
 |
 |
 |
 |
 |
Artigo
|
|
| Dois novos alcaloides azafenantreno de Anaxagorea dolichocarpa Sprague & Sandwith Two new azaphenanthrene alkaloids from Anaxagorea dolichocarpa Sprague & Sandwith |
|
Kaio A. SalesI; Anderson A. V. PinheiroI; Diego I. A. F. AraújoI; Rodrigo S. de AndradeI; Maria de Fátima AgraII; Marianna V. SobralI; Hemerson I. F. MagalhãesI; Valgrícia M. de SousaI; Raimundo Braz-FilhoIII; Marcelo S. da SilvaI; Josean F. TavaresI,*
I. Centro de Ciências da Saúde, Universidade Federal da Paraíba, 58051-900 João Pessoa - PB, Brasil Recebido em 01/11/2019 *e-mail: josean@ltf.ufpb.br A chemical investigation of Anaxagorea dolichocarpa Sprague & Sandwith, a member of Annonaceae family, was carried out. The ethanolic extract from the roots of this plant led, by chromatography tecniches, to isolation of the new azaphenanthrene alkaloids dolichocarpine (1) and 9-methoxyeupolauramine (2), besides the known alkaloids eupolauramine (3), 3-methoxyeupolauridine (4), eupolauramine (5) and 4-methylsampangine (6). The structures of isolated compounds were established by 1D and 2D NMR, HRESIMS, tandem MSn and IR data. The cytotoxicity of compounds 1 - 5 was evaluated against HCT-116 (human colorectal carcinoma) and L929 (murine fibroblast) cell lines. INTRODUÇÃO Annonaceae é uma família de árvores floríferas, arbustos e cipós com distribuição pantropical, sendo composta por 110 gêneros e aproximadamente 2.430 espécies.1,2 No Brasil apresenta ampla distribuição, com cerca de 390 espécies encontradas principalmente na Região Amazônica e Mata Atlântica.3,4 As espécies da família são conhecidas pela biossíntese de alcaloides e os estudos demonstraram considerável diversidade estrutural, além de notáveis atividades biológicas para esses metabólitos secundários.5,6 Com relação ao gênero, Anaxagorea compreende 26 espécies,7 ocorrendo nas regiões neotropical e paleotropical, e no Brasil é encontrado principalmente no Norte e Nordeste.8-10 Das espécies do gênero investigadas até o presente momento, foram identificados principalmente alcaloides, terpenoides, xantonas, flavonoides e lignoides.11-16 Ao passo que em Anaxagorea dolichocarpa Sprague & Sandwith, estudos fitoquímicos prévios permitiram o isolamento de alcaloides azafenantrenos e aporfínicos.11,17 Diversos compostos dessas duas subclasses de alcaloides já foram testados em ensaios de atividade citotóxica e antitumoral, alguns deles apresentando resultados positivos, sendo por isso considerados promissores.5,11 Historicamente, produtos naturais obtidos de plantas representam a principal fonte de novos fármacos, além de fornecerem protótipos para a síntese de compostos farmacologicamente ativos, particularmente com atividade anticâncer. Considerando que o câncer é uma das doenças com as mais altas taxas de morbidade e mortalidade no mundo, há um considerável interesse científico e comercial na descoberta de novas drogas a partir de fontes naturais.18 Então, como continuidade dos estudos do nosso grupo de pesquisa com a família Annonaceae, este trabalho buscou isolar e identificar novos compostos de A. dolichocarpa por meio de métodos cromatográficos e espectroscópicos, respectivamente, e ainda avaliar a citotoxicidade dessas substâncias.
RESULTADOS E DISCUSSÃO O estudo químico do extrato etanólico das raízes de A. dolichocarpa levou ao isolamento de seis compostos (1 - 6) (Figura 1), sendo esse o primeiro relato das substâncias 1 e 2 na literatura. Suas estruturas químicas foram determinadas com base na análise dos dados espectroscópicos de RMN uni e bidimensionais, espectrometria de massa de alta resolução com ionização por electrospray (EMAR-IES) e comparação com dados da literatura. Os dados de RMN de 1H e 13C dos compostos 1 e 2 estão compilados na Tabela 1.
