
 |
 |
 |
 |
 |
Revisão
|
|
| Evolução dirigida de enzimas: pequenas modificações, melhores biocatalisadores Directed evolution of enzymes: small changes, better biocatalysts |
|
Iris S. Teixeira; Cintia D. F. Milagrea*
Instituto de Química, Universidade Estadual Paulista, 14800-060 Araraquara - SP, Brasil Recebido em 05/11/2019 *e-mail: cintia.milagre@unesp.br Biocatalysis is now a mature field, both in the laboratory and industrial scale. This approach counts on enzyme high selectivity, biodegradability, elegant control over the outcome of reaction conditions and as a possible solution to address some challenges in Green Chemistry faced by synthetic organic chemists. However, many wild-type ready-to-use enzymes were not designed to accommodate the organic substrates needed by today's demands. Or they do not fit a predefined optimum process condition or even, useful reactions are not accessible because there is no enzyme counterpart for metallo- and organocatalysts. This review will give a brief introduction on the protein engineering tools (directed evolution, rational design, and semi-rational design) and will focus on directed evolution of enzymes, their impact in chemistry with examples from production of commodity chemicals to pharmaceutical intermediates and its potential as a tool to achieve greener chemistry criteria will be discussed. INTRODUÇÃO O planeta Terra está ameaçado por nós, seres humanos. O consumo pouco consciente dos recursos naturais que são limitados, aliado à crescente geração de resíduos, muitas vezes tóxicos, coloca em risco a vida e a biodiversidade de todos os seres que habitam este planeta. Desse desequilíbrio sem precedentes na história da Terra, surgiu a Química Verde, que preconiza a prevenção da geração de resíduos e não simplesmente o manejo dos resíduos já gerados e o uso eficiente dos recursos atualmente disponíveis.1-6 Para atender às demandas atuais da sociedade e das legislações em vigor, o setor industrial tem passado por uma mudança de paradigma na manufatura de seus produtos e está cada vez mais atento às questões relacionadas à sustentabilidade ambiental de seus processos químicos vigentes e daqueles que ainda estão por vir. No cenário da síntese orgânica, os químicos dispõem de um arsenal de catalisadores para utilizar em suas rotas sintéticas como, por exemplo, os catalisadores metálicos, organometálicos, organocatalisadores, catalisadores de transferência de fase e biocatalisadores, com cada um deles oferecendo vantagens e desvantagens.7-11 Do ponto de vista da sustentabilidade ambiental, o uso de biocatalisadores traz inúmeras vantagens, como pode ser observado na Figura 1, mas isso não é uma prerrogativa apenas da biocatálise.12 Realizar reações em condições catalíticas, independentemente do tipo de catalisador empregado, é por si só uma vantagem sobre as reações em condições estequiométricas, conforme preconizado no 9º princípio da Química Verde (9 - Catálise: utilizar catalisadores que ajudem a aumentar a seletividade das reações, diminuir a geração de resíduos, reduzir os tempos reacionais e demandas de energia).
 Figura 1. A relação entre a Biocatálise e os 12 princípios da Química Verde
Diante dessa variedade de opções de catalisadores, por que escolher os biocatalisadores dentre os demais? As enzimas são os catalisadores que a natureza desenvolveu ao longo de bilhões de anos e que permitem que a vida seja possível, através de rotas biossintéticas eficientes e seguras. Entretanto, elas evoluíram para catalisar determinadas reações com substratos naturais específicos e em condições reacionais fisiológicas, características essas muito diferentes daquelas muitas vezes exigidas para a produção, em escala industrial, dos compostos orgânicos de interesse comercial. Por exemplo, para que uma enzima seja utilizada industrialmente, ela precisa se manter ativa em elevadas concentrações de substrato e produto (geralmente 50-400 g L-1).13 Esses valores excedem em média cerca de cem vezes as condições naturais. Além disso, as enzimas são conhecidas por utilizarem água como solvente; entretanto, muitos compostos orgânicos utilizados como substratos são hidrofóbicos. Nesses casos, é necessário adicionar pequenas quantidades de solventes orgânicos (cossolventes) para auxiliar na solubilização do substrato e, para tanto, as enzimas precisam ser tolerantes a estes solventes orgânicos. As enzimas utilizadas industrialmente ainda precisam tolerar as elevadas concentrações salinas, a heterogeneidade da temperatura, pH e concentrações de substrato e produto nos reatores, em função das limitações de transferência de massa e calor em misturas em grande escala, dentre vários outros fatores operacionais. Portanto, é essencial que metodologias para a produção de enzimas mais robustas sejam desenvolvidas, só assim o uso de biocatalisadores em escala industrial continuará a se expandir.14,15 A engenharia de proteínas consiste na modificação de proteínas, geralmente através da substituição de resíduos de aminoácidos por outros, visando a melhoria das propriedades dessa proteína. Atualmente, os avanços científicos na área de engenharia de proteínas permitem que seja possível modular ou mesmo criar novos biocatalisadores adequados para aplicações industriais (Quadros 1 e 2).16,17

FERRAMENTAS PARA MODIFICAÇÃO DE ENZIMAS As estratégias disponíveis para a modificação de enzimas podem ser divididas entre três categorias, sendo elas evolução dirigida, design racional e design semirracional, como ilustrado na Figura 2. Essas estratégias diferem entre si principalmente em dois aspectos: o grau de necessidade de conhecimento prévio da estrutura tridimensional da enzima e do número de variantes enzimáticos gerados.20 Esse último fator é importante visto que implicará diretamente em maior ou menor demanda nas etapas de ensaios de triagens enzimáticas para seleção dos candidatos mais promissores.
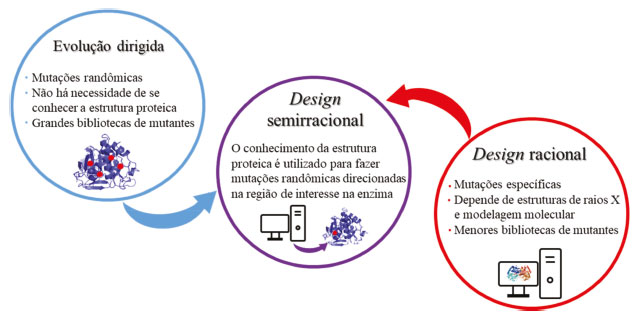 Figura 2. Estratégias para obtenção de modificações nas enzimas: evolução dirigida, design semirracional e design racional
Nas técnicas de evolução dirigida são realizadas mutações randômicas na sequência primária de aminoácidos da enzima.20 Como essas mutações são introduzidas de forma aleatória, essa abordagem possui como principal vantagem a não necessidade de conhecimentos prévios da estrutura terciária da proteína, como por exemplo, estruturas de raios X.21 Cada mutante recém-criado pela técnica de evolução dirigida é submetido a ensaios de triagem enzimática para a detecção da propriedade de interesse. Em seguida, os melhores hits são selecionados e submetidos a novos ciclos de mutações, a fim de aperfeiçoar a característica escolhida, como, por exemplo, atividade enzimática, termoestabilidade, seletividade, dentre outras.22 Uma vez que essa técnica gera bibliotecas com um grande número de enzimas mutantes, tipicamente números da ordem de 103-106, as etapas de triagem e seleção dos melhores mutantes demandam grandes esforços.23 O design racional, por sua vez, é baseado em mutações pontuais e específicas em resíduos de aminoácidos que se sabe ou se espera que estejam diretamente envolvidos nas funções que se deseja otimizar.24 Essa abordagem depende de conhecimento prévio da estrutura terciária da enzima, fornecido por estruturas geradas a partir de cristalografia de raios X. Além disso, ainda são necessárias técnicas de bioinformática, modelagem molecular e química computacional para que possam ser previstas as relações entre estrutura-atividade da enzima para a criação dos melhores mutantes.25 Devido a essa abordagem ser mais pontual, ela leva à criação de bibliotecas com menor número de mutantes e que, portanto, podem ser triados mais rapidamente. Ambas as estratégias descritas acima, design racional e evolução dirigida, possuem limitações. No caso da evolução dirigida, o tamanho das bibliotecas de mutantes dificulta a triagem para escolha das melhores variantes enzimáticas, e no caso do design racional, as informações necessárias nem sempre estão disponíveis. Uma combinação de ambas as estratégias é, muitas das vezes, a melhor alternativa para superar as limitações de cada uma delas.20 No design semirracional são utilizadas informações estruturais conhecidas, assim como algoritmos, para predizer quais substituições de aminoácidos serão benéficas, e essas informações são utilizadas para uma randomização direcionada em certas áreas alvo da proteína.26 As discussões a seguir focarão na estratégia de evolução dirigida de enzimas e exemplos ilustrando o seu potencial de aplicações. Evolução dirigida (mutagênese randômica) As estratégias de evolução dirigida, também conhecida como mutagênese randômica, mimetizam uma evolução darwiniana in vitro, usando ciclos de mutagênese, recombinação e seleção ou triagem da enzima com as propriedades desejadas (Figura 3).21
