
 |
 |
 |
 |
 |
Artigo
|
|
| Dinâmica molecular reativa da abertura do anel epóxi em reações com poliamina Reactive molecular dynamics of the epoxy ring-opening in polyamine reactions |
|
Ellen Cristine Araújo Rosa*; Rene Francisco Boschi Gonçalves; Marcela Galizia Domingues; José Atílio Fritz Fidel Rocco
Departamento de Química, Instituto Tecnológico de Aeronáutica, 12228-900 São José dos Campos - SP, Brasil Recebido em 09/09/2019 *e-mail: ellen@ita.br The molecular dynamics simulations were used to study the epoxy ring-opening in the determination of kinetic parameters, with a temperature range from 1500 to 2500 K in a 20 x 20 x 20 Å unit cell containing 15 molecules of C2H4O (ethylene oxide) and 35 molecules of CH6N2 (methanediamine). The activation energy values for epoxy and diamine was 66 and 92 kJ mol-1, respectively. The simulation showed epoxy ring breakage in some of the molecules, but mainly the release of ammonia by diamine. It was observed that the activation energy involved in diamine consumption for ammonia formation is higher than for the epoxy ring opening. The results of the epoxy ring-opening study show that the polymerization occurs slowly, which leads to high computational simulation values. INTRODUÇÃO As resinas epóxi são formadas a partir de um oligômero contendo pelo menos dois grupos epóxidos e um extensor de cadeia, normalmente composto de amina ou diácido.1 Podem ser amplamente utilizadas em revestimentos de proteção, adesivos, materiais de embalagem eletrônica, compósitos de alto desempenho, tintas etc. São polímeros termofixos com alta rigidez, estabilidade dimensional, resistência mecânica, química e ambiental, processabilidade versátil e relação favorável custo-benefício.2 Em aplicações práticas, é necessário que ocorra um processo de cura para transformar resinas epóxi de monômeros ou oligômeros em macromolécula altamente reticulada na presença de um extensor de cadeia através de um mecanismo de abertura do anel epóxi.2 Dessa forma, é possível carregar, aditivar ou homogeneizar de maneira adequada os sistemas, anteriormente de transformá-los em um sólido. Uma característica distintiva deste processo consiste na reação que envolve a abertura do anel epóxi e a adição de moléculas e reagentes, simbolizados da seguinte forma:3 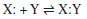 Em que X e Y são as espécies doadoras e receptoras de elétrons, respectivamente. Em condições básicas ou neutras, todas as reações de abertura do anel são essencialmente similares e envolvem o ataque nucleofílico em um dos átomos de carbono do anel epóxi, como no caso de um extensor de cadeia de amina: Os compostos epoxídicos possuem uma ampla gama de aplicações técnicas devido as suas excelentes propriedades mecânicas, elétricas, isolantes e adesivas. Como os produtos de epóxi na aplicação final exigem uma reação com extensor de cadeia (geralmente uma amina), a elucidação do mecanismo de reação é de especial interesse,4 conforme indicado por muitos estudos cinéticos nas últimas décadas,5-7 algum destes citados a seguir. Unger et al.5 estudaram as resinas epóxi por simulação computacional e foi demonstrado o procedimento de cura influencia a estrutura final da rede e, consequentemente, as propriedades finais do material. Acocella et al.6 examinaram a influência de diferentes nanofiltros à base de grafite nas reações de abertura do anel epóxido, induzidas por diamina para o éter diglicidílico do bisfenol. Os resultados mostraram que estes nanofiltros influenciam significativamente nas reações de abertura do anel epóxido, reduzindo a temperatura e/ou tempo de reticulação de resinas epóxi. Sbirrazzuoli et al.7 estudaram sistemas de cura epóxi com diferentes proporções de amina. O sistema estequiométrico com amina mostrou uma dependência decrescente da energia de ativação e em excesso de amina, a reação de cura é controlada pela amina primária. Pham8 propôs um modelo cinético de epóxi-amina baseado em química quântica. Os cálculos mostraram que esse modelo cinético pode ser estendido a outros sistemas por meio de catálise alcoólica para diferentes aminas e epóxis. A reação geral ocorre por meio de um ataque nucleofílico do nitrogênio da amina no carbono terminal da função epóxi. O mecanismo é conhecido por ser um SN2 tipo II, na qual a velocidade de reação obedece a cinética de segunda ordem (Figura 1).4
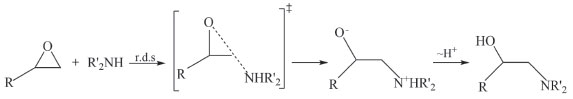 Figura 1. Mecanismo geral para cura com epóxi-amina4
Os compostos de amina estão entre os agentes de cura epoxídicos mais antigos e usados. As aminas primárias (R-NH2) e secundárias (R-NH-R') sofrem as reações SN2 (Figura 2a) e são altamente reativas com epóxis. Dependendo dos substituintes, estas aminas podem ser divididas em três grupos principais: aminas alifáticas, cicloalifáticas e aromáticas. Entre estes grupos, as aminas aromáticas produzem resinas curadas com altas propriedades de resistência química e térmica. Entretanto, exigem longos ciclos de cura em alta temperatura (~ 150 ºC).8,9 Dessa forma, aplicações práticas demandam mais tempo e, consequentemente, são mais custosas.