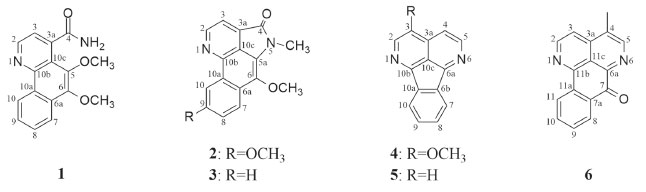 Figura 1. Alcaloides isolados de A. dolichocarpa
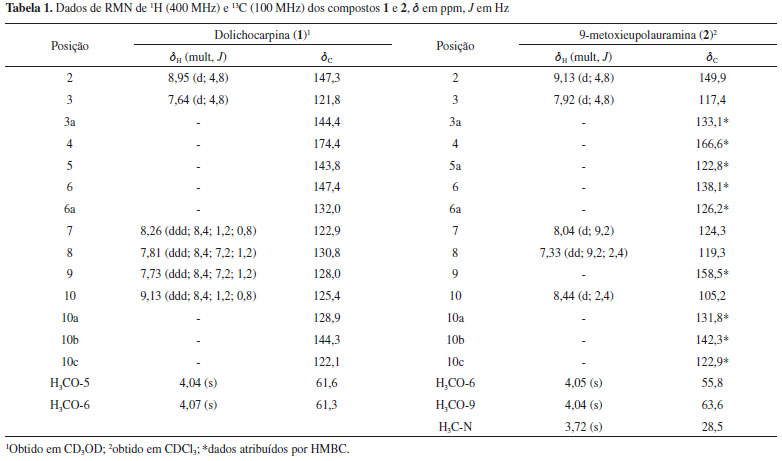
O composto 1 foi isolado como cristais amarelados. O espectro de EMAR-IES mostrou o pico do íon [M+H]+ com m/z 283,1073 (calcd. para C16H15N2O3, 283,1077), compatível com a fórmula molecular C16H15N2O3. O espectro de infravermelho mostrou absorção de duas bandas em 3306 e 3139 cm-1 de estiramento N-H2, banda de carbonila de amida em 1646 cm-1, absorções em 1618 e 1582 cm-1 de C=C de aromático, além do sinal em 1055 cm-1 de estiramento C-O. O espectro de RMN de 1H (CD3OD, 400 MHz) mostrou seis sinais de prótons aromáticos, sendo dois dupletos em δH 8,95 (d; J = 4,8 Hz) e δH 7,64 (d; J = 4,8 Hz), sugestivos para os prótons H-2 e H-3 de um anel piridínico do esqueleto azaaporfinoide,19,20 e também quatro duplos dupletos duplos compatíveis com um sistema aromático ABCD, sendo um mais desprotegido em δH 9,13 (ddd; J = 8,4; 1,2; 0,8 Hz) característico do próton H-10 desse tipo de esqueleto, um em δH 8,26 (ddd; J = 8,4; 1,2; 0,8 Hz) atribuído ao H-7 e os outros dois em δH 7,81 (ddd; J = 8,4; 7,2; 1,2 Hz) e 7,73 (ddd; J = 8,4; 7,2; 1,2 Hz) assinalados para H-8 e H-9, respectivamente. Além desses sinais, foram ainda observados dois simpletos, ambos integrando para três prótons, em δH 4,07 (s) e δH 4,04 (s), correspondentes a duas metoxilas. No espectro de 13C (CD3OD, 100 MHz) foram observados dezesseis sinais. Desses, seis denotam ser de carbonos metínicos aromáticos e dois de grupos metoxila, confirmando dessa forma os dados observados no espectro de RMN de 1H. Observou-se também um sinal característico de um grupo carbonila e mais sete sinais indicativos de carbonos aromáticos não hidrogenados. A análise desses dados e do espectro de correlações HSQC permitiram atribuir os sinais dos carbonos metínicos em δC 147,3 e 121,8 aos carbonos C-2 e C-3, o primeiro mais desprotegido por estar ligado a um heteroátomo, e em δC 122,9, 130,8, 128,0 e 125,4 aos carbonos C-7, C-8, C-9 e C-10, respectivamente. Os demais carbonos foram definidos com auxílio das correlações a longa distância (2J e 3J) observadas no experimento HMBC (Figura 2), que permitiu também confirmar as demais atribuições já sugeridas. A correlação dos sinais em δH 8,95 (H-2) com δC 121,8 (C-3), 144,4 (C-3a), 144,3 (C-10b) e em δH 7,64 (H-3) com δC 147,3 (C-2), 174,4 (C-4), 122,1 (C-10c) estabeleceu os valores dos carbonos C-3a, C-10b e C-10c, bem como ratificou a posição da carbonila em C-4. A correlação dos sinais em δH 7,81 (H-8) e δH 9,13 (H-10) com δC 132,0 (C-6a) e em δH 8,26 (H-7) e δH 7,73 (H-9) com δC 128,9 (C-10a) determinou o deslocamento dos carbonos C-6a e C-10a. Além dessa correlação, o sinal em δH 8,26 (H-7), assim como o δH 4,07 (H3CO-6), correlacionaram-se com δC 147,4 (C-6), permitindo dessa forma atribuir o deslocamento de C-6, substituído, portanto, por um grupo