
 |
 |
 |
 |
 |
Revisão
|
|
| Imobilização enzimática: conceito e efeitos na proteólise Enzyme immobilization: concepts and effects on proteolysis |
|
Izadora L. FurlaniI; Bruno S. AmaralII; Regina V. OliveiraI; Quezia B. CassI,*
I. Departamento de Química, Universidade Federal de São Carlos, 13565-905 São Carlos - SP, Brasil Recebido em 25/11/2019 *e-mail: qcass@ufscar.br In recent years, great advances have been observed in the proteomic area regarding the different approaches adopted as top-down, middle-down and bottom-up. Regarding middle-down and bottom-up approaches, a previous step for protein digestion is necessary for further analysis of the generated peptides. Classical digestion protocols employing proteases are usually carried out in solution, which can lead to a number of disadvantages such as high digestion time, enzymatic autolysis and impossibility to reuse the enzymes. In view of these drawbacks, different approaches have been studied and applied for improving enzymatic efficiency, inhibition of autolysis, reduction of digestion time, and to minimize the number of missed cleavages. In the literature, great efforts have been made in order to achieve maximum efficiency in enzymatic digestion, with emphasis on proteases immobilization. In this review, concepts related to protein digestion, immobilization techniques, as well as the main support used for enzyme immobilization are discussed considering new tools for proteomic analysis. INTRODUÇÃO A elevada importância das proteínas nas vias metabólicas e o papel que essas desempenham nas funções fisiológicas das células, conduziu à necessidade de procedimentos analíticos específicos para a caracterização estrutural, análise funcional e conhecimento das modificações pós-traducionais das proteínas.1-3 A abordagem proteômica pode variar de estudos abrangentes, os quais auxiliam na identificação e caracterização de uma ampla variedade de proteínas presentes em amostras complexas, até as abordagens direcionadas, empregadas em estudos de proteínas específicas.1,2,4-6 A proteômica surgiu nos anos 70 com a aplicação e aperfeiçoamentos da técnica de eletroforese bidimensional em gel (2D - PAGE), a partir da qual foi possível realizar separações de proteínas de amostras complexas.7-9 No entanto, a análise detalhada com a identificação dessas proteínas só foi possível alguns anos depois com a introdução da espectrometria de massas.7,8,10,11 Durante anos a abordagem SDS-PAGE foi amplamente utilizada em proteômica, entretanto, essa técnica apresentava algumas limitações como a baixa reprodutibilidade, principalmente quando se tratava de proteínas hidrofóbicas, básicas e com massas moleculares muito elevadas ou muito pequenas. Além disso, a quantificação de proteínas era realizada por análise de "spots" proteicos (ou bandas) detectados por vários métodos de coloração (azul de Coomassie, nitrato de prata ou compostos fluorescentes), que representam as diferentes proteínas separadas (ou formas proteicas) e com diferentes intensidades de coloração de acordo com a abundância relativa. A necessidade de manipulação, com o corte das bandas do gel, também pode interferir na análise.7,9,11 Com isso, estratégias alternativas para as análises proteômicas foram desenvolvidas. Atualmente com os avanços nas técnicas de espectrometria de massas juntamente com as ferramentas de bioinformática, abordagens como top-down, middle-down e bottom-up podem ser facilmente empregadas em análises proteômicas de acordo com os objetivos do estudo (Figura 1).1,2,5,6
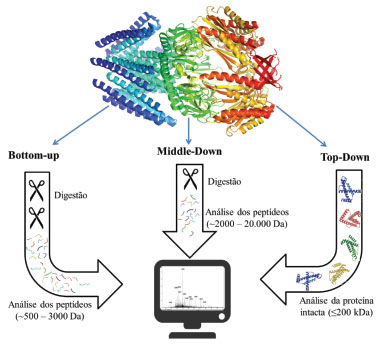 Figura 1. Abordagens proteômicas: Bottom-up, Middle-Down e Top-Down
Na abordagem top-down, as proteínas são analisadas intactas (proteínas com massa molar de até 200 kDa) seguidas da fragmentação utilizando dissociação induzida por colisão (CID), dissociação por captura de elétrons (ECD), dissociação por transferência de elétrons (ETD) ou MALDI (do inglês, matrix assisted laser desorption/ionization).5,6,12 Esse último é capaz de preservar as modificações pós-traducionais das proteínas em análise. O fato de as proteínas serem analisadas intactas é vantajoso por preservar todas as informações da biomolécula, incluindo conhecimento completo da sequência de aminoácidos e completa caracterização de suas proteoformas (diferentes formas moleculares nas quais o produto proteico de um único gene pode ser encontrado).13 No entanto, devido à complexidade das proteínas, a detecção requer espectrômetros de massas de alta resolução e exatidão de massas para separar e atribuir picos espectrais provenientes de um espectro complexo contendo múltiplas proteoformas ou espectros de fragmentos contendo inúmeros íons fragmentos.1,5,6,10 Na estratégia bottom-up, o estudo proteômico é realizado através da análise de peptídeos (de 500 Da a 3 kDa) obtidos da digestão proteica. Este procedimento proporciona elevada sensibilidade e reprodutibilidade mesmo para misturas complexas de proteínas, sendo a abordagem mais utilizada em análises proteômicas.2,4-6,14,15 A identificação da proteína original ocorre através do peptide mass fingerprint (PMF), em que a relação m/z e, consequentemente, a massa molecular dos peptídeos é obtida e, adicionalmente, obtém-se os espectros de fragmentação (MS/MS). Juntas essas informações são usadas para correlacionar os dados experimentais com proteínas em bancos de dados, por exemplo, Mascot® e Sequest®. Já em middle-down o tamanho das proteínas é reduzido através da sua digestão parcial (de 3 a 20 kDa). Esta abordagem combina o melhor da top-down com a bottom-up e é utilizada principalmente na identificação de modificações pós-traducionais em peptídeos obtidos que são mais longos do que aqueles do bottom-up e que se aproximam em tamanho de pequenas proteínas intactas, como os experimentos em top-down. Essa estratégia é muito empregada na caracterização de anticorpos monoclonais bioterapêuticos.2,6 A digestão de proteínas é um procedimento importante no preparo das amostras nas abordagens middle-down e bottom-up e apresenta grande influência na elucidação de proteínas, uma vez que a análise de peptídeos costuma apresentar maior sensibilidade frente à análise de proteínas intactas. Com isso, a digestão de proteínas tem sido melhorada com o desenvolvimento de novas técnicas que permitam promover um aumento do rendimento, da reprodutibilidade e da otimização do tempo de digestão.4,6,16,17 Em estudos proteômicos buscam-se por digestões que promovam elevado índice de clivagem proteolítica e elevada quantidade de peptídeos, ou seja, que ocorra de maneira específica e em acordo com as características da enzima ou do reagente utilizado para a digestão da proteína de interesse.18 Existem duas principais técnicas para digestão de proteínas, sendo elas a digestão não-enzimática, que faz uso de reagentes químicos e são principalmente realizados em solução ou em gel e a digestão enzimática, que envolve enzimas proteolíticas.6,19,20 Na digestão não-enzimática, também conhecida como clivagem química, são utilizados principalmente soluções ácidas diluídas como: ácido fórmico, ácido clorídrico e ácido acético, os quais atuam especificamente em um único local de clivagem, o aminoácido aspartato.19-22 Além dos ácidos, pode-se também utilizar outros reagentes químicos tais como: brometo de cianogênio (CNBr) específico para o aminoácido metionina, 2-nitro-5-tiocianobenzoato (NTCB) específico para o aminoácido cisteína e hidroxilamina específico para os aminoácidos aspargina e glicina.23-25 Esse tipo de digestão pode levar a obtenção de peptídeos de alta massa molecular, adequados para a abordagem proteômica do tipo middle-down.6 A digestão enzimática é a mais utilizada em análises proteômicas e envolve a utilização de enzimas proteolíticas como tripsina, quimiotripsina, carboxipeptidase ou alcalase, sendo tripsina a mais amplamente empregada em análises proteômicas com abordagem bottom-up. Isso ocorre pois trata-se de uma enzima de baixo custo e facilmente encontrada em grandes quantidades, além de possuir elevada especificidade em catalisar a hidrólise em regiões C-terminais dos resíduos de lisina e arginina, exceto quando estes estão próximos a resíduos de prolina, a qual possui um anel aromático, ocasionando elevado impedimento estérico e dificultando o alcance da enzima ao aminoácido específico de clivagem.26-28 Como a principal desvantagem é a auto-clivagem, atualmente algumas empresas comercializam esta enzima com modificações pós-traducionais que levam a uma elevada eficiência digestiva e a uma resistência na autólise.6,29-31 Outra alternativa para se alcançar uma digestão proteica eficiente, com uma grande cobertura sequencial da proteína digerida, é a mistura de enzimas proteolíticas. A estratégia mais adotada para isto está na combinação de uma exopeptidase, com outras enzimas que desempenham o papel de endopeptidase.32 As endopeptidases possuem seletividade para um único resíduo de aminoácido (C-lisina, N-lisina, C-glutamina, C-arginina ou N-aspartato) e atuam preferencialmente nas regiões internas da cadeia polipeptídica, enquanto as exopeptidases atuam nas regiões finais da cadeia polipeptídica. Esta combinação pode auxiliar na digestão de amostras complexas como, por exemplo, plasma e no sequenciamento proteômico completo.32-35 A digestão clássica de proteínas é realizada com a enzima em solução. No entanto, a mistura pós-reação pode conter fragmentos de proteínas provenientes da autólise da protease empregada. Quando a enzima proteolítica se encontra em elevadas concentrações, os fragmentos proteicos da autodigestão enzimática dificultam a interpretação do espectro de massas devido à menor intensidade dos sinais dos peptídeos da proteína digerida em comparação com os