com sondas específicas (criossondas).165,166 A presença de macromoléculas e traços de água também limitam a técnica de RMN, sendo fundamental a seleção e otimização de sequências de pulso e os respectivos parâmetros experimentais para a sua supressão. Sequências que realizam a supressão do solvente ou sinal da água são comumente selecionadas para aumentar o ganho (gain), permitindo, consequentemente, maior e melhor detecção e análise de metabólitos minoritários. A supressão do sinal da água é realizada pelas sequências do tipo WATERGATE (Gradient-tailored excitation for single-quantum NMR),167 pré-saturação e excitation sculpting water suppression.168 Para minimizar a presença de sinais alargados no espectro de ressonância devido ao baixo tempo de relaxação do T2 e difusão rotacional limitada de macromoléculas, sequências como 1D nuclear Overhauser effect spectroscopy com pré-saturação (1D NOESY-presat)169,170 e 1D Carr-Purcell-Meiboom-Gill (CPMG)171 podem ser empregadas. Em 1D NOESY-PRESAT, a sequência de pré-saturação causa supressão do sinal da água enquanto a CPMG remove sinais alargados de proteínas, diminuindo o alargamento dos sinais. Além dos experimentos realizados para o núcleo de hidrogênio (monodimensionais), existem sequências que ajudam na identificação dos metabólitos e determinação de suas conectividades. J-resolved,172 correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), heteronuclear multiple bond correlation (HMBC) e heteronuclear single quantum correlation (HSQC)173,174 informam de maneira detalhada a disposição e correlação dos sistemas de spin, sendo atualmente utilizadas para fins de elucidação estrutural em estudos metabolômicos. Ferramentas computacionais e workflows em metabolômica Na última década, um grande número de pacotes vem sendo relatados envolvendo os estudos metabolômicos. Tais ferramentas podem fornecer guias operacionais automatizados e padronizados para o pré-processamento de dados (por exemplo, alinhamento, deconvolução e normalização de dados), análises estatísticas uni e multivariadas, identificação metabólica e ferramentas de visualização, como a modelagem por redes metabólicas. A Sociedade de Metabolômica publicou em 2017, em sua revista (Metabolomics), um trabalho avaliando as principais ferramentas metabolômicas utilizadas pelos grupos de pesquisa que pertencem a sociedade.175 Esses pacotes e serviços são mostradas na Tabela 4.
PRÉ-PROCESSAMENTO DOS DADOS O pré-processamento dos dados é uma etapa intermediária entre a aquisição dos dados obtidos através da metodologia analítica escolhida e a análise estatística. Ela visa melhorar a qualidade dos sinais e reduzir a interferência de ruídos, tornando os dados comparáveis entre si. A rotina de pré-processamento é necessária para garantir a detecção e a quantificação de picos de alta qualidade, sendo o seu maior objetivo a transformação dos dados em uma matriz robusta, eliminando alterações que acontecem devido às variações na amostra e no equipamento, como diferença na concentração de sais, temperatura e pH.199-202 Algumas funções de pré-processamento são específicas para cada técnica analítica, como é o caso do faseamento e referenciamento nos espectros de RMN. Outras, entretanto, são gerais para todas as matrizes de dados, podendo aplicar diferentes funções e algoritmos para otimização do resultado. A Figura 4 mostra um fluxograma descrevendo algumas das etapas de processamento que serão discutidas nas próximas seções.
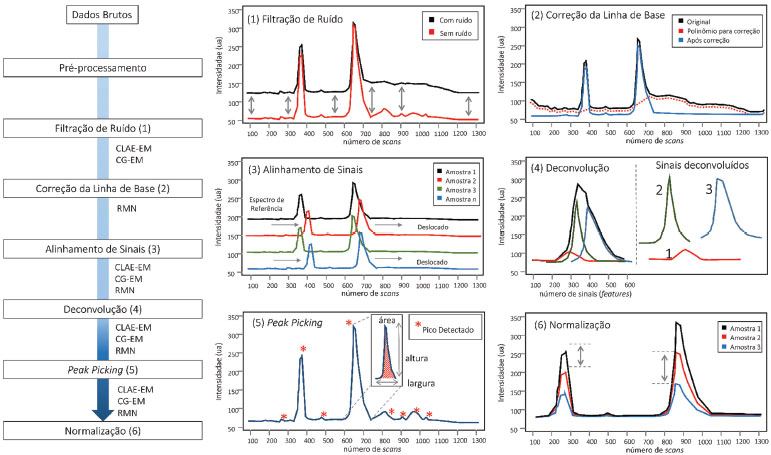 Figura 4. Fluxograma das possíveis etapas de processamento de matrizes biológicas em estudos metabolômicos
Pré-processamento de dados cromatográficos e de espectrometria de massas Os dados de CLAE-EM ou CG-EM são um conjunto de vetores gravados durante pequenos intervalos de tempo sucessivos, em que cada ponto consiste em valores de m/z e intensidade. Uma vez que cada instrumento fornece dados em diferentes formatos, diferentes ferramentas são utilizadas para conversão dos dados brutos.203-205 A rotina de pré-processamento para dados de CLAE-EM ou CG-EM é baseada na detecção de picos e, portanto, requer funções robustas e reprodutivas para as três dimensões dos dados, ou seja, tempo, m/z e intensidade. Os algoritmos devem ser capazes de detectar picos mesmo com baixa razão sinal/ruído (S/N), simultaneamente filtrando qualquer interferência.141 Um pré-processamento inadequado resulta em um conjunto de dados que exibe desvios substanciais, impossibilitando a conclusão dos dados químicos e biológicos.206 Filtração de ruído (denoiesing) Devido à complexidade do perfil químico e a baixa reprodutibilidade dos instrumentos cromatográficos, diversas funções de pré-processamento foram desenvolvidas para garantir o bom alinhamento e resolução dos picos em análises de CLAE-EM. Dentre essas, a filtração ou subtração da linha de fundo acontece no domínio cromatográfico e visa diminuir o ruído proveniente de interferências instrumentais,207 Figura 4. As funções mais comumente utilizadas para a diminuição do ruído são a filtração combinada (matched filtration),141 que se baseia na aplicação de um filtro cujo coeficientes são iguais à extensão da forma do sinal, ou a filtração pela mediana em uma janela de tamanho determinado. Independente da função, recomenda-se que a filtração seja baseada em um espectro de massa em que não haja sinais de interesse (linha de base ou linha somente com ruído), evitando a remoção de sinais relativos às moléculas minoritárias ou fracamente ionizáveis. Alguns softwares disponíveis integram os algoritmos de filtragem e detecção de pico em uma única função, como, por exemplo, o pacote de XCMS, que usa a filtração combinada baseada na segunda derivada da função Gaussiana,141 o pacote de apLCMS, que realiza a filtração dos dados através do padrão de distribuição dos pontos208 e o software MAVEN.209 MZMine,140 Analyst,151 OpenMS210 e MetAlign,211 oferecem, além da filtração, diferentes funções de redução de ruído e smoothing, incluindo filtro Gaussian, Savitzky Golay e algoritmos de correção da linha de base.212,213 O Matched Filtration with Experimental Noise Determination (MEND)214 é uma função de filtração combinada utilizada em dados de CLAE-EM e explora as razões massa/carga (m/z) de áreas com ruído aleatório para melhorar o algoritmo de filtração. Para tanto, o algoritmo determina o ruído característico em região sem eluição de picos no cromatograma (linha de base) para, em seguida, correlacionar os m/z desse ruído com outras regiões do cromatograma, diminuindo drasticamente o ruído aleatório e a distorção da forma do pico no EM. Alinhamento Durante a aquisição dos dados de CLAE/CG-EM algumas variáveis aleatórias, como pH, mudanças na temperatura, vazão ou concentração da fase móvel, presença de sais, efeito de matrizes, degradação da coluna cromatográfica entre outras, podem levar a variações não-lineares na eluição de um mesmo analito ao longo do tempo. Na prática, essas variações causam alterações indesejadas nas etapas subsequentes de análise dos dados, devendo ser minimizadas por algoritmos de alinhamento,6,141 como pode ser visualizado na Figura 4. De maneira simples, os algoritmos de alinhamento podem ser divididos em duas classes: o alinhamento baseado em uma referência (feature-based approaches), ou seja, valores estabelecidos de tempos de retenção para determinado metabólito presente nas amostras e, o segundo, conhecido como profile-based approaches, em que o alinhamento é realizado durante a detecção dos picos, estimando a variabilidade de sinais semelhantes ao longo dos cromatogramas de todas as amostras de uma matriz.207,215 Existem diversas funções para o processo de alinhamento, destacando a Dynamic Time Warping (DTW),216 Correlation Optimized Warping (COW),217 Recursive Alignment Fast Fourier Transform (RAFFT)218 e o Peak Alignment by Fast Fourier Transform (PAFFT).218 Essas funções estão disponíveis em praticamente todos os softwares de análise de dados de CLAE-EM, como o XCMS,141 SpecAlign,219 RANSAC,220 OpenMS,210,221 msInspect,222 SpecArray,223 XAlign,224 MetAlign211 e MZMine 2.140 O COW é um dos principais métodos de alinhamento em CLAE-EM, pois utiliza programação dinâmica como função alvo.216,217 Nesse algoritmo, os cromatogramas são divididos em segmentos de mesmo tamanho e, baseado em uma referência cromatográfica, os outros cromatogramas são alinhados por funções de estiramento e compressão linear.218,222,225 Detecção de picos (deconvolução, peak picking, deisotoping and gap filling) A finalidade da detecção de um sinal é extrair informações robustas para a elucidação e quantificação dos metabólitos em uma amostra. Idealmente, um método de detecção deve identificar sinais verdadeiros e evitar falsos positivos, reduzindo a complexidade dos dados e tornando a análise viável.212,226 Durante a detecção dos picos, diferentes processos podem ser sistematicamente realizados, incluindo: (i) a determinação automática ou manual de sinais de interesse por algoritmos de peak picking,227,228 (ii) a aplicação de algoritmos de deconvolução em sinais que eluíram simultaneamente durante a corrida,229-233 (iii) o deisotoping ou identificação dos sinais isotópicos, removendo informações redundantes207,234,235 e o (iv) gap filling ou recuperação de sinais fracos, que refere-se a recuperação de sinais que não foram detectados durante o peak picking devido à baixa intensidade, falta de qualidade da forma do pico ou erro do algoritmo de detecção. A detecção de picos por algoritmos de peak picking permite a identificação de um sinal em determinado tempo de retenção ou valor de m/z, indicando a sua altura, que é a máxima intensidade dos pontos, e sua área, definida como a soma das intensidades dos pontos (Figura 4). As funções para essa identificação utilizam diferentes estratégias, destacando o F-score (algoritmo centroidPicker) do MZmine 2,236 análise da largura do pico cromatográfico por centWave237 e binning nos dados por função matched filter,141 podendo ser facilmente realizadas em softwares de processamento de dados como Open MS,210,221 apLCMS,208 MAVEN,209 VIPER,238 MZmine,140 Sirius,239,240 Decon2LS,240,241 e MS-Dial.149,150 Para a identificação dos sinais isotópicos, os softwares mais recomendados são ApLCMS,208 msInspect,235 XCMS,141 DeconTools,241 e MetaboAnalyst,151 sendo essa etapa especialmente importante para a análise de peptídeos e proteínas. Mais comumente, os algoritmos integram sinais de íons originados da mesma molécula, originando um único sinal de massa monoisotópica.207 Por exemplo, o software DeconTools realiza a identificação dos sinais isotópicos através de três etapas que consistem na (i) identificação do padrão de isotopia, (ii) predição da carga com base na distância entre os picos e (iii) comparação dos dados experimentais com os padrões teóricos, gerados pela média dos isótopos. MZmine 2 é o software mais utilizado para detecção dos dados espectrais de massas.140 Sua função de detecção de picos é dividida em (i) construção do cromatograma, que cria uma lista de massas em cada ponto tempo de retenção e (ii) deconvolução dos picos por quatro algoritmos distintos. Em ambos os passos, fica disponível um módulo de visualização, que pode ser utilizado para otimizar os parâmetros manualmente a qualquer momento.212 Normalização A normalização remove variações sistemáticas não desejadas entre amostras e permite a comparação quantitativa (absoluta ou proporcional) das amostras. Para estudos em matrizes naturais, como micro-organismos e plantas, essa etapa é particularmente relevante, pois as variações de concentração podem interferir na interpretação química e biológica das análises multivariadas.242 A eliminação do efeito da variabilidade pela normalização permite, por exemplo, a comparação entre sinais de duas amostras preparadas em concentrações distintas. Contudo é necessário o uso de um sinal de referência para a normalização de diferentes amostras.243 A metodologia mais utilizada para normalizar dados de CLAE-EM é através de fatores de escala, como valores de média ou mediana, ajustando as intensidades de acordo com o conjunto de dados. Outros modelos mais rigorosos também podem ser aplicados, como é o caso dos métodos de regressão,243 a utilização de padrões internos244 ou o uso de uma amostra (controle de qualidade), isso é, uma mistura que contém os metabólitos de todas as amostras analisadas.245 Para a metabolômica destacamos as seguintes formas de normalização: (1) Adição de padrão(ões) interno/externo: nessa normalização, um padrão de concentração conhecida é adicionado em cada amostra. Esse procedimento pode ser realizado de duas formas: (a) inclusão de um padrão interno no solvente extrator para avaliação do processo de amostragem, extração e análise ou (b) na etapa de introdução da amostra no instrumento. Esse último visa apenas corrigir as variações instrumentais; (2) Normalização por área total: o fator de normalização aplicado a cada sinal da amostra (intensidade dos picos em CLAE-EM, CG-EM ou sinais de RMN) é calculado pela soma de todas as informações (sinais) correspondentes daquela amostra; (3) Normalização por quociente probabilístico: esse método leva em consideração a relação probabilística dos sinais de uma amostra frente a uma referência, ou seja, cada sinal de uma amostra é dividido pelo correspondente sinal de amostra referência gerando um quociente. Os valores resultantes são arranjados num histograma revelando qual é o quociente com maior frequência nas amostras, sendo esse valor escolhido como fator de normalização; (4) Normalização Quantil: esse método exige que todas as amostras de um conjunto amostral tenham a mesma intensidade/área. A diferença desse método aos demais é que não existe um fator único de normalização. Inicialmente, os dados (tabela de dados - CLAE-EM, CG-EM ou RMN) são ordenados dos menores para os maiores valores. Em seguida é calculado um valor médio ou mediana de cada sinal (de cada coluna de dados) e atribuído aos valores das amostras. Esse processo ocorre para cada sinal da amostra. Os dados são reordenados originalmente e a tabela de dados normalizada. É importante ter em mente que a escolha dos métodos depende da origem dos dados. No pré-processamento, os dados devem ser analisados em diferentes fontes de normalização para definir a melhor opção. O MZmine140 e SuperHirn246 são os softwares mais utilizados para normalização de dados. O SuperHirn utiliza uma versão modificada do método padrão de normalização por tendência central247 e mostra resultados satisfatórios para normalização de conjunto de dados de matrizes de produtos naturais. Pré-processamento de dados de RMN Pré-processamento do decaimento indutivo livre (FID) O decaimento indutivo livre (FID) adquirido durante experimento de RMN contém toda informação espectral da amostra, incluindo sinais dos compostos presentes em solução e ruído instrumental. Esses