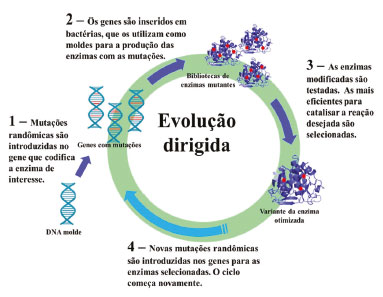 Figura 3. Fluxo de trabalho durante o desenvolvimento de processos de evolução dirigida
Entre as técnicas de evolução dirigida, as mais comumente utilizadas são as de reação da cadeia da polimerase propensa a erros (epPCR, error-prone polymerase chain reaction), embaralhamento de DNA (DNA shuffling) e mutagênese por saturação. O desenvolvimento da técnica de epPCR no final dos anos 1980 e início dos anos 1990 foi um importante marco na história da evolução dirigida.27 Essa técnica consiste em uma reação em cadeia da polimerase (PCR) propensa a erros, ou seja, serão introduzidas mutações pontuais aleatórias nos genes clonados durante a PCR.28 Para que os erros aleatórios aconteçam são realizadas algumas modificações nas condições reacionais que resultem no aumento da taxa de erros na inserção dos oligonucleotídeos durante a síntese de DNA pela enzima DNA polimerase. Tudo isso sem afetar significativamente o nível de amplificação alcançado na PCR.28 Entre as modificações nas condições reacionais mais utilizadas podemos citar o aumento na concentração de magnésio, suplementação do meio reacional com manganês ou ainda o uso de análogos mutagênicos dos desoxirribonucleotídeos trifosfatos (dNTPs). Com essas modificações a fidelidade no pareamento das bases é diminuída, aumentando as taxas de mutação em 10-4~10-3 por base replicada por ciclo.29 A técnica DNA shuffling foi desenvolvida no início dos anos 1990 por Stemmer e é baseada em recombinação de sequências de DNA.30 Um pool de genes homólogos com diferentes mutações pontuais é fragmentado com uma enzima DNase e, então, é permitido que os fragmentos se anelem um ao outro de maneira randômica em uma PCR sem adição de primers.31 Outra técnica bastante utilizada é a mutagênese por saturação, também conhecida como mutagênese cassete, e que consiste na randomização de aminoácidos em uma ou mais posições da estrutura primária da enzima. Neste caso, o códon ou conjunto de códons responsável pela produção de um determinado aminoácido é substituído de forma que nas posições pré-selecionadas para a mutação exista uma variante para cada um dos 20 aminoácidos proteinogênicos.32 As técnicas de evolução dirigida revolucionaram a forma como as enzimas podem ser moduladas. Atualmente, estas técnicas são usadas em centenas de laboratórios e empresas espalhadas pelo mundo, para produzir uma variedade de compostos como, por exemplo, detergentes, biocombustíveis, ingredientes ativos de medicamentos, vacinas e no desenvolvimento de novos materiais. As enzimas geradas a partir da evolução dirigida têm substituído reagentes químicos tóxicos em muitos processos industriais e contribuído para o desenvolvimento de uma química mais ambientalmente sustentável. O potencial de benefícios e retorno para a sociedade possibilitados pelo desenvolvimento destas técnicas é evidenciado pelo Prêmio Nobel de Química 2018, que laureou Frances H. Arnold com metade do prêmio pelas suas contribuições na área de "Evolução Dirigida de Enzimas" e a outra metade que foi dividida entre George P. Smith e Sir Gregory P. Winter "pelo desenvolvimento da tecnologia phage display de peptídeos e anticorpos".33,34 A seguir serão abordados alguns exemplos de como as enzimas têm sido utilizadas em escala industrial para a produção de ingredientes ativos farmacêuticos, na produção de biocombustíveis, na valorização da biomassa e as novas perspectivas geradas a partir da criação de enzimas que catalisam reações conhecidas pelos químicos orgânicos sintéticos, porém inéditas nos sistemas naturais. A intenção deste artigo de revisão é apresentar alguns exemplos pontuais do que vem acontecendo em termos de otimização de biocatalisadores através de metodologias de evolução dirigida de enzimas e que possibilitam o seu uso em escala industrial. Não há qualquer pretensão de realização de uma análise exaustiva dos inúmeros exemplos bem-sucedidos descritos na literatura.35,36 Para facilitar a compreensão, a Figura 4 apresenta um guia de como as mutações devem ser lidas. Cada aminoácido será identificado com sua respectiva abreviatura no sistema de uma letra. A primeira letra (aquela que aparece à esquerda) é referente ao resíduo de aminoácido na enzima selvagem, ou seja, antes de sofrer qualquer mutação. O número que aparece entre as duas letras indica a posição que o resíduo de aminoácido ocupa na estrutura primária da enzima e, por fim, a última letra (à direita) indica o resíduo de aminoácido que substituiu o resíduo original. No caso da Figura 4, lê-se: o resíduo de alanina da posição 284 foi substituído por um resíduo de prolina.
 Figura 4. Guia de como ler as mutações introduzidas nas enzimas selvagens
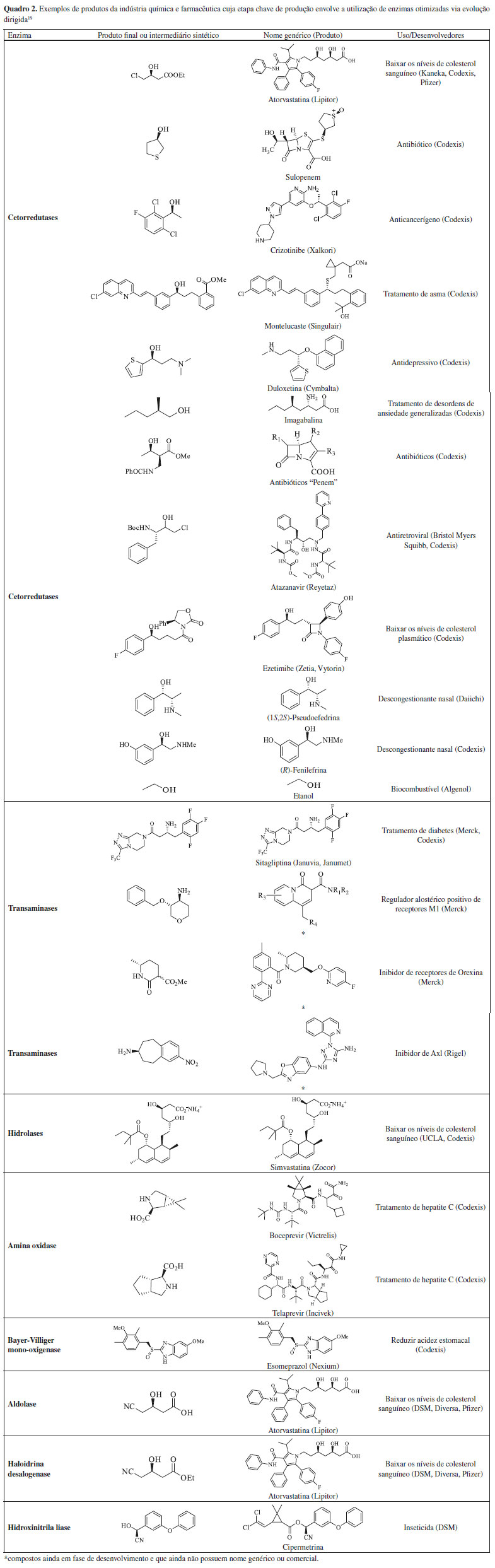
EXEMPLOS DE APLICAÇÕES DE ENZIMAS MODIFICADAS POR EVOLUÇÃO DIRIGIDA No início da implementação da biocatálise, quer fosse em escala laboratorial, piloto ou industrial, era necessário projetar processos que respeitassem as propriedades intrínsecas do biocatalisador. As principais limitações apresentadas pela maioria das enzimas selvagens, ou seja, de ocorrência natural, eram sua baixa termoestabilidade, atividade catalítica, seletividade, tolerância a solventes orgânicos e elevada especificidade de substrato. As conquistas alcançadas a partir do advento da engenharia de proteínas facilitaram significativamente a vida dos químicos e engenheiros químicos devido à possibilidade de se desenhar enzimas que atendessem às necessidades dos processos industriais.33 Centenas de processos biocatalíticos, alguns deles exemplificados no Quadro 2, operam em escala industrial comprovando a sua viabilidade e exequibilidade econômico-ambiental.37 Dentre esses processos biocatalíticos, a maior parte está concentrada na indústria farmacêutica, embora na última década tenha havido um crescente interesse em uma série de outras áreas de aplicação.38 Evolução dirigida de enzimas na indústria farmacêutica Síntese da atorvastatina A atorvastatina (Quadro 2), utilizada para diminuir os níveis de colesterol sanguíneo, é um dos medicamentos mais comercializados e, ao longo dos últimos anos tem se mantido na lista dos cinco medicamentos mais consumidos no mundo, rendendo à Pfizer 2 bilhões de dólares em 2019.39-41 Um dos grupos farmacofóricos da atorvastatina é sua cadeia lateral 1 que contém dois centros estereogênicos adjacentes (Esquema 1). As empresas Kaneka, Ciba, Dow Pharma (antiga Diversa) e Codexis além de inúmeros grupos de pesquisa acadêmicos voltaram seus esforços para a síntese da cadeia lateral da atorvastatina usando catalisadores químicos clássicos e biocatalisadores. Há várias abordagens de rotas biocatalíticas que utilizam diferentes classes de enzimas, provenientes tanto de linhagens selvagens quanto de linhagens modificadas geneticamente.42,43 Serão apresentados a seguir três exemplos que foram selecionados por dois motivos: (i) utilizam classes de enzimas diferentes e (ii) são as metodologias de manufatura em escala industrial (Esquema 1).