sinais de autólise registrados.4,36 Por outro lado, ao utilizar uma baixa concentração de enzima tem-se uma redução na razão enzima/substrato, ocasionando tempos de digestão muito longos (12 - 24 horas).3,37-40 Com o objetivo de acelerar a proteólise, algumas estratégias têm sido adotadas como: a aplicação de micro-ondas,41,42 ultrassom,43 infravermelho,44,45 aumento na pressão do sistema,46-48 aumento na temperatura49,50 e emprego de solventes orgânicos.46 No entanto, a imobilização da enzima proteolítica em um suporte (IMERs - Immobilized Enzyme Reactors) para realizar a digestão de proteínas é uma estratégia que têm sido amplamente utilizada. A enzima imobilizada em um suporte sólido pode atribuir diversas vantagens em relação à digestão clássica realizada em solução, como por exemplo, o aumento da estabilidade térmica, resistência a desnaturação proteica por solventes químicos e uma elevada razão enzima/substrato devido à maior área superficial, o que reduz o tempo requerido de digestão.1,3,6,32,51 Além disso, a enzima imobilizada pode ser removida do meio reacional antes da análise por MS e pode ser facilmente recuperada para reuso. Com a maior estabilidade química obtida do processo de imobilização há uma redução/eliminação do processo de auto-clivagem, permitindo espectros de massas com um número menor de interferentes.6,14 A reprodutibilidade da digestão realizada com a enzima imobilizada também é aumentada quando comparada com ensaios em solução, o que propicia uma identificação acurada de proteínas.3,40 Diferentes técnicas de imobilização podem ser empregadas, no entanto, é importante destacar que com a imobilização a enzima sofre mudanças nas propriedades químicas, bioquímicas, mecânicas e cinéticas.52,53 A atividade enzimática será afetada principalmente pelo método de imobilização selecionado, quantidade e pureza da enzima utilizada.53 A funcionalização do suporte é um dos fatores importantes a ser considerado para a imobilização da enzima. O modo de ligação entre os grupos reativos do suporte e os resíduos de aminoácidos que não fazem parte do sítio ativo são de extrema importância para a preservação da estrutura terciária da enzima, a fim de maximizar a estabilidade e a reatividade frente à sua imobilização.40,52,54 Assim, esta revisão apresenta um levantamento bibliográfico dos últimos dez anos sobre os principais avanços e inovações dos suportes utilizados para imobilização de tripsina, bem como o papel desses biorreatores em proteoma, empregados no modo de digestão on-line e off-line.
DIGESTÃO PROTEICA ON-LINE Para aumentar a produtividade em análises proteômicas uma abordagem promissora é a digestão proteica on-line.4,48,55,56 Essa estratégia é baseada na criação de um reator enzimático de vazão contínua e minimiza o manuseio com as amostras, eliminando fontes de erros.4,48,55-58 No entanto, vale destacar que a abordagem on-line não dispensa o tratamento da amostra com reagentes químicos (redução, alquilação, etc.) ou solventes orgânicos previamente à digestão. Embora a ideia de biorreatores enzimáticos não seja nova, a expectativa de facilidades operacionais, a busca por aprimoramento no processo de imobilização e aumento na produtividade e reprodutibilidade dos resultados é constante.4,40,52,58-60 Na abordagem on-line, os IMERs podem ser utilizados como pré ou pós-coluna analítica em um sistema de cromatografia líquida. Contudo, a grande maioria dos trabalhos publicados os utilizam como pré-coluna.3,4,40,61 Nos cromatógrafos, os IMERs são usados como pré-colunas através do uso de válvulas de desvio. Para eluição, solventes orgânicos, tampões em diferentes forças iônicas e pH são utilizados objetivando a transferência dos peptídeos sem a desnaturação do IMER. Também se utilizam colunas de captura - entre o IMER e a coluna analítica - para extrair e concentrar os produtos da digestão antes da transferência para a coluna analítica.3,40,58,61 Como pós-coluna, os analitos são inicialmente separados em uma coluna analítica e apenas as bandas cromatográficas contendo as proteínas de interesse são seletivamente transferidas para os IMERs através de válvulas de acoplamento. As vantagens dessa configuração residem na possibilidade de diminuir a presença de interferências e a maior capacidade reprodutiva dos ensaios, mesmo em casos de conversão incompleta ou instabilidade do produto da reação. As desvantagens se devem ao fato de possível alargamento das bandas cromatográficas; efeitos de diluição da amostra e a necessidade de ajustes e modificações nas condições cromatográficas na coluna analítica para que se tornem compatíveis com a etapa de reação enzimática.40,58,61
DIGESTÃO PROTEICA OFF-LINE O modo de digestão off-line pode ser subdividido em dois tipos, sendo eles: 1) a digestão realizada em solução com a enzima livre e 2) a digestão realizada com a enzima imobilizada em um suporte, seguido da análise dos peptídeos por espectrometria de massas.3,39,40,62 Nessa revisão não será abordada a digestão com a enzima livre em solução. Para o procedimento com enzima imobilizada vários suportes têm sido descritos na literatura tais como, partículas magnéticas,63,64 microchips,16 sílica mesoporosa,65-67 nanofibras68,69 dentre outros.68-71 O emprego destes suportes apresenta vantagens devido à elevada área superficial, o que garante a imobilização de grande quantidade de enzima; fácil preparo e manuseio em relação à separação dos peptídeos obtidos; redução no tempo de digestão e aumento na estabilidade da enzima.16,63-68 Assim como na abordagem on-line, os métodos off-line também necessitam de um pré-tratamento da amostra antes do contato com a protease. Como a abordagem off-line exige maior manipulação das amostras, as possíveis contaminações são inerentes aos métodos, o que é uma desvantagem para este tipo de digestão.72,73
TRIPSINA: ESTRUTURA E FUNÇÃO O tripsinogênio é a forma inativa da tripsina, o qual é produzido e excretado pelo pâncreas e, ao chegar no intestino delgado é convertido em tripsina ativa.74-76 A tripsina consiste em uma única cadeia polipeptídica de 223 aminoácidos e o sítio ativo inclui His46 e Ser183. Para o caso da trispina bovina a massa molecular é de 23,3 kDa e possui ponto isoelétrico (pI) de 10,1 - 10,5 para a forma bovina. Para uma maior eficiência catalítica, o pH de atividade varia de 7,0 a 9,0 e a temperatura ótima de 10 a 65 °C, devendo ser levado em consideração a fonte da enzima obtida.74,77,78 Além de apresentar elevada especificidade em catalisar a hidrólise em regiões C-terminais dos resíduos de lisina e arginina, quando estes não estão próximos à resíduos de prolina, a tripsina também hidrolisa as ligações de éster e amida de alguns substratos sintéticos, sendo os mais conhecidos o cloridrato de éster etílico de Nα-benzoil-L-arginina (BAEE) e o cloridrato de Nα-benzoil-L-arginina 4-nitroanilida (BAPNA).24,76,77 É uma enzima comercial e seus principais fornecedores são Biocompare,79 Creative Enzymes,80 Scripps Laboratories,81 Sigma-Aldrich,82 Promega,83 dentre outros.84,85 No que diz respeito à digestão tríptica de proteínas, a diferença na conformação entre a enzima e o substrato pode produzir diferentes produtos de digestão, mesmo com a aplicação de um protocolo idêntico.18 De modo geral, a tripsina seleciona sítios para hidrólise com base em duas principais características relacionadas com a estrutura do substrato: i) presença de aminoácidos básicos na estrutura primária e ii) estruturas bem enoveladas a qual orienta os estágios iniciais da proteólise e produz influência no número de clivagens perdidas.18,86-88 Os principais sítios de clivagem ocorrem em regiões pouco rígidas e preferencialmente na superfície da proteína. Aminoácidos básicos fortemente associados à estrutura do substrato, ou pertencentes à estrutura secundária não são bons candidatos a sofrerem digestão pela tripsina. Há ainda uma dificuldade na proteólise devido ao "mascaramento" de aminoácidos básicos, o que ocorre principalmente em proteínas que possuem maior número de ligações de dissulfeto, arranjos específicos em sua estrutura secundária e, consequentemente, forte enovelamento. Esses fatores resultam na dificuldade de acesso da tripsina ao sítio de clivagem dos aminoácidos básicos e dessa forma a ocorrência de maior número de clivagens perdidas fica iminente.18,87,88 Quando os aminoácidos básicos entram em contato com o sítio ativo da tripsina, grupos amina em sua cadeia lateral, juntamente com os resíduos ao lado da ligação peptídica, fornecem informações necessárias para o acoplamento. Além disso, resíduos de aminoácidos mais distantes também podem modular a ligação, porém mais fracamente. O mecanismo de clivagem da tripsina é semelhante as demais serino proteases e envolve uma tríade catalítica composta por His-57, Asp-102 e Ser-195.18,89-91 O mecanismo de clivagem descreve que a His-57 abstrai um próton da Ser-195 para formar um nucleófilo alcóxido e, ao mesmo tempo, a interação eletrostática de Asp-102 adjacente a His-57 auxilia na aceitação do próton. A combinação dessa tríade faz com que haja um aumento na nucleofilicidade da Ser-195, facilitando seu ataque a ligação peptídica do C-terminal de um resíduo de lisina ou arginina. A Ser-195 é auxiliada pela His-57 no ataque ao grupo carbonila da proteína, produzindo um intermediário tetraédrico. Esse intermediário se rompe e tem-se a formação de um intermediário acil-enzima e o produto N-terminal. A dissociação deste produto juntamente com a introdução de uma molécula de água proporciona o ataque ao grupo carboxila do substrato, gerando o produto C-terminal e regenerando a enzima, o que torna possível catálises subsequentes.18,89,91-93 A Figura 2 exemplifica o mecanismo de clivagem que ocorre com as serinas proteases.