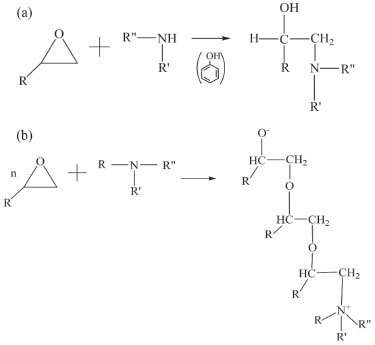 Figura 2. (a) Reação de aminas primárias e secundárias. (b) Reação de aminas terciárias9
As aminas cicloalifáticas resultam em produtos com resistência térmica e tenacidade superiores às das aminas alifáticas, porém com custo mais elevado. Por outro lado, as aminas terciárias (R-NR'R") causam as polimerizações de epóxi como mostrado na Figura 2b.8,9 Devido à sua importância dos efeitos dos substituintes sobre a reatividade, o processo de cura com epóxi-amina vêm sendo objeto de muitos estudos.4,7-11 Entretanto, ainda existe dificuldade em entender seu mecanismo e limitações. As resinas são polímeros termofixos caracterizados pelo seu processo de cura irreversível, no qual sofre alteração de consistência do material, de pré-polímero de líquido solúvel para sólido. As propriedades físicas da formação da rede altamente ligada dependem da estrutura da rede subjacente da molécula, influenciada, principalmente, pelos monômeros constituintes e pela reação de polimerização.5 Como os mecanismos para reações de cura do epóxi ainda não foram totalmente elucidados, modelos cinéticos são usados para aproximar mecanismos empíricos ou semiempíricos.8 Desse modo, os cálculos teóricos contribuem no entendimento em nível molecular do mecanismo da reação de cura da epóxi-amina. Embora as metodologias computacionais atuais não permitam modelar uma reação em condição real de poliadição na fase condensada, a análise dos dados teóricos pode fornecer informações valiosas sobre a química desta reação,4 tais como: examinar as superfícies de energia potencial das reações em fase gasosa e em diferentes solventes, variação do número de espécies dos reagentes e produtos, parâmetros cinéticos, entre outros. Geralmente, as propriedades finais dos materiais epóxi curados são determinadas, principalmente, pela estrutura da rede, que depende do tipo de reação e do estágio resultante do processo de cura. Portanto, é fundamental estudar o mecanismo de reação, a abertura do anel epoxídico e os comportamentos de cura dos sistemas epóxi.11 Como existem muitos monômeros constituintes para resinas epóxi e extensor de cadeia que permitem uma enorme variedade de combinações, a seleção de resinas epóxi de alto desempenho a partir de experimentos pode ser demorada, cara e de baixa eficiência, além de as correlações entre estrutura molecular e propriedades macroscópicas de redes de resina epóxi serem de difícil análise. Assim, a Dinâmica Molecular (MD) pode reduzir o ciclo de desenvolvimento de novas formulações, otimizando e reduzindo os custos na previsão das propriedades dos materiais da resina epóxi.12 As simulações de Dinâmica Molecular Reativa (RMD) fornecem baixo custo computacional e permitem estimar as interações entre as espécies presentes nos limites de simulação. O ReaxFF é um campo de força criado para simular reações químicas, em que as ligações são quebradas e formadas diversas vezes durante a simulação. Existem vários métodos de simulação computacional que envolvem dinâmica molecular. Dentre estes, foi usado o pacote computacional LAAMPS (Large-scale Atomic/Molecular Massively Parallel Simulator)13 para estudar a abertura do anel epoxídico por meio da simulação de dinâmica molecular reativa do sistema contendo óxido de etileno e metanodiamina, visto que o primeiro passo dessa reação de polimerização decorre, inicialmente, na abertura do anel epóxido e, em seguida, a cura com esse extensor de cadeia.