metoxila. A localização da segunda metoxila foi definida por meio da correlação do sinal em δH 4,04 (H3CO-5) com o sinal em δC 143,8 (C-5). Para confirmar o grupo NH2 na estrutura química, a amostra foi submetida a análise de RMN de 1H em CDCl3, na qual pôde-se observar dois sinais largos em δH 5,74 e 5,87 que desapareceram ao adicionar-se D2O (Figuras 9S e 10S). Assim, com base nesses dados, foi possível propor para 1 a estrutura mostrada na Figura 1, um novo alcaloide azafenantreno denominado dolichocarpina. Sua análise por EMn apresentou os íons indicados na Figura 2S e a proposta de fragmentação desses íons pode ser observada na Figura 3.21,22
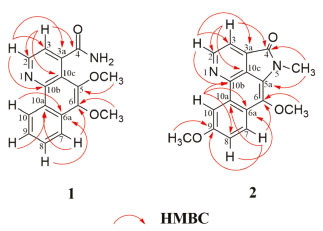 Figura 2. Correlações HMBC de 1 e 2
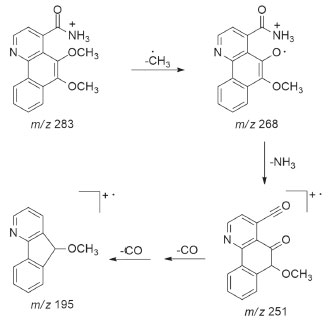 Figura 3. Proposta de fragmentação de EMn de 1
O composto 2 foi isolado como cristais amarelados. O espectro de EMAR-IES mostrou o pico do íon [M+H]+ com m/z 295,1078 (calcd. para C17H15N2O3, 295,1077), compatível com a fórmula molecular C17H15N2O3. Os dados de RMN dessa substância foram comparados com os dados do alcaloide eupolauramina, isolado previamente de A. dolichocarpa e novamente neste trabalho (composto 3),11 como também com outros relatados na literatura.20 O espectro de RMN de 1H (CDCl3, 400 MHz) apresentou semelhança com o da eupolauramina (3), observando-se porém um sinal de grupo metoxila a mais. Além dessa diferença, o espectro revelou a ausência de um sinal de próton aromático e alteração na multiplicidade dos sinais, apresentando um dupleto duplo e dois dupletos no lugar dos quatro duplos dupletos duplos, sugerindo dessa maneira uma substituição no anel D do esqueleto eupolaumarina. Os sinais em δH 7,33 (dd; J = 9,2; 2,4 Hz), δH 8,04 (d; J = 9,2 Hz) e δH 8,44 (d; J = 2,4 Hz) sugerem substituição no C-9, devido à ausência do sinal mais desprotegido na região próxima de 9,0 ppm atribuído ao H-10, que pode ter sofrido um efeito de proteção orto da metoxila na posição vizinha (C-9), podendo-se então atribuir esses sinais para H-8, H-7 e H-10, respectivamente. No espectro de RMN de 13C foi possível observar apenas oito sinais. Dentre eles se destaca o sinal de metoxila adicional em δC 55,8 e o de um carbono aromático mais protegido em δC 105,2, o qual apresentou correlação no espectro HSQC com o sinal em δH 8,44, atribuindo-se, por conseguinte, esse sinal ao carbono C-10. Os carbonos que não tiveram seus sinais observados no espectro de RMN de 13C (CDCl3, 100 MHz) foram assinalados após análise do espectro de correlações HMBC (Figura 2). Esse experimento permitiu também confirmar a posição da metoxila em C-9 por meio da correlação do sinal em δC 158,5 (C-9) com δH 4,04 (H3CO-9) e 8,04 (H-7), e deste último com δC 138,1 (C-6). Assim, o composto 2, pôde ser identificado como um novo alcaloide azafenantreno, denominado 9-metoxieupolauramina. Composto 3: RMN de 1H (CDCl3, 400 MHz), δ em ppm (mult.; J