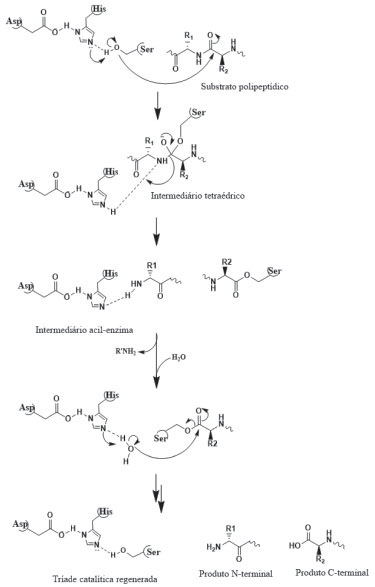 Figura 2. Mecanismo de clivagem das serinas proteases93
A presença do aminoácido aspartato, localizado no sítio ativo da enzima, é responsável por atrair e estabilizar a lisina ou arginina carregada positivamente, sendo responsável pela especificidade da enzima. Isso atribui à tripsina a característica de clivar predominantemente proteínas na extremidade C-terminal dos aminoácidos lisina ou arginina, exceto quando esses aminoácidos estão próximos à resíduos de prolina ou demais grupos volumosos. Além disso, a introdução de cargas negativas à proteína ocasionaria repulsão eletrostática pelo Asp-189, dificultando o acesso de aminoácidos básicos ao sítio de clivagem.18,89,91 Resumidamente, uma pequena mudança na estrutura primária da proteína pode acarretar mudanças em sua conformação e, portanto, diferença na digestão tríptica. Desta forma, é viável considerar que há certa diferença nos peptídeos obtidos através da digestão com a enzima livre e com a enzima imobilizada, principalmente devido às diferenças nas condições de reação e nas conformações estruturais da enzima e do substrato.4,56,94 Mesmo que com a utilização de IMERs a digestão possa ser alcançada em pouco tempo devido à alta concentração de tripsina, as clivagens perdidas ainda são inevitáveis.18
TÉCNICAS EMPREGADAS NA IMOBILIZAÇÃO A imobilização de biomoléculas envolve a ligação de um alvo de origem biológica (enzima, anticorpo, DNA etc.) na superfície de uma matriz insolúvel (suporte).39,53,95 Os métodos de imobilização de proteínas exploram as propriedades físico-químicas das cadeias laterais dos aminoácidos que a compõe, como a acidez, basicidade e polaridade. Assim, o método de imobilização baseia-se nos diferentes tipos de ligações e/ou interações que podem ser feitas com o suporte.39,52,95 A escolha do método de imobilização deve levar em consideração desde parâmetros da atividade global dos IMERs, as características de regeneração e desativação, o custo do processo e a toxicidade dos reagentes, até as propriedades finais desejadas para a enzima imobilizada.96 De modo geral, a imobilização de proteínas pode ser subdividida em métodos químicos e físicos. Os métodos químicos envolvem a formação de ligações covalentes obtidas pela ligação de éter, tio-éter, amida ou carbamato entre a enzima e o suporte ou ainda por meio de ligações cruzadas, a qual as proteínas são imobilizadas a partir da reação direta com um agente reticulante. Os métodos físicos são caracterizados por ligações mais fracas, como ligações de hidrogênio, ligação por afinidade, forças de Van der Waals, interações hidrofóbicas e ligação iônica entre a enzima e o suporte. As proteínas podem também ser encapsuladas/aprisionadas no suporte. A Figura 3 exemplifica os tipos de imobilização utilizados.52,61
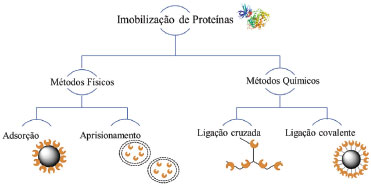 Figura 3. Métodos de imobilização de biomoléculas
É importante ressaltar a dificuldade em encontrar trabalhos comparando diferentes métodos de imobilização, uma vez que a maioria dos autores compara o desempenho da enzima imobilizada em relação à enzima livre. Adsorção física A imobilização de enzimas por meio do método de adsorção física foi uma das primeiras técnicas reportadas.97 Trata-se de um método simples e reversível de imobilização, podendo ocorrer por meio de interações hidrofóbicas, ligações de hidrogênio e forças de Van der Waals. As principais vantagens da imobilização por adsorção estão na facilidade e simplicidade da técnica que permite a imobilização sob condições brandas, a possibilidade de alta retenção da atividade enzimática visto que não há grandes modificações estruturais na biomolécula e, a reversibilidade que permite a reutilização dos suportes.98 No entanto, como desvantagem tem-se a aleatoriedade da interação enzima-suporte e a possibilidade de dessorção por meio de variações de temperatura, pH e força iônica.98 Como alternativa para contornar essas desvantagens, diversos métodos foram desenvolvidos nos últimos anos, como por exemplo a modificação química do suporte e a utilização de agentes reticulantes.98,99 Sun e colaboradores100 imobilizaram a tripsina em nanopartículas de sílica hidrofóbicas revestidas com celulose através do método de adsorção física. Para isso, nanopartículas de sílica foram revestidas com tris-O-fenilcarbamoil celulose. Para imobilização, uma solução de tripsina em pH 6,1 e uma razão de 13% em relação ao suporte foi usada. Em comparação com a enzima livre, a enzima imobilizada apresentou maior resistência a processos térmicos e condições desnaturantes, além de múltiplas reutilizações. A aplicabilidade do biorreator desenvolvido foi demonstrada mediante a digestão de albumina sérica bovina (BSA) e citocromo c, com coberturas sequenciais de 88,3 e 62,5% respectivamente, para o biorreator e de 84,7 e 66,3% para a enzima livre. O tempo de digestão foi de 20 horas para ambos os ensaios. Os autores relatam que a eficiência de digestão do biorreator é comparável com a enzima livre, uma vez que apresentaram coberturas sequenciais muito similares podendo facilmente ser aplicado como estratégia na reutilização e aumento na estabilidade enzimática para estudos proteômicos. Outro modo de contornar as possíveis dessorções proteica é a estratégia de adsorção por troca iônica, a qual é baseada nos princípios de interações iônicas entre a enzima e o suporte, onde as atrações eletrostáticas são provenientes de cargas opostas presentes na superfície do suporte com a enzima.52,61 As resinas de troca iônica adsorvem facilmente as enzimas devido as suas propriedades polares. Tanto as resinas de troca catiônica, tal como a carboximetilcelulose, quanto as resinas de troca aniônica, como N-N-dietilaminoetilcelulose (DEAE celulose), são utilizadas industrialmente. Embora as forças de ligação sejam mais fortes do que as forças envolvidas na adsorção física simples, a imobilização por troca iônica é suscetível à presença de outros íons. Como consequência, a consideração da força iônica, tampão de imobilização e o pH são fatores importantes a serem considerados para a imobilização por troca iônica e para a prevenção da dessorção da enzima.51,52,98,101-103 Aprisionamento Aprisionamento é definido como um método físico e irreversível de imobilização no qual a enzima fica confinada no suporte. A imobilização por aprisionamento requer a síntese da rede polimérica na presença da enzima. À medida que a polimerização se processa, a matriz polimérica se forma em torno da enzima, confinando-a em sua estrutura.52,61,99 Este método permite realizar modificações com o material de encapsulação, de modo que proporcione ao suporte condições ótimas de imobilização como pH, polaridade e afinidade. Os polímeros mais utilizados como matriz são o alginato, colágeno, poliacrilamida, gelatina, borracha de silicone, poliuretano e álcool polivinílico com grupo estirilpirídio.52 A vantagem da utilização desse método é que o aprisionamento/encapsulamento protege a enzima do contato direto com o meio reacional e, desta forma, minimiza a inativação da enzima por solventes orgânicos. É um dos métodos mais simples de imobilização, além disso, pode ser utilizado para imobilizar um ou mais tipos de proteínas.39,51,98 Liu e colaboradores104 sintetizaram nanopartículas de sílica mesoporosa modulada com 272 mg de ácido tânico para controle dos poros. A superfície externa foi modificada com 3-aminopropil-trimetóxisilano (APTMS). As nanopartículas obtidas foram caracterizadas por métodos físicos e apresentou: diâmetro de 201 ± 11 nm, volume dos poros de 0,63 cm3 g-1 e tamanho de 6,2 nm. A atividade enzimática foi avaliada frente a hidrólise do substrato BAPNA e monitorada via espectrofotômetro com comprimento de onda de 410 nm. A atividade ótima da enzima imobilizada foi alcançada com a utilização de 153 µg do biorreator, em pH 7,9 e temperatura de 40,4 °C. Em comparação com a enzima livre, a enzima encapsulada demonstrou maior estabilidade térmica e de armazenamento, podendo ser reutilizada diversas vezes. Nesse estudo, o objetivo