METODOLOGIA As simulações de dinâmica molecular reativa foram realizadas empregando o software LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) e o campo de forças ReaxFF. Esse campo de força reativa divide a energia do sistema em várias contribuições parciais de energia, como demonstrado pela Eq. (1):14,15 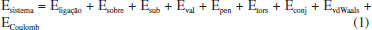 Em que: • Eligação representa a energia de ligação; • Esobre e Esub representa o átomo sob-coordenado com excesso e falta na contribuição de energia, respectivamente; • Eval, Epen e Etors são os termos da energia do ângulo de valência, penalidade de energia (estabilidade do sistema) e energia do ângulo de torsão, respectivamente; • Econj, EvdWaals e ECoulumb representam os efeitos de conjugação à energia molecular, interação de van der Waals e interação Coulômbica, respectivamente. A distância interatômica do ReaxFF é obtida a partir da ordem de ligação e a energia de ligação entre o par de átomos, de acordo com a Eq. (6). Nessa equação é possível observar a contribuição da ligação sigma (pbo,1 e pbo,2), da primeira e segunda ligação pi (pbo,3 e pbo,4) e (pbo,5 e pbo,6), respectivamente.1 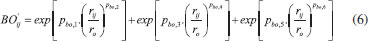 As ordens de ligação BO'ij são, então, corrigidas por excesso de coordenação e por ordens de ligação residuais de 1-3 em ângulos de valência. Uma ordem de ligação corrigida (BOij) é usada para calcular as energias de ligação:14 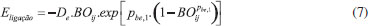 O ReaxFF também considera o excesso e a falta de coordenação, os termos do ângulo de valência (contribuição de energia dos termos do ângulo de valência tende a zero quando BO é zero), os ângulos de torção (para BO → 0 e BO > 1), a energia molecular, as interações não ligadas de Van der Waals (interações repulsivas a curtas distâncias interatômicas devido às principais energias de ortogonalização e a atração de Pauli a longas distâncias devido à dispersão, para todos os átomos) e as interações de Coulomb (ajustadas para sobreposição orbital entre os átomos a distâncias próximas com um potencial Coulomb blindado).14 A simulação de modelagem molecular corresponde às seguintes etapas: (1) geração de modelo desvinculado; (2) inicialização; (3) reticulação e (4) equilíbrio final do sistema. A primeira etapa envolve a geração inicial de um modelo desvinculado aleatório a partir de monômeros separados e moléculas de extensor de cadeia. Aplica-se uma otimização de energia ao modelo inicial para minimizar a energia interna. A reticulação é iniciada aquecendo a caixa de simulação até a temperatura de cura desejada e o equilíbrio ocorre antes de iniciar a reticulação. No procedimento real, o número de ligações é formado em cada etapa de reticulação, seguido por um tempo de flexibilização para permitir que o sistema reaja às alterações da estrutura em rede. Dependendo do método específico, é formada somente a ligação com a menor distância reativa ou todas as ligações dentro de um determinado limite de reação.5 Os campos de força usados no ReaxFF foram otimizados usando uma técnica de busca sucessiva de um parâmetro, no qual dados obtidos de cálculos com a Teoria do Funcional de Densidade (DFT) foram utilizados. Calores de formação com erro absoluto de até 4,0 kcal mol-1 foram reproduzidas com emprego desta técnica. Em cada etapa da MD, esse campo de força atualiza as ordens de títulos e fornece o caminho para as ligações se formarem e quebrarem durante o curso da simulação. O ReaxFF pode reproduzir com precisão todos os dados relevantes de mecânica quântica, bem como fornecer descrições atomísticas de várias reações químicas complexas.14 A simulação da reação entre a resina epoxídica e a diamina foi realizada em uma célula unitária de dimensões 20 x 20 x 20 Å, contendo 15 moléculas de C2H4O (óxido de etileno ou epóxi-etano) e 35 moléculas de CH6N2 (metanodiamina). O sistema foi minimizado utilizando dinâmica molecular de baixa temperatura (5 K). Após a minimização, para a fase de produção foi usado o ensemble do tipo canônico (NVT), no qual o número de moléculas (N), volume da caixa (V) e temperatura (T) são mantidos constantes. A dinâmica molecular reativa envolve, geralmente, reações rápidas. O intervalo de tempo de 0,1 femtosegundos (fs) foi usado, enquanto o tempo total de simulação foi de 255 ps para a temperatura selecionada de 1500 K (temperatura elevada para acelerar as interações intermoleculares e reações químicas). As temperaturas foram controladas pelo termostato Berendsen, com temperatura de amortecimento constante de 100 fs. O sistema molecular é apresentado na Figura 3.