ruídos, que são aleatoriamente ocasionados pela oscilação térmica das bobinas e interferentes eletrônicos, causam considerável diminuição de sensibilidade, tornando necessária a realização de etapas que melhorem a qualidade do espectro e a detecção metabólica. Durante a aquisição de um FID, a informação espectral dos metabólitos decai com o tempo, enquanto o ruído permanece na mesma intensidade durante toda a evolução da sequência de pulso. Isso é importante, pois, quando o tempo de aquisição (AQ) é significativamente maior do que o tempo de relaxamento (T1), a contribuição do ruído torna-se muito intensa, diminuindo a relação entre o sinal e ruído (<S/N), dificultando a detecção de sinais minoritários. Diversas funções podem ser realizadas ao FID para aumento da qualidade espectral, sendo as mais comuns o truncamento, zero filling e apodização. O truncamento é uma função que remove todos os pontos finais do decaimento, aumentando a razão entre o sinal e o ruído no espectro. Porém, como nem toda informação removida pertence aos sinais aleatórios, isso pode levar uma diminuição da resolução digital, sendo recomendável a realização concomitante de zero-filling. Essa função corrige a diminuição do número de pontos do truncamento pela adição de pontos de intensidade nula (I = 0) ao fim do decaimento, melhorando a relação do sinal/ruído sem diminuição significativa da resolução digital. Em geral, esses procedimentos visam aumentar a resolução aparente e a visualização das constantes de acoplamento.248 Ao contrário do truncamento e zero filling, a apodização não envolve manipulação direta do FID. Nesse caso, o decaimento é multiplicado por uma função, levando a uma melhora na razão S/N ou na resolução espectral (nunca em ambas). Entre as funções mais utilizadas estão: apodização exponencial, gaussiana, dupla apodização (DM), sino sine (MS), entre outras,248 sendo a otimização realizada através de avaliações individuais. Referenciamento e faseamento Análises metabolômicas que envolvem dados de RMN passam pelo cálculo da transformada de Fourier (FT). Esse procedimento converte os dados de RMN no domínio tempo em domínio de frequência. Isso é fundamental para o processo metabolômico na discriminação do número de frequências (3 ou mais sinais diferentes) que estão presentes em um sinal no domínio do tempo. Após a FT é realizado o referenciamento (ou calibração), que visa a corrigir variações globais dos deslocamentos químicos. Essa função é de extrema importância para análises metabolômicas pois permite a correta comparação entre os dados analíticos de diferentes amostras. A calibração é comumente realizada por adição de um padrão interno de deslocamento conhecido à solução, como o tetrametilsilano (TMS) e hexametilsiloxano (HMDSO), ou pelo deslocamento químico do sinal residual do solvente deuterado utilizado na análise.248 O domínio de frequência, ou espectro, também deve ser faseado, visando garantir a visualização de linhas espectrais com fases puras (100% absortivas), Figura 5A. A diferença de fases em um espectro acontece principalmente devido a variações do ponto de origem do FID, sendo corrigida pela alteração (manual ou automática) das fases de primeira e segunda ordem. A primeira, conhecida como PH0, é independente da frequência e é invariável para todos os sinais em uma amostra, independente do offset. Já a segunda, também chamada de PH1, é dependente da frequência e faz o ajuste fino dos sinais, garantindo formato absortivo até em deslocamentos distantes do pivot point.248
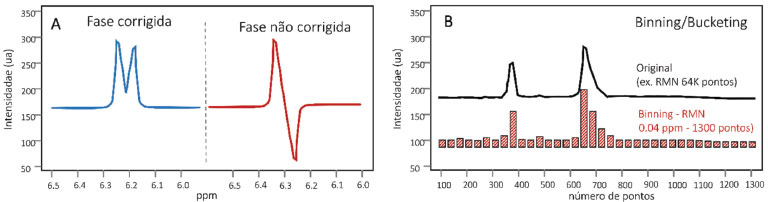 Figura 5. (A) Faseamento do domínio de frequência ou espectro, visando garantir a visualização de linhas espectrais com fases puras (100% absortivas); (B) Redução da dimensionalidade dos dados por bucketing/bining
Correção da linha de base (smoothing) Distorções da linha de base em espectros unidimensionais são comumente originados pela alteração dos primeiros pontos do FID, que adicionam modulações de baixa frequência ao longo de todo o espectro. A correção dessas distorções é uma etapa essencial ao pré-processamento de dados de RMN pois a variação das intensidades pode resultar em resultados quimiométricos e quantitativos equivocados, dificultando a análise biológica e a identificação de biomarcadores.249 A correção da linha de base na metabolômica é feita diretamente no espectro, podendo ser realizada por diferentes algoritmos de acordo com as características das amostras. Os algoritmos constroem uma linha base teórica e subtraem os espectros reais para minimizar as distorções, apresentando variações somente na maneira na qual o cálculo e modelagem da linha base são realizados (Figura 4). A maioria das funções são baseadas no ajuste polinomial interativo (iterative polynomial fitting),250 porém, novos algoritmos vêm sendo desenvolvidos para superar obstáculos dessa estratégia, como a necessidade de intervenção do usuário e a tendência de alta variação de sinais em ambientes com baixa razão S/N. Esses algoritmos incluem procedimentos robustos de predição, destacando o locally weighted scatterplot smoothing (Lowess fit),249 penalized least squares (airPLS),251 Mínimos Quadrados Assimétricos,252 B-splines253 ou aplicação de modelos de mistura.254 O airPLS é outro algoritmo extensamente aplicado em metabolômica de plantas e atua através da mudança repetitivamente dos pesos das somas dos erros ao Quadrado (SSE) entre a linha de base corrigida e a original. Os pesos da SSE, obtidos de forma adaptada, utiliza a diferença entre a linha corrigida e a original, garantindo ajuste e correção fina das distorções.251 Alinhamento Um dos problemas mais frustrantes na avaliação dos perfis metabólicos por RMN de hidrogênio é a presença de picos deslocados em diferentes amostras que são referentes ao mesmo analito. Essas variações, como similarmente ocorre para CLAE, acontecem devido às variações aleatórias das amostras (pH e efeitos de matrizes) e do instrumento, dificultando a descoberta de tendências entre os espectros.255,256 Dentre os algoritmos utilizados para correção dessas mudanças locais, destacam-se o Partial Linear Fit (PLF),256 Peak Alignment by Genetic Algorithm (PAGA),165 Peak Alignment using Reduced Set (PARS),257 Peak Alignment by Beam Search (PABS),258 Peak Alignment by PCA (PAPCA),259 Fuzzy Warping (FW),260 Generlized Fuzzy Hought Transform (GFHT),261 Recursive Segment wise Peak Alignment (RSPA),262 Progressive Consensus Alignment of NMR Spectra (PCANS),263 Hierarchical Cluster-based Peak Alignment (CluPA),264 e Bayesian approach for alignment (BAA).265 Interval COrrelated Shifting (ICOShift) é a função de alinhamento mais utilizada em metabolômica para dados de RMN.266 Nesse algoritmo, os espectros são divididos em diferentes segmentos e alinhados segundo um espectro de referência (média ou variância do conjunto amostral pode também ser utilizado). O alinhamento ocorre pelo cálculo da correlação cruzada entre os segmentos, através de uma rápida transformação matemática (fast Fourrier transformation), permitindo alinhamento simultâneo de todas as regiões. É importante mencionar que o alinhamento, por vezes, pode afetar a área dos picos, podendo influenciar negativamente as etapas de análise e quantificação de metabólitos. Portanto, para estudos quantitativos, recomenda-se unicamente o uso da correção de fase. Binning/bucketing Historicamente, a metabolômica por RMN é realizada pela quantificação ou avaliação das integrais dos sinais de interesse. Porém, esses protocolos são de difícil automação em matrizes com um grande número de pontos e não podem ser usados em regiões de coalescência entre sinais, dificultando a aplicação dessa técnica em dados complexos. A fim de reduzir esses efeitos, abordagens de binning ou bucketing foram introduzidas para reduzir a dimensionalidade dos dados. Especificamente, intervalos de dados são somados permitindo a simplificação, comparação espectral e redução da interferência do efeito de matrizes e ruídos e provenientes de fenômenos de segunda ordem201 (Figura 5B). O binning era inicialmente realizado por segmentação equidistante, dividindo os espectros em regiões uniformemente espaçadas e larguras pré-determinadas.200,201,267 Segundo o protocolo de Kim et al.,72 a largura padrão em metabolômica é de 0,04 ppm, sendo esse valor ponderado de acordo com a resolução e a boa distribuição dos sinais e suas multiplicidades. Entretanto, essa função ainda apresenta algumas limitações, como dividir um sinal em duas regiões distintas. Atualmente, as funções de binning permitem utilizar diferentes limites de largura, como por exemplo no binning inteligente e adaptativo (AI-binning),268 Gaussian binning,269 adaptive binning using wavelet transform270 e o dynamic adaptive binning,271 gerando melhores resultados. O binning oferece uma metodologia consistente para a formação de modelos de análise quimiométrica classificatória e na avaliação de matrizes na identificação de biomarcadores. Porém, deve-se ter em mente que a segmentação reduz significativamente a resolução espectral, devendo ter seu uso ponderado de acordo com a natureza dos dados. Deconvolução A coalescência de sinais é uma séria limitação analítica da RMN, dificultando sua aplicação na análise metabolômica em plantas. Nesse contexto, algoritmos de deconvolução exercem um papel crucial na "separação metabólica" como ocorre no caso das técnicas hifenadas como a CLAE-EM ou CG-EM. Diversas funções de deconvolução foram desenvolvidas para a identificação de metabólitos majoritários e minoritários, mesmo em áreas altamente sobrepostas. Deconvolução por Bayesian Automated Metabolite Analyzer for NMR (BATMAN),272 Global Spectrum Deconvolution (GSD, MestreNova)273,274 e Statistical Total Correlation Spectroscopy (STOCSY)275 são algumas das funções disponíveis na literatura. O STOCSY auxilia na identificação de biomarcadores pois avalia a correlação dos sinais em uma matriz de dados, criando um pseudo-espectro bidimensional que mostra as conectividades entre os sinais. Esse método é muito vantajoso para análise de misturas complexas, pois avalia a covariância (aumento ou diminuição da concentração) de metabólitos de uma mesma via biosintética e também a correlação espectral (avaliação se os mesmos sinais estão presentes em diferentes amostras), garantindo a deconvolução de um mesmo sistema de spin, ou seja, de moléculas estruturalmente semelhantes. Sua aplicação, associada ou não a análises quimiométricas, oferece uma plataforma robusta para análise metabolômica, sendo aplicada a diferentes espécies e famílias de plantas.275-277 Normalização A normalização em metabolômica de RMN consiste na multiplicação de cada espectro de RMN por uma constante, sendo esta computada por diferentes algoritmos.201 O método padrão é a normalização por área total (como já discutido previamente),278 porém, outros métodos também podem ser aplicados de acordo com a complexidade das amostras, como a normalização por padrão interno, por quociente probabilístico (PQ),279 por histograma,280 ou por agrupamento de clusters (group aggregating normalization - GAN).281 É importante ressaltar que uma normalização inadequada pode trazer sérios problemas nas etapas subsequentes, como as análises quimiométricas, levando à resultados desastrosos.199 Todas as etapas do pré-processamento previamente descritas podem ser realizadas em diferentes pacotes e serviços, como PERCH,282 TopSpin (Bruker), Chenomx NMR Suite,283 MestReNova,284 algoritmo AutoFit,174 assim como as ferramentas descritas na Tabela 4. Esses programas possuem funções referentes não só ao pré-tratamento, mas também ao tratamento quimiométrico das funções que auxiliam a elucidação estrutural e quantificação metabólica.
PROCESSAMENTO DOS DADOS Centralização e escalonamento dos dados O uso de ferramentas quimiométricas na análise dos dados permite a avaliação discriminante entre amostras e variáveis por meio projeções matemáticas, como é o caso da análise de componentes principais (PCA) e abordagens relacionadas, bem como a criação sistemática de modelos que facilitem a interpretação química, biológica e ecológica de conjuntos amostrais.285 No entanto, a análise de dados de RMN e EM por meio de técnicas de projeção de variáveis latentes como a PCA, PLS e outras, toma em consideração o espectro ou perfil médio e, qualquer tipo de variação biológica sutil nos dados é, muitas vezes, disfarçada. Assim, o uso de técnicas de centralização e escalonamento é uma prática comum em metabolômica e considera diferentes formas de análise de dados. A centralização na média, ou seja, procedimento em que a média de um sinal (intensidade de um íon, ou sinal de RMN) obtida a partir do conjunto de amostras é subtraída de cada sinal das amostras, é realizada afim de remover a compensação dos dados e focar na variação biológica, como as similaridades e discrepâncias entre as amostras.286 Apesar da centralização dos dados ser importante, o uso exclusivo desse tratamento matemático pode não ser suficiente para a determinação de discriminantes uma vez que sinais abundantes (metabólitos) irão predominar no ajuste do modelo. Consequentemente, acrescentar diferentes formas de escalonamento é crucial para a extração adequada de informações em um estudo metabolômico. A seguir são descritas as principais estratégias de escalonamento aplicadas em metabolômica.285 (1) Auto-escalonamento: nesse método a média (centralização) e o desvio padrão de um sinal (intensidade de RMN ou íon) são calculados e, em seguida, o sinal da amostra é subtraído da média e dividido pelo desvio padrão. A função do auto-escalonamento é dar pesos iguais a todos sinais da amostra. Assim metabólitos minoritários e majoritários irão contribuir igualmente para o ajuste. A desvantagem desse método é que ruído e sinais indesejados também vão contribuir para o modelo. Além disso, os erros de medida serão valorizados, como a falta de repetibilidade, linearidade como ocorre para os dados de EM de baixa qualidade. (2) Escalonamento por Pareto: esse método é semelhante ao auto-escalonamento, mas após a centralização na média os dados são divididos pela raiz quadrada do desvio padrão. O Pareto oferece um compromisso entre o escalonamento centrado na média e o auto-escalonamento uma vez que os sinais majoritários são menos predominantes quando comparados a centralização na média. No entanto, Pareto ainda sofre as desvantagens de se concentrar nos metabólitos majoritários. (3) Escalonamento por estabilidade de variável: cada sinal autoescalado é dividido pelo coeficiente de variação, razão do desvio padrão pela média. Nessa abordagem, o foco está nos sinais mais estáveis, ou seja, sinais importantes devem possuir baixos coeficientes de variação (desvio padrão relativo) e dessa forma são mais estáveis. A Figura 6 mostra o efeito do escalonamento centrado na média, auto-escalonamento e Pareto para um conjunto de dados.