dos autores foi realizar a imobilização da tripsina via método de encapsulação e avaliar sua atividade em comparação com a enzima livre. Avaliação de digestão de proteínas não foi relatada. As limitações causadas pela imobilização via aprisionamento referem-se especialmente a problemas de transferência de massa para o sítio ativo da enzima, as quais tornam-se ainda mais pronunciadas quando o interesse da imobilização não é biocatalítico e sim proteômico. Essa limitação é mais expressiva quanto maior for a massa molecular da proteína a ser clivada. Em contrapartida, suportes com poros maiores podem levar a uma ineficiência na imobilização, promovendo perda de enzima durante a utilização, baixa capacidade de carga e desgaste do suporte durante o uso. Dessa forma, o tamanho do poro do material usado para o aprisionamento é um fator importante a ser considerado para uma boa imobilização.52,99 Outra desvantagem é que muitos percursores utilizados na etapa de polimerização podem ocasionar inativação das enzimas.98 Ligação cruzada A técnica de ligação cruzada foi inicialmente descrita para promover cristais de proteínas a fim de se realizar estudos estruturais por raio-X. É um método de imobilização irreversível que não requer o uso de suporte.52,61 As enzimas são ligadas umas às outras, ou às proteínas inativas, formando uma estrutura tridimensional complexa.61 Tecnicamente, são formadas ligações cruzadas intermoleculares entre a enzima e reagentes bi- ou multifuncionais com finalidade de torná-las insolúvel ao meio reacional. Os agentes de reticulação são moléculas que possuem ao menos duas extremidades reativas que se ligam a grupos específicos de aminoácidos da enzima. Um dos reagentes mais utilizados para aplicação desta técnica é o glutaraldeído, por ser econômico e facilmente obtido em grandes volumes.52,53,99 É uma técnica ainda laboriosa e lenta e, além disso, pode ocorrer uma perda de 50% da atividade enzimática, baixa estabilidade mecânica e pouca reprodutibilidade, especialmente quando utilizada uma grande quantidade de enzima.61,99 Como alternativa aos métodos convencionais, foram desenvolvidos métodos por ligação cruzada empregando enzimas cristalinas (cross-linked enzyme crystals - CLECs) e agregados enzimáticos reticulados (cross-liked enzyme aggregates - CLEAs).51,61,98,99 No caso de CLECs, as enzimas sofrem a precipitação controlada para formação de microcristais, seguido da reticulação. Após a formação da ligação cruzada, os CLECs se tornam insolúveis em solvente orgânico e soluções tampão o que permite sua aplicação em diferentes meios reacionais sem que ocorra significativas perdas da atividade catalítica. Apesar desta abordagem apresentar uma elevada retenção da atividade catalítica, sua principal desvantagem está na necessidade da cristalização da enzima antes do processo de reticulação, requerendo que a enzima esteja na sua forma pura, além de ser um processo laborioso e complexo.51,98,99,105 Para o caso dos CLEAs, ocorre inicialmente a precipitação da enzima por meio de sais, ácidos, solventes orgânicos e polímeros não iônicos, seguido então da reticulação. Nesse caso, não há exigências quanto a pureza da enzima. Assim como no caso dos CLECs, essa técnica também apresenta como principal vantagem a elevada atividade catalítica. Além disso, tem sido realizada para a imobilização de diferentes tipos de enzimas, para catálise de múltiplas reações ou em processos em série.39,51,98,99 Em ambas as técnicas, ainda há a necessidade de estudos mais aprofundados com respeito a morfologia adquirida após o processo de reticulação, dada a possível formação de grandes aglomerados que resultam, muitas vezes, em dificuldades difusionais do substrato e do produto no ambiente catalítico.51,98,99 Como aplicação, Mageed e colaboradores106 desenvolveram um CLEA de tripsina (CLEA-Try) através da agregação com quitosana, preparado sob condições suaves utilizando 1% v/v de glutaraldeído como agente reticulante. Parâmetros cinéticos foram mensurados utilizando BAPNA como substrato, obtendo KM de 1,89 nmol L-1 para a enzima imobilizada. A imobilização da enzima aumentou a estabilidade térmica, sendo obtida atividade residual de 64% a 60 °C, enquanto que com a enzima livre a atividade residual foi de apenas 10%, o que evidencia o aumento na rigidez da estrutura enzimática através da reticulação, com isso modificações conformacionais e perda da atividade catalítica são evitadas com o aumento da temperatura. CLEA é um método simples e que dispensa a necessidade de suportes, podendo ser utilizada em diversas aplicações biotecnológicas. Ligação covalente É um dos métodos mais empregados para a imobilização de enzimas com aplicação em estudos proteômicos e baseia-se na formação de ligações covalentes entre os grupos funcionais presentes na superfície do suporte e os grupos funcionais dos resíduos de aminoácidos da enzima [(-NH2) da lisina e arginina; (-CO2H) do ácido aspártico e ácido glutâmico; (-OH) da serina e treonina e o grupo (-SH) da cisteína].51,52,61,98,105 A atividade da enzima imobilizada covalentemente irá depender do tamanho e forma do suporte, natureza do método de acoplamento, composição do material e condições específicas durante a imobilização. A maior atividade enzimática ocorre quando os aminoácidos do sítio ativo não estão envolvidos na ligação com o suporte.51,52,98,107 Na imobilização covalente, são usuais o emprego de reagentes espaçadores que têm como finalidade deixar a enzima em maior contato com o meio reacional e sua real necessidade irá depender do tamanho do grupo funcional adicionado ao suporte.39,98 Suportes funcionalizados como, por exemplo, 3-glicidoxipropiltrimetóxisilano (GPTMS) não requerem uso de espaçadores.73 A reatividade dos suportes epóxidos-derivados permitem a imobilização diretamente com os grupos amina, tióis e hidroxilas dos resíduos de aminoácidos das enzimas fazendo com que ocorra a abertura de anel, criando ligações secundárias de amina, tioéter ou éter, respectivamente. A Figura 4 apresenta uma reação típica de derivação do suporte para imobilização covalente de enzimas utilizando GPTMS.73
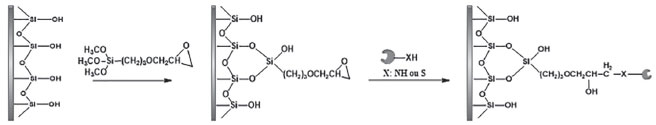 Figura 4. Reação de funcionalização do suporte com GPTMS, seguido da imobilização por ligação covalente da enzima73
Outro exemplo de imobilização de enzimas por ligação covalente está na funcionalização do suporte com 3-aminopropiltrietoxissilano (APTS), ou outro reagente funcionalizador, seguido então da reação com carbonato de N-N'-disuccinimidila (DSC) como agente ativador ou outros grupos de cadeias carbônicas longas entre o suporte e a enzima para aumentar a hidrofobicidade do biorreator. A Figura 5 (a) exemplifica a reação de funcionalização do suporte com APTS seguido da adição de DSC e da imobilização enzimática. O grupo carboxila introduzido na superfície do suporte com a utilização do reagente DSC é altamente susceptível as reações com os grupos nucleofílicos da enzima.61,73 Outro reagente frequentemente utilizado como espaçador após a funcionalização do suporte é o glutaraldeído, o qual irá introduzir grupos carbonila ao suporte, tornando-os susceptíveis a reações com os grupos nucleofílicos da enzima, como mostra a Figura 5 (b). No caso de formação de bases de Schiff, podem-se utilizar reagentes como cianoboroidreto de sódio, tetraboroidreto de sódio e boroidreto de sódio para redução das ligações iminas formadas, indicado em casos que se trabalhe em meios ácidos.61
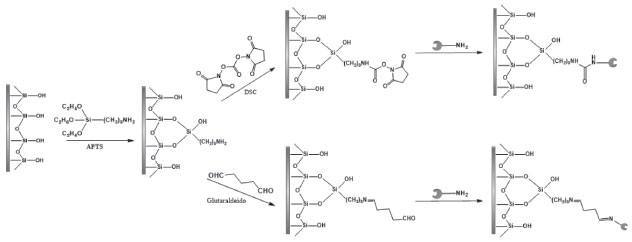 Figura 5. Funcionalização do suporte com APTS. Utilização de DSC (a) e glutaraldeído (b) como espaçador, seguido da imobilização da enzima de forma covalente