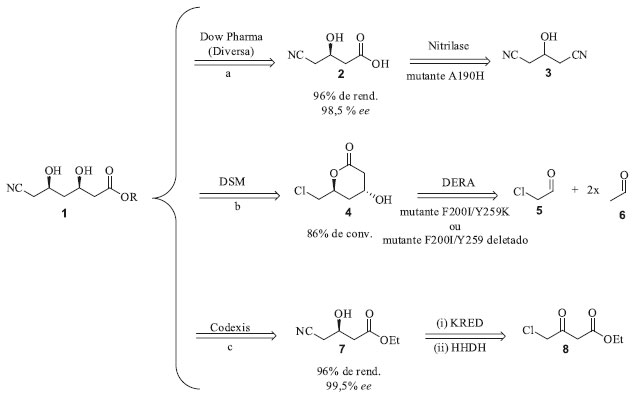 Esquema 1. Etapas biocatalíticas para a síntese industrial da cadeia lateral da atorvastatina
O primeiro caso a ser abordado envolve a etapa sintética utilizada pela companhia Dow Pharma para a produção do intermediário quiral ácido (R)-3-ciano-3-hidroxibutírico 2, que é baseada na dessimetrização do substrato pró-quiral 3-hidroxiglutaronitrila 3 catalisada pela enzima nitrilase (E.C. 3.5.5.1), conforme apresentado no Esquema 1a.44,45 O início desse trabalho se deu pela triagem de bibliotecas genômicas preparadas a partir de amostras isoladas do meio ambiente, das quais foram descobertas aproximadamente 200 nitrilases capazes de hidrolisar nitrilas diretamente nos respectivos ácidos carboxílicos em condições brandas de reação e de forma seletiva. Dentre essas, a nitrilase mais eficiente foi capaz de utilizar uma concentração inicial de 3 igual a 100 mmol L-1, com conversão total no produto esperado e 95% de ee após 24 horas. Entretanto, essa concentração inicial de substrato não é aplicável a processos industriais. Nos experimentos que se seguiram foi observado que o aumento da concentração inicial de substrato (0,5; 1,0; 2,0 e 3 mol L-1) resultou na diminuição do ee (92,1; 90,7; 89,2 e 87,6 respectivamente). De modo a contornar esses problemas, essa nitrilase selvagem foi submetida a ciclos de evolução dirigida utilizando a metodologia de mutagênese por saturação. A sequência primária dessa nitrilase era formada por 330 aminoácidos, resultando, portanto, em uma biblioteca contendo 10.528 enzimas mutantes. Os ensaios enzimáticos para cada mutante foram realizados em triplicata, totalizando em 31.584 análises. O ensaio de triagem enzimática de alta eficiência utilizado pela Dow Pharma consistiu no monitoramento das reações enzimáticas por espectrometria de massas, utilizando o substrato quiral (R)-3 enantiomericamente puro e marcado isotopicamente com 15N. Os candidatos mais promissores identificados nessa primeira triagem foram reavaliados por cromatografia a gás utilizando o substrato 3 não marcado isotopicamente. Esses estudos identificaram que os resíduos de aminoácidos A190 e F191 eram os responsáveis chave por influenciar a enantiosseletividade da reação. Dentre os vários mutantes gerados a partir de substituições nesses resíduos, o mutante A190H se mostrou o mais ativo e enantiosseletivo, catalisando a dessimetrização enantiosseletiva da dinitrila 3 nas seguintes condições reacionais: nitrilase A190H (30 mg) e a dinitrila 3 (3 mol L-1, 9 mmol, 1 g) dissolvidos em 2,03 mL de tampão fosfato. Após 15 horas de reação sob agitação a 20 °C foi recuperado o produto (R)-2 (1,1 g, 96% de rendimento e 98,5% ee) com produtividade volumétrica de 619 g L-1 dia. A evolução dirigida deste biocatalisador propiciou uma melhora considerável em relação à enzima selvagem que só era capaz de fornecer o produto com 88% ee, após 24 horas de reação e com concentração inicial do substrato 3 igual a 2,25 mol L-1. Essa mutação resultou em um aumento de 10% na enantiosseletividade, e possibilitou que a reação ocorresse com concentração inicial de substrato igual a 3 mol L-1, o que é relevante do ponto de vista industrial. Apesar da estratégia de mutagênese por saturação levar à construção de bibliotecas muito grandes e demandar elevados esforços na triagem dos clones, ela foi fundamental para a otimização deste biocatalisador, uma vez que as nitrilases disponíveis nos bancos de dados daquela época não continham o aminoácido histidina na posição 190 e, portanto, a utilização de técnicas como epPCR e recombinação gênica não levariam a esse mutante mais ativo. O segundo exemplo de produção biocatalítica da atorvastatina envolve a estratégia utilizada pela DSM Pharma Chemicals que utiliza aldolases, mais especificamente a DERA (aldolase 2-desoxi-D-ribose-5-fosfato, E.C. 4.1.2.4) (Esquema 1b). Essa aldolase catalisa a formação do (3R,5S)-6-cloro-2,4,6-tridesoxi-hexapiranosídeo através da formação de uma nova ligação C-C resultante da reação aldólica assimétrica tandem do cloro-acetaldeído 5 com 2 mols de acetaldeído 6, dois compostos de baixo custo e disponíveis em grande escala.46 As barreiras que tiveram de ser superadas pela DSM para a produção industrial deste intermediário foram a baixa afinidade da enzima DERA pelo substrato 5 e, a inativação rápida e irreversível da enzima também por 5, em concentrações de relevância industrial. Em primeiro lugar foi realizada a evolução dirigida da DERA visando aumentar a aceitação desta enzima pelo substrato 5 através da técnica epPCR e que, consequentemente levou a introdução de mutações pontuais e aleatórias. Em seguida as melhores variantes foram subclonadas através de recombinações sítio específicas. Através dessa abordagem, foram obtidos no total 10.000 enzimas mutantes DERA que foram avaliados quanto à sua resistência ao substrato não-natural 5. Destes, 63 mutantes foram identificados com atividades superiores à enzima selvagem sendo que o melhor mutante (DERAVar9) carregava uma única mutação F200I, e que resultou em um aumento de atividade de 14 vezes. A análise das estruturas de raios X da DERA e os clones de variantes de DERA revelaram que os resíduos F200, I166 e M185 juntos formavam um pequeno cluster hidrofóbico próximo às cadeias laterais dos resíduos K167 e K201 pertencentes ao sítio ativo da enzima e responsáveis pela produtividade catalítica e resistência a 5. Além disso, os experimentos de mutação sítio dirigidas que combinaram a substituição de F200I com a deleção Y259 na região C-terminal ou a substituição Y259K mostraram que essas mutações agem de modo sinérgico. Esse mutante foi utilizado para a síntese estereosseletiva do (3R,5S)-6-cloro-2,4,6-tridesoxi-hexapiranosideo 4 a partir de 5 e 6, em condições reacionais de relevância industrial. O terceiro exemplo de produção biocatalítica da atorvastatina apresenta a primeira estratégia adotada pela Codexis e que utilizou a técnica de DNA shuffling para aumentar a estabilidade e atividade de duas enzimas, cetorredutase (KRED) e haloidrina desidrogenase (HHDH), responsáveis por catalisar a redução assimétrica de 8 gerando o intermediário 4-cloroacetoacetato de etila e subsequente substituição do cloro pelo grupo ciano para formação do intermediário sintético (S)-3-hidroxi-4-cianoacetoacetato de etila 7 (Esquema 1c).47 Após quatro ciclos de DNA shuffling foi obtido um clone de KRED com atividade 7 vezes maior que a da enzima selvagem e retenção da enantiosseletividade > 99,5%. Em relação à enzima HHDH foi necessário otimizar