 Figura 6. Comportamento dos dados relativos as diferentes técnicas de centralização e escalonamento dos dados: (A) sem tratamento; (B) centrado na média; (C) auto-escalado e (D) Pareto
Na Figura 6A, a maioria dos sinais parece em baixa abundância. Na Figura 6B, a centralização na média, levou os sinais sob uma distribuição em torno de zero. Isso é importante quando diferentes conjuntos de dados são levados em consideração (EM e RMN juntos); no entanto, os sinais abundantes continuam predominantes. Na Figura 6C foi aplicado um auto-escalonamento. Sinais da linha de base são superestimados. Na Figura 6D foi aplicado o escalonamento do tipo Pareto. Nesse tipo de escalonamento existe um compromisso entre a modelagem centrado na média e auto-escalado. Transformação de dados Os dados das plataformas de RMN e EM estão sujeitos a várias fontes de ruídos heterocedásticos, em que a contribuição do ruído é função da intensidade do sinal, o que gera um viés nos dados e consequentemente afeta as análises quimiométricas subsequentes.201,285 Assim, os dados precisam ser transformados para que a estrutura do ruído seja homocedástica (uniformemente distribuída). Além disso, a distribuição de probabilidades dos dados pode apresentar obliquidade, ou seja, distorção de um dos lados da cauda das probabilidades normais, e necessitar de alguma correção antes das análises estatísticas. As transformações de dados corrigem a heterocedasticidade e obliquidade pode ter um efeito de pseudo escalonamento pois a diferença entre sinais abundantes e minoritários são diminuídas. Vale ressaltar que esse tipo de tratamento não descarta a aplicação dos métodos de escalonamento.201,285 A seguir são mostrados alguns métodos de transformação de dados para estudos metabolômicos: (1) Transformação log: o logaritmo de cada sinal/elemento é calculado e substituído aos dados originais. No caso de valores entre 0 e 1 é adicionado o valor de 1 em cada elemento (sinal) antes da operação matemática. As transformações log visam converter os ruídos multiplicativos em ruídos aditivos. (2) Transformação Glog: apesar de similar a operação logarítmica essa transformação é aplicada a log (3) Transformação Potência: nesse tipo de modelo os valores são transformados com base em potenciações. Embora ele não converta os ruídos multiplicativos pelos aditivos, o feito é similar as transformações log. Análises quimiométricas De maneira geral, a necessidade por análises quimiométricas (multivariadas) surgiu por volta de três décadas atrás como consequência do desenvolvimento da instrumentação analítica (velocidade de aquisição, sensibilidade e estabilidade) e foi impulsionado pelo aumento da capacidade computacional. O grande volume de dados produzido em estudos metabolômicos torna a visualização e análise de dados muito difícil por abordagens univariadas, como por exemplo, modelagens por regressão múltipla, onde o fator de colinearidade de variáveis limita sua utilização (necessidade de relação linear entre as variáveis).285 Existem diferentes métodos multivariados para a modelagem de dados. Eles podem ser não-supervisionados (nenhuma atribuição é feita sobre os dados), oferecendo uma visão global do comportamento ou, abordagens supervisionadas, em que as amostras são definidas em classes ou associadas a um valor de atributo, por exemplo, uma atividade biológica. Em geral, os métodos multivariados representam as amostras como pontos dentro de um espaço de variáveis. Nesse caso, a principal vantagem é que as amostras são projetadas em um espaço de menor dimensionalidade - componentes ou variáveis latentes - tal como uma linha, um plano ou um hiperplano (projeção n-1 de planos, por exemplo, 4D sendo projetadas em 3D). Métodos lineares baseados em projeções são populares em metabolômica pela simplicidade da interpretação e assim são usados para o entendimento de eventos biológicos. No entanto, métodos não lineares como redes neurais, máquina de vetores de suporte, random forest são menos utilizados em metabolômica, sendo restritos a predição de variáveis dentro contexto de classificação.285 Análise não-supervisionada (análise exploratória) Análises não-supervisionadas visam o entendimento global de toda a natureza do dado, bem como a detecção de tendências, padrões ou agrupamentos. Na metabolômica de plantas, a uma visão geral da flutuação metabólica permite a identificação de fatores discriminantes entre os grupos estudados através de métodos exploratórios. Existem diversos métodos não-supervisionados, sendo a Análise de Componentes Principais (PCA) e a Análise de Agrupamentos Hierárquicos (HCA) os mais utilizados. Dentre os softwares disponíveis para esse tipo de processamento, destaca-se MATLAB (Mathworks, EUA), com as funções PLS Toolbox287 e TOMCAT,288 Amix (Bruker, EUA), Biodiversity Pro (BD Pro., Reino Unido) e Statistical Software and Data Analysis (XLSTAT, Excel, EUA). Análise de componentes principais (PCA) A metabolômica de produtos naturais realiza o reconhecimento de processos metabólicos dinâmicos através de estudos quimiométricos comparativos, sendo o PCA a ferramenta não-supervisionada mais empregada. Em geral, essa técnica reduz as dimensões originais de um conjunto de dados numéricos através das componentes principais (PCs), convertendo dados multidimensionais em um modelo plano com dimensões reduzidas.225,289 As PCs são combinações lineares das variáveis originais e explicam o máximo de valor de variância que não foi contabilizado nas PCs anteriores. Basicamente, PCs são eixos ortogonais entre si, construídos em ordem decrescente da variância, sendo sua principal função a determinação da dimensionalidade intrínseca do conjunto de dados.225 Durante seu processo, a PCA converte o conjunto de dados originais em duas matrizes distintas, conhecidas como matriz de scores e matriz de loadings. Os scores são definidos como as novas coordenadas das amostras, sendo seu gráfico uma representação de como estas interagem entre si em um espaço n-dimensional. Já o loading descreve a maneira como as variáveis dos dados originais estão combinadas nas PCs, indicando quais possuem maior contribuição para a formação de determinada PC.225 Diferentes algoritmos são utilizados para a decomposição das matrizes complexas, a exemplo do algoritmo Nonlinear Iterative Partial Least Square (Nipals),290,291 que aplica método iterativo dos mínimos quadrados parciais não linear, ou Singular Value Decomposition (SDV), que realiza a decomposição da matriz em matrizes de score e loading de maneira singular.292,293 Na análise de perfis metabólicos, a composição química de cada amostra influencia as PCs, permitindo separação dessas em um espaço multidimensional, observado no gráfico de scores, sendo a razão química dessa separação observada no gráfico de loadings. Como amostras similares permanecem próximas no espaço, através dessa análise, é possível a detecção de padrões e observação de tendências, garantindo discriminação entre grupos amostrais. Adicionalmente à exploração não-tendenciosa dos dados, um aspecto importante do PCA é permitir a detecção de outliers, que são definidos como amostras dispostas além do intervalo de confiança definido para análise quimiométrica, prejudicando análises de quadrados parciais supervisionadas. Para a análise de matrizes biológicas complexas, a detecção de outliers é um fator limitante para a interpretação química, ecológica e biológica dos sistemas.294 Apesar de eficaz, deve-se ter em mente que a PCA não é capaz de determinar todas as amostras discrepantes, pois durante sua projeção, outliers ortogonais são agrupados com a maioria, impedindo sua visualização. Atualmente, diferentes estratégias vêm sido desenvolvidas para a determinação de outliers, sendo a robust-PCA a mais comumente empregada.295 Para produtos naturais, as matrizes comumente empregadas no PCA contêm dados espectroscópicos ou espectrométricos, como CLAE-DAD, CLAE-DAD-EM ou 1H-RMN, sendo aplicado principalmente para discriminação de amostras submetidas a diferentes condições genéticas e ambientais. Análise de agrupamento hierárquico (HCA) O HCA é outro método não-supervisionado comumente empregado na análise de perfis metabólicos e consiste no agrupamento de amostras de acordo com a sua similaridade. Nessa análise, é necessário o input das funções (a) métrica, que determina a distância utilizada para o cálculo da similaridade, como euclidiana, minkowski e mahalanobis, e (b) de conexão entre as amostras, como completo, médio, mediana ou Ward. A escolha das funções influencia drasticamente o resultado da HCA, comumente disposto em dendrograma de árvore.296,297 Na prática, a variação das funções de conexão e agrupamento podem resultar em dendrogramas completamente distintos devido a forma como a similaridade é calculada e, como consequência, a forma como a amostras são agrupadas. Uma analogia interessante seria uma análise de um grupo populacional: enquanto uma função agrupa as pessoas baseado no sexo, outras funções podem agrupá-los baseado na cor do cabelo, tamanho ou peso, formando grupos distintos no dendrograma de acordo com os inputs da função.297 Para análises de matrizes naturais muito complexas, como CLAE-UV, TIC ou 1H RMN, além dos procedimentos de pré-processamento, é recomendado a realização de redução dos dados (binning ou bucketing) para garantir uma análise mais reprodutível e precisa, visto que matrizes não normalizadas ou com muito ruído podem erroneamente agrupar amostras que apresentam pouca similaridade química.298 A análise hierárquica tem sido extensamente empregada na química de produtos naturais para a quimiotaxonomia de amostras, permitindo a classificação genética de espécies de plantas e micro-organismos baseados no perfil químico de cada espécie.299 Apesar de simples, a HCA apresenta como principal desvantagem não informar a razão química dos agrupamentos, não revelando, portanto, os sinais químicos (metabólitos) responsáveis pelo agrupamento de determinada amostra. Para tanto, é necessária a realização da HCA bidimensional (clustergram), em que é possível observar as principais variáveis responsáveis pelo agrupamento, como intensidade de um sinal em determinado deslocamento químico ou tempo de retenção. Análise supervisionada A análise supervisionada tem sido amplamente utilizada na descoberta de biomarcadores e metabólitos bioativos.300-302 Nesse processo, a matriz de dados é obrigatoriamente comparada a um modelo, que deve ser robusto e estatístico, sendo este criado através de dados biológicos, taxonômicos ou químicos. Durante a análise supervisionada, a exploração das variáveis do sistema visa forçar a separação baseado no modelo, produzindo discriminação tendenciosa das amostras. No agrupamento de indivíduos suscetíveis ou resistentes à determinada doença, emprega-se estratégias classificatórias, enquanto na predição de concentração de metabólitos ou expressão do nível genético, é mais comum a utilização de métodos de regressão. O objetivo principal dos métodos supervisionados é obter uma correlação entre as variáveis químicas e um conjunto de dados (modelo) usado como base para previsão. Dependendo do objetivo do estudo e tipo de cálculo aplicado nas análises supervisionadas, diferentes ferramentas podem ser usadas para discriminação, classificação ou regressão, sendo o Partial Least Squares (PLS), Orthogonal Partial Least Squares - Discriminant Analysis (OPLS-DA), k-nearest neighbors (K-NN) e o Soft Independente Modeling of Class Analogy (SIMCA). Diversos softwares permitem a inserção de dados químicos para as análises supervisionadas e outros comandos de pré-processamento, incluindo AMIX (Bruker), SIMCA Software (Umetrics), PLS Toolbox (Eigenvector Research Inc, EUA). A PLS-DA é a técnica discriminatória mais utilizada, sendo primeiramente desenvolvida para buscar soluções para os problemas de regressão.303,304 O método consiste em um vetor Y que representa os valores das classes em relação ao vetor X (conjunto de dados). O modelo gerado tem como parâmetros o R2, que mede a adequação do modelo para o conjunto de dados de treino, enquanto Q2 e PRESS são utilizados para avaliar o capacidade de previsão do modelo (validação), sendo recomendado o uso de dupla validação cruzada305 juntamente com o número de erros de classificação e a área sob a curva de ROC como estatísticas de diagnóstico. Outros algoritmos similares podem ser encontrados em conjunto, como é o caso de SPLS-DA306 e OPLS-DA.307 É muito importante ressaltar que durante a análise supervisionada, a modelagem é a etapa mais crítica do processo, sendo comumente atribuídas aos resultados biológicos (IC50, MIC, etc) ou outras características das amostras, como espécies, tecidos ou dados abióticos (local de coleta, temperatura, humidade, altitude, etc). Especificamente para a identificação de biomarcadores, o modelo deve ser cuidadosamente selecionado, garantindo assim, a validação e correta interpretação dos dados. Isso é especialmente válido para PLS-DA e OPLS-DA, pois ambos forçam a separação dos dados em grupos experimentais. Para esses casos, a PCA pode ser estrategicamente usada como forma de modelagem, garantindo a validação cruzada e a análise estatística de todas as variáveis complexas do seu conjunto de dados. Em metabolômica, o conjunto de dados comumente empregado para o processamento apresentam natureza espectroscópica ou espectrométrica.303-305,308-312 Isolamento e identificação de substâncias de origem natural em estudos metabolômicos A identificação não-ambígua das substâncias presentes em espécies vegetais é uma tarefa que está intimamente relacionada aos estudos metabolômicos. Tanto para compor as bases de dados de moléculas oriundas dos metabolismos primário e secundário, quanto para identificar novas substâncias de interesse elencadas nos estudos envolvendo matrizes de origem natural, ou mesmo para que testes e ensaios biológicos sejam realizados, a posse dessas substâncias é imprescindível. Obtê-las é um trabalho muitas vezes tedioso e complexo, e requer uma estrutura de pesquisa multidisciplinar que permita a condução de procedimentos fitoquímicos para a extração (que pode ser feita em micro- ou larga escala), fracionamento dos extratos brutos obtidos, isolamento, purificação e determinação estrutural dessas moléculas. Por ser um tema muito amplo e com diversas aplicações práticas, optamos, aqui, falar brevemente sobre as etapas principais relacionadas à tais estudos fitoquímicos, visto que a literatura possui uma vasta gama de referências bibliográficas que explicam e elucidam esses procedimentos. O leitor que se interessar e precisar de maiores informações poderá recorrer aos livros de cromatografia,313,314 farmacognosia,315,316 análise orgânica317,318 e outras tantas fontes especializadas nas diversas classes de metabólitos de interesse. Uma das grandes dificuldades em se estabelecer protocolos e de se trabalhar com tais moléculas, principalmente aquelas relacionadas ao metabolismo secundário, está relacionada, em parte, à alta complexidade da matriz biológica em questão, o que requer, logo num primeiro momento, a extração dessas substâncias, o que é geralmente realizada com a utilização de solventes de diferentes polaridades (geralmente misturas hidroalcoólicas, metanol, acetato de etila ou clorofórmio, dentre outros possíveis), assistidas ou não por temperatura, agitação, ou ultrassom, dentre as mais comuns. No entanto, dependendo da polaridade do solvente extrator, uma ou outra classe de metabólitos será extraída de forma majoritária. Extratos polares, por exemplo, são ricos em metabólitos primários, principalmente açúcares e aminácidos, enquanto que extratos de baixa polaridade são ricos em substâncias lipofílicas e pigmentos.319 Misturas hidroalcoólicas em diferentes concentrações são o ponto de partida para a extração da maioria das moléculas de alta a média polaridade, que pode incluir flavonoides, cumarinas, glicosídeos, alcaloides, diterpenos e saponinas, dentre outros. Além disso, estratégias de alcalinização ou acidificação, seguidos por procedimentos de partição utilizando solventes orgânicos, são muito úteis quando se quer extrair prioritariamente determinada classe de metabólitos, tais como os alcalóides de diferentes classes.315 Uma outra técnica de extração, que evita o emprego extensivo de solventes, é a extração com fluido supercrítico, que se baseia