A estabilidade da ligação covalente evita perdas da enzima com a utilização do biorreator e aumenta a estabilidade térmica.52,53,61 No entanto, ligações multipontuais podem gerar rigidez, bem como alterar a estrutura terciária nativa da enzima, resultando na diminuição da atividade enzimática quando comparado aos resultados dos ensaios em solução.52,52,53,61,105 Fatores que podem melhorar a reatividade e proporcionar maior interação enzima-suporte devem ser considerados: 1 - tempo de reação para que haja uma interação efetiva entre a enzima e o suporte; 2 - temperatura moderadamente elevada para favorecer a interação enzima-suporte e aumentar a possibilidade de ligações entre eles; 3 - escolha de soluções tampão que não interfiram na etapa de imobilização; 4 - presença de inibidores ou outros protetores de proteína que possam minimizar a perda de atividade enzimática com a imobilização e na incubação com o suporte durante o processo de imobilização em pH alcalino.51,98,99,101
SUPORTES PARA IMOBILIZAÇÃO Com o interesse e aplicabilidade cada vez maior no uso de enzimas imobilizadas, novos avanços nas técnicas e suportes para imobilização vêm sendo desenvolvidos. No que se refere ao suporte adequado para imobilização, embora não haja um suporte universal para todas as enzimas e aplicações, alguns fatores devem ser considerados quanto à sua escolha, dentre eles: possuir uma superfície de contato adequada, porosidade (de acordo com a aplicação), evitar perdas da enzima por difusão, não ser um suporte solúvel, possuir alta afinidade com a proteína, disponibilidade de grupos funcionais reativos, estabilidade mecânica, rigidez, possibilidade de reuso, baixa toxicidade e, sempre que possível, apresentar baixo custo.52,53,64,99,105 Atualmente diversos suportes oferecem propriedades físicas, químicas e morfológicas que podem afetar diretamente a imobilização da enzima e suas características catalíticas. Para a superfície do suporte é fundamental que haja um balanço entre os grupos funcionais hidrofílicos/hidrofóbicos, uma vez que uma presença mínima de moléculas de água no microambiente faz-se necessária para a preservação estrutural da enzima.52 Já o tamanho e distribuição dos poros têm influência direta no rendimento e em efeitos difusionais causados pela transferência de massa entre o meio líquido e os biocatalisadores imobilizados.52,73,98,99 Os suportes podem ser classificados como orgânicos e inorgânicos, de acordo com suas características químicas, e podem ser subdivididos em naturais ou sintéticos. Suportes naturais possuem facilidade na degradação sem causar danos ao meio ambiente, além de possuírem baixo custo. Os suportes sintéticos, por sua vez, podem ser combinados com uma variedade de propriedades físicas e químicas de tal modo a formar um suporte ideal para uma aplicação específica.51,52,99 No que se refere a imobilização de tripsina, vários suportes têm sido relatados na literatura, como sílicas, monolitos a base de polímeros ou sílica, partículas magnéticas, grafeno, quitosana, dentre outros.15,16,57 O desenvolvimento de novos suportes com alto desempenho e extensa cobertura para imobilização de proteases tem sido investigados para emprego em estudos proteômicos.3,105 Dentre os suportes, diversos formatos também são relatados e aplicados nos estudos on-line, off-line, ou ainda visando a aceleração na etapa de digestão proteica.17,53,72 Nesse contexto, apresentamos uma breve descrição de trabalhos selecionados onde diferentes suportes e/ou formatos foram utilizados para imobilização da tripsina com aplicação em proteólise. Ge e colaboradores17 realizaram a imobilização covalente da tripsina em quitosana na superfície externa de mini lâmpadas incandescentes. Ao acender a lâmpada contendo a tripsina imobilizada e imersa em solução proteica, a radiação emitida acelerou a digestão das proteínas com redução do tempo de reação de 12 horas para 5 minutos. O método foi avaliado em proteínas modelo como hemoglobina, citocromo c, lisozima e ovalbumina. As coberturas sequenciais obtidas foram de 91%, 77%, 80% e 52%, respectivamente. As análises foram realizadas por MALDI-TOF MS. De acordo com os autores, a biocompatibilidade do revestimento de quitosana no bulbo das lâmpadas proporcionou um ambiente suave, de modo que a desnaturação e a autólise enzimática fossem minimizadas. O minirreator de lâmpada incandescente de tripsina imobilizada pode ser uma estratégia promissora para a digestão eficiente e mapeamento de peptídeos. Além disso, a facilidade, simplicidade, eficiência e o baixo custo indicam boa perspectiva na identificação de proteínas.17 Bao e colaboradores16 empregaram óxido de grafeno para preparar um revestimento biocompatível nos canais de microchips de poli(metilmetacrilato) através de solução sol-gel para a imobilização covalente da tripsina. Os microchips preparados foram acoplados ao sistema MALDI-TOF MS para identificação e mapeamento de peptídeos. A viabilidade e desempenho do microchip foram demonstrados pela digestão de hemoglobina, citocromo c, mioglobina e ovalbumina com tempo de digestão de 5 segundos e cobertura sequencial de 95%, 76%, 69% e 55%, respectivamente. De acordo com os autores, os resultados evidenciaram que essa abordagem proporcionou um excelente ambiente enzimático, de modo que a autólise e desnaturação da tripsina fossem evitadas. Ainda, a digestão realizada em um curto intervalo de tempo minimizou o consumo de reagentes.16 Cao e colaboradores108 realizaram a imobilização da tripsina em dois tipos de nanopartículas magnéticas de Fe3O4, uma delas revestida com ouro e outra revestida com Cu2+. A aplicação do ensaio foi realizada com a mioglobina para ambos os biorreatores, enquanto as digestões da albumina sérica bovina (BSA) e do citocromo c foram feitas somente no biorreator revestido com ouro.108 De acordo com os autores, a digestão da mioglobina foi alcançada em 15 minutos com 11 peptídeos identificados e cobertura sequencial de 68,8% para o biorreator revestido com Cu2+ e, para o biorreator revestido com ouro, 15 peptídeos identificados com cobertura sequencial de 93,8%. Para a digestão com citocromo c e BSA foram identificados 10 peptídeos com cobertura sequencial de 90,9% para o citocromo c e 33 peptídeos com cobertura sequencial de 54,1% para a BSA. Além disso, a imobilização da tripsina nas nanopartículas de ouro através das ligações Au-S e Au-NH2 permitiram a regeneração das partículas pela remoção da tripsina utilizando boroidreto de sódio.108 Naldi e colaboradores57 realizaram a imobilização covalente da tripsina em uma coluna de 5,0 × 5,2 mm d.i preenchida com monolito à base de sílica (tryp-IMER). O diâmetro do poro (0,60 µm), assim como a superfície do monolito, foram avaliados para evitar adsorção da proteína, alargamento de pico e perda da amostra, além da busca por uma maior eficiência na digestão, mesmo para proteínas de alta massa molecular. O estudo foi realizado on-line através de um sistema cromatográfico multidimensional que incluiu o tryp-IMER, uma coluna C18 para concentração dos peptídeos digeridos e uma coluna analítica C4 acoplada a um espectrômetro de massas de alta resolução (ESI-QTOF). Os autores empregaram uma mistura de proteínas com diferentes tamanhos e pontos isoelétricos para avaliar a eficiência do modelo desenvolvido, sendo elas: citocromo c, ovalbumina, albumina sérica bovina, albumina sérica humana e mioglobina. Como resultados, foi alcançada uma rápida digestão com um tempo de aproximadamente 90 segundos e, com exceção da mioglobina a qual não é relatada a cobertura sequencial dos peptídeos, as demais apresentaram cobertura sequencial de 37,5%; 32,5%; 24% e 31,2%, respectivamente. A despeito das baixas taxas de cobertura, os autores concluíram que o biorreator preparado apresentou eficiência na digestão proteica, especialmente considerando a redução do tempo de digestão associado ao baixo custo de preparo do mesmo. A Tabela 1 apresenta uma variedade de suportes e formas usadas para imobilização da tripsina em abordagens proteômica, com ênfase nos suportes e formatos utilizados para imobilização, sendo a imobilização covalente a principal técnica utilizada. Os tempos de digestão alcançados com os IMERs de tripsina produzidos são muito menores (10 s a 6 h) quando comparados à digestão tríptica em solução (5-24 h). O efeito no percentual de coberturas obtidas, com valores que alcançam 99%, é também um atrativo do uso desses IMERs em proteólise.