tanto sua baixa atividade e estabilidade frente a reação de cianação quanto sua elevada inibição pelo produto 7. Após inúmeros ciclos iterativos de DNA shuffling, com incorporação de 38 mutações no total, a inibição pelo produto foi contornada e a atividade enzimática aumentou em > 2.500 vezes quando comparada à enzima selvagem. Esse processo enzimático teve sua escala aumentada para reatores de 2.000 L, com o aumento de 1.000 vezes para a produtividade volumétrica da reação de biorredução e 4.000 vezes para a reação de cianação. Esse processo da Codexis é operado pela companhia Lonza para manufatura do intermediário sintético 7 que é utilizado pela Pfizer para a produção da atorvastatina de cálcio. O desenvolvimento destes novos biocatalisadores foi responsável pela condução da reação em condições muito mais sustentáveis (Fator E = 5,8 sem incluir a água e 18 se a água é incluída) que as utilizadas anteriormente e culminou com a premiação da Codexis no Presidential Green Chemistry Challenge: 2006, categoria Greener Reaction Conditions Award.48 Infelizmente os dados das sequências dos melhores mutantes não foram disponibilizados pela Codexis, até o momento dessa publicação. Síntese da sitagliptina Um dos processos biocatalíticos vigentes industrialmente mais bem documentado na literatura científica e que utiliza biocatalisadores modificados através de evolução dirigida é a manufatura da sitagliptina (Quadro 2).49 A sitagliptina é um fármaco usado no tratamento de diabetes tipo II, comercializada sob o nome de Januvia® e Janumet® pela Merck.50 Em uma colaboração com a empresa Codexis, foram realizadas mutações no sítio catalítico da enzima ω-transaminase (ATA-117) de Arthrobacter sp. de modo que ela aceitasse substratos volumosos em seu sítio catalítico. As ω-transaminases são responsáveis por catalisar reações de aminação redutiva de cetonas e aldeídos na presença de grupos doadores de amino. O sítio catalítico de uma transaminase é constituído de dois bolsões, um maior e outro menor, e estudos de ancoragem molecular indicaram que a enzima ATA-117 não seria capaz de se ligar à cetona precursora prositagliptina 9 (Esquema 2) devido a impedimentos estéricos no bolsão menor e potenciais interações indesejadas no bolsão maior.50
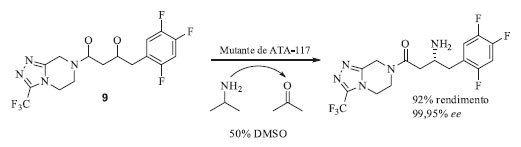 Esquema 2. Etapa biocatalítica na síntese da sitagliptina
Ao todo, foram necessárias 27 mutações, de modo a acomodar a cetona precursora prositagliptina no sítio catalítico da enzima.51 As 27 mutações foram obtidas através de triagens de várias bibliotecas que incluíam o aumento do bolsão menor baseado na estrutura do substrato (4 mutações), mutagênese por saturação do bolsão maior (5 mutações), nova mutagênese por saturação do bolsão menor (1 mutação), mutagênese por saturação de posições fora dos bolsões de ligação do substrato (2 mutações), mutações baseadas em bibliotecas de homologia (10 mutações) e mutagênese aleatória (5 mutações). Os ciclos iniciais de mutação focaram na ligação da cetona precursora prositagliptina ao sítio ativo e sua conversão na sitagliptina. Em seguida, os pesquisadores se concentraram em aumentar a atividade enzimática nas condições do processo industrial, elevando a concentração do substrato de 2 para 100 g L-1, a concentração de isopropilamina (doador de grupo amino) de 0,5 para 1 mol L-1, o cossolvente DMSO de 5 para 50%, o pH de 7,5 para 8,5, e a temperatura de 22 para 45 °C.51 Nas condições reacionais ótimas, a enzima mutante converteu 200 g L-1 da cetona prositagliptina em sitagliptina com ee > 99,95%, usando 6 g L-1 de biocatalisador e 50% de DMSO, em uma etapa com rendimento de 92%. O catalisador quiral a base de ródio (Rh[Josiphos]) utilizado anteriormente para síntese da sitagliptina demandava pressões elevadas durante a hidrogenação (17 atm), além da necessidade de uma etapa de tratamento para remoção de traços de Rh, seguida por uma etapa de recristalização diastereoisomérica para aumentar o ee de 97% para ee > 99%. Quando comparado com o processo anterior foi observado que o processo biocatalítico resultou no aumento de 10% no rendimento global, aumento de 56% na produtividade (kg L-1 por dia), utilizando o maquinário já existente na empresa, e eliminação de 19% na formação total de resíduos.50,51 Em 2010 a Codexis recebeu novamente a premiação Presidential Green Chemistry Challenge: 2010, categoria Greener Reaction Conditions Award e em 2012 a agência norte-americana FDA (Food and Drug Administration) aprovou a implementação do processo biocatalítico para a manufatura do Januvia pela Merck.52,53 Este é outro exemplo de como a tecnologia de evolução dirigida de enzimas pode fornecer intermediários farmacêuticos bem controlados e com custos vantajosos. Evolução dirigida de enzimas na bioeconomia A utilização de fontes renováveis de matéria-prima é um dos pilares da Química Verde (3º princípio) e também da Economia Circular, que estão intimamente inter-relacionadas e que permitirão a construção de capital econômico, natural e social.54-57 Na manufatura, principalmente de commodities químicas e de combustíveis, existe uma necessidade pela substituição do uso dos recursos fósseis não renováveis, como petróleo, carvão e gás natural, por alternativas mais sustentáveis e de origem renovável.58,59 Nesse contexto, a aplicação de biomassa vegetal como matéria prima torna-se um substituto ideal aos recursos fósseis, já que ela é renovável e contribuirá para reduzir o impacto ambiental gerado pela indústria petroquímica. A biomassa utilizada pode ser de primeira geração, como lavouras de cana-de-açúcar, milho, soja e outras commodities alimentícias, ou de segunda geração, fazendo-se uso do material lignocelulósico, que é um resíduo da agroindústria. Essa última alternativa é ainda mais vantajosa pois agrega valor ao que antes era descartado como resíduo, principalmente em um país como o nosso em que a agroindústria desempenha papel chave na balança econômica.59-61 Produção de álcoois O Brasil é o segundo maior produtor de bioetanol do mundo e a produção brasileira desse combustível usando como matéria-prima a cana-de-açúcar ultrapassa seu consumo como combustível, assim outras fontes para seu uso são incentivadas.15,61,62 De fato, o melhor uso para o álcool seria como material de partida para obtenção de outros compostos químicos de maior complexidade.63 A versatilidade do etanol consiste no fato de que ele é facilmente convertido a outros blocos de construção, como etileno, butadieno, acetato de etila e acetaldeído, só para citar alguns (Esquema 3).