na utilização de uma substância em que os estados líquidos e gasosos possuem mesma densidade, o que é possível quando a matéria se encontra acima da temperatura e pressão críticas. Dentre os fluidos mais utilizados, o CO2 tem sido o de escolha devido ao seu baixo custo, por ser relativamente não-tóxico, não-inflamável, além de ser facilmente removido após a extração.320-323 Uma vez obtidos, os extratos deverão ser concentrados e/ou secos e posteriormente fracionados, procedimento que visa diminuir a complexidade do extrato bruto obtido e enriquecê-lo em determinada classe de substância. Dentre as técnicas mais empregadas e, dependendo da estrutura física do laboratório existente e da quantidade de substância desejada, o pesquisador poderá optar pela realização da extração ou partição líquido-líquido, ou pela extração em fase sólida utilizando resinas de diferentes afinidades, como por exemplo fase normal, fase reversa, ou troca iônica.322 A utilização da cromatografia em coluna, à média ou baixa pressão é também uma estratégia muito útil quando se deseja diminuir a complexidade dos extratos brutos obtidos, permitindo a obtenção de frações enriquecidas nos metabólitos de interesse. Finalmente, para o isolamento e purificação, a cromatografia líquida de alta eficiência, em fase normal ou reversa, ou mesmo empregando fases estacionárias contendo ligantes específicos para o isolamento de determinado composto, é a técnica mais utilizada, tanto para o isolamento de substâncias em escala analítica, quanto em escalas semi-preparativa e preparativa. É interessante pontuar que, nos estudos metabolômicos, muitas vezes, a condição cromatográfica desenvolvida (gradiente, temperatura, fase estacionária, composição e vazão da fase móvel, etc) permite a separação das substâncias com alto grau de resolução e seletividade. Tendo isto em mente, a transferência de método de escala analítica para escala semi-preparativa ou preparativa, se bem calculada e planejada, permite manter os índices de resolução e seletividade, favorecendo assim, o isolamento da substância de interesse com alto grau de confiabilidade. O escalonamento de métodos otimizados em escala analítica para as escalas semi-preparativa e preparativa poderá ser feito utilizando cálculos matemáticos, porém, existem alguns softwares livres e comerciais que facilitam a transposição de métodos analíticos.324 É especialmente interessante destacar que, em sistemas modernos de CLAE-EFS-RMN, é possível coletar o pico de interesse em um cartucho de extração em fase sólida após sucessivas injeções, o qual pode ser diretamente eluído e submetido à RMN para a determinação estrutural. Tais sistemas podem, ainda, acoplar a detecção por espectrometria de massas, aumentando ainda mais a confiabilidade e o sucesso no isolamento e identificação do pico de interesse, o que é muito útil, dentre outras finalidades, quando se deseja obter informações fidedignas oriundas da execução de bioensaios.319 Nesse sentido e, de forma geral, uma vez isoladas e purificadas, a identificação dessas moléculas é feita através da combinação de diferentes técnicas, principalmente a ressonância magnética nuclear (RMN) e a espectrometria de massas (EM). Técnicas espectrométricas na região do ultravioleta (UV) e infravermelho (IV) são também muito úteis para a caracterização dos grupos funcionais presentes. Além disso, a utilização de métodos espectroscópicos quirópticos tais como dicroísmo circular eletrônico e vibracional (ECD e VCD, respectivamente), atividade óptica Raman (ROA) e a cristalografia de raios X são técnicas importantes para a determinação da configuração absoluta.325 É interessante destacar que a elucidação estrutural de produtos de origem natural era uma tarefa demasiadamente difícil e demorada até cerca de 50 a 60 anos atrás, uma vez que exigia a condução de inúmeros procedimentos de reações sintéticas e de degradação, para que ao final, tais dados em conjunto permitissem a determinação da estrutura química da molécula de interesse. No entanto e, felizmente, o desenvolvimento e a disponibilização comercial de novas técnicas e instrumentação espectroscópica revolucionou a área, de tal forma que hoje, estruturas químicas podem ser determinadas em menos de 24 horas e com uma quantidade diminuta de amostra, de cerca de 1 mg às frações de microgramas. Isto foi possível graças às constantes e necessárias melhorias das técnicas utilizadas em elucidação estrutural, em especial a espectrometria de massas, a difração em raios-x e a ressonância magnética nuclear, por exemplo, tanto em termos de sensibilidade e resolução, como no desenvolvimento de processadores com maior poder de computação.326 Estratégias para anotação e/ou identificação de produtos naturais em metabolômica A natureza diversificada e complexa dos metabólitos secundários, resultado da multiplicidade de seus quimiotipos e isomerias e a ampla faixa dinâmica de concentrações, consiste em um grande desafio para desenvolvimento de métodos robustos e eficazes para automação das etapas de anotação e identificação de substâncias.327 As comunidades científicas, atentas às inúmeras dificuldades em se trabalhar com tais substâncias, têm proposto medidas alternativas e racionais para o processo de anotação molecular, visando flexibilizar o refinamento da anotação de acordo com os tipos e objetivos de cada estudo metabolômico. Existem várias classificações para anotação de compostos. No entanto, a maioria leva em consideração o grau de confiabilidade da anotação, ou seja, o preenchimento de requisitos durante esse processo. A comunidade Metabolites Standard Initiaves (MSI) é um exemplo de iniciativa que visa a racionalização dos procedimentos de anotação e considera diferentes níveis de confiabilidade.328 Após revisão da publicação original de 2007, as anotações de substâncias para estudos metabolômicos foram classificadas em cinco diferentes categorias. O primeiro nível ou chamado de Nível 0 (ou identificação não-ambígua), ocorre quando toda a elucidação molecular tridimensional (incluindo, se for o caso, a definição dos centros estereogênicos) é realizada a partir do isolamento do composto interesse a partir da fonte de origem biológica. Esse nível corresponde à identificação mais completa possível e requer conhecimento e excelência em todas as etapas que envolvem a determinação da substância de interesse. Desta forma, a identificação de substâncias em nível 0 consiste em um trabalho complexo que abrange uma estrutura de pesquisa multidisciplinar para a condução de laboriosos, porém necessários, procedimentos fitoquímicos, que contemplam desde a extração (que pode ser feita em micro- ou larga escala), fracionamento, isolamento e purificação, até a determinação estrutural das moléculas isoladas, conforme já descrito anteriomente. Tipicamente, a identificação em Nível 0 é necessária em casos onde a estrutura de interesse é inédita ou quando quantidade de dados espectroscópicos disponíveis na literatura é insuficiente para a sua elucidação. No entanto, existem situações em que substância de interesse já é conhecida ou derivado de um composto previamente isolado e elucidado. Nesse sentido, a comparação de dados duas ou mais abordagens analíticas, para determinação estrutural (EM e RMN), com dados espectrais de padrões autênticos leva à anotação em Nível 1. O segundo nível de anotação (Nível 1) se dá pela comparação de análises espectroscópicas e espectrométricas (elucidação) com um padrão autêntico, num nível de confiabilidade estrutural bidimensional. Para esse nível, além da presenca de padrões autênticos, os métodos analíticos requerem a utilização de pelo menos duas dimensões ortogonais. Isso pode ser conseguido a partir de uma etapa de separação cromatográfica (por exemplo CG ou CLAE), que fornecerá dados de tempos de retenção, seguida de uma etapa de detecção espectral, que pode variar de acordo com o tipo de estudo ou a natureza das molélulas de interesse. Tais métodos de detecção incluem, principalmente, a espectrometria de massas com diferentes fontes de ionização e analisadores e a ressonância magnética nuclear (de hidrogênio ou carbono, mono ou dimensional, dependendo do arranjo de das probes utilizadas, como já mencionado anteriormente).327,328 O terceiro e quarto níveis (Nível 2 e Nível 3, respectivamente) ocorre pela análise de dados espectrométricos ou espectroscópicos sem a comparação com um padrão autêntico. Muitas vezes, em estudos preliminares ou em etapas intermediárias de estudos metabolômicos, a determinação em Nível 2 ou 3 pode ser suficiente para o prosseguimento de novos estudos. Em Nível 2 é possível determinar a possível estrutura da substância em questão, sem uso de padrão de referência. No entanto, é ainda necessário a obtenção de dados em duas dimensões ortogonais, para que esses possam ser comparados com dados descritos na literatura ou em bases de dados, através da análise de informações de diagnóstico que permitem, inclusive, excluir possíveis candidatos. Em Nível 3, é possível determinar ao menos a classe da substância em questão, ou até mesmo assinalar a estrutura mais provável, sem a possibilidade de diferenciar isômeros, por exemplo. Nesse caso, um ou mais candidatos podem ser relacionados ao sinal analítico, o qual pode ser obtido através do uso de uma única abordagem analítica, não necessariamente com respostas ortogonais. Finalmente, o quinto e último nível (Nível 4) consiste na ausência completa informações acerca do metabólito e fornece informações acerca da simples presença e/ ou ausência. Em todos os casos, a elucidação estrutural de substâncias é ainda uma ciência manual e laboriosa dependendo de habilidades específicas e experiência do espectroscopista. Nesse sentido, diversas áreas de fronteira entre as ciências da vida e computacionais têm-se esforçado para desenvolvimentos de ferramentas que possam auxiliar o processo de anotação molecular. De modo geral, essas ferramentas podem ser classificadas dentro duas categorias abrangentes: (i) aquelas em que é utilizada comparação com dados espectrais, isso é, dados da amostra de interesse são analisados frente à dados presentes em bibliotecas espectrais; enquanto que (ii) a segunda categoria consiste na predição de sinais, ou seja, um algoritmo é formulado a partir de um conjunto de dados teóricos e/ou experimentais para o reconhecimento de sinais analíticos visando sua relação com estruturas moleculares. Em ambos os casos, os bancos de dados espectrais de substâncias são essenciais para a anotação e/ou identificação dos metabólitos, sendo que tanto a qualidade dos dados, como a quantidade de espectros disponíveis nesses bancos de dados são fatores críticos para a performance dos algoritmos de identificação.329,330 A Tabela 5, mostra diversas dessas ferramentas de anotação apontando as abordagens utilizadas e as técnicas analíticas relacionadas.
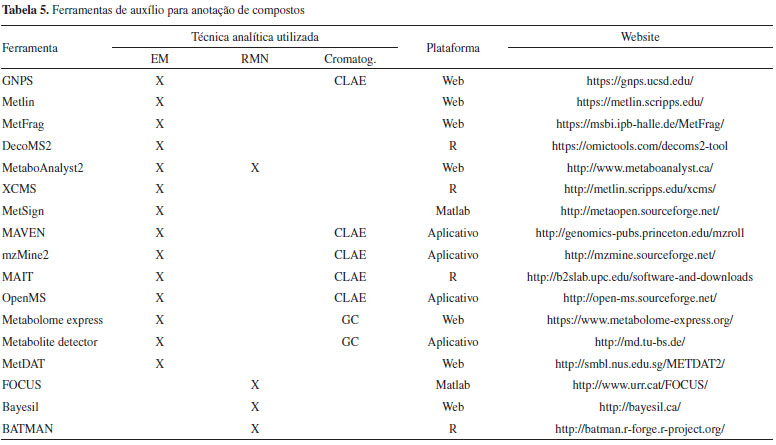
CONSIDERAÇÕES FINAIS E FUTURAS PERSPECTIVAS A metabolômica de plantas progrediu de um conceito ambicioso do início do século XXI para uma estratégia científica valiosa e rápida que fornece uma visão global da organização e canalização molecular. Essa evolução, observada principalmente na última década, ocorreu devido ao desenvolvimento sinérgico de tecnologias analíticas e ferramentas de bioinformática. Em espectrometria de massas, o desenvolvimento de instrumentos de alta resolução e sua combinação com métodos cromatográficos rápidos e eficientes representaram avanços importantes. Já para a RMN, o aumento das forças de campos magnéticos e o advento de sondas criogênicas aumentaram ainda mais os limites de detecção e contribuíram para melhora da resolução espectral. Atualmente, a metabolômica pode ser considerada um campo maduro e representa um dos ramos das tecnologias "ômicas" que compõem a biologia de sistemas, abordando questões biológicas pela avaliação de dados, o que traz várias vantagens em comparação com a abordagem clássica orientada por hipóteses. Apesar dos crescentes avanços nos últimos anos, a extensão e cobertura do metaboloma em plantas ainda está longe de ser alcançada. Uma das principais dificuldades da metabolômica é a identificação inequívoca dos inúmeros sinais detectados (features) que podem conter informações importantes sobre um sistema biológico. Nesse sentido, é essencial a criação e acesso a bases de dados públicas de compostos, vias metabólicas, espectros e procedimentos experimentais assim como estabelecer protocolos padronizados que normalizem e facilitem a troca de informações entre laboratórios. O desenvolvimento e a coordenação de bancos de dados provavelmente constituirão a base para a extração de informações biológicas importantes. Neste contexto, o desenvolvimento de recursos humanos com expertise em isolamento, purificação, análise e determinação estrutural de moléculas orgânicas é ainda essencial, visto que os bancos de dados utilizados nos estudos metabolômicos dependem da inserção de informações químicas e estruturais que só podem ser validadas com dados experimentais obtidos a partir da substância isolada. Além disso, mesmo que as técnicas modernas em cromatografia, espectrometria de massas e ressonância magnética nuclear sejam capazes de fornecer informações relacionadas aos metabólitos de origem vegetal mesmo sem a necessidade de isolamento prévio, ou por meio de identificações tentativas, a confirmação da identidade (identificação inequívoca), definição dos centros estereogênicos, ou determinação da configuração absoluta ainda requer a substância isolada da matriz vegetal. O mesmo pode-se dizer sobre posteriores estudos envolvendo análise quantitativa, determinação da relação estrutura-atividade, validação de estudos in silico, ou de possíveis ensaios biológicos com os marcadores ou substâncias de interesse, que só é possível com a utilização de padrões autênticos ou de substâncias isoladas. Para quaisquer finalidades em que se requer a substância isolada da matriz vegetal, algumas estratégias modernas para o isolamento de moléculas-alvo elencadas em estudos metabolômicos ou bioguiados para a descoberta de novos alvos terapêuticos (desde microescala até escala semipreparativa) foram elencadas no artigo de revisão de Wolfender e colaboradores.319 De fato, os autores corroboram com a necessidade de se obter tais substâncias para que todo o processo de estudo envolvendo produtos de origem natural sejam completados e validados. Outra questão crucial é a forma de extração da informação a partir dos dados brutos. A natureza multivariada dos dados requer protocolos de processamento robustos e abordagens específicas para extração de informações valiosas, muitas vezes ocultas, em dados metabolômicos de alta dimensionalidade. Nesse sentido, as ferramentas quimiométricas tem um papel central na interpretação dos dados e na inferência das estruturas que governam os fenômenos biológicos. A combinação de dados de EM e RMN (entre outros) fornecem uma visão mais abrangente, convergindo características comuns de diferentes conjuntos de dados. No entanto, a quimiometria não pode ser encarada como uma "caixa preta", e o entendimento dos procedimentos que envolvem análise dos dados brutos é necessário para evitar problemas na interpretação dos resultados. Por fim, é interessante destacar que, por conta da enorme diversidade de micromoléculas oriundas do metabolismo vegetal, a metabolômica, assim como as outras ciências ômicas tais como a proteômica e a genômica de plantas continuam em constante desenvolvimento. A integração entre elas, da mesma forma, é uma realidade que vem ao encontro da necessidade se avançar no entendimento de como ocorrem as interações entre as plantas e o meio ambiente, incluindo tembém os estudos de atividades biológicas de derivados de origem vegetal, sejam eles isolados, na forma de frações enriquecidas ou na forma de extratos brutos. Como perspectiva, os próximos anos serão marcados por trabalhos em que a metabolômica será usada de forma integrada às outras abordagens ômicas (genômica, transcriptômica e proteômica), chamada de fusão inter-ômica.331 No caso de estudos intra-ômicos (uso exclusivo da abordagem metabolômica) prevê-se o desenvolvimento de mecanismos específicos em maior nível de compartimentalização. Estudos em vacúolos, mitocôndrias ou até mesmo células isoladas332 conduzirá a metabolômica a novos rumos.