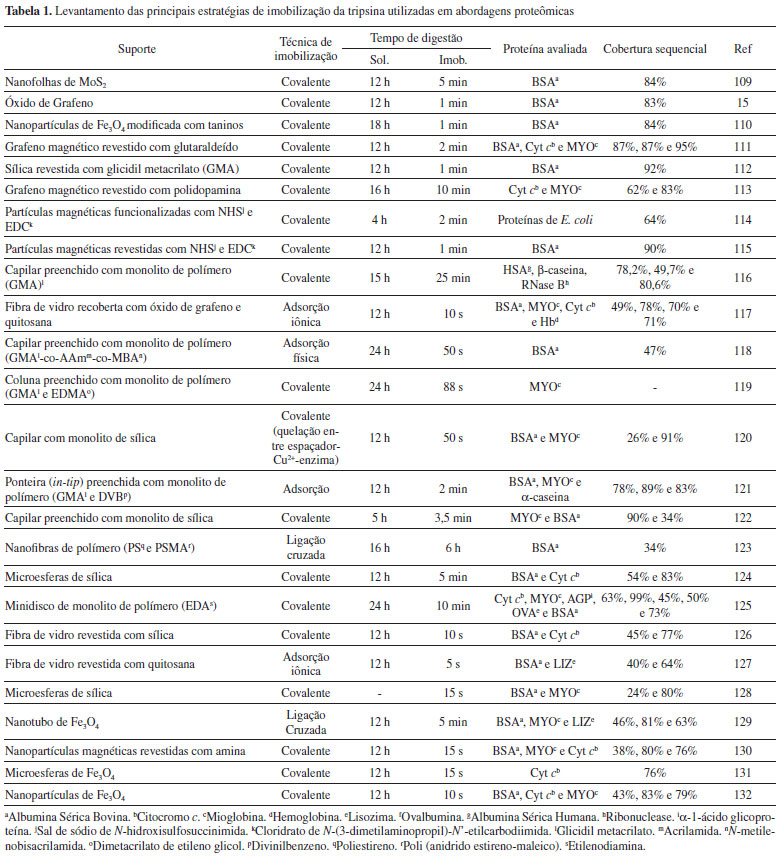
Em geral, os trabalhos com tripsina imobilizada comparam as digestões feitas com emprego dos IMERs àquelas obtidas com a enzima livre, e não a eficiência da digestão proteica realizada com tripsina imobilizada em diferentes suportes. No entanto, Sun e colaboradores114 reportaram que o suporte ideal para a digestão com tripsina deve ser hidrofílico e neutro. Também chamaram a atenção para perda de peptídeos básicos devido a interação destes peptídeos com as partículas magnéticas funcionalizadas com grupos carboxilas. Fan e colaboradores133 imobilizaram tripsina através da preparação de nanopartículas magnéticas revestidas de duas diferentes formas, uma com polímero hidrofóbico (baseado no glicidil metacrilato, GMA-tripsina) e outra com polímero hidrofílico (baseado no glicidil metacrilato-Glucosamina, GMA-G-tripsina). Os autores também observaram um efeito significativo na escolha dos suportes em relação as proteínas e peptídeos identificados a partir da digestão de extratos proteicos de leveduras. O uso da GMA-G-tripsina resultou na identificação de uma porcentagem mais alta de proteínas hidrofílicas, enquanto a digestão com GMA-tripsina digere mais lentamente as proteínas hidrofóbicas. Esses dados sugerem que a química da superfície do suporte pode influenciar o processo de digestão tríptica. Para averiguar o efeito da superfície, Shen e Sun134 realizaram análises proteômica de E. coli (bottom-up) em três diferentes formas: com tripsina em solução, com tripsina imobilizada em partículas magnéticas carregadas negativamente e também neutras (funcionalizadas com amina e com grupo carboxila). Eles observaram que os suportes neutros e carregados negativamente não afetaram significativamente a digestão, comparado à enzima livre. Ademais, a digestão por 15 min com tripsina imobilizada em suporte negativamente modificado teve desempenho comparável à feita com enzima livre com 12 h de digestão. Os autores destacam também que a enzima imobilizada não levou a nenhum artefato causado pela autodigestão.
PERSPECTIVAS FUTURAS E CONCLUSÃO As diversas estratégias desenvolvidas para digestão de proteínas envolvendo tripsina imobilizada apresentam, de modo geral, menor tempo de digestão, maior número de peptídeos sendo alcançados e baixos valores de clivagens perdidas. As condições de digestão dependem não somente dos IMERs produzidos, mas também das estruturas das proteínas em digestão e, como notado por Regnier e Kim,17 a digestão de uma proteína por tripsina, em uma determinada condição, ainda não é uma ciência exata. É importante destacar que diferentes tipos de imobilização de tripsina (suporte, reação, formato, etc.) podem levar a diferentes características de digestão, com consequente impacto na identificação da relação proteína/peptídeos. No entanto, espera-se que a automação e, consequentemente, a maior reprodutibilidade dos processos de digestão realizados com IMERs de tripsina, levem a uma menor variação entre as condições de digestão proteica. Alta produtividade nos processos proteômicos tem sido observada. Devido ao elevado número de amostras, alta produtividade é desejada, especialmente em abordagens clínicas. O desenvolvimento de plataformas integradas de digestão e análise tem aumentado a capacidade de identificação de proteínas de baixa abundância em amostras de pequenos volumes. Assim, espera-se que novos suportes biocompatíveis que facilitem o manuseio e automação dos IMERs nos processos de digestão possam ser desenvolvidos e comercializados.
AGRADECIMENTOS À Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) pelo apoio financeiro (2013/01710-1 e 2014/50249-8) e pelas bolsas de iniciação científica (2015/25755-0) e de doutorado (2018/03035-3) concedidas. Ao Conselho Nacional de Pesquisa (CNPq) pela bolsa de doutorado. A Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - (CAPES) - Código financeiro 001, e a GSK, pelos apoios financeiros.