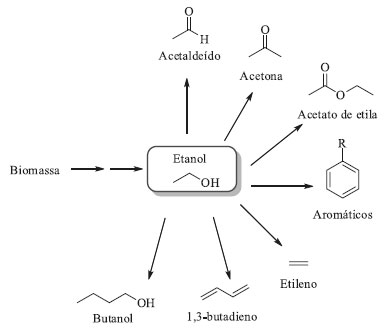 Esquema 3. Algumas moléculas produzidas a partir do etanol
Meinhold e colaboradores desenvolveram uma enzima mono-oxigenase citocromo P450BM3 para obtenção de etanol a partir do etano.64 A reação proposta pelos pesquisadores, oxidação do etano a etanol, não é catalisada naturalmente pela enzima BM-3 ou por nenhuma das enzimas da família das mono-oxigenases P450. Usando mutagênese por saturação, um dos mutantes obtidos, nomeado como 35-E11, foi capaz de produzir etanol com um número total de turnover (TTN) de 250, em uma taxa de nmol de etanol formado por minuto por nmol de enzima de 0,4 min-1. Além do etanol foi possível produzir propanol, a partir do propano, com um TTN de 6000, e converter octano a octanol com um valor de TTN de 8000. O octanol foi obtido com boa regiosseletividade, sendo 89% de octan-2-ol, e boa enantiosseletividade: 65% do isômero (S). Essa variante contou com um total de 17 mutações, sendo três delas no sítio ativo: A78F, A82S e A328F, todas elas trocando um resíduo de alanina por um aminoácido que contivesse uma cadeia lateral mais volumosa, capaz de reduzir o tamanho do sítio ativo e direcionar os alcanos pequenos mais próximo ao grupo heme, durante a catálise enzimática.64 Evolução dirigida de enzimas para aplicação em "reações não-naturais" Como vimos nas seções anteriores, a evolução dirigida de enzimas se tornou uma ferramenta importante para melhorar as propriedades intrínsecas dos biocatalisadores e que possibilitaram sua aplicação em processos industriais, atendendo às restrições operacionais exigidas pelos mesmos. Do ponto de vista retrossintético, existe um arsenal de biocatalisadores disponíveis e capazes de catalisar vários tipos de reações orgânicas tais como clivagem e formação de novas ligações C-C, C-heteroátomo, oxidações e reduções, dentre outras.65-67 Apesar de todas estas possibilidades, ainda existem várias reações orgânicas para as quais não existem biocatalisadores que possam substituir os catalisadores químicos. Apesar da reconhecida habilidade das enzimas para catalisar reações específicas, existe um grande número de enzimas capazes de se ligar a substratos diferentes e também catalisar reações diferentes de suas reações naturais, ainda que em baixos níveis de atividade. Essa capacidade das enzimas de catalisarem reações diferentes das quais elas foram especializadas é conhecida por promiscuidade enzimática.68 Aproveitando a promiscuidade enzimática e impulsionado pelos conhecimentos sobre mecanismos de reações orgânicas, atualmente é possível modificar enzimas com atividades já conhecidas para que as mesmas catalisem reações ditas "não-naturais", ou seja, aquelas reações que ainda não foram observadas ocorrendo na natureza.69 Não se trata da criação de novos sítios catalíticos a partir do zero, e sim do aproveitamento da existência de sítios catalíticos já existentes e ampliação do fornecimento de substratos que nunca estariam disponíveis naturalmente.70,71 Por exemplo, as enzimas do tipo mono-oxigenases P450 são metaloproteínas dependentes de ferro heme e são capazes de transferir espécies de oxigênio reativo aos seus substratos. Será que essas enzimas poderiam ser modeladas para que também pudessem transferir espécies reativas de carbono e nitrogênio, como os carbenos e nitrenos? Após a realização de vários experimentos a resposta é que sim. Essas reações, desconhecidas no mundo biológico, tornaram-se possíveis quando os pesquisadores forneceram às enzimas precursores sintéticos de carbenos e diazo e modularam catalisadores metálicos a partir de cofatores tipo ferro heme.72 Essa possibilidade propiciada pela evolução dirigida de enzimas abriu um novo caminho para a química orgânica sintética que jamais seria imaginado há apenas algumas décadas atrás. Reações de ciclopropanação As enzimas pertencentes à classe das mono-oxigenases dependentes de ferro, em especial as do citocromo P450 (mono-oxigenases P450) são conhecidas por sua habilidade de catalisar reações de oxidação em ligações C-H não ativadas utilizando oxigênio molecular. Por exemplo, a reação entre as espécies ferro-oxo de alta valência presentes nas mono-oxigenases P450 e que são os agentes oxidantes ativos, reagem com olefinas promovendo a transferência do átomo de oxigênio (transferência de oxeno) para a ligação dupla C-C, formando um epóxido. Por analogia, a formação de espécies ferro-carbenoides e ferro-nitrenos de alta valência deveriam conduzir à formação de ciclopropanos e aziridinas, respectivamente (Esquema 4).
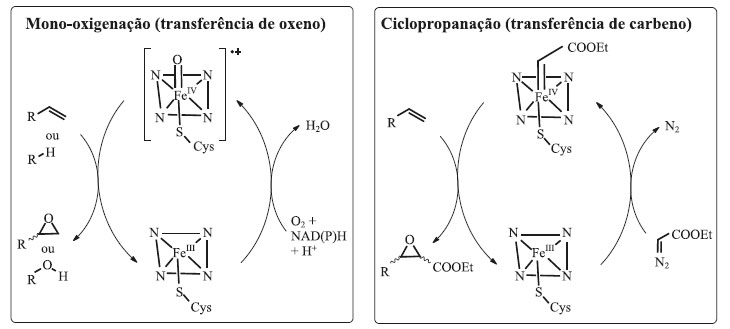 Esquema 4. Reações naturais e não-naturais catalisadas por enzimas derivadas de mono-oxigenases P450. A) Reação de transferência de oxeno catalisada por mono-oxigenases P450 selvagem. B) Reação de transferência de carbeno catalisada por mono-oxigenases P450 modificadas através de técnicas de evolução dirigida
Recentemente, Arnold e colaboradores foram pioneiros ao divulgar exemplos notáveis em que novos biocatalisadores, produzidos através de técnicas de evolução dirigida, foram capazes de catalisar reações de ciclopropanação e aminação C-H. Isso foi possível devido à promiscuidade enzimática das mono-oxigenases P450 e no conhecimento detalhado do mecanismo químico destas reações orgânicas que guiaram a escolha das mutações de aminoácidos na enzima. O Esquema 5 apresenta alguns exemplos de reações não-naturais catalisadas por enzimas derivadas de mono-oxigenases P450 através de evolução dirigida. Alguns destes exemplos serão discutidos a seguir.
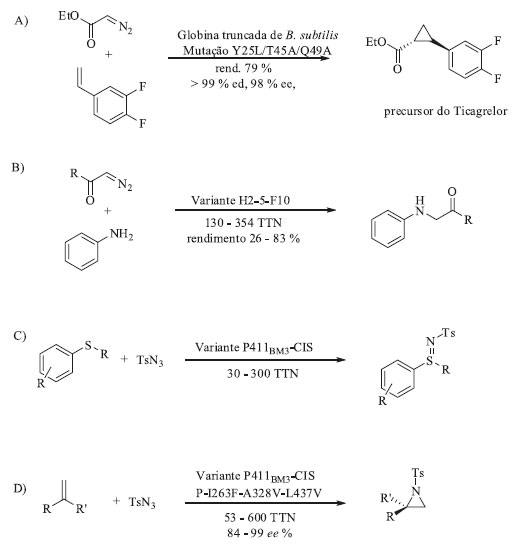 Esquema 5. Reações não-naturais catalisadas por enzimas derivadas de mono-oxigenases P450 através de engenharia de proteínas e evolução dirigida. A) Reação de ciclopropanação;73,74 B) Reação de inserção N-H;75 C) Reação de sulfimidação;76 D) Reação de aziridinação77 e E) Reação de aminação C-H78,79
O desenvolvimento de biocatalisadores para a reação de ciclopropanação começou com a mono-oxigenase selvagem extraída de Bacillus megaterium (P450BM3). Essa enzima mostrou habilidade de catalisar, ainda que com baixa eficiência (5 TON) e baixas diastero- e enantiosseletividades, a ciclopropanação do estireno, ao invés da epoxidação, ao utilizar o diazoacetato de etila (EDA) como reagente precursor para a ciclopropanação. Em todas as mono-oxigenases P450 selvagens, o ferro heme presente no sítio ativo da enzima está ligado axialmente ao átomo de enxofre de um resíduo de cisteína. A espécie oxidante ativa é um cátion radicalar porfirínico Fe(IV)-oxo formado pela reação entre o Fe(II) presente na enzima, o oxigênio molecular e dois equivalentes de hidreto provenientes do cofator NAD(P)H. Para as reações de ciclopropanação vários carbenos de diazoesteres foram selecionados e o carbeno oriundo dos intermediários metalocarbenoides foram transferidos à ligação dupla C-C. Utilizando estas informações como ponto de partida, iniciou-se a evolução dirigida desta enzima em busca de uma variante mais ativa. A primeira etapa envolveu a triagem de uma série de noventa e duas enzimas mutantes de P450BM3, escolhidas por sua diversidade em relação à atividade enzimática para catalisar a reação entre o estireno e o EDA em condições anaeróbicas e suas