AGRADECIMENTOS Este trabalho foi financiado pela Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), [Processos No. 2014/50265-3; No. 2015/50488-5; No. 2013/07600-3; No. 2014/50926-0; No. 2016/13292-8, 2019/15889-0 (ACP); No. 2014/05935-0 (DMS); No. 2017/06446-2 (DMS); No. 2017/17850-8 (DRP); 2017/19702-6, 2019/08477-7 (PCPB)] e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq
REFERENCES 1. Hall, R. D. Em Biology of Plant Metabolomics; Hall, R. D., ed.; Blackwell Publishing: Oxford, 2011, cap. 1. 2. Nicholson, J. K.; Mol. Syst. Biol. 2006, 2, Article 52. 3. Holmes, E.; Wilson, I. D.; Nicholson, J. K.; Cell 2008, 134, 714. 4. Calvin, M.; Am. Sci. 1975, 63, 169. 5. Croteau, R.; Kutchan, T. M.; Lewis, N. G. Em Biochemistry & Molecular Biology of Plants; Buchanan, B., Gruissem, W., Jones, R., eds.; American Society of Plant Physiologists: Rockville, 2000, cap 24. 6. Dewick, P. M.; Medicinal Natural Products: A Biosynthetic Approach, 3rd ed., Wiley: Chichester, 2009. 7. Weng, J.-K.; Philippe, R. N.; Noel, J. P.; Science 2012, 336, 1667. 8. Pilon, A. C.; Paez-Garcia, A.; Pavarini, D. P.; Scott, M. T. Em Mass Spectrometry in Chemical Biology: Evolving Applications; Lopes, N. P., Silva, R. R., eds.; Royal Society of Chemistry: London, 2018, cap 9. 9. Oliver, S.G.; Winson, M. K.; Kell, D. B.; Baganz, F.; Trends Biotechnol. 1998, 16, 373. 10. Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R. N.; Willmitzer, L.; Nat. Biotechnol. 2000, 18, 1157. 11. Belinato, J. R.; Bazioli, J. M.; Sussulini, A.; Augusto, F.; Fill, T. P.; Quim. Nova 2019, 42, 546. 12. Canuto, G. A. B.; Costa, J. L.; Cruz, P. L. R.; Souza, A. R. L.; Faccio, A. T.; Klassen, A.; Rodrigues, K. T.; Tavares, M. F. M.; Quim. Nova 2018, 41, 75. 13. Funari, C. S.; Castro-Gamboa, I.; Cavalheiro, A. J.; Bolzani, V. S.; Quim. Nova 2013, 36, 1605. 14. Wolfender, J.-L.; Marti, G.; Queiroz, E. F.; Curr. Org. Chem. 2010, 14, 1808. 15. Moco, S.; Bino, R. J.; De Vos, R. C. H.; Vervoort, J.; Trends Anal. Chem. 2007, 26, 855. 16. Kim, H. K.; Choi, Y. H.; Verpoorte, R.; Trends Biotechnol. 2011, 29, 267. 17. Schripsema, J. Phytochem. Anal. 2010, 21, 14.18. Costa, C.; Maraschin, M.; Rocha, M.; Comput. Methods Programs Biomed. 2016, 129, 117. 19. Guizellini, F. C.; Marcheafave, G. G.; Rakocevic, M.; Bruns, R. E.; Scarminio, I. S.; Soares, P. K.; Food Res. Int. 2018, 113, 9. 20. Yamaguchi, L. F.; Freitas, G. C.; Yoshida, N. C.; Silva, R. A.; Gaia, A. M.; Silva, A. M.; Scotti, M. T.; Emerenciano, V. P.; Guimarães, E. F.; Floh, E. I. S.; J. Braz. Chem. Soc. 2011, 22, 2371. 21. Martins, C. A. F.; Piantavini, M. S.; Ribeiro, R. P.; Amano, E.; Dal Prá, B. V; Pontarolo, R.; J. Braz. Chem. Soc. 2015, 26, 365. 22. Hoffmann, J. F.; Carvalho, I. R.; Barbieri, R. L.; Rombaldi, C. V; Chaves, F. C.; J. Agric. Food Chem. 2017, 65, 523. 23. Leme, G. M.; Coutinho, I. D.; Creste, S.; Hojo, O.; Carneiro, R. L.; Bolzani, V. S.; Cavalheiro, A. J.; Anal. Methods 2014, 6, 7781. 24. Vargas, L. H. G.; Rodrigues Neto, J. C.; de Aquino Ribeiro, J. A.; Ricci-Silva, M. E.; Souza, M. T.; Rodrigues, C. M.; de Oliveira, A. E.; Abdelnur, P. V.; Metabolomics 2016, 12, 153. 25. Coutinho, I. D.; Baker, J. M.; Ward, J. L.; Beale, M. H.; Creste, S.; Cavalheiro, A. J.; J. Agric. Food Chem. 2016, 64, 4198. 26. Hantao, L. W.; Aleme, H. G.; Passador, M. M.; Furtado, E. L.; Ribeiro, F. A. L.; Poppi, R. J.; Augusto, F. J.; Chromatogr. A 2013, 1279, 86. 27. Fraige, K.; González-Fernández, R.; Carrilho, E.; Jorrín-Novo, J. V. J.; Proteomics 2015, 113, 206. 28. Uarrota, V. G.; Moresco, R.; Coelho, B.; Nunes, E. C.; Peruch, L. A. M.; Neubert, E. O.; Rocha, M.; Maraschin, M.; Food Chem. 2014, 161, 67. 29. Martins, L. R. R.; Pereira-Filho, E. R.; Cass, Q. B.; Anal. Bioanal. Chem. 2011, 400, 469. 30. Garrett, R.; Schmidt, E. M.; Pereira, L. F. P.; Kitzberger, C. S. G.; Scholz, M. B. S.; Eberlin, M. N.; Rezende, C. M.; LWT - Food Sci. Technol. 2013, 50, 496. 31. Miranda, A. M.; Carioca, A. A. F.; Steluti, J.; da Silva, I. D. C. G.; Fisberg, R. M.; Marchioni, D. M.; Clin. Nutr. 2017, 36, 1635. 32. Prado, R. M.; Porto, C.; Nunes, E.; Aguiar, C. L.; Pilau, E. J.; mSystems 2018, 3, 2. 33. Pontes, J. G. M.; Ohashi, W. Y.; Brasil, A. J. M.; Filgueiras, P. R.; Espíndola, A. P. D. M.; Silva, J. S.; Poppi, R. J.; Coletta-Filho, H. D.; Tasic, L.; ChemistrySelect 2016, 1, 1176. 34. Cardoso, S.; Maraschin, M.; Peruch, L. A. M.; Rocha, M.; Pereira, A.; Journal of Integrative Bioinformatics 2017, 14, 1. 35. Bittencourt, M. L. F.; Ribeiro, P. R.; Franco, R. L. P.; Hilhorst, H. W. M.; de Castro, R. D.; Fernandez, L. G.; Food Res. Int. 2015, 76, 449. 36. Dembogurski, D. S. O.; Trentin, D. S.; Boaretto, A. G.; Rigo, G. V; da Silva, R. C.; Tasca, T.; Macedo, A. J.; Carollo, C. A.; Silva, D. B.; Food Res. Int. 2018, 111, 661. 37. Chagas-Paula, D. A.; Oliveira, T. B.; Zhang, T.; Edrada-Ebel, R.; Da Costa, F. B.; Planta Med. 2015, 81, 450. 38. Pilatti, F. K.; Ramlov, F.; Schmidt, E. C.; Costa, C.; Oliveira, E. R.; Bauer, C. M.; Rocha, M.; Bouzon, Z. L.; Maraschin, M.; Mar. Pollut. Bull. 2017, 114, 831. 39. Budzinski, I. G. F.; Moon, D.; Morosini, J. S.; Lindén, P.; Bragatto, J.; Moritz, T.; Labate, C. A.; BMC Plant Biol. 2016, 16, 1. 40. Sampaio, B. L.; Edrada-Ebel, R.; Da Costa, F. B.; Sci. Rep. 2016, 6, 1. 41. Azevedo, R. A.; Mazzafera, P.; Ann. Appl. Biol. 2013, 163, 319. 42. Schaker, P. D. C.; Peters, L. P.; Cataldi, T. R.; Labate, C. A.; Caldana, C.; Monteiro-Vitorello, C. B.; Front. Plant Sci. 2017, 8, 1. 43. Padilla-González, G. F.; Diazgranados, M.; Da Costa, F. B.; Sci. Rep. 2017, 7, 1. 44. dos Santos, V. S.; Macedo, F. A.; do Vale, J. S.; Silva, D. B.; Carollo, C. A.; Metabolomics 2017, 13, 1. 45. Richards, L. A.; Dyer, L. A.; Forister, M. L.; Smilanich, A. M.; Dodson, C. D.; Leonard, M. D.; Jeffrey, C. S.; Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10973. 45. Smilde, A. K.; Westerhuis, J. A.; Hoefsloot, H. C. J.; Bijlsma, S.; Rubingh, C. M.; Vis, D. J.; Jellema, R. H.; Pijl, H.; Roelfsema, F.; van der Greef, J.; Metabolomics 2010, 6, 3. 47. Dunn, W. B.; Broadhurst, D. I.; Atherton, H. J.; Goodacre, R.; Griffin, J. L.; Chem. Soc. Rev. 2011, 40, 387. 48. Müller, P.; Parmigiani, G.; Robert, C.; Rousseau, J. J.; Am. Stat. Assoc. 2004, 99, 990. 49. Nyamundanda, G.; Gormley, I. C.; Fan, Y.; Gallagher, W. M.; Brennan, L.; BMC Bioinformatics 2013, 14, 338. 50. Tibshirani, R.; BMC Bioinformatics 2006, 7, 106. 51. Liu, P.; Hwang, J. T. G.; Bioinformatics 2007, 23, 739. 52. Lin, W.-J.; Hsueh, H.-M.; Chen, J. J.; BMC Bioinformatics 2010, 11, 48. 53. Bruns, R.; Scarminio, I.; de Barros Neto, B.; Statistical Design - Chemometrics, 2nd ed., Elsevier Science: Amsterdam, 2006. 54. Okazaki, K.; Shinano, T.; Oka, N.; Takebe, M.; Soil Sci. Plant Nutr. 2012, 58, 696. 55. Bracarense, A. A. P.; Takahashi, J. A.; Braz. J. Microbiol. 2014, 45, 313. 56. Pimenta, E. F.; Vita-Marques, A. M.; Tininis, A.; Seleghim, M. H. R.; Sette, L. D.; Veloso, K.; Ferreira, A. G.; Williams, D. E.; Patrick, B. O.; Dalisay, D. S.; J. Nat. Prod. 2010, 73, 1821. 57. Abdel-Fattah, Y. R.; El Enshasy, H.; Anwar, M.; Omar, H.; Abolmagd, E.; Abou Zahra, R.; J. Microbiol. Biotechnol. 2007, 17, 1930. 58. Pereira Filho, E. R.; Planejamento Fatorial Em Química: Maximizando a Obtenção de Resultados, 1a ed., EDUFSCAR: São Carlos, 2015. 59. Pilon, A. C.; Carnevale Neto, F.; Freire, R. T.; Cardoso, P.; Carneiro, R. L.; Bolzani, V. S.; Castro-Gamboa, I.; J. Sep. Sci. 2016, 39, 1023. 60. Bueno, P. C. P.; Pereira, F. M. V.; Torres, R. B.; Cavalheiro, A. J.; J. Sep. Sci. 2015, 38, 1649. 61. Zhang, X.; Chen, J.; Mao, M.; Guo, H.; Dai, Y.; Int. J. Biol. Macromol. 2014, 67, 318. 62. Ramandi, N. F.; Najafi, N. M.; Raofie, F.; Ghasemi, E.; J. Food Sci. 2011, 76, 1262. 63. Izadiyan, P.; Hemmateenejad, B.; Food Chem. 2016, 190, 864. 64. Rai, A.; Mohanty, B.; Bhargava, R.; Food Chem. 2016, 192, 647. 65. Asati, A.; Satyanarayana, G. N. V; Patel, D. K.; J. Chromatogr. A 2017, 1513, 157. 66. MATLAB, Version R2018a, Mathworks, MA, USA, 2018, disponível em https://www.mathworks.com/products/matlab.html, acessada em março de 2020. 67. Minitab Statistical Software, Version 18, Minitab Inc, PA, USA, 2018, disponível em http://www.minitab.com/en-us/products/minitab, acessada em março de 2020. 68. Piruette, Version 4.5, Infometrix, WA, USA, 2018, disponível em https://infometrix.com/pirouette/, acessada em março de 2020. 69. Pereira, F. M. V; Pereira-Filho, E. R.; Quim. Nova 2018, 41, 1061. 70. Jezus, R. A.; Prado, V. M. J.; Pinto, V. S.; Silva, V. R.; Santos, L. S.; Nogueira, P. C. L.; Navickiene, S.; Pereira-Filho, E. R.; Blank, A. F.; Bezerra, D. P.; Soarea, M. B. P.; Seid. C.; Cardoso, C. L.; Moraes, V. R. S.; J. Braz. Chem. Soc. 2019, 30, 978. 71. Teahan, O.; Gamble, S.; Holmes, E.; Waxman, J.; Nicholson, J. K.; Bevan, C.; Keun, H. C.; Anal. Chem. 2006, 78, 4307. 72. Kim, H. K.; Choi, Y. H.; Verpoorte, R.; Nat. Protoc. 2010, 5, 536. 73. Biais, B.; Bernillon, S.; Deborde, C.; Cabasson, C.; Rolin, D.; Tadmor, Y.; Burger, J.; Schaffer, A. A.; Moing, A. Em Plant Metabolomics: Methods in Molecular Biology (Methods and Protocols); Hardy, N. W., Hall, R. D., eds.; Humana Press: Totowa, 2012, cap. 4. 74. Scotti, M. T.; Herrera-Acevedo, C.; Oliveira, T. B.; Costa, R. P. O.; Santos, S. Y. K. O.; Konno, Y.; Rodrigues, R. P.; Scotti, L.; Da-Costa, F. B.; Molecules 2018, 23, 1. 75. Queiroz, O.; Ann. Rev. Plant Physiol. 1974, 25, 115. 76. Espinoza, C.; Degenkolbe, T.; Caldana, C.; Zuther, E.; Willmitzer, L.; Hincha, D. K.; Hannah, M. A.; PLoS One 2010, 5. 77. Urbanczyk-Wochniak, E.; Baxter, C.; Kolbe, A.; Kopka, J.; Sweetlove, L. J.; Fernie, A. R.; Planta 2005, 221, 891. 78. Kim, H. K.; Verpoorte, R.; Phytochem. Anal. 2010, 21, 4. 79. Koning, W. d; Dam, K.; Anal. Biochem. 1992, 204, 118. 80. Hajjaj, H.; Blanc, P. J.; Goma, G.; Franc, J.; FEMS Microbiol. Lett. 1998, 164, 1. 81. Teng, Q.; Huang, W.; Collette, T. W.; Ekman, D. R.; Tan, C.; Metabolomics 2009, 5, 199. 82. Mushtaq, M. Y.; Choi, Y. H.; Verpoorte, R.; Wilson, E. G.; Phytochem. Anal. 2014, 25, 291. 83. Rangarajan, R.; Ghosh, P.; Isotopes Environ. Health Stud. 2011, 47, 498. 84. Harbourne, N.; Marete, E.; Jacquier, J. C.; O'Riordan, D.; LWT - Food Sci. Technol. 2009, 42, 1468. 85. Lin, C. Y.; Wu, H.; Tjeerdema, R. S.; Viant, M. R.; Metabolomics 2007, 3, 55. 