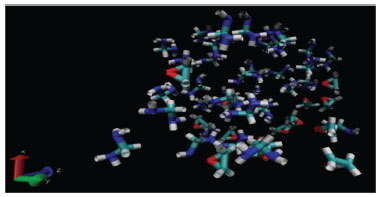 Figura 3. Célula unitária contendo 15 moléculas de óxido de etileno e 35 moléculas de metanodiamina. As terminações com cores azul claro, vermelhas, brancas e azul escuro representam respectivamente os átomos de carbono, oxigênio, hidrogênio e nitrogênio
As simulações de dinâmica molecular reativa também permitem o cálculo da energia de ativação e fator de frequência do processo reacional envolvido. Para tanto, foram selecionadas as temperaturas de 1500, 2000, 2300 e 2500 K (simulações isotérmicas) para avaliação do consumo de epóxi (abertura do anel epoxídico) e diamina (liberação de amônia). Com base no decaimento do número de reagentes ao longo do tempo, utilizou-se a hipótese de primeira ordem e a equação de Arrhenius foi aplicada para determinação dos parâmetros cinéticos.
RESULTADOS E DISCUSSÃO A simulação de dinâmica molecular reativa do sistema contendo óxido de etileno e metanodiamina, mantido à 1500 K, apresentou a quebra do anel epóxi em algumas das moléculas, mas principalmente, a liberação de amônia pela diamina. Nesse processo, ocorre uma abstração de hidrogênio de uma molécula de diamina pelo átomo de nitrogênio de outra, sendo, em geral, retirado o hidrogênio ligado ao átomo de carbono (ligação menos intensa). Dessa forma, o átomo de nitrogênio (agora ligado ao carbono e mais três hidrogênios) desprende-se para a formação de amônia: 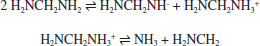 A abertura do anel do óxido de etileno pode ser realizada através de diferentes mecanismos, incluindo espécies de propagação aniônicas e catiônicas. Para a preparação de estruturas poliméricas bem definidas, o mecanismo aniônico é desejado, uma vez que o processo ocorre a partir de reagentes puros. Em contraste, a polimerização de abertura do anel catiônico para o óxido de etileno pode ser realizada através do método de monômero ativado catiônico ao invés da ativação no final da cadeia, de modo que as reações de transferência de cadeia levam à formação de oligômeros cíclicos.16 A ocorrência de reações de transferência de cadeira deve-se ao caráter nucleofílico relativamente alto dos átomos de oxigênio na cadeia polimérica, que podem reagir com a extremidade da cadeia catiônica ao invés do monômero livre em solução.16 A Figura 4 apresenta o sistema molecular após 200 ps do início da simulação. Nota-se claramente o rompimento da ligação C-O no anel epóxi e o desprendimento de moléculas de amônia. Também foi possível observar o início da reação de polimerização do sistema, com a reação entre o epóxi e a metanodiamina: 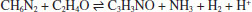
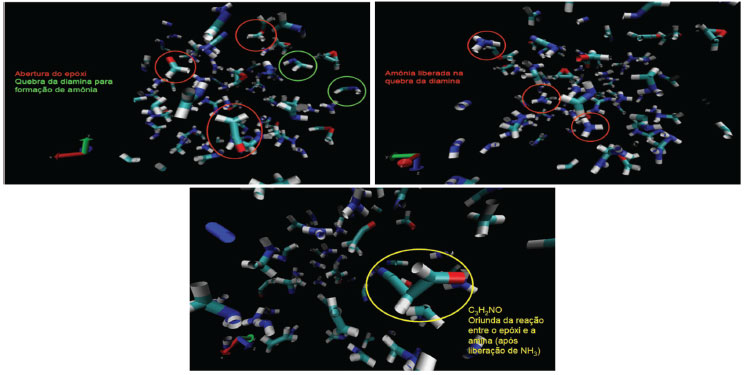 Figura 4. Sistema molecular após 200 ps do início da simulação (temperatura constante de 1500 K)
A reação apresentada acima foi construída com base na observação conforme a variação do sistema. Obviamente, um processo como esse não ocorre em uma única etapa, mas no sistema foi observada a formação e consumo de hidrogênio gasoso e molecular, o que subsidia a hipótese. A polimerização ocorre segundo um mecanismo reacional lento, o que acarreta a um tempo maior de simulação e, consequentemente, a modelagem completa do processo geraria um alto custo computacional. O tempo total das simulações apresentadas foi de, aproximadamente, 80 horas cada (2,5 milhões de iterações por simulação). A simulação da polimerização total do sistema levaria cerca de 50 vezes mais tempo, tendo em vista o estado do sistema após 80 horas de cálculo, mesmo com a temperatura elevada aplicada. A simulação à temperatura ambiente aumentaria o custo de simulação exponencialmente. Nas Figuras 5 e 6 estão representadas as variações dos reagentes e principais produtos (no caso, intermediários) durante a simulação, respectivamente.