diferentes sequências primárias de aminoácidos.73 Em seguida, foram selecionados os dez mutantes mais promissores. Cinco desses dez apresentaram aumento na atividade de ciclopropanação (TTN, número total de turnovers, >100, diastereoseletividade cis:trans variando na faixa de 9:91 a 60:40 e enantiosseletividade de até 95%) quando comparadas à enzima selvagem P450BM3. Além disso foi possível observar que a diastereoseletividade cis:trans poderia ser modulada. Por exemplo, o mutante denominado H2-5-F10 e que contém 16 substituições de aminoácidos favorece a formação do produto de trans-ciclopropanação (cis:trans 16:84) com 63% de eetrans enquanto o mutante denominado H2-A10 apresentou uma diasterosseletividade reversa, favorecendo a formação do produto de cis-ciclopropanação (cis:trans 60:40) com elevado excesso enantiomérico, 95% de eecis. A partir dos mutantes H2-A10 e H2-5-F10 descobriu-se que a mutação T268A resultou em uma variante 60 vezes mais ativa e com excelente seletividade para formação do cis-ciclopropano. A mutação T268A é o elemento chave para a atividade de ciclopropanação. Essa mutação sozinha é responsável por converter a enzima inativa 9-10A-TS-F87V no catalisador ativo 9-10A-TS-F87V-T268A, conhecido como BM3-cis, apresenta 199 TTN com diasterosseletividade para formação do produto cis (cis:trans 71:29), com enantiosseletividade >90% para ambos os diastereoisomêros e ela é tão estável quanto a enzima selvagem. Por outro lado, a adição de I263A ao mutante BM3-cis reverte a diastereosseletividade (cis:trans 19:81). Além disso, a substituição da cisteína axial ligada ao ferro por uma serina gerou um mutante com TON maior que 67.000 e que pode ser usado na forma de célula íntegra. Não há relatos de nenhuma mono-oxigenase natural contendo essa mutação, uma vez que a substituição da cisteína por serina elimina completamente a atividade mono-oxigenase. As variantes contendo esta mutação passaram a ser denominadas P411 em função do deslocamento do λ de 450 nm para 411 nm do pico de CO. Essas reações de ciclopropanação tiveram o escopo de substrato avaliado e puderam ser empregadas na síntese de intermediários sintéticos importantes na indústria farmacêutica como o Levomilnaciprano,80 um antidepressivo inibidor seletivo da recaptação de serotonina e norepinefrina e o Ticagreclor,81 inibidor de agregação plaquetária utilizado para a prevenção de eventos trombóticos como, por exemplo, o AVC (acidente vascular cerebral) (Esquema 5a). Além das mono-oxigenases P450, outras enzimas podem ser usadas como ponto de partida para esse fim. Fasano e colaboradores utilizaram mioglobinas modificadas geneticamente via evolução dirigida e as mesmas também foram capazes de catalisar a transferência de carbenos em reações de ciclopropanação.82 Partindo do pressuposto que espécies nitrenóides são equivalentes sintéticos das carbenóides e que as reações baseadas na transferência de carbeno poderiam ser expandidas para transferência de nitrenos, a biblioteca de variantes de P411 derivadas da mono-oxigenase P450 de Bacillus megaterium construída por Arnold e colaboradores também foram avaliadas quanto à sua capacidade de catalisar outras reações não-naturais tais como: aminação de ligações C-H, sulfimidação e aziridinação (Esquema 5b, 5c e 5d, respectivamente). Novas mutações foram realizadas para cada uma dessas reações resultando em catalisadores eficientes para cada uma delas. Uma vez demonstrada a capacidade das variantes de P411 catalisarem as reações de transferência de nitrenos e, inspirados no trabalho de Dawson e colaboradores que em 1985 reportaram a inserção de nitrenos em ligações C-H não ativadas catalisadas por uma mono-oxigenase P450, Arnold e colaboradores partiram para a avaliação de variantes capazes de realizar a aminação C-H benzílica. De posse dos resultados positivos, esses pesquisadores verificaram que as aminações C-H benzílicas foram também régio- e enantiosseletivas, dependendo das mutações inseridas.83-85 Esses resultados sugerem que as possibilidades para a evolução de biocatalisadores para reações não-naturais são enormes e a literatura já começa a apresentar os primeiros resultados promissores que descrevem a inserção de carbenos em ligações Si-H e B-H, catalisadas por enzimas do tipo heme.86,87 Formação de ligação C-Si Em 2017, Arnold e colaboradores descreveram a primeira reação de formação de ligação C-Si catalisada enzimaticamente através da evolução dirigida de uma enzima citocromo c.86 Eles descobriram que proteínas heme catalisam a formação de compostos organosilício sob condições fisiológicas via inserção de carbeno na ligação Si-H, tanto in vivo quanto in vitro. Inicialmente mono-oxigenases citocromo c foram triadas em busca de atividade catalítica para a reação entre o fenildimetilsilano e 2-diazopropanoato de etila (Esquema 6). O melhor resultado foi obtido com a enzima citocromo c de Rhodothermus marinus (Rma cyt c) que forneceu o produto desejado com 97% ee. Utilizando mutagênese sítio dirigida para a Rma cyt c, após 3 mutações (V75T/M100D/M103E) foi possível aumentar o TTN da transferência de carbeno em 40 vezes.
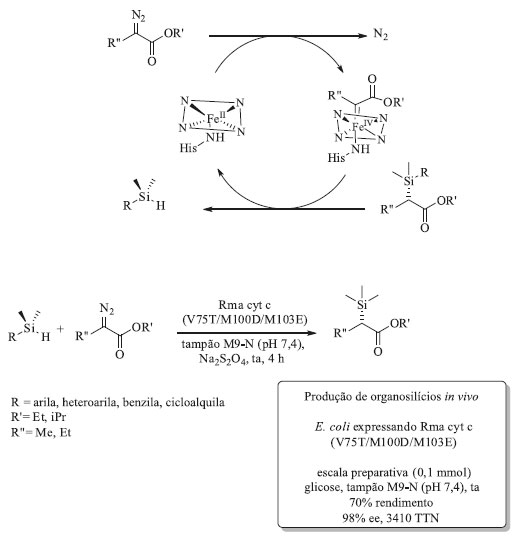 Esquema 6. Ciclo catalítico e formação biocatalítica de ligação C-Si catalisada pelo mutante Rma cyt c (V75T/M100D/M103E)
Essa reação foi testada para 20 substratos diferentes (arila, heteroarila, benzila e cicloalquila), com TTN variando entre 47 a 8210 e enantiosseletividade de moderadas a excelentes. Nos substratos contendo ligações C-C duplas ou triplas não foi observada atividade de ciclopropanação ou ciclopropenação, demonstrando a quimiosseletivade deste mutante (Rma cyt c). Para o substrato nitrogenado dimetilsililanilina observou-se um aumento de 29 vezes na quimiosseletidade de formação de ligação C-Si, em detrimento da formação de ligação C-N, para esse mutante quando comparado com a enzima selvagem. Além das reações realizadas com a enzima isolada, foi possível realizar esta reação com células íntegras de E. coli expressando o triplo mutante de Rma cyt c em uma escala de 0,1 mmol e rendimento de 70% do produto isolado, demonstrando que os organosilanos são estáveis in vivo e possuem permeabilidade na célula de E. coli. Comparado aos catalisadores químicos tradicionais, esse biocatalisador é superior em termos de TTN. Além disso, esses dados norteiam o caminho para novas reações de incorporação de átomos de silício em compostos orgânicos polifuncionalizados sem a necessidade de proteção/desproteção de grupos funcionais. Reações de desalogenação enantiosseletiva de halolactonas As cetorredutases (KREDs) são enzimas que naturalmente catalisam reações de redução de aldeídos e cetonas para os respectivos álcoois e a reação inversa. Emmanuel e colaboradores demonstraram que, sob irradiação de luz azul, a enzima KRED atuou na catálise de reação de desalogenação enantiosseletiva de halolactonas.88 As halolactonas foram escolhidas como substrato para essa reação pois elas são capazes de se ligar ao sítio ativo da enzima, sem sofrerem redução da ligação C=O.89 Em uma triagem inicial, usando vinte cetorredutases provenientes de um kit adquirido da empresa Codexis, apenas três enzimas, (KRED-4, KRED-12 e KRED-14) forneceram o produto desejado com bons rendimentos e excessos enantioméricos.88 Os pesquisadores, então, em contato com a Codexis, constataram que várias KREDs do kit eram mutantes derivados da cetorredutase de Lactobacillus kefiri (LKADH), que após a mutação Y190 passa a aceitar substratos com maior impedimento estérico. Possuindo como ponto de partida a cetorredutase selvagem de Lactobacillus kefiri foram realizadas mutações sítio dirigidas, na qual o mutante Y190C aumentou a atividade da enzima. O melhor mutante desenvolvido continha uma mutação tripla (E154F/F147L/Y190C), sendo que esse foi capaz de catalisar a reação modelo (Esquema 7) gerando o produto desejado com 72% de rendimento e 98% de ee.88