REFERÊNCIAS 1. Resing, K. A.; Ahn, N. G.; FEBS Lett. 2005, 579, 885. 2. Moradian, A.; Kalli, A.; Sweredoski, M. J.; Hess, S.; Proteomics 2014, 14, 489. 3. Ma, J.; Zhang, L.; Liang, Z.; Shan, Y.; Zhang, Y.; TrAC, Trends Anal. Chem. 2011, 30, 691. 4. Moore, S.; Hess, S.; Jorgenson, J.; J. Chromatogr A 2016, 1476, 1. 5. Barbosa, E. B.; Vidotto, A.; Polachini, G. M.; Henrique, T.; Marqui, A. B. T. d.; Rev. Assoc. Med. Bras. 2012, 58, 366. 6. Switzar, L.; Giera, M.; Niessen, W. M. A.; J. Proteome Res. 2013, 12, 1067. 7. O'Farrell, P. H.; J. Biol. Chem. 1975, 250, 4007. 8. Rabilloud, T.; Lelong, C.; J. Proteomics 2011, 74, 1829. 9. Kim, H.; Eliuk, S.; Deshane, J.; Meleth, S.; Sanderson, T.; Pinner, A.; Robinson, G.; Wilson, L.; Kirk, M.; Barnes, S.; Methods Mol. Biol. 2007, 371, 349. 10. Karpievitch, Y. V.; Polpitiya, A. D.; Anderson, G. A.; Smith, R. D.; Dabney, A. R.; Ann. Appl. Stat. 2010, 4, 1797. 11. Zhou, H.; Ning, Z.; Wang, F.; Seebun, D.; Figeys, D.; FEBS J. 2011, 278, 3796. 12. Toby, T. K.; Fornelli, L.; Kelleher, N. L.; Annu. Rev. Anal. Chem. 2016, 9, 499. 13. Smith, L. M.; Kelleher, N. L.; Nat. Methods 2013, 10, 186. 14. Sun, J.; Hu, K.; Liu, Y.; Pan, Y.; Yang, Y.; J. Chromatogr. B 2017, 942, 9. 15. Guo, C.; Zhao, X.; Zhang, W.; Bai, H.; Qin, W.; Song, H.; Qian, X.; Anal. Bioanal. Chem. 2017, 409, 4741. 16. Bao, H.; Zhang, L.; Chen, G.; J. Chromatogr. A 2013, 1310, 74. 17. Ge, H.; Bao, H.; Zhang, L.; Chen, G.; Anal. Chim. Acta 2014, 845, 77. 18. Regnier, F. E.; Kim, J.; Bioanalysis 2014, 6, 2685. 19. Basile, F.; Hauser, N.; Anal. Chem. 2011, 83, 359. 20. Chen, L. C.; Kinoshita, M.; Noda, M.; Ninomiya, S.; Hiraoka, K.; J. Am. Soc. Mass Spectrom. 2015, 26, 1085. 21. Swatkoski, S.; Gutierrez, P.; Ginter, J.; Petrov, A.; Dinman, J. D.; Edwards, N.; Fenselau, C.; Journal of Proteome 2007, 6, 4525. 22. Lo, L. H.; Huang, T. L.; Shiea, J.; Rapid Commun. Mass Spectrom. 2009, 23, 589. 23. Tang, H. Y.; Speicher, D. W.; Anal. Biochem. 2004, 334, 48. 24. Li, A.; Sowder, R. C.; Henderson, L. E.; Moore, S. P.; Garfinkel, D. J.; Fisher, R. J.; Anal. Chem. 2001, 73, 5395. 25. Patwa, T. H.; Wang, Y.; Simeone, D. M.; Lubman, D. M.; J. Proteome Res. 2008, 7, 2553. 26. Eatemadi, A.; Aiyelabegan, H. T.; Negahdari, B.; Mazlomi, M. A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E.; Biomed. Pharmacother. 2017, 86, 221. 27. Kim, H. J.; Jan, C.-H.; J. Mol. Liq. 2016, 222, 596. 28. Min, W.; Cui, S.; Wang, W.; Chen, J.; Hu, Z.; Anal. Biochem. 2013, 438, 32. 29. Finehout, E. J.; Cantor, J. R.; Lee, K. H.; Proteomics 2005, 5, 2319. 30. Syal, K.; Tadala, R.; Protein Pept. Lett. 2015, 22, 372. 31. Freije, J. R.; Mulder, P. P.; Werkman, W.; Rieux, L.; Niederlander, H. A.; Verpoorte, E.; Bischoff, R.; J. Proteome Res. 2005, 4, 1805. 32. Meyer, B.; Papasotiriou, D. G.; Karas, M.; Amino Acids 2011, 41, 291. 33. Swaney, D. L.; Wenger, C. D.; Coon, J. J.; J. Proteome Res. 2010, 9, 1323. 34. Choudhary, G.; Wu, S. L.; Shieh, P.; Hancock, W. S.; J. Proteome Res. 2003, 2, 59. 35. Meller, K.; Pomastowski, P.; Grzywinski, D.; Szumski, M.; Buszewski, B.; J. Chromatogr. A 2016, 1440, 45. 36. Weston, L. A.; Bauer, K. M.; Hummon, A. B.; Anal. Methods 2013, 5, 4615. 37. Meller, K.; Pomastowski, P.; Szumski, M.; Buszewski, B.; J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1043, 128. 38. Safdar, M.; Spross, J.; Janis, J.; J. Chromatogr. A 2014, 1324, 1. 39. Cao, S.; Xu, P.; Ma, Y.; Yao, X.; Yao, Y.; Zong, M.; Li, X.; Lou, W.; Chin. J. Catal. 2016, 37, 1814. 40. Ma, J.; Zhang, L.; Liang, Z.; Zhang, W.; Zhang, Y.; Anal. Chim. Acta 2009, 632, 1. 41. Segu, Z. M.; Hammad, L. A.; Mechref, Y.; Rapid Commun. Mass Spectrom. 2010, 24, 3461. 42. Hua, L.; Low, T. Y.; Sze, S. K.; Proteomics 2006, 6, 586. 43. Lopez-Ferrer, D.; Capelo, J. L.; Vazquez, J.; J. Proteome Res. 2005, 4, 1569. 44. Wang, S.; Zhang, L.; Yang, P.; Chen, G.; Proteomics 2008, 8, 2579 45. Wang, S.; Liu, T.; Zhang, L.; Chen, G.; J. Proteome Res. 2008, 7, 5049. 46. Lopez-Ferrer, D.; Petritis, K.; Hixson, K. K.; Heibeck, T. H.; Moore, R. J.; Belov, M. E.; Camp 2nd, D. G.; Smith, R. D.; J. Proteome Res. 2008, 7, 3276. 47. Yang, H. J.; Hong, J.; Lee, S.; Shin, S.; Kim, J.; Rapid Commun. Mass Spectrom. 2010, 24, 901. 48. Jones, L. M.; Zhang, H.; Vidavsky, I.; Gross, M. L.; Anal. Chem. 2010, 82, 1171. 49. Park, Z. Y.; Russell, D. H.; Anal. Chem. 2000, 72, 2667. 50. Bark, S. J.; Muster, N.; Yates 3rd, J. R.; Siuzdak, G.; J. Am. Chem. Soc. 2001, 123, 1774. 51. Cao, L.; Curr. Opin. Chem. Biol. 2005, 9, 217. 52. Mohamad, N. R.; Marzuki, N. H. C.; Buang, N. A.; Huyop, F.; Wahab, R. A.; Biotechnol. Biotechnol. Equip. 2015, 29, 205. 53. Es, I.; Vieira, J. D.; Amaral, A. C.; Appl. Microbiol. Biotechnol. 2015, 99, 2065. 54. Kecskemeti, A.; Gaspar, A.; Talanta 2018, 180, 211. 55. Brandtzaeg, O. K.; Roen, B. T.; Enger, S.; Lundanes, E.; Wilson, S. R.; Anal. Chem. 2017, 89, 8667. 56. Kim, J.-H.; Inerowicz, D.; Hedrick, V.; Regnier, F.; Anal. Chem. 2013, 85, 8039. 57. Naldi, M.; Cernigoj, U.; Strancar, A.; Bartolini, M.; Talanta 2017, 167, 143. 58. Zhang, S.; Yuan, H.; Zhao, B.; Zhang, L.; Zhang, Y.; Anal. Chim. Acta 2018, 1000, 172. 59. Ma, J.; Zhang, L.; Liang, Z.; Zhang, W.; Zhang, Y.; Anal. Chim. Acta 2009, 632, 1. 60. Monzo, A.; Sperling, E.; Guttman, A.; TrAC, Trends Anal. Chem. 2009, 28, 854. 61. Cardoso, C. L.; de Moraes, M. C.; Cass, Q. B.; Quim. Nova 2009, 32, 175. 62. Hettiarachchy, N. S.; Feliz, D. J.; Edwards, J. S.; Horax, R. Em Proteins in Food Processing (Second Edition); Yada, R. Y., ed.; Woodhead Publishing: Cambridge, 2018, cap. 21. 63. Maciel, J. C.; Andrad, P. L.; Neri, D. F. M.; Carvalho Jr., L. B.; Cardoso, C. A.; J. Magn. Magn. Mater. 2012, 324, 1312. 64. Li, Y.; Xu, X.; Deng, C.; Yang, P.; Zhang, X.; J. Proteome Res. 2007, 6, 3849. 65. Karimi, B.; Emadi, S.; Safari, A. A.; Kermanian, M.; RSC Adv. 2014, 4, 4387. 66. Gustafsson, H.; Thorn, C.; Holmberg, K.; Colloids Surf., B 2011, 87, 464. 67. Malvi, B.; Gupta, S. S.; Chem. Commun. (Cambridge, U. K.) 2012, 48, 7853. 68. Lee, B.; Kim, C. B.; Chang, S. M.; Kim, S. H.; Na, B. H.; Park, I. Y.; Lee, J.; Hyeon, T.; Lee, H.; Lee, S.-W.; Kim, J.; Chem. Eng. J. 2016, 288, 770. 69. Srbová, J.; Slováková, M.; Křípalová, Z.; Zárská, M.; Spačková, M.; Stránská, D.; Bílková, Z.; React. Funct. Polym. 2016, 104, 38. 