em Hz; H): 9,16 (d; 4,8; H-2), 9,01 (ddd; 8,0; 1,6; 0,8; H-10), 8,13 (ddd; 8,0; 1,2; 0,8; H-7), 7,91 (d; 4,8; H-3), 7,73 (ddd; 8,0; 6,8; 1,6; H-8), 7,67 (ddd; 8,0; 6,8; 1,2; H-9), 4,06 (s; H3CO-6), 3,73 (s; H3C-N). RMN de 13C (CDCl3, 100 MHz), δ em ppm: 166,8 (C-4), 150,1 (C-2), 142,5 (C-10b), 132,6 (C-6a), 129,6 (C-8), 126,8 (C-9), 124,9 (C-5a), 124,4 (C-10), 122,8 (C-7), 117,2 (C-3), 63,5 (H3CO-6), 28,5 (H3C-N). EMAR-IES: m/z 265,0979 [M+H]+ (calcd. para C16H13N2O2, 265,0972). Dados coerentes com os relatados na literatura para eupolauramina,11 já isolado em A. dolichocarpa. Composto 4: RMN de 1H (CDCl3, 400 MHz), δ em ppm (mult.; J em Hz; H): 8,72 (d; 6,0; H-5), 8,15 (s; H-2), 7,98 (ddd; 6,8; 1,2; 0,8; H-10), 7,91 (ddd; 6,8; 1,2; 0,8; H-7), 7,61 (d; 6,0; H-4), 7,45 (dt; 7,6; 1,2; H-9), 7,40 (dt; 7,6; 1,6; H-8), 4,08 (s; H3CO-3). RMN de 13C (CDCl3, 100 MHz), δ em ppm (alguns dados atribuídos por HMBC): 161,6 (C-6a), 155,0 (C-10b), 150,5 (C-3), 149,2 (C-5), 131,1 (C-9), 129,9 (C-8), 129,4 (C-2), 128,5 (C-3a), 122,6 (C-10), 121,7 (C-7), 113,5 (C-4), 56,4 (CH3O-3). EMAR-IES: m/z 235,0872 [M+H]+ (calcd. para C15H11N2O, 235,0866). Dados compatíveis com os reportados para 3-metoxieupolauridina,23 isolado pela primeira vez na família Annonaceae. Composto 5: RMN de 1H (CDCl3, 400 MHz), δ em ppm (mult.; J em Hz; H): 8,69 (d; 6,0; H-2/H-5), 7,99 (dd; 8,4; 2,0 H-10/H-7), 7,46 (dd; 8,4; 2,0; H-8/H-9), 7,42 (d; 6,0; H-3/H-4). RMN de 13C (CDCl3, 100 MHz), δ em ppm: 162,7 (C-6a/C-10b), 149,7 (C-2/C-5), 139,6 (C-6b/C-10a), 135,3 (C-3a), 131,4 (C-8/C-9), 122,9 (C-7/C-10), 120,9 (C-10c), 117,6 (C-3/C-4). EMAR-IES: m/z 205,0762 [M+H]+ (calcd. para C14H9N2, 205,0760). Dados semelhantes com os relatados na literatura para eupolauridina,15,24 relatado pela primeira vez na espécie A. dolichocarpa. Composto 6: RMN de 1H (CDCl3, 400 MHz), δ em ppm (mult.; J em Hz; H): 8,95 (d; 0,8; H-5), 8,89 (d; 6,0; H-2), 8,83 (ddd; 8,0; 1,2; 0,8; H-11), 8,45 (ddd; 8,0; 1,6; 0,8; H-8), 7,81 (ddd; 8,0; 7,6; 1,6; H-10), 7,79 (d; 6,0; H-3), 7,67 (ddd; 8,0; 7,6; 1,2; H-9), 2,76 (d; 0,8; CH3-4). EMAR-IES: m/z 247,0875 [M+H]+ (calcd. para C16H11N2O, 247,0866). Dados compatíveis com os relatados na literatura para 4-metilsampangina,25 relatado pela primeira vez como produto natural. Os compostos 2 e 4 inibiram significativamente a proliferação de células HCT-116 com percentual de inibição de 30,22 ± 2,63% e 32,42 ± 0,53%, respectivamente (Figura 4). Assim, comparando-se as estruturas químicas de 2 com 3 e de 4 com 5, observou-se nos dois casos uma substituição por um grupo metoxila em 2 e 4, o que permitiu sugerir que essa substituição aumentou a atividade citotóxica desses compostos para células tumorais da linhagem HCT-116. O estudo de citotoxicidade em linhagem não tumoral de fibroblasto de camundongo L929 mostrou que os compostos 2 - 5 podem reduzir a proliferação celular, o que indica citotoxicidade.