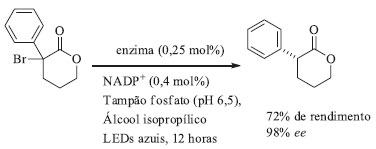 Esquema 7. Desalogenação enatiosseletiva biocatalítica de halolactona catalisada por cetorredutase
Foi proposto que a catálise desta reação não-natural ocorre por mecanismo radicalar: o cofator fotoexcitado gera um intermediário radicalar quiral que atua como fonte de hidrogênio, e isso só é possível se o cofator NADP(H) estiver firmemente ligado ao sítio ativo da enzima e for excitado com luz azul.88 Partindo desse princípio, outras transformações dependentes de NAD(P)H ou cofatores análogos como, por exemplo, os cofatores dependentes de flavina (FAD -dinucleotídio de adenina e flavina, FMN - mononucletotideo de flavina e seus análogos artificiais) podem ser previstas para um futuro próximo. Reação de eliminação de Kemp Rothlisberger e colaboradores utilizaram oito liases, dentre elas uma aldolase, para a realização de estudos computacionais de novo a fim de criar uma enzima capaz de catalisar a reação de eliminação de Kemp (Esquema 8).90 Para os estudos computacionais foram levadas em consideração as informações sobre o mecanismo da reação: por exemplo, a etapa chave de uma eliminação de Kemp é a desprotonação do carbono por uma base. Analisando as cadeias laterais dos vinte aminoácidos proteinogênicos, eles escolheram duas possibilidades para atuar como base nesse mecanismo: a primeira delas seria um grupo carboxilato de um aspartato ou glutamato e, a segunda delas, um imidazol de uma histidina, polarizado com um aspartato ou glutamato.90
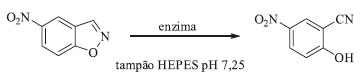 Esquema 8. Reação de eliminação de Kemp biocatalítica
Após a otimização dos resíduos de aminoácidos necessários para a catálise da reação, foram realizados estudos computacionais visando a escolha de um esqueleto proteico para acomodar esses aminoácidos. Foram construídas 59 variantes, sendo que 8 dessas variantes exibiram um baixo nível de atividade para a reação de eliminação de Kemp. Os melhores candidatos obtidos a partir dos estudos computacionais passaram por experimentos de evolução dirigida (vários ciclos de mutagênese e DNA shuffling) até chegar em um novo biocatalisador para essa reação não-natural. As mutações inseridas por meio de evolução dirigida se encontravam em posições adjacentes ao sítio ativo proposto nos estudos computacionais anteriores e complementaram a evolução desse biocatalisador, resultando em um aumento de >200 vezes na constante kcat/Km.90 Outros exemplos de reações não-naturais catalisadas por enzimas que passaram por evolução dirigida podem ser encontrados nos artigos de revisão publicados recentemente.91,92
CONSIDERAÇÕES FINAIS E PERSPECTIVAS Espera-se que o desenvolvimento da Química passe pela busca progressiva por reações e/ou processos mais sustentáveis, tanto ambiental quanto economicamente. Esses dois fatores, na realidade, estão entrelaçados, visto que não há como existir a implementação de reações e processos mais verdes se eles não forem economicamente competitivos. Como foi visto nas sessões anteriores, o uso de biocatalisadores traz inúmeras vantagens do ponto de vista da sustentabilidade ambiental, sendo uma estratégia valiosa a ser considerada. As ferramentas para o desenvolvimento de metodologias biocatalíticas melhoraram consideravelmente na última década e os avanços contínuos na área de biologia molecular sinalizam que haverá melhorias adicionais. Tudo indica que os químicos orgânicos sintéticos capazes de usar o potencial que os biocatalisadores oferecem terão uma clara vantagem sobre aqueles limitados a métodos não biológicos. Isso se deve ao fato de que as habilidades de lidar com a geração de novos problemas na interface entre a química e a biologia como, por exemplo, aqueles decorrentes da necessidade de usar matérias-primas renováveis, exigirão conhecimentos mínimos nas áreas de conhecimento envolvidas. Entretanto, a disponibilidade comercial de enzimas (selvagens e mutantes) com atividade catalítica elevada, em grandes quantidades e preços acessíveis é condição sine qua non para a democratização do uso dos biocatalisadores. Do ponto de vista estratégico, o maior entrave atual dentro da área de engenharia de proteínas permanece sendo a necessidade de melhoria das metodologias de evolução dirigida. Entre os desafios a serem superados, podem ser citados a construção de bibliotecas de mutantes menores e mais eficientes, o desenvolvimento de ensaios de triagens enzimáticas de alto desempenho rápidos e eficazes além de avanços na área de bioinformática. Esses fatores combinados são importantes para que o tempo empregado na evolução de biocatalisadores adequados seja compatível com o tempo exigido pelo setor industrial. A biocatálise será uma estratégia importante e se tornará uma das principais tecnologias para a fabricação de produtos químicos nas próximas décadas. O próximo grande desafio dependerá do conhecimento, visão e inspiração dos químicos para criar reações que nem a biologia e nem a química sintética conquistaram, assim a evolução dirigida nos levará onde a biologia nunca foi. No Brasil existem vários grupos de pesquisa consolidados na área de biocatálise, entretanto, ainda é necessário um grande avanço na área de engenharia de proteínas, em especial utilizando técnicas de evolução dirigida de enzimas, bem como investimentos e incentivos das agências de fomento apoiando o desenvolvimento de projetos na área de biocatálise e na formação de recursos humanos especializados para atuarem na área quimioenzimática.93,94
AGRADECIMENTOS As autoras expressam seus agradecimentos à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), GlaxoSmithKline e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo financiamento dos projetos em rede CERSusChem (Proc. 2014/50249-8) e INCTBioNat (Proc. 465637/2014-0). O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - (Código de Financiamento 001). Agradecemos ainda à CAPES pela manutenção do Portal de Periódicos. Dedicamos este trabalho ao Prof. J. Augusto R. Rodrigues (in memoriam) pelas suas contribuições na área de Biocatálise no Brasil.