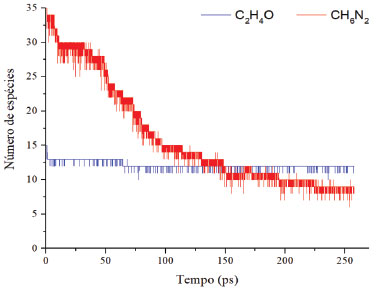 Figura 5. Variação do número de espécies dos reagentes em função do tempo
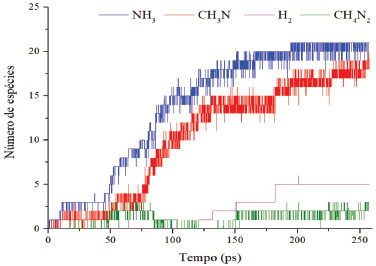 Figura 6. Variação do número de espécies dos produtos em função do tempo
Durante a simulação, o número de moléculas de diamina decresce rapidamente em relação ao epóxi. Essa redução deve-se às reações para liberação de amônia, enquanto o epóxi sofre uma pequena redução, embora continue decaindo na presença das espécies do sistema. Apenas a nível de comparação, a velocidade média de decaimento da diamina é de, aproximadamente, 8,3x10-16 mol s-1, ou 0,1 molécula ps-1, sendo assim uma velocidade bastante baixa, conforme esperado para uma polimerização. No entanto, a adição de catalisadores metálicos tende a acelerar substancialmente o processo.17 A quebra das moléculas de diamina é seguida pela formação de amônia e de metanoamina, conforme esperado e supracitado. Uma molécula que aparece diversas vezes no sistema é o CH4N2, resultado da abstração de dois hidrogênios ligados ao átomo de carbono da molécula, gerando NH3. Por CH4N2 estar no equilíbrio, ocorre sua formação e consumo; o processo de abstração de hidrogênio é reversível. Durante o processo, também é gerado hidrogênio molecular, possivelmente devido às reações entre o epóxi e a amina, indicando a fase de propagação da reação em cadeia de polimerização. O processo de cura do sistema epóxi com um extensor de cadeia amina pode abrir o anel epoxídico. Quanto às adições das aminas na reação, a amina primária leva ao aumento da cadeia polimérica e a amina secundária na formação de cadeias ramificadas, eterificação do anel epóxi com grupo hidroxila pendente e a homopolimerização do grupo epóxi não reagido a temperatura mais alta na ausência de funcionalidade ativa de N-H. Em tese, podem ocorrer as reações acima mencionadas, simultaneamente ou em diferentes estágios do processo de cura, dependendo da reatividade relativa dos componentes e da temperatura do processo.11 Como o sistema é mantido sob altas temperaturas, as colisões são mais eficazes e causam rupturas de ligações externas, sendo produzidos fragmentos de C2H4, CH6N2. Na formação dos produtos durante a simulação, a amônia é produzida em maior quantidade quando comparada aos demais componentes (Figura 6), o que leva a reação ao meio básico. Sendo assim, possivelmente o ataque nucleofílico na abertura do anel do óxido etileno ocorre via propagação catiônica. As simulações isotérmicas para cálculo dos parâmetros de Arrhenius foram realizadas (tempo total de 50 ps). Com base nos resultados, foi possível determinar os parâmetros cinéticos para a reação de abertura do anel epoxídico e para a produção de amônia da diamina. Os gráficos resultantes da linearização dos dados são apresentados nas Figuras 7 e 8 a seguir. A energia de ativação da abertura do anel epóxi é dada a partir do coeficiente angular da reta multiplicado pela constante universal dos gases. Quanto ao extensor de cadeia, o valor da energia de ativação consiste na liberação de amônia pela diamina. O coeficiente de correlação obtido foi acima de 0,95 para os dois casos, validando, portanto, a hipótese de processo de primeira ordem. A Tabela 1 apresenta os parâmetros de Arrhenius para as duas espécies.