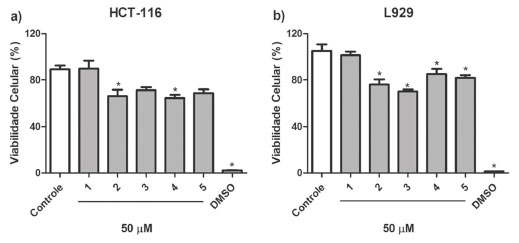 Figura 4. Citotoxicidade dos compostos 1 - 5 em linhagens de células HCT-116 e L929, após 72 h de tratamento. Os valores representam a média ± erro padrão de quatro replicatas na concentração de 50 μmol L-1 (*p < 0,05 comparado ao grupo controle)
CONCLUSÃO O estudo do extrato etanólico das raízes de A. dolichocarpa levou ao isolamento, por cromatografia líquida de média pressão e cromatografia líquida de alta eficiência, dos novos alcaloides azafenantrenos dolichocarpina (1) e 9-metoxieupolauramina (2), além dos alcaloides eupolauramina (3), já isolado na espécie, 3-metoxieupolauridina (4), novo na família, eupolauramina (5), novo na espécie e 4-metilsampangina (6), relatado pela primeira vez como produto natural. O estudo da atividade citotóxica mostrou que os compostos 2 - 5 induziram citotoxicidade em linhagem celular não tumoral murina L929 e que, em linhagem tumoral HCT-116, apenas os compostos 2 e 4 foram capazes de inibir a proliferação celular.
PARTE EXPERIMENTAL Procedimentos experimentais gerais As separações por cromatografia líquida de média pressão (CLMP) foram feitas utilizando como fase estacionária sílica gel (40-63 µm, 230-400 mesh, SiliCycle) e um equipamento Buchi modelo Sepacore flash system X-50. O software desse equipamento permitiu analisar as frações coletadas, auxiliando desse modo no agrupamento das amostras. As frações eluídas foram também analisadas por cromatografia de camada delgada (CCD) utilizando-se placas de alumínio pré-revestidas com sílica gel F254 (SiliCycle), revelando-se com reagente de Dragendorff e exposição à luz UV (254 e 366 nm), e agrupadas de acordo com a similaridade de eluição. A cromatografia líquida de alta eficiência (CLAE) analítica foi realizada no instrumento Shimadzu Prominence equipado com uma bomba de solvente binária LC-20AT, auto-injetor SIL-20A, sistema degaseificador DGU-20A, detector DAD SPD-M20A, sistema de controle CBM-20A e as colunas de fase reversa Kromasil 100-10-C18 (250 mm × 4,6 mm preenchida com partículas de 10 μm) ou ACE 5 C18 (250 mm × 4,6 mm e partículas de 5 µm). As frações foram purificadas por CLAE na escala semi-preparativa e preparativa. Para a primeira técnica foi utilizado um equipamento Shimadzu composto por bomba LC-10AD vp, válvula solenoide FCV-10AL vp, injetor manual Rheodyne, desgaseificador DGU-14A, detector UV-vis SPD-10A vp, controlador de sistema SLC-10A vp e uma coluna semi-preparativa Venusil XBP C18 (250 mm × 10 mm e partículas de 10 μm). No caso da escala preparativa a purificação foi realizada em um Shimadzu contendo duas bombas LC-6AD, injetor manual Rheodyne, detector DAD SPD-M10A vp, controlador de sistema SLC-10A vp e coluna preparativa ACE 5 C18 (250 mm × 21,2 mm e partículas de 5 μm). O fluxo empregado para as purificações foi de 3,5 mL min-1 para a escala semipreparativa e 8,0 mL min-1 para a preparativa, realizando-se, para ambas, injeções de 100 μL de amostra para cada corrida cromatográfica. Os espectros de massa de alta resolução