86. Lee, D. Y.; Fiehn, O.; Plant Methods 2008, 4, 7. 87. Villas-Bôas, S. G.; Roessner, U.; Hansen, M. A. E.; Smedsgaard, J.; Nielsen, J.; Metabolome Analysis: An Introduction; John Wiley & Sons, Inc.: Hoboken, 2007. 88. Junge, K.; Eicken, H.; Swanson, B. D.; Deming, J. W.; Cryobiology 2006, 52, 417. 89. Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R. N.; Willmitzer, L.; Plant J. 2000, 23, 131. 90. Chemat, F.; Vian, M. A.; Cravotto, G.; Int. J. Mol. Sci. 2012, 13, 8615. 91. Nahar, L.; Sarker, S. D. Em Natural Products Isolation; Sarker, S. D., Nahar, L., eds.; Humana Press: Totowa, NJ, 2012, cap. 3. 92. Owczarek, K.; Szczepanska, N.; Plotka-Wasylka, J.; Rutkowska, M.; Shyshchak, O.; Bratychak, M.; Namiesnik, J.; Chem. Technol. 2016, 10. 93. Dai, Y.; Witkamp, G. J.; Verpoorte, R.; Choi, Y. H.; Anal. Chem. 2013, 85, 6272. 94. Bajkacz, S.; Adamek, J.; Talanta 2017, 168, 329. 95. Maharjan, R. P.; Ferenci, T.; Anal. Biochem. 2003, 313, 145. 96. Jiye, A.; Trygg, J.; Gullberg, J.; Johansson, A. I.; Jonsson, P.; Antti, H.; Marklund, S. L.; Moritz, T.; Anal. Chem. 2005, 77, 8086. 97. Gullberg, J.; Jonsson, P.; Nordström, A.; Sjöström, M.; Moritz, T.; Anal. Biochem. 2004, 331, 283. 98. Schripsema, J.; Phytochem. Anal. 2010, 21, 14. 99. Maltese, F.; van der Kooy, F.; Verpoorte, R.; Nat. Prod. Commun. 2009, 4, 447. 100. Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A. R.; Nat. Protoc. 2006, 1, 387. 101. Higgs, R. E.; Zahn, J. A.; Gygi, J. D.; Hilton, M. D.; Appl. Environ. Microbiol. 2001, 67, 371. 102. De Vos, R. C. H.; Moco, S.; Lommen, A.; Keurentjes, J. J. B.; Bino, R. J.; Hall, R. D.; Nat. Protoc. 2007, 2, 778. 103. Vorst, O.; de Vos, C. H. R.; Lommen, A.; Staps, R. V.; Visser, R. G. F.; Bino, R. J.; Hall, R. D.; Metabolomics 2005, 1, 169. 104. Keurentjes, J. J. B.; Fu, J.; de Vos, C. H. R.; Lommen, A.; Hall, R. D.; Bino, R. J.; van der Plas, L. H. W.; Jansen, R. C.; Vreugdenhil, D.; Koornneef, M.; Nat. Genet. 2006, 38, 842. 105. Moco, S.; Bino, R. J.; Vorst, O.; Verhoeven, H. A.; Groot, J. D.; van Beek, T. A.; Vervoort, J.; Vos, R. D.; Plant Physiol. 2006, 141, 1205. 106. Vinatoru, M.; Ultrason. Sonochem. 2001, 8, 303. 107. Knapp, D.; Handbook of Analytical Derivatization Reactions, 1st ed., John Wiley & Sons: Hoboken, 1979. 108. Arruda, M. A. Z.; Santelli, R. E.; Quim. Nova 1997, 20, 599. 109. Frias, C. F.; Gramacho, S. A.; Pineiro, M.; Quim. Nova 2014, 37, 176. 110. Söderholm, S. L.; Damm, M.; Kappe, C. O.; Mol. Divers. 2010, 14, 869. 111. Garda, J.; Badiale-Furlong, E.; Quim. Nova 2008, 31, 270. 112. Schummer, C.; Delhomme, O.; Appenzeller, B. M. R.; Wennig, R.; Millet, M.; Talanta 2009, 77, 1473. 113. Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X.; Analyst 2012, 137, 293. 114. Maier, T. V.; Schmitt-Kopplin, P. Em Capillary Electrophoresis Methods and Protocols; Schmitt-Kopplin, P., ed.; Humana Press: New York, 2016, cap. 20. 115. García, A.; Godzien, J.; López-Gonzálvez, A.; Barbas, C.; Bioanalysis 2017, 9, 99. 116. Coralie, A.; Serge, H.; Khadidja, R.; Olivier, T.; Grégory, G.-J.; J. Planar Chromatogr. 2014, 27. 117. Fichou, D.; Ristivojevic, P.; Morlock, G. E.; Anal. Chem. 2016, 88, 12494. 118. Ogegbo, O. L.; Eyob, S.; Parmar, S.; Wang, Z.-T.; Bligh, S. W. A.; Anal. Methods 2012, 4, 2522. 119. Agnolet, S.; Jaroszewski, J. W.; Verpoorte, R.; Staerk, D.; Metabolomics 2010, 6, 292. 120. Simirgiotis, M. J.; Quispe, C.; Mocan, A.; Villatoro, J. M.; Areche, C.; Bórquez, J.; Sepúlveda, B.; Echiburu-Chau, C.; Braz. J. Pharmacogn. 2017, 27, 179. 121. Cevallos-Cevallos, J. M.; Reyes-De-Corcuera, J. I.; Etxeberria, E.; Danyluk, M. D.; Rodrick, G. E.; Trends Food Sci. Technol. 2009, 20, 557. 122. Wolfender, J.-L.; Rudaz, S.; Choi, Y. H.; Kim, H. K.; Curr. Med. Chem. 2013, 20, 1056. 123. Okazaki, Y.; Saito, K.; Plant Biotechnol. Rep. 2012, 6, 1. 124. Watanabe, M.; Kusano, M.; Oikawa, A.; Fukushima, A.; Noji, M.; Saito, K.; Plant Physiol. 2007, 146, 310. 125. Lisec, J.; Meyer, R. C.; Steinfath, M.; Redestig, H.; Becher, M.; Witucka-Wall, H.; Fiehn, O.; Törjék, O.; Selbig, J.; Altmann, T.; Willmitzer, L.; Plant J. 2008, 53, 960. 126. Valli, M.; Russo, H. M.; Pilon, A. C.; Pinto, M. E. F.; Dias, N. B.; Freire, R. T.; Castro-Gamboa, I.; Bolzani V. S.; Phys. Sci. Rev. (2019), doi:10.1515/psr-2018-0167. 127. Griffiths, J.; Anal. Chem. 2008, 80, 5678. 128. Fiehn, O.; Curr. Protoc. Mol. Biol. (2017), doi:10.1002/0471142727.mb3004s114. 129. Kind, T.; Wohlgemuth, G.; Lee, D. Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O.; Anal. Chem. 2009, 81, 10038. 130. Hummel, J.; Strehmel, N.; Beolling, C.; Schmidt, S.; Walther, D.; Kopka, J. Em The Handbook of Plant Metabolomics; Weckwerth, W., Kahl, G., eds.; Wiley-VCH Verlag GmbH & Co.: Hoboken, 2013, cap. 18. 131. Guijas, C.; Montenegro-Burke, J. R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A. E.; Wolan, D.W.; Spilker, M. E.; Benton, H. P.; Siuzdak G.; Anal. Chem. 2018, 90, 3156. 132. Carnevale Neto, F.; Pilon, A. C.; Selegato, D. M.; Freire, R. T.; Gu, H.; Raftery, D.; Lopes, N. P.; Castro-Gamboa, I.; Front. Mol. Biosci. 2016, 3, 59. 133. Ernst, M.; Silva, D. B.; Silva, R. R.; Vêncio, R. Z. N.; Lopes, N. P.; Nat. Prod. Rep. 2014, 31, 784. 134. Dunn, W. B.; Broadhurst, D. I.; Atherton, H. J.; Goodacre, R.; Griffin, J. L.; Chem. Soc. Rev. 2011, 40, 387-426. 135. Guaratini, T.; Vessecchi, R.; Pinto, E.; Colepicolo, P.; Lopes, N. P.; J. Mass Spectrom. 2005, 40, 963. 136. Zhou, B.; Xiao, J. F.; Tuli, L.; Ressom, H. W.; Mol. Biosyst. 2012, 8, 470. 137. Vessecchi, R.; Gozzo, F. C; Dörr, F. A.; Murgu, M.; Lebre, D. T.; Abreu, R.; Bustillos, O. V.; Riveros, J. M.; Lopes, N. P.; Quim. Nova 2011, 34, 1875. 138. Vessecchi, R.; Galembeck, S. E.; Lopes, N. P.; Nascimento, P. G. B. D.; Crotti, A. E. M.; Quim. Nova 2008, 31, 840. 139. Lopes, N. P.; Stark, C. B. W.; Hong, H.; Gates, P. J.; Staunton, J.; Analyst 2001, 126, 1630. 140. Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M.; BMC Bioinformatics 2010, 11, 1471. 141. Smith, C. A.; Want, E. J.; O'Maille, G.; Abagyan, R.; Siuzdak, G.; Anal. Chem. 2006, 78, 779. 142. Vinaixa, M.; Schymanski, E. L.; Neumann, S.; Navarro, M.; Salek, R. M.; Yanes, O.; TrAC - Trends Anal. Chem. 2016, 78, 23. 143. Wang, M.; Carver, J. J.; Phelan, V. V.; Sanchez, L. M.; Garg, N.; Peng, Y.; Nguyen, D. D.; Watrous, J.; Kapono, C. A.; Luzzatto-Knaan, T.; Porto, C.; Bouslimani, A.; Melnik, A. V.; Meehan, M. J.; Liu, W.-T.; Crüsemann, M.; Boudreau, P. D.; Esquenazi, E.; Sandoval-Calderón, M.; Kersten, R. D.; Pace, L. A.; Quinn, R. A.; Duncan, K. R.; Hsu, C.-C.; Floros, D. J.; Gavilan, R. G.; Kleigrewe, K.; Northen, T.; Dutton, R. J.; Parrot, D.; Carlson, E. E.; Aigle, B.; Michelsen, C. F.; Jelsbak, L.; Sohlenkamp, C.; Pevzner, P.; Edlund, A.; McLean, J.; Piel, J.; Murphy, B. T.; Gerwick, L.; Liaw, C.-C.; Yang, Y.-L.; Humpf, H.-U.; Maansson, M.; Keyzers, R. A.; Sims, A. C.; Johnson, A. R.; Sidebottom, A. M.; Sedio, B. E.; Klitgaard, A.; Larson, C. B.; Boya P, C. A.; Torres-Mendoza, D.; Gonzalez, D. J.; Silva, D. B.; Marques, L. M.; Demarque, D. P.; Pociute, E.; O'Neill, E. C.; Briand, E.; Helfrich, E. J N.; Granatosky, E. A.; Glukhov, E.; Ryffel, F.; Houson, H.; Mohimani, H.; Kharbush, J. J.; Zeng, Y.; Vorholt, J. A.; Kurita, K. L.; Charusanti, P.; McPhail, K. L.; Nielsen, K. F.; Vuong, L.; Elfeki, M.; Traxler, M. F.; Engene, N.; Koyama, N.; Vining, O. B.; Baric, R.; Silva, R. R.; Mascuch, S. J.; Tomasi, S.; Jenkins, S.; Macherla, V.; Hoffman, T.; Agarwal, V.; Williams, P. G.; Dai, J.; Neupane, R.; Gurr, J.; Rodríguez, A. M. C.; Lamsa, A.; Zhang, C.; Dorrestein, K.; Duggan, B. M.; Almaliti, J.; Allard, P.-M.; Phapale, P.; Nothias, L.-F.; Alexandrov, T.; Litaudon, M.; Wolfender, J.-L.; Kyle, J. E.; Metz, T. O.; Peryea, T.; Nguyen, D.-T.; VanLeer, D.; Shinn, P.; Jadhav, A.; Müller, R.; Waters, K. M.; Shi, W.; Liu, X.; Zhang, L.; Knight, R.; Jensen, P. R.; Palsson, B. Ø.; Pogliano, K.; Linington, R. G.; Gutiérrez, M.; Lopes, N. P.; Gerwick, W. H.; Moore, B. S.; Dorrestein, P. C.; Bandeira, N.; Nat. Biotechnol. 2016, 34, 828. 144. Silva, R.; Lopes, N. P.; Silva, D. B.; Planta Med. 2016, 82, 671. 145. Pavarini, D. P.; Da Silva, D. B.; Carollo, C. A.; Portella, A. P. F.; Latansio-Aidar, S. R.; Cavalin, P. O.; Oliveira, V. C.; Rosado, B. H. P.; Aidar, M. P. M.; Bolzani, V. S.; Lopes, N. P.; Joly, C. A.; J. Mass Spectrom. 2012, 47, 1482. 146. Ernst, M.; Silva, D. B.; Silva, R. R.; Monge, M.; Semir, J.; Vêncio, R. Z. N.; Lopes, N. P.; Planta Med. 2014, 80, SL2. 147. Silva, D. B.; Turatti, I. C. C.; Gouveia, D. R.; Ernst, M.; Teixeira, S. P.; Lopes, N. P.; Sci. Rep. 2014, 4, 4309. 148. Soares, M. S.; da Silva, D. F.; Forim, M. R.; da Silva, M. F.; Fernandes, J. B.; Vieira, P. C.; Silva, D. B.; Lopes, N. P.; de Carvalho, S. A.; de Souza, A. A.; Machado, M. A.; Phytochemistry 2015, 115, 161. 149. Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W.; Anal. Chim. Acta 2018, 1044, 199. 150. Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; Vandergheynst, J.; Fiehn, O.; Arita, M.; Nat. Methods 2015, 12, 523. 151. Xia, J.; Psychogios, N.; Young, N.; Wishart, D. S.; Nucleic Acids Res. 2009, 37, 652. 152. Bronsema, K. J.; Bischoff, R.; Van De Merbel, N. C.; Anal. Chem. 2013, 85, 9528. 153. Sargent, M.; Guide to Achieving Reliable Quantitative LC-MS Measurements, 1st ed., RSC Analytical Methods Commitee, 2013. 154. Dahal, U. P.; Jones, J. P.; Davis, J.; Rock, D.; Drug. Metab. Dispos. 2011, 39, 2355. 155. Markley, J. L.; Brüschweiler, R.; Edison, A. S.; Eghbalnia, H. R.; Powers, R.; Raftery, D.; Wishart, D. S.; Curr. Opin. Biotechnol. 2017, 43, 34. 156. Akoka, S.; Barantin, L.; Trierweiler, M.; Anal. Chem. 1999, 71, 2554. 157. Farrant, R. D.; Hollerton, J. C.; Lynn, S. M.; Provera, S.; Sidebottom, P. J.; Upton, R. J.; Magn. Reson. Chem. 2010, 48, 753. 158. Pauli, G. F.; Jaki, B. U.; Lankin, D. C.; J. Nat. Prod. 2005, 68, 133. 159. Giraudeau, P.; Silvestre, V.; Akoka, S.; Metabolomics 2015, 11, 1041. 160. Pauli, G. F.; Gödecke, T.; Jaki, B. U.; Lankin, D. C.; J. Nat. Prod. 2012, 75, 834. 161. Pauli, G. F.; Chen, S. N.; Simmler, C.; Lankin, D. C.; Gödecke, T.; Jaki, B. U.; Friesen, J. B.; McAlpine, J. B.; Napolitano, J. G.; J. Med. Chem. 2014, 57, 9220. 162. Gödecke, T.; Napolitano, J. G.; Rodríguez-Brasco, M. F.; Chen, S.