70. Gkantzou, E.; Patila, M.; Stamatis, H.; Catalysts 2018, 8, 282. 71. Meller, K.; Szumski, M.; Buszewski, B.; Sens. Actuators, B 2019, 244, 84. 72. Zheng, Y. Z.; DeMarco, M. L.; Clinical Mass Spectrometry 2017, 6, 1. 73. Li, D.; Teoh, W. Y.; Gooding, J. J.; Selomulya, C.; Adv. Funct. Mater. 2010, 20, 1767. 74. Chen, J.-M.; Radisky, E. S.; Férec, C. Em Handbook of Proteolytic Enzymes; Rawling, N. D.; Salvesen, G., eds.; Elsevier Ltd.: Amsterdam, 2013, cap. 576. 75. Gong, J. S.; Li, W.; Zhang, D. D.; Xie, M. F.; Yang, B.; Zhang, R. X.; Li, H.; Lu, Z. M.; Xu, Z. H.; Shi, J. S.; Int. J. Mol. Sci. 2015, 16, 30061. 76. Baird, T. T.; Craik, C. S. Em Encyclopedia of Genetics; Brenner, S.; Miller, J., eds.; Academic Press: Cambridge, 2001. 77. https://www.sigmaaldrich.com/technical-documents/articles/biology/trypsin.html, acessada em Março 2020. 78. Gudmundsdóttir, Á.; Stefansson, B.; Bjarnason, J. B. Em Handbook of Proteolytic Enzymes; Rawling, N. D.; Salvesen, G., eds.; Elsevier Ltd.: Amsterdam, 2013, cap. 580. 79. https://www.biocompare.com/, acessada em Março 2020. 80. https://www.creative-enzymes.com/, acessada em Março 2020. 81. https://scrippslabs.com/, acessada em Março 2020. 82. https://www.sigmaaldrich.com/brazil.html, acessada em Março 2020. 83. https://www.promega.com.br/en/, acessada em Março 2020. 84. https://www.thermofisher.com/br/en/home.html, acessada em Março 2020. 85. https://www.labcorp.com/tests/010355/trypsin, acessada em Março 2020. 86. Lauer-Fields, J. L.; Minond, D.; Sritharan, T.; Kashiwagi, M.; Nagase, H.; Fields, G. B.; J. Biol. Chem. 2007, 282, 142. 87. Hubbard, S. J.; Biochim. Biophys. Acta 1998, 1382, 191. 88. Ellison, D.; Hinton, J.; Hubbard, S. J.; Beynon, R. J.; Protein Sci. 1995, 4, 1337. 89. Nosrati, G. R.; Houk, K. N.; Biochemistry 2012, 51, 7321. 90. Madian, A. G.; Hindupur, J.; Hulleman, J. D.; Diaz-Maldonado, N.; Mishra, V. R.; Guigard, E.; Kay, C. M.; Rochet, J. C.; Regnier, F. E.; Mol. Cell Proteomics 2012, 11, M111.010892. 91. Nosrati, G. R.; Houk, K. N.; Protein Sci. 2012, 21, 697. 92. Drag, M.; Salvesen, G. S.; Nat. Rev. Drug Discov. 2010, 9, 690. 93. Raney, K. D.; Sharma, S. D.; Moustafa, I. M.; Cameron, C. E.; J. Biol. Chem. 2010, 285, 22725. 94. Rivera-Burgos, D.; Regnier, F. E.; Anal. Chem 2012, 84, 7021. 95. Lei, Z.; Gao, C.; Chen, L.; He, Y.; Ma, W.; Lin, Z.; J. Mater. Chem. B 2018, 11, 1581. 96. Mateo, C.; Abian, O.; Fernandez-Lorente, G.; Pedroche, J.; Fernandez-Lafuente, R.; Guisan, J. M.; Tam, A.; Daminati, M.; Biotechnol. Prog. 2002, 18, 629. 97. Nelson, J. M.; Griffin, E. G.; J. Am. Chem. Soc. 1916, 38, 1109. 98. Cao, L.; Carrier-bound Immobilized Enzymes: Principles, Application and Design; Wiley-VCH: New York, 2005. 99. Sheldon, R. A.; Adv. Synth. Catal. 2017, 349, 1289. 100. Sun, X.; Cai, X.; Wang, R. Q.; Xiao, J.; Anal. Biochem. 2015, 477, 21. 101. Mateo, C.; Palomo, J. M.; Fernandez-Lorente, G.; Guisan, J. M.; Fernandez-Lafuente, R.; Enzyme Microb. Technol. 2007, 40, 1451. 102. Bastida, A.; Sabuquillo, P.; Armisen, P.; Fernández-Lafuente, R.; Huguet, J.; Guisán, J. M.; Biotechnology and Bioengineering; Wiley Online Library: New York, 1998. 103. Fernandez-Lafuente, R.; Armisen, P.; Sabuquillo, P.; Fernandez-Lorente, G.; Guisan, J. M.; Chem. Phys. Lipids 1998, 93, 185. 104. Liu, J.; Liu, Y.; Jin, D.; Meng, M.; Jiang, Y.; Ni, L.; Liu, Z.; Solid State Sci. 2019, 89, 15. 105. Datta, S.; Christena, L. R.; Rajaram, Y. R. S.; 3 Biotech. 2013, 3, 1. 106. Mageed, H. A.; Ezz, N. A.; Radwan, R.; BioTechnologia 2019, 100, 301. 107. Chuang, C.-H.; Lin, Y.-C.; Chen, W.-L.; Chen, Y.-H.; Chen, Y.-X.; Chen, C.-M.; Shiu, H. W.; Chang, L.-Y.; Chen, C.-H.; Chen, C.-H.; Biosens. Bioelectron. 2016, 78, 213. 108. Cao, Y.; Wen, L.; Svec, F.; Tan, T.; Lv, Y.; Chem. Eng. J. 2016, 286, 272 109. Xia, C.; Wang, H.; Jiao, F.; Gao, F.; Wu, Q.; Shen, Y.; Zhang, Y.; Qian, X.; Talanta 2018, 179, 393. 110. Atacan, K.; Cakiroglu, B.; Ozacar, M.; Colloids Surf., B 2017, 156, 9. 111. Wang, H.; Jiao, F.; Gao, F.; Zhao, X.; Zhao, Y.; Shen, Y.; Zhang, Y.; Qian, X.; Anal. Bioanal. Chem. 2017, 409, 2179. 112. Jiao, F.; Zhai, R.; Huang, J.; Zhang, Y.; Zhang, Y.; Qian, X.; RSC Adv. 2016, 87, 84113. 113. Shi, C.; Deng, C.; Li, Y.; Zhang, X.; Yang, P.; Proteomics 2014, 14, 1457. 114. Sun, L.; Zhu, G.; Yan, X.; Mou, S.; Dovichi, N. J.; J. Chromatogr. A 2014, 1337, 40. 115. Sun, L.; Li, Y.; Yang, P.; Zhu, G.; Dovichi, N. J.; J. Chromatogr. A 2012, 1220, 68. 116. Calleri, E.; Temporini, C.; Gasparrini, F.; Simone, P.; Villani, C.; Ciogli, A.; Massolini, G.; J. Chromatogr. A 2011, 1218, 8937. 117. Bao, H.; Chen, Q.; Zhang, L.; Chen, G.; Analyst 2011, 136, 5190. 118. Wu, S.; Zhang, L.; Yang, K.; Liang, Z.; Zhang, Y.; Anal. Bioanal. Chem. 2012, 402, 703. 119. Yao, C.; Qi, L.; Hu, W.; Wang, F.; Yang, G.; Anal. Chim. Acta 2011, 692, 131. 120. Ma, J.; Hou, C.; Liang, Y.; Wang, T.; Liang, Z.; Zhang, L.; Zhang, Y.; Proteomics 2011, 11, 991. 121. Hahn, H. W.; Rainer, M.; Ringer, T.; Huck, C. W.; Bonn, G. K.; J. Proteome Res. 2009, 8, 4225. 122. Ma, J.; Liu, J.; Sun, L.; Gao, L.; Liang, Z.; Zhang, L.; Zhang, Y.; Anal. Chem. 2009, 81, 6534. 123. Kim, B. C.; Lopez-Ferrer, D.; Lee, S. M.; Ahn, H. K.; Nair, S.; Kim, S. H.; Kim, B. S.; Petritis, K.; Camp, D. G.; Grate, J. W.; Smith, R. D.; Koo, Y. M.; Gu, M. B.; Kim, J.; Proteomics 2009, 9, 1893. 124. Bao, H.; Lui, T.; Zhang, L.; Chen, G.; Proteomics 2009, 9, 1114. 125. Nicoli, R.; Rudaz, S.; Stella, C.; Veuthey, J. L.; J. Chromatogr. A 2009, 1216, 2695. 126. Liu, T.; Wang, S.; Chen, G.; Talanta 2009, 77, 1767. 127. Wang, S.; Chen, Z.; Yang, P.; Chen, G.; Proteomics 2008, 8, 1785. 128. Lin, S.; Yao, G.; Qi, D.; Li, Y.; Deng, C.; Yang, P.; Zhang, X.; Anal. Chem. 2008, 80, 3655. 129. Wang, S.; Bao, H.; Yang, P.; Chen, G.; Anal. Chim. Acta 2018, 612, 182. 130. Lin, S.; Yun, D.; Qi, D.; Deng, C.; Li, Y.; Zhang, X.; J. Proteome Res. 2008, 7, 1297. 131. Lin, S.; Lin, Z.; Yao, G.; Deng, C.; Yang, P.; Zhang, X.; Rapid. Commun. Mass Spectrom. 2007, 21, 3910. 132. Liu, J.; Lin, S.; Qi, D.; Deng, C.; Yang, P.; Zhang, X.; J. Chromatogr. A 2007, 1176, 169. 133. Fan, C.; Shi, Z.; Pan, Y.; Song, Z.; Zhang, W.; Zhao, X.; Tian, F.; Peng, B.; Qin, W.; Cai, Y.; Qian, X.; Anal. Chem. 2014, 86, 1452. 134. Shen, X.; Sun, L.; Proteomics 2018, 18, 1700432. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access