com ionização por electrospray (EMAR-IES) e de baixa resolução em tandem (MSn) foram obtidos em equipamentos Bruker modelos micrOTOF II e Ion Trap-amaZonX, respectivamente, os dois operando no modo positivo. Os espectros de RMN de 1H, 13C e bidimensionais foram adquiridos no espectrômetro Bruker AVANCE III HD (400 MHz e 100 MHz para 1H e 13C, respectivamente) usando CDCl3 e CD3OD como solvente e CHCl3 (δH 7,24 e δC 77,0) e CH3OH (δH 3,30 e δC 49,0) residual como padrão interno. As análises de infravermelho foram realizadas nos espectrômetros Rayleigh WQF-510 FT-IR e PerkinElmer Frontier FT-IR, com pastilhas de KBr e número de onda medido em cm-1. Material vegetal As raízes de Anaxagorea dolichocarpa Sprague & Sandwith foram coletadas em Cruz do Espírito Santo, Paraíba, Brasil em dezembro de 2010. O registro de acesso ao Sistema Nacional de Gestão do Patrimônio Genético e do Conhecimento Tradicional Associado (SisGen) foi obtido sob o número AE4B71A. A identificação botânica foi realizada pela Drª Maria de Fátima Agra e uma exsicata encontra-se depositada no Herbário Prof. Lauro Pires Xavier (JPB), no Centro de Ciências Exatas e da Natureza, registrada como AGRA & GÓES 5543. Extração e isolamento As raízes de A. dolichocarpa foram secas em estufa de ar circulante a 45 °C por 92 h e em seguida trituradas produzindo 700,0 g de pó. Esse material foi, então, submetido ao processo de extração por maceração com etanol 95% por 72 h, em cinco repetições. A solução extrativa obtida foi concentrada em evaporador rotativo a 45 °C, resultando em 77,6 g de extrato etanólico bruto (EEB). Uma alíquota de 76,1 g do EEB foi inicialmente submetida a uma extração com hexano, para separação dos constituintes com baixa polaridade, permanecendo em contato com o solvente, em agitação mecânica, por cerca de 5 minutos. A seguir o material foi filtrado e o resíduo insolúvel pesando 66,4 g foi submetido ao procedimento de extração ácido-base para alcaloides,26 fornecendo 2,3 g de fração alcaloídica (FA). Uma amostra da FA (2,1 g) foi fracionada por CLMP, usando sílica gel previamente tratada com NaHCO3 10%,26 e gradientes dos solventes Hex-CHCl3 (100:0 → 0:100), CHCl3-AcOEt (100:0 → 0:100) e AcOEt:MeOH (100:0 → 3:7), resultando em 116 frações de 50 mL que após análise em CCD foram agrupadas em 20 (FA1-FA20). A fração FA2 (95,0 mg), eluída em Hex-CHCl3 (8:2), foi submetida a um novo fracionamento por CLMP, com sílica gel tratada com NaHCO3 10%, e gradiente de Hex-CHCl3 (100:0 → 0:100) e CHCl3-AcOEt (100:0 → 0:100), fornecendo 9 subfrações (FA2A-FA2I). A subfração FA2D (15,8 mg), obtida em Hex-CHCl3 (95:5), foi purificada por CLAE semi-preparativa com sistema gradiente de água (0,1% de ácido fórmico) e MeOH (95:5 → 0:100, em 60 minutos) a 254 nm, dando origem ao composto 5 (2,0 mg), eluído em 74% de MeOH. A subfração FA2E (23,4 mg), também obtida em Hex-CHCl3 (95:5), foi purificada por CLAE semi-preparativa usando o seguinte gradiente de eluição: solvente A = água (0,1% de ácido fórmico); solvente B = MeOH; sistema de eluição = 0-40 min (20-70% de B); 40-50 min (70% de B); e 50-65 min (70-100% de B); a 254 nm; fornecendo as substâncias 2 (0,6 mg), 3 (4,7 mg) e 4 (1,5 mg), eluídas em 80%, 72% e 70% de B, respectivamente. A fração FA7 (197,3 mg), eluída em Hex-CHCl3 (1:9), foi purificada por CLAE preparativa com o gradiente de eluição: solvente A = água (0,1% de ácido fórmico); solvente B = MeOH; sistema de eluição = 0-45 min (0-65% de B); 45-55 min (65% de B); e 55-90 min (65-100% de B); a 254 nm; isolando 6 (1,5 mg), obtido em 65% de B. A fração FA8 (295,5 mg), eluída em CHCl3-AcOEt (9:1), foi separada em 11 frações (FA8A-FA8M) por CLMP, com sílica gel tratada com NaHCO3 10%, e gradiente de Hex-CHCl3 (100:0 → 0:100), CHCl3-AcOEt (100:0 → 0:100) e AcOEt:MeOH (100:0 → 3:7). A subfração FA8G (121,4 mg), eluída em CHCl3, foi submetida a CLAE semi-preparativa com o gradiente de eluição: solvente A = água (0,1% de ácido fórmico); solvente B = MeOH; sistema de eluição = 0-20 min (30-40% de B); 20-65 min (40% de B); e 65-100 min (40-100% de B); a 254 nm; obtendo-se o composto 1 (3,6 mg), obtido em 45% de B. Avaliação da citotoxicidade Para a avaliação da citotoxicidade dos compostos 1 - 5 foram utilizadas as linhagens de células HCT-116 (carcinoma colorretal humano) e L929 (fibroblastos murinos não-tumorais). As células foram cultivadas em meio RPMI 1640 suplementado com 10% de soro bovino fetal, 100 U mL-1 de penicilina e 100 μg mL-1 de estreptomicina a 37 °C em uma atmosfera umidificada de 5% CO2. As células foram semeadas em placas de 96 poços em uma densidade de 3 × 105 células/poço. Após um período de 24 h, as células foram incubadas com os compostos 1 - 5 (50 µmol L-1) dissolvidos em DMSO (0,4%), por 72 h. Então, o sobrenadante foi descartado e a solução de MTT [brometo de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio] (5 mg mL-1) foi adicionada e incubada por mais 3 h. O sal de formazam depositado foi dissolvido em dodecil sulfato de sódio (SDS) (100 μL).27 O controle positivo foi o DMSO (20%). A densidade ótica foi medida em um leitor de microplacas (Synergy HT, BioTek®) no comprimento de onda de 570 nm. Os resultados foram expressos como média ± e.p.m. (erro padrão da média) de quatro replicatas e foram comparados por análise de variância (ANOVA - one way), seguido do pós-teste de Tukey. Os resultados foram considerados significativos quando p < 0,05.
MATERIAL SUPLEMENTAR Os espectros no IV, de RMN uni e bidimensionais, EMAR-IES e de baixa resolução MSn estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem às agências de Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (código de financiamento 001) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo suporte financeiro e bolsas de pesquisa, ao Laboratório Multiusuário de Caracterização e Análise (LMCA-UFPB) e ao Centro Analítico de Instrumentação da Universidade de São Paulo (Central Analítica IQ-USP) pela aquisição dos espectros, e à Rede Norte-Nordeste de Fitoprodutos (INCT-RENNOFITO) pela colaboração.