-N.; Jaki, B. U.; Lankin, D. C.; Pauli, G. F.; Phytochem. Anal. 2013, 24, 581. 163. Maniara, G.; Rajamoorthi, K.; Rajan, S.; Stockton, G. W.; Anal. Chem. 1998, 70, 4921. 164. Malz, F.; Jancke, H.; J. Pharm. Biomed. Anal. 2005, 38, 813. 165. Forshed, J.; Schuppe-Koistinen, I.; Jacobsson, S. P.; Anal. Chim. Acta 2003, 487, 189. 166. Schey, K. L.; Luther, J. M.; Rose, K. L.; Magn. Reson. Chem. 2015, 53, 1043. 167. Piotto, M.; Saudek, V.; Sklenar, V.; J. Biomol. NMR 1992, 2, 661. 168. Hwang, T. L.; Shaka, A. J.; J. Magn. Reson. - Ser. A 1995, 112, 275. 169. Neuhaus, D.; Ismail, I. M.; Chung, C. W. A.; J. Magn. Reson. - Ser. A 1996, 118, 256. 170. McKay, R. T.; Concepts Magn. Reson., Part A 2011, 38A, 197. 171. Le Guennec, A.; Tayyari, F.; Edison, A. S.; Anal. Chem. 2017, 89, 8582. 172. Huang, Y.; Zhang, Z.; Chen, H.; Feng, J.; Cai, S.; Chen, Z.; Sci. Rep. 2015, 5, 1. 173. Breton, R. C.; Reynolds, W. F.; Nat. Prod. Rep. 2013, 30, 501. 174. Mercier, P.; Lewis, M. J.; Chang, D.; Baker, D.; Wishart, D. S.; J. Biomol. NMR 2011, 49, 307. 175. Weber, R. J. M.; Lawson, T. N.; Salek, R. M.; Ebbels, T. M. D.; Glen, R. C.; Goodacre, R.; Griffin, J. L.; Haug, K.; Koulman, A.; Moreno, P.; Ralser, M.; Steinbeck, C. ; Dunn, W. B.; Viant, M. R.; Metabolomics 2017, 13, 1. 176. Wolstencroft, K.; Haines, R.; Fellows, D.; Williams, A.; Withers, D.; Owen, S.; Soiland-Reyes, S.; Dunlop, I.; Nenadic, A.; Fisher, P.; Bhagat, J.; Belhajjame, K.; Bacall, F.; Hardisty, A.; Nieva de la Hidalga, A.; Balcazar Vargas, M. P.; Sufi, S.; Goble, C.; Nucleic Acids Res. 2013, 41, 557. 177. Aiche, S.; Sachsenberg, T.; Kenar, E.; Walzer, M.; Wiswedel, B.; Kristl, T.; Boyles, M.; Duschl, A.; Huber, C. G.; Berthold, M. R.; Reinert, K.; Kohlbacher, O.; Proteomics 2015, 15, 1443. 178. Haug, K.; Salek, R. M.; Conesa, P.; Hastings, J.; De Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; Maguire, E.; González-Beltrán, A.; Sansone, S.-A.; Griffin, J. L.; Steinbeck, C.; Nucleic Acids Res. 2013, 41(D1), 781. 179. Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K. S.; Sumner, S.; Subramaniam, S.; Nucleic Acids Res. 2016, 44(D1), 463. 180. Kind, T.; Fiehn, O.; BMC Bioinformatics 2007, 8, 1. 181. Skogerson, K.; Wohlgemuth, G.; Barupal, D. K.; Fiehn, O.; BMC Bioinformatics 2011, 12, 1. 182. Watanabe, K.; Yasugi, E.; Oshima, M.; Trends Glycosci. Glycotechnol. 2000, 12, 175. 183. Brown, M.; Dunn, W. B.; Dobson, P.; Patel, Y.; Winder, C. L.; Francis-Mcintyre, S.; Begley, P.; Carroll, K.; Broadhurst, D.; Tseng, A.; Swainston, N.; Spasic, I.; Goodacre, R.; Kell, D. B.; Analyst 2009, 134, 1322. 184. Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; Oda, Y.; Kakazu, Y.; Kusano, M.; Tohge, T.; Matsuda, F.; Sawada, Y.; Hirai M. Y.; Nakanishi, H.; Ikeda, K.; Akimoto, N.; Maoka, T.; Takahashi, H.; Ara, T.; Sakurai, N.; Suzuki, H.; Shibata, D.; Neumann, S.; Iida, T.; Tanaka, K.; Funatsu, K.; Matsuura, F.; Soga, T.; Taguchi, R.; Saito, K.; Nishioka T.; J. Mass Spectrom. 2010, 45, 703. 185. Ruttkies, C.; Schymanski, E. L.; Wolf, S.; Hollender, J.; Neumann, S.; J. Cheminform. 2016, 8, 1. 186. Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmüller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; Bioinformatics 2005, 21, 1635. 187. Willcott, M. R.; J. Am. Chem. Soc. 2009, 131, 13180. 188. Günther, U. L.; Ludwig, C.; Rüterjans, H.; J. Magn. Reson. 2000, 145, 201. 189. Lewis, I. A.; Schommer, S. C.; Markley, J. L.; Magn. Reson. Chem. 2009, 47, S123. 190. Ulrich, E. L.; Akutsu, H.; Doreleijers, J. F.; Harano, Y.; Ioannidis, Y. E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; Nakatani, E.; Schulte, C. F.; Tolmie, D. E.; Kent Wenger, R.; Yao, H.; Markley, J. L.; Nucleic Acids Res. 2008, 36, 402. 191. Ellinger, J. J.; Chylla, R. A.; Ulrich, E. L.; Markley, J. L.; Curr Metabolomics 2013, 1, 1. 192. Xia, J.; Bjorndahl, T. C.; Tang, P.; Wishart, D. S.; BMC Bioinformatics 2008, 9, 1. 193. López-Pérez, J. L.; Therón, R.; del Olmo, E.; Díaz, D.; Bioinformatics 2007, 23, 3256. 194. Steinbeck, C.; Kuhn, S.; Phytochemistry 2004, 65, 2711. 195. Seiler, K. P.; George, G. A.; Happ, M. P.; Bodycombe, N. E.; Carrinski, H. A.; Norton, S.; Brudz, S.; Sullivan, J. P.; Muhlich, J.; Serrano, M.; Ferraiolo, P.; Tolliday, N. J.; Schreiber, S. L.; Clemons, P. A.; Nucleic Acids Res. 2008, 36, 351. 196. de Matos, P.; Alcántara, R.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Spiteri, I.; Turner, S.; Steinbeck, C.; Nucleic Acids Res. 2009, 38, 249. 197. Pilon, A. C.; Valli, M.; Dametto, A. C.; Pinto, M. E. F.; Freire, R. T.; Castro-Gamboa, I.; Andricopulo, A. D.; Bolzani, V. S.; Sci. Rep. 2017, 7, 1. 198. Kim, S.; Thiessen, P. A.; Bolton, E. E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B. A.; Wang, J.; Yu, B.; Zhang, J.; Bryant, S. H.; Nucleic Acids Res. 2016, 44(D1), D1202. 199. Ebbels, T. M. D.; Lindon, J. C.; Muireann, C.; Methods Mol. Biol. 2011, 708, 365. 200. De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Rietzschel, E. R.; De Buyzere, M. L.; Langlois, M. R.; Bekaert, S.; Martins, J. C.; Van Criekinge, W.; Anal. Bioanal. Chem. 2010, 398, 1781. 201. Craig, A.; Cloarec, O.; Holmes, E.; Nicholson, J. K.; Lindon, J. C.; Anal. Chem. 2006, 78, 2262. 202. Zhang, S.; Zheng, C.; Lanza, I. R.; Nair, K. S.; Raftery, D.; Vitek, O.; Anal. Chem. 2009, 81, 6080. 203. Deutsch, E.; Proteomics 2008, 8, 2776. 204. Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W. H.; Römpp, A.; Neumann, S.; Pizarro, A. D.; Montecchi-Palazzi, L.; Tasman, N.; Coleman, M.; Reisinger, F.; Souda, P.; Hermjakob, H.; Binz, P. A.; Deutsch E. W.; Mol. Cell. Proteomics 2011, 10, R110.000133. 205. Deutsch, E. W.; Curr. Protoc. Protein Sci. 2010, 60, 319. 206. Baggerly, K. A.; Morris, J. S.; Wang, J.; Gold, D.; Xiao, L. C.; Coombes, K. R.; Proteomics 2003, 3, 1667. 207. Tsai, T.-H.; Wang, M.; Ressom, H. W.; Methods Mol. Biol. 2016, 1362, 63. 208. Yu, T.; Park, Y.; Johnson, J. M.; Jones, D. P.; Bioinformatics 2009, 25, 1930. 209. Melamud, E.; Vastag, L.; Rabinowitz, J. D.; Anal. Chem. 2010, 82, 9818. 210. Sturm, M.; Bertsch, A.; Gröpl, C.; Hildebrandt, A.; Hussong, R.; Lange, E.; Pfeifer, N.; Schulz-Trieglaff, O.; Zerck, A.; Reinert, K.; Kohlbacher, O.; BMC Bioinformatics 2008, 9, Article 163. 211. Lommen, A.; Anal. Chem. 2009, 81, 3079. 212. Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Oresic, M.; Chemom. Intell. Lab. Syst. 2011, 108, 23. 213. Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Current Bioinformatics 2012, 7, 96. 214. Andreev, V. P.; Rejtar, T.; Chen, H. S.; Moskovets, E. V; Ivanov, A. R.; Karger, B. L.; Anal. Chem. 2003, 75, 6314. 215. Vandenbogaert, M.; Li-Thiao-Té, S.; Kaltenbach, H. M.; Zhang, R.; Aittokallio, T.; Schwikowski, B.; Proteomics 2008, 8, 650. 216. Tomasi, G.; Van Den Berg, F.; Andersson, C.; J. Chemom. 2004, 18, 231. 217. Nielsen, N. P. V; Carstensen, J. M.; Smedsgaard, J.; J. Chromatogr. A 1998, 805, 17. 218. Wong, J. W. H.; Durante, C.; Cartwright, H. M.; Anal. Chem. 2005, 77, 5655. 219. Wong, J. W. H.; Cagney, G.; Cartwright, H. M.; Bioinformatics 2005, 21, 2088. 220. Fischler, M. A.; Bolles, R. C.; Commun. ACM 1981, 24, 381. 221. Röst, H. L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H. C.; Gutenbrunner, P.; Kenar, E.; Kenar, E.; Liang, X.; Nahnsen, S.; Nilse, L.; Pfeuffer, J.; Rosenberger, G.; Rurik, M.; Schmitt, U.; Veit, J.; Walzer, M.; Wojnar, D.; Wolski, W. E.; Schilling, O.; Choudhary, J. S.; Malmström, L.; Aebersold, R.; Reinert, K.; Kohlbacher, O.; Nat. Methods 2016, 13, 741. 222. Bellew, M.; Coram, M.; Fitzgibbon, M.; Igra, M.; Randolph, T.; Wang, P.; May, D.; Eng, J.; Fang, R.; Lin, C.; Chen, J.; Goodlett, D.; Whiteaker, J.; Paulovich, A.; McIntosh, M.; Bioinformatics 2006, 22, 1902. 223. Li, X.; Yi, E. C.; Kemp, C. J.; Zhang, H.; Aebersold, R.; Mol. Cell. Proteomics 2005, 4, 1328. 224. Zhang, X.; Asara, J. M.; Adamec, J.; Ouzzani, M.; Elmagarmid, A. K.; Bioinformatics 2005, 21, 4054. 225. Smolinska, A.; Blanchet, L.; Buydens, L. M. C.; Wijmenga, S. S.; Anal. Chim. Acta 2012, 750, 82. 226. Katajamaa, M.; Oresic, M.; J. Chromatogr. A 2007, 1158, 318. 227. Rafiei, A.; Sleno, L.; Rapid Commun. Mass Spectrom. 2014, 29, 119. 228. Brodsky, L.; Moussaieff, A.; Shahaf, N.; Aharoni, A.; Rogachev, I.; Anal. Chem. 2010, 82, 9177. 229. Vemula, H.; Kitase, Y.; Ayon, N. J.; Bonewald, L.; Gutheil, W. G.; Anal. Biochem. 2017, 516, 75. 230. Yu, T.; Peng, H.; BMC Bioinformatics 2010, 11, 559. 231. Torres-Lapasió, J. R.; García-Alvarez-Coque, M. C.; Baeza-Baeza, J. J.; Anal. Chim. Acta 1997, 348, 187. 232. Youn, D. Y.; Yun, S. J.; Jung, K. H.; J. Chromatogr. A 1992, 591, 19. 233. Caballero, R. D.; García-Alvarez-Coque, M. C.; Baeza-Baeza, J. J.; J. Chromatogr. A 2002, 954(1-2), 59. 234. Yuan, Z.; Shi, J.; Lin, W.; Chen, B.; Wu, F. X.; Adv. Bioinformatics 2011, 2011, ID 210805. 235. Tuli, L.; Tsai, T.-H.; Varghese, R. S.; Cheema, A.; Ressom, H. W.; The 2010 IEEE International Conference on Bioinformatics and Biomedical Technology 2010, 10, 67. 236. Katajamaa, M.; Oresic, M.; BMC Bioinformatics, 2005, 6, 1. 237. Tautenhahn, R.; Bottcher, C.; Neumann, S.; BMC Bioinformatics 2008, 9, 1. 238. Monroe, M. E.; Tolic, N.; Jaitly, N.; Shaw, J. L.; Adkins, J. N.; Smith, R. D.; Bioinformatics 2007, 23, 2021. 239. Böcker, S.; Letzel, M. C.; Lipták, Z.; Pervukhin, A.; Bioinformatics 2009, 25, 218. 240. Horn, D. M.; Zubarev, R. A.; McLafferty, F. W.; J. Am. Soc. Mass Spectrom. 2000, 11, 320. 241. Jaitly, N.; Mayampurath, A.; Littlefield, K.; Adkins, J. N.; Anderson, G. A.; Smith, R. D.; BMC Bioinformatics 2009, 10, 1. 242. Nicholson, J. K.; Wilson, I. D.; Prog. Nucl. Magn. Reson. Spectrosc. 1989, 21, 449. 243. Callister, S. J.; Barry, R. C.; Adkins, J. N.; Johnson, E. T.; Qian, W.; Webb-Robertson, B.-J. M.; Smith, R. D.; Lipton, M. S.; J. Proteome Res. 2007, 5, 277. 244. Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Oresic, M.; BMC Bioinformatics 2007, 8, 1. 245. Dunn, W. B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-Mcintyre, S.; Anderson, N.; Brown, M.; Knowles, J. D.; Halsall, A.; Haselden, J. N.; Nicholls, A. W.; Wilson, I. D.; Kell, D. B.; Goodacre, R.; Human Serum Metabolome (HUSERMET) Consortium; Nat. Protoc. 2011, 6, 1060. 246. Mueller, L. N.; Rinner, O.; Schmidt, A.; Letarte, S.; Bodenmiller, B.; Brusniak, M. Y.; Vitek, O.; Aebersold, R.; Müller, M.; Proteomics 2007, 7, 3470. 