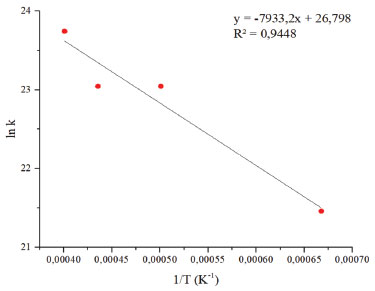 Figura 7. Curvas de Arrhenius para consumo das moléculas de epóxi
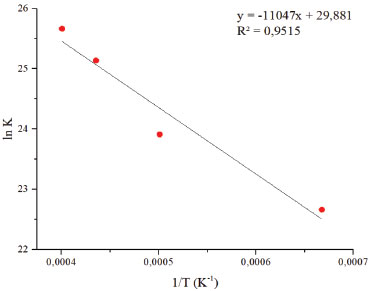 Figura 8. Curvas de Arrhenius para consumo das moléculas de diamina
As curas epóxi podem ocorrer em várias etapas e, consequentemente, com diferentes energias de ativação. Infelizmente, o uso desses modelos não parece ser muito eficaz na detecção adequada da complexidade das reações de cura. Nesse caso, pode-se empregar métodos isoconversionais sem assumir uma forma particular do modelo de reação e avaliar uma energia de ativação efetiva como uma função da extensão da reação. Se mudanças no mecanismo de cura estiverem associadas a mudanças na energia de ativação, elas podem ser detectadas usando métodos isoconversionais.7 Sendo assim, os valores teóricos da simulação (LAMMPS) estão de acordo com a literatura.2,8,12 Wu et al. estudaram experimentalmente os sistemas de epóxi-amina por análise térmica. Os valores da energia de ativação encontrados no seu estudo para processo de cura foram de 69,7 e 88,7 kJ mol-1, respectivamente. O valor de Ea 69,7 kJ mol-1 do primeiro estágio encontra-se na faixa característica de polimerizações de epóxi-amina, provavelmente indicando que o mecanismo principal consiste na abertura do anel da reação. O segundo estágio do processo de polimerização, com valor de 88,7 kJ mol-1, correspondente à cura do epóxi com o grupo amina.11 Nota-se que a energia de ativação envolvida no consumo de diamina para formação de amônia é maior que para a abertura do anel epoxídico. Tal comportamento é esperado, pois o anel epoxídico concentra muita tensão e a ruptura do anel favorece termodinamicamente a liberação de uma espécie com menor energia em seu estado fundamental. O procedimento de cura das resinas epóxi influencia na estrutura final da rede e, portanto, nas propriedades do material. Assim, uma representação precisa do processo de reticulação é crucial para entender o mecanismo subjacente e derivar as propriedades do material com simulações MD.5 Os resultados do estudo da abertura do anel epóxi mostram que a polimerização ocorre lentamente, o que acarreta alto custo computacional de simulação.