REFERÊNCIAS 1. Guo, X.; Tang, C. C.; Thomas, D. C.; Couvreur, T. L. P.; Saunders, R. M. K.; Sci. Rep. 2017, 7, 1. 2. Couvreur, T. L. P.; Helmstetter, A. J.; Koenen, E. J. M.; Bethune, K.; Brandão, R. D.; Little, S. A.; Sauquet, H.; Erkens, R. H. J.; Front. Plant Sci. 2019, 9, 1941. 3. Lopes, J. C.; Mello-Silva, R.; Rev. Bras. Frutic. 2014, 36, 125. 4. Lobão, A. Q.; de Araujo, D. S. D.; Kurtz, B. C.; Rodriguésia 2005, 56, 85. 5. Lúcio, A. S. S. C.; Almeida, J. R. G. S.; Da-Cunha, E. V. L.; Tavares, J. F.; Barbosa Filho, J. M. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., ed.; Academic Press: San Diego, 2015, vol. 74, pp. 233-409. 6. González-Esquinca, A. R.; De-La-Cruz-Chacón, I.; Castro-Moreno, M.; Orozco-Castillo, J. A.; Riley-Saldaña, C. A.; Rev. Bras. Frutic. 2014, 36, 01. 7. Teichert, H.; Dötterl, S.; Gottsberger, G.; Plant Syst. Evol. 2011, 291, 25. 8. Maas, P. J. M.; Westra, L. Y. T.; Bot. Jahrb. Syst. 1984, 105, 73. 9. Maas, P. J. M.; Westra, L. Y. T.; Bot. Jahrb. Syst. 1985, 105, 145. 10. Scharaschkin, T.; Doyle, J. A.; Syst. Bot. 2005, 30, 712. 11. Lúcio, A. S. S. C.; Almeida, J. R. G. S.; Barbosa-Filho, J. M.; Pita, J. C. L. R.; Branco, M. V. S. C.; Melo, M. F. F.; Agra, M. F.; Da-Cunha, E. V. L.; Da Silva, M. S.; Tavares, J. F.; Molecules 2011, 16, 7125. 12. Pinheiro, R. S.; Rabelo, S. V.; De Oliveira, A. P.; Guimarães, A. L.; De Moraes-Filho, M. O.; Da Costa, M. P.; Pessoa, C. Ó.; Lúcio, A. S. S. C.; Almeida, J. R. G. S.; Trop. J. Pharm. Res. 2016, 15, 793. 13. Sabphon, C.; Temkitthawon, P.; Ingkaninan, K.; Sawasdee, P.; Nat. Prod. Commun. 2015, 10, 301. 14. de Díaz, A. M. P.; Phytochemistry 1997, 44, 345. 15. Husain, K.; Zakaria, S. M.; Lajis, N. H.; Shaari, K.; Ismail, I. S.; Israf, D. A.; Paetz, C.; Phytochem. Lett. 2012, 5, 788. 16. Almeida, J. R. G. S.; Lúcio, A. S. S. C.; Barbosa-Filho, J. M.; Agra, M. F.; da Silva, M. S.; da Cunha, E. V. L.; Uchoa, D. E. A.; Braz-Filho, R.; Biochem. Syst. Ecol. 2007, 35, 456. 17. Hocquemiller, R.; Rasamizafy, S.; Moretti, C.; Jacquemin, H.; Cavé, A.; Planta Med. 1981, 41, 48. 18. Amaral, R. G.; dos Santos, S. A.; Andrade, L. N.; Severino, P.; Carvalho, A. A.; Clin. Oncol. 2019, 4, 1562. 19. Guinaudeau, H.; Leboeuf, M.; Cavé, A.; J. Nat. Prod. 1994, 57, 1033. 20. Taylor, W.; Aust. J. Chem. 1984, 37, 1095. 21. da Silva, F. M. A.; Koolen, H. H. F.; de Almeida, R. A.; de Souza, A. D. L.; Pinheiro, M. L. B.; Costa, E. V.; Quim. Nova 2012, 35, 944. 22. Stévigny, C.; Jiwan, J. H.; Rozenberg, R.; de Hoffmann, E.; Quetin-Leclercq, J.; Rapid Commun. Mass Spectrom. 2004, 18, 523. 23. Carroll, A.; Taylor, W.; Aust. J. Chem. 1991, 44, 1615. 24. Hang, N. T. M.; Oanh, N. T. T.; Hue, C. T.; Tung, T. H.; Thoa, H. T.; Thanh, L. N.; Giap, T. H.; Dung, N. A.; Van Hung, N.; Van Minh, C.; Vietnam J. Chem. 2015, 53, 73. 25. Hufford, C. D.; Zjawiony, J. K.; Srivastava, A. R.; Clark, A. M.; Heterocycles 1994, 39, 779. 26. Costa, E. V.; Pinheiro, M. L. B.; Xavier, C. M.; Silva, J. R. A.; Amaral, A. C. F.; Souza, A. D. L.; Barison, A.; Campos, F. R.; Ferreira, A. G.; Machado, G. M. C.; Leon, L. L. P.; J. Nat. Prod. 2006, 69, 292. 27. Mosmann, T.; J. Immunol. Methods 1983, 65, 55. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access