247. Selegato, D. M.; Freire, R. T.; Pilon, A. C.; Biasetto, C. R.; Oliveira, H. C.; Abreu, L. M.; Araujo, A. R.; Bolzani, V. S.; Castro-Gamboa I.; Magn. Reson. Chem. 2019, 57, 458. 248. Hoch, J. C.; Stern, A.; NMR Data Processing, 1st ed. Wiley-Liss: New York, 1996. 249. Xi, Y.; Rocke, D. M.; BMC Bioinformatics 2008, 9, 1. 250. Gan, F.; Ruan, G.; Mo, J.; Chemom. Intell. Lab. Syst. 2006, 82, 59. 251. Zhang, Z. M.; Chen, S.; Liang, Y. Z.; Analyst 2010, 135, 1138. 252. Eilers, P. H. C.; Anal. Chem. 2003, 75, 3631. 253. Eilers, P. H. C.; Marx, B. D.; Stat. Sci. 1996, 11, 89. 254. de Rooi, J. J.; Eilers, P. H. C.; Chemom. Intell. Lab. Syst. 2012, 117, 56. 255. Defernez, M.; Colquhoun, I. J.; Phytochemistry 2003, 62, 1009. 256. Vogels, J. T. W. E.; Tas, A. C.; Venekamp, J.; van der Greef, J.; J. Chemom. 1996, 10, 425. 257. Torgrip, R. J. O.; Åberg, M.; Karlberg, B.; Jacobsson, S. P.; J. Chemom. 2003, 17, 573. 258. Lee, G. C.; Woodruff, D. L.; Anal. Chim. Acta 2004, 513, 413. 259. Stoyanova, R.; Nicholls, A. W.; Nicholson, J. K.; Lindon, J. C.; Brown, T. R.; J. Magn. Reson. 2004, 170, 329. 260. Walczak, B.; Wu, W.; Chemom. Intell. Lab. Syst. 2005, 77, 173. 261. Csenki, L.; Alm, E.; Torgrip, R. J. O.; Åberg, K. M.; Nord, L. I.; Schuppe-Koistinen, I.; Lindberg, J.; Anal. Bioanal. Chem. 2007, 389, 875. 262. Veselkov, K. A.; Lindon, J. C.; Ebbels, T. M. D.; Crockford, D.; Volynkin, V. V; Holmes, E.; Davies, D. B.; Nicholson, J. K.; Anal. Chem. 2009, 81, 56. 263. Staab, J. M.; O'Connell, T. M.; Gomez, S. M.; BMC Bioinformatics 2010, 11, 123. 264. Vu, T. N.; Valkenborg, D.; Smets, K.; Verwaest, K. A.; Dommisse, R.; Lemière, F.; Verschoren, A.; Goethals, B.; Laukens, K.; BMC Bioinformatics 2011, 12, 405. 265. Kim, S. B.; Wang, Z.; Hiremath, B.; Ann. Oper. Res. 2010, 174, 19. 266. Savorani, F.; Tomasi, G.; Engelsen, S. B.; J. Magn. Reson. 2010, 202, 190. 267. Izquierdo-García, J. L.; Villa, P.; Kyriazis, A.; Del Puerto-Nevado, L.; Pérez-Rial, S.; Rodriguez, I.; Hernandez, N.; Ruiz-Cabello, J.; Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 263. 268. De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Tsiporkova, E.; Rietzschel, E. R.; De Buyzere, M. L.; Gillebert, T. C.; Bekaert, S.; Martins, J. C.; Van Criekinge, W.; Anal. Chem. 2008, 80, 3783. 269. Anderson, P. E.; Reo, N. V; DelRaso, N. J.; Doom, T. E.; Raymer, M. L.; Metabolomics 2008, 4, 261. 270. Davis, R. A.; Charlton, A. J.; Godward, J.; Jones, S. A.; Harrison, M.; Wilson, J. C.; Chemom. Intell. Lab. Syst. 2007, 85, 144. 271. Anderson, P. E.; Mahle, D. A.; Doom, T. E.; Reo, N. V; DelRaso, N. J.; Raymer, M. L.; Metabolomics 2011, 7, 179. 272. Hao, J.; Liebeke, M.; Astle, W.; De Iorio, M.; Bundy, J. G.; Ebbels, T. M. D.; Nat. Protoc. 2014, 9, 1416. 273. Cobas, C.; Sykora, S.; Resumos da 50th ENC Conference; Asilomar, USA, 2009. 274. Cobas, C.; Seoane, F.; Sýkora, S.; Resumos da SMASH Conference; Santa Fé, USA, 2008. 275. Cloarec, O.; Dumas, M.; Craig, A.; Barton, R. H.; Trygg, J.; Hudson, J.; Blancher, C.; Gauguier, D.; Lindon, J. C.; Holmes, E.; Nicholson, J.; Anal. Chem. 2005, 77, 1282. 276. Blaise, B. J.; Navratil, V.; Domange, C.; Shintu, L.; Dumas, M. E.; Elena-Herrmann, B.; Emsley, L.; Toulhoat, P.; J. Proteome Res. 2010, 9, 4513. 277. Dona, A. C.; Kyriakides, M.; Scott, F.; Shephard, E. A.; Varshavi, D.; Veselkov, K.; Everett, J. R.; Comput. Struct. Biotechnol. J. 2016, 14, 135. 278. Spraul, M.; Neidig, P.; Klauck, U.; Kessler, P.; Holmes, E.; Nicholson, J. K.; Sweatman, B. C.; Salman, S. R.; Farrant, R. D.; Rahr, E.; Beddell, C. R.; Lindon, J. C.; J. Pharm. Biomed. Anal. 1994, 12, 1215. 279. Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H.; Anal. Chem. 2006, 78, 4281. 280. Torgrip, R. J. O.; Åberg, K. M.; Alm, E.; Schuppe-Koistinen, I.; Lindberg, J.; Metabolomics 2008, 4, 114. 281. Dong, J.; Cheng, K. K.; Xu, J.; Chen, Z.; Griffin, J. L.; Chemom. Intell. Lab. Syst. 2011, 108, 123. 282. PERCH NMR Software. PERCH Solutions Ltd, Finland, 2018, disponível em http://new.perchsolutions.com/index.php?id=34, acessada em março de 2020 283. Chenomx NMR Suite. Version 8.2. CHENOMX Inc, Canada, 2018, disponível em https://www.chenomx.com/software, acessada em março de 2020. 284. Mestrenova. Version 12.0.2. Mestrelab Research, Spain, 2018, disponível em http://mestrelab.com/download/mnova, acessada em março de 2020. 285. Sussulini, A.; Metabolomics: From Fundamentals to Clinical Applications, 1st ed., Springer: New York, 2017. 286. van den Berg, R. A.; Hoefsloot, H. C. J.; Westerhuis, J. A.; Smilde, A. K.; van der Werf, M. J.; BMC Genomics 2006, 7, 1. 287. Eigenvector Research Inc.: Manson, WA, USA. 288. Daszykowski, M.; Serneels, S.; Kaczmarek, K.; Van Espen, P.; Croux, C.; Walczak, B.; Chemom. Intell. Lab. Syst. 2007, 85, 269. 289. Worley, B.; Powers, R.; Curr. Metabolomics 2016, 4, 97. 290. Martens, H.; Martens, M.; Multivariate Analysis of Quality: An Introduction, 1st ed., John Wiley & Sons: New York, 2001. 291. Andrecut, M. J.; Comput. Biol. 2008, 16, 1593. 292. Jolliffe, I. T.; Principal Component Analysis, 1st ed., Springer-Verlag: New York, 2002. 293. Wall, M. E.; Rechtsteiner, A.; Rocha, L. M. Em A Practical Approach to Microarray Data Analysis; Berrar, D. P., Dubitzky, W., Granzow, M., eds.; Springer: New York, 2003, cap. 5. 294. Liu, M.; Grkovic, T.; Liu, X.; Han, J.; Zhang, L.; Quinn, R. J.; Synth. Syst. Biotechnol. 2017, 2, 276. 295. Candes, E. J.; Li, X.; Ma, Y.; Wright, J.; J. ACM 2011, 58, 1. 296. da Costa, D.; Silva, C.; Pinheiro, A.; Frommenwiler, D.; Arruda, M.; Guilhon, G.; Alves, C.; Arruda, A.; da Silva, M.; Molecules 2016, 21, 569. 297. Duda, R. O.; Hart, P. E.; Stork, D. G.; Pattern Classification; John Wiley & Sons: New York, 2000. 298. Mazzei, P.; Spaccini, R.; Francesca, N.; Moschetti, G.; Piccolo, A.; J. Agric. Food Chem. 2013, 61, 10816. 299. Liu, Z.; Liu, Y.; Liu, C.; Song, Z.; Li, Q.; Zha, Q.; Lu, C.; Wang, C.; Ning, Z.; Zhang, Y.; Tian, C.; Lu, A.; Chem. Cent. J. 2013, 7, 1. 300. Marzetti, E.; Landi, F.; Marini, F.; Cesari, M.; Buford, T. W.; Manini, T. M.; Onder, G.; Pahor, M.; Bernabei, R.; Leeuwenburgh, C.; Calvani, R.; Front. Med. 2014, 1, 1. 301. Velagapudi, V. R.; Hezaveh, R.; Reigstad, C. S.; Gopalacharyulu, P.; Yetukuri, L.; Islam, S.; Felin, J.; Perkins, R.; Borén, J.; Oresic, M.; Bäckhed, F.; J. Lipid Res. 2010, 51, 1101. 302. Wang, X.; Zhang, A.; Han, Y.; Wang, P.; Sun, H.; Song, G.; Dong, T.; Yuan, Y.; Yuan, X.; Zhang, M.; Xie, N.; Zhang, H.; Dong, H.; Dong, W.; Mol. Cell. Proteomics 2012, 11, 370. 303. Nguyen, D. V; Rocke, D. M.; Bioinformatics 2002, 18, 1216. 304. Boulesteix, A.-L.; Stat. Appl. Genet. Mol. Biol. 2004, 3, 1. 305. Szymanska, E.; Saccenti, E.; Smilde, A. K.; Westerhuis, J. A.; Metabolomics 2012, 8, 3. 306. Chung, D.; Keles, S.; Stat. Appl. Genet. Mol. Biol. 2010, 9, 1. 307. Bylesjo, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J. K.; Holmes, E.; Trygg, J.; J. Chemom. 2006, 20, 341. 308. Marhuenda-Egea, F. C.; Gonsálvez-Álvarez, R.; Martínez-Sabater, E.; Lledó, B.; Ten, J.; Bernabeu, R.; Metabolomics 2011, 7, 247. 309. Guo, Q.; Sidhu, J. K.; Ebbels, T. M. D.; Rana, F.; Spurgeon, D. J.; Svendsen, C.; Stürzenbaum, S. R.; Kille, P.; Morgan, A. J.; Bundy, J. G.; Metabolomics 2009, 5, 72. 310. Holmes, E.; Nicholls, A. W.; Lindon, J. C.; Connor, S. C.; Connelly, J. C.; Haselden, J. N.; Damment, S. J. P.; Spraul, M.; Neidig, P.; Nicholson, J. K.; Chem. Res. Toxicol. 2000, 13, 471. 311. Tsugawa, H.; Tsujimoto, Y.; Arita, M.; Bamba, T.; Fukusaki, E.; BMC Bioinformatics, 2011, 12, 131 . 312. Shi, H.; Paolucci, U.; Vigneau-Callahan, K. E.; Milbury, P. E.; Matson, W. R.; Kristal, B. S.; Omics 2004, 8, 197. 313. Snyder, L. R.; Kirkland, J. J.; Glajch, J. L.; Practical HPLC Method Development, 2a ed., Wiley: New York, 1997, cap. 15. 314. Snyder, L. R.; Kirkland, J. J.; Dolan, J. W.; Introduction to Modern Liquid Chromatography, John Wiley & Sons: Hoboken, 2010. cap. 4. 315. Simões C. M. O.; Schenkel, E. P.; Gosmann, G.; Mello, J. C. P.; Mentz, L. A.; Petrovick, P. R.; Farmacognosia: da Planta ao Medicamento, 6ª ed., Editora da Universidade Federal do Rio Grande do Sul: Porto Alegre, 2007. 316. Evans W. C.; Trease and Evans' Pharmacognosy, 14th ed., WB Saunders Company: London, 1996. 317. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Vyvyan, J. A.; Introduction to Spectroscopy, 5th ed., Cengage Learning: Stamford, 2014. 318. Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric Identification of Organic Compounds, 8th ed., Wiley: New York, 2014. 319. Wolfender, J-L.; Litaudon, M.; Touboul. D.; Queiroz. E. F.; Nat. Prod. Rep. 2019, 36, 855. 320. Carrilho, E.; Tavares, M. C. H.; Lanças, F. M.; Quim. Nova 2001, 24, 509. 321. Lang, Q.; Wai, C. M.; Talanta 2001, 53, 771. 322. Queiroz, S. C. N.; Collins, C. H.; Jardim, I. C. S. F.; Quim. Nova 2001, 24, 68. 323. Machado, B. A. S.; Pereira, C. G.; Nunes, S. B.; Padilha, F. F.; Umsza-Guez, M. A.; Sep. Sci. Technol. 2013, 48, 2741. 324. Guillarme, D.; Nguyen, D. T. T.; Rudaz, S.; Wolfender, J.-L.; Eur. J. Pharm. Biopharm. 2008, 68, 430. 325. Batista Jr, J. M.; Blanch, E. W.; Bolzani, V. S.; Nat. Prod. Rep. 2015, 32, 1280. 326. Breton, R. C.; Reynolds, W. F.; Nat. Prod. Rep. 2013, 30, 501. 327. Krastanov, A.; Biotech. Biotechnol. Equip. 2010, 24, 1537. 328. Blazenovic, I.; Kind, T.; Ji, J.; Fiehn, O.; Metabolites 2018, 8, 1. 329. Alonso, A.; Marsal, S.; Julià, A.; Frontiers in Bioengineering and Biotechnology 2015, 3, 23. 330. Pilon, A. C.; Gu, H.; Raftery, D.; Bolzani, V. S.; Castro-Gamboa, I.; Lopes, N. P.; Carnevale Neto, F.; Anal. Chem. 2019, 91, 10413. 331. Brunetti, A. E.; Carnevale Neto, F.; Vera, M. C.; Taboada, C.; Pavarini, D. P.; Bauermeister, A.; Lopes, N. P.; Chem. Soc. Rev. 2018, 47, 1574. 332. Zenobi, R.; Science 2013, 342, 1201. # Alan C. Pilon e Denise M. Selegato contribuiram igualmente para o trabalho e são co-autores. |
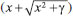 , onde x são os dados originais e γ é um parâmetro de transformação. Nesse tipo de tratamento, somente a variação biológica será predominante após a transformação.
, onde x são os dados originais e γ é um parâmetro de transformação. Nesse tipo de tratamento, somente a variação biológica será predominante após a transformação.On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access