CONCLUSÃO A simulação de dinâmica molecular reativa (LAMMPS) permitiu observar o mecanismo de polimerização das resinas epóxi-amina, baseado no sistema de óxido de etileno e metanodiamina, no qual foi verificado a quebra do anel epóxi em algumas das moléculas e a liberação de amônia pela diamina. A partir do decaimento do número de reagentes ao longo do tempo, foi possível a análise de parâmetros cinéticos, como a energia de ativação para a abertura do anel epoxídico e a liberação de amônia pela diamina, com valores de 66 e 92 kJ mol-1, respectivamente. O valor encontrado na abertura do anel epoxídico mostra que essa é a etapa lenta do processo de polimerização. Entretanto, é desejável o estudo de simulações desta polimerização deste processo na presença de catalisadores metálicos para investigar o aumento da velocidade de reação. Foi observado, também, que o ataque nucleofílico na abertura do anel do óxido etileno ocorre em meio catiônico. Dessa forma, os resultados da simulação são promissores no entendimento de mecanismo das resinas epóxi, e indicam que a cura interfere diretamente na estrutura final do material.
AGRADECIMENTOS ANEEL - Agência Nacional de Energia Elétrica e EDF Norte-Fluminense.
REFERÊNCIAS 1. Fink, J. K.; Reactive Polymers: Fundamentals and Applications, William Andrey Publishing: Norwich, 2005, p. 139. 2. Wan, J.; Li, C.; Bu, Z. Y.; Xu, C. J.; Li, B. G.; Fan, H.; Chem. Eng. J. 2012, 188, 160. 3. May, C.; Epoxy Resins: Chemistry and Technology, 2nd ed., Marcel Dekker: New York, 1988. 4. Ehlers, J. E.; Rondan, N. G.; Huynh, L. K.; Pham, H.; Marks, M.; Truong, T. N.; Macromolecules 2007, 40, 4370. 5. Unger, R.; Braun, U.; Fankhänel, J.; Daum, B.; Arash, B.; Rolfes, R.; Comput. Mater. Sci. 2019, 161, 223. 6. Acocella, M. R.; Corcione, C. E.; Giuri, A.; Maggio, M.; Maffezzoli, A.; Guerra, G.; RSC Adv. 2016, 6, 23858. 7. Sbirrazzuoli, N.; Vyazovkin, S.; Mititelu, A.; Sladic, C.; Vincent, L.; Macromol. Chem. Phys. 2003, 204, 1815. 8. Pham, M. P.; Ph.D. Thesis, University of Utah, United States of America, 2011. 9. Okumoto, S.; Yamabe, S.; J. Comput. Chem. 2003, 24, 244. 10. Wan, J.; Li, C.; Bu, Z. Y.; Xu, C. J.; Li, B. G.; Fan, H.; Chem. Eng. J. 2012, 188, 160. 11. Wu, F.; Zhou, X.; Yu, X.; RSC Adv. 2018, 8, 8248. 12. Fu, K.; Xie, Q.; Lü, F.; Duan, Q.; Wang, X.; Zhu, Q.; Huang, Z.; Polymers (Basel) 2019, 11, 1. 13. LAMMPS Molecular Dynamics Simulator, disponível em https://lammps.sandia.gov/, acessado em March 2020. 14. Gonçalves, R. F. B.; Iha, K.; Rocco, J. A. F. F.; Quim. Nova 2018, 41, 507. 15. van Duin, A. C. T.; Dasgupta, S.; Lorant, F.; Goddard, W. A.; J. Phys. Chem. A 2001, 105, 9396. 16. Handbook of ring-opening polymerization; Dubois, P., Coulembier, O., Raquez, J. M., eds.; Wiley-VCH: New York, 2009. 17. Vara, J. A.; Dave, P. N.; Ram, V. R.; Nano-Struct. Nano-Objects 2019, 20, 100372. |
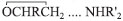 . Sob condições ácidas, a adição da maioria dos nucleófilos é acelerada consideravelmente por ácidos ou espécies eletrofílicas devido à formação reversível do ácido conjugado mais reativo do epóxido, como um ácido de Lewis como BF3:
. Sob condições ácidas, a adição da maioria dos nucleófilos é acelerada consideravelmente por ácidos ou espécies eletrofílicas devido à formação reversível do ácido conjugado mais reativo do epóxido, como um ácido de Lewis como BF3: 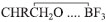 .3
.3On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access