REFERÊNCIAS 1. Anastas, P. T.; Warner, J.; Green Chemistry: Theory and Practice, Oxford University Press: Oxford, 1988, cap. 1. 2. Química Verde: fundamentos e aplicações; Corrêa, A. G., Zuin, V. G., eds.; EdUFSCar: São Carlos, 2009, cap. 1. 3. Prado, A. G. S.; Quim. Nova 2003, 26, 738. 4. Silva, F. M.; Lacerda, P. S. B.; Jones Junior, J.; Quim. Nova 2005, 28, 103. 5. Farias, L. A.; Fávaro, D. I. T.; Quim. Nova 2011, 34, 1089. 6. Sousa-Aguiar, E. F.; de Almeida, J. M. A. R.; Romano, P. N.; Fernandes, R. P.; Carvalho, Y.; Quim. Nova 2014, 37, 1. 7. Sheldon, R. A.; Arends, I.; Hanefeld, U.; Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2008. 8. Braga, A. L.; Lüdke, D. S.; Schneider, P. H.; Andrade, L. H.; Paixão, M. W.; Quim. Nova 2013, 36, 1591. 9. Bernardo-Gusmão, K.; Pergher, S. B. C.; dos Santos, E. N.; Quim. Nova 2017, 40, 650. 10. Finelli, F. G.; Santos, B. M. S.; Frota, L. C. R. M.; Quim. Nova 2019, 42, 425. 11. Sheldon, R. A.; Brady, D.; Chem. Commun. 2018, 54, 6088. 12. Sheldon, R. A.; Woodley, J. M.; Chem. Rev. 2018, 118, 801. 13. Woodley, J. M.; Curr. Opin. Chem. Biol. 2013, 17, 310. 14. Sheldon, R. A.; Pereira, P. C.; Chem. Soc. Rev. 2017, 46, 2678. 15. Oliveira, L. G.; Mantovani, S. M.; Quim. Nova 2009, 32, 742. 16. Arnold, F. H.; Angew. Chem., Int. Ed. 2018, 57, 4143. 17. Woodley, J. M.; Phil. Trans. R. Soc. A 2017, 376, 20170062. 18. https://www.ncbi.nlm.nih.gov/genome, acessada em Abril 2020. 19. Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K.; Nature 2012, 485, 185. 20. Behrens, G. A.; Hummel, A.; Padhi, S. K.; Schätzle, S.; Bornscheuer, U. T.; Adv. Synth. Catal. 2011, 353, 2191. 21. Otten, L. G.; Hollmann, F.; Arends, I. W. C. E.; Cell Press 2009, 28, 46. 22. Bloom, J. D.; Arnold, F. H.; Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 9995. 23. Reetz, M. T.; Krebs, G. P. L.; C. R. Chim. 2011, 14, 811. 24. Martínez, R.; Schwaneberg, U.; Biol. Res. 2013, 46, 395. 25. Kumar, A.; Singh, S.; Crit. Rev. Biotechnol. 2013, 33, 365. 26. Cheng, F.; Zhu, L.; Schwaneberg, U.; Chem. Commun. 2015, 51, 9760. 27. Bornscheuer, U. T.; Hauer, B.; Jaeger, K. E.; Schwaneberg, U.; Angew. Chem., Int. Ed. 2019, 58, 36. 28. Cadwell, R. C.; Joyce, G. F.; PCR Methods Appl. 1992, 2, 28. 29. Packer, M. S.; Liu, D. R.; Nat. Rev. Genet. 2015, 16, 379. 30. Stemmer, W. P. C.; Nature 1994, 370, 389. 31. Stemmer, W. P. C.; Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 10747. 32. Reetz, M. T.; Carballeira, J. D.; Nat. Protoc. 2007, 4, 891. 33. https://www.nobelprize.org/prizes/chemistry/2018/press-release, acessada em Abril 2020. 34. Arnold, F. H.; Angew. Chem., Int. Ed. 2019, 58, 2. 35. Sheldon, R. A.; Brady, D.; ChemSusChem 2019, 12, 2859. 36. Alcántara, A. R.; Catalysts 2019, 9, 792. 37. Wenda, S.; Illner, S.; Mell, A.; Kragl, U.; Green Chem. 2011, 13, 3007. 38. Abdelraheem, E. M. M.; Busch, H.; Hanefeld, U.; Tonin, F.; React. Chem. Eng. 2019, 4, 1878. 39. Muller, M.; Angew. Chem., Int. Ed. 2005, 44, 362. 40. https://www.fiercepharma.com/pharma/from-old-behemoth-lipitor-to-new-king-humira-u-s-best-selling-drugs-over-25-years, acessada em Abril 2020. 41. https://www.axios.com/lipitor-pfizer-drug-patent-sales-2019-6937cdfb-47f1-46bc-8cf0-39e6b88e235e.html, acessada em Abril 2020. 42. Liljeblad, A.; Kallinen, A.; Kanerva, L. T.; Curr. Org. Synth. 2009, 6, 362. 43. Luo, Y.; Chen, Y.; Ma, H.; Tian, Z.; Zhang, Y.; Zhang, J.; Sci. Rep. 2017, 7, 42074. 44. DeSantis, G.; Zhu, Z.; Greenberg, W. A.; Wong, K.; Chaplin, J.; Hanson, S. R.; Farwell, B.; Nicholson, L. W.; Rand, C. L.; Weiner, D. P.; Robertson, D. E.; Burk, M. J.; J. Am. Chem. Soc. 2002, 124, 9024. 45. DeSantis, G.; Wong, K.; Farwell, B.; Chatman, K.; Zhu, Z.; Tomlinson, G.; Huang, H.; Tan, X.; Bibbs, L.; Chen, P.; Kretz, K.; Burk, M. J.; J. Am. Chem. Soc. 2003, 125, 11476. 46. Jennewein, S.; Schurmann, M.; Wolberg, M.; Hilker, I.; Luiten, R.; Wubbolts, M.; Mink, D.; Biotechnol. J. 2006, 1, 537. 47. Ma, S. K.; Gruber, J.; Davis, C.; Newman, L.; Gray, D.; Wang, A.; Grate, J.; Huisman, G. W.; Sheldon, R. A.; Green Chem. 2010, 12, 81. 48. https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2006-greener-reaction-conditions-award, acessada em Abril 2020. 49. Rosenthal, K.; Lütz, S.; Current Opinion in Green and Sustainable Chemistry 2018, 11, 58. 50. Patel, R. N.; Bioorg. Med. Chem. 2018, 26, 1252. 51. Savile, C. K.; Janey, J. M.; Mundorff, E. C.; Moore, J. C.; Tam, S.; Jarvis, W. R.; Colbeck, J. C.; Krebber, A.; Fleitz, F. J.; Brands, J.; Devine, P. N.; Huisman, G. W.; Hughes, G. J.; Science 2010, 329, 305. 52. https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2010-greener-reaction-conditions-award, acessada em Abril 2020. 53. http://www.pharmafile.com/news/172417/fda-approves-greener-process-januvia-production, acessada em Abril 2020. 54. Linder, M.; Green Chem. Lett. Rev. 2017, 10, 428. 55. Keijer, T.; Bakker, V.; Slootweg, J. C.; Nature Chem. 2019, 11, 190. 56. Ferreira, P. G.; da Silva, F. C.; Ferreira, V. F.; Rev. Virtual Quim. 2017, 9, 452. 57. Sheldon, R. A.; Green Chem. 2017, 19, 18. 58. Sheldon, R. A.; Green Chem. 2014, 16, 950. 59. Sheldon, R. A.; J. R. Soc., Interface 2016, 13, 20160087. 60. Pellis, A.; Cantone, S.; Ebert, C.; Gardossi, L.; New Biotechnol. 2018, 40, 154. 61. Sheldon, R. A.; J. Mol. Catal. A: Chem. 2016, 422, 3. 62. Gallo, J. M. R.; Bueno, J. M. C.; Schuchardt, U.; J. Braz. Chem. Soc. 2014, 25, 2229. 63. Rass-Hansen, J.; Falsig, H.; Jørgensen, B.; Christensen, C. H.; J. Chem. Technol. Biotechnol. 2007, 82, 329. 64. Meinhold, P.; Peters, M. W.; Chen, M. M. Y.; Takahashi, K.; Arnold, F. H.; ChemBioChem 2005, 6, 1765. 65. Souza, R. O. M. A.; Miranda, L. S. M.; Bornscheuer, U. T.; Chem. - Eur. J. 2017, 23, 12040. 66. Honig, M.; Sondermann, P.; Turner, N. J.; Carreira, E. M.; Angew. Chem., Int. Ed. 2017, 56, 8942. 67. Turner, N. J.; O'Reilly, E.; Nat. Chem. Biol. 2013, 9, 285. 68. Gupta, R. D.; Sustain. Chem. Process. 2016, 4, 2. 69. Renata, H.; Wang, Z. J.; Arnold, F. H.; Angew. Chem., Int. Ed. 2015, 54, 3351. 70. Bartlett, G. J.; Borkakoti, N.; Thronton, J. M.; J. Mol. Biol. 2003, 331, 829. 71. Arnold, F. H.; Q. Rev. Biophys. 2015, 48, 404. 72. Arnold, F. H.; Angew. Chem., Int. Ed. 2019, 58, 14420. 73. Coelho, P. S.; Wang, Z. J.; Ener, M. E.; Baril, S. A.; Kannan, A.; Arnold, F. H.; Nat. Chem. Biol. 2013, 9, 485. 74. Coelho, P. S.; Brustad, E. M.; Kannan, A.; Arnold, F. H.; Science 2013, 339, 307. 75. Wang, Z. J.; Peck, N. E.; Renata, H.; Arnold, F. H.; Chem. Sci. 2014, 5, 598. 76. Farwell, C. C.; McIntosh, J. A.; Arnold, F. H.; J. Am. Chem. Soc. 2017, 136, 8766. 77. Farwell, C. C.; Zhang, R. K.; McIntosh, J. A.; Hyster, T. K.; Arnold, F. H.; ACS Cent. Sci. 2015, 1, 89. 78. McIntosh, J. A.; Coelho, P. S.; Farwell, C. C.; Wang, Z. J.; Lewis, J. C.; Brown, T. R.; Arnold, F. H.; Angew. Chem., Int. Ed. 2013, 52, 9309. 79. Singh, R.; Bordeaux, M.; Fasan, R.; ACS Catal. 2014, 4, 546. 80. Wang, Z. J.; Renata, H.; Peck, N. E.; Farwell, C. C.; Coelho, P. S.; Arnold, F. H.; Angew. Chem., Int. Ed. 2014, 53, 6810. 81. Hernandez, K. E.; Renata, H.; Lewis, R. D.; Kan, S. B. J.; Zhang, C.; Forte, J.; Rozzell, D.; McIntosh, J. A.; Arnold, F. H.; ACS Catal. 2016, 6, 7810. 82. Tinoco, A.; Steck, V.; Tyagi, V.; Fasan, R.; J. Am. Chem. Soc. 2014, 139, 5293. 83. Svastits, W.; Dawson, J. H.; Breslow, R.; Gellman, S.; J. Am. Chem. Soc. 1985, 107, 6427. 84. Hyster, T. K.; Farwell, C. C.; Buller, A. R.; McIntosh. J. A.; Arnold, F. H.; J. Am. Chem. Soc. 2014, 136, 15505. 85. Prier, C. K.; Zhang, R. K.; Arnold, F. H.; Nat. Chem. 2017, 9, 629. 86. Kan, S. B. J.; Lewis, R. D.; Chen, K.; Arnold, F. H.; Science 2016, 354, 1048. 87. Kan, S. B. J.; Huang, X.; Gumulya, Y.; Chen, K.; Arnold, F. H.; Nature 2017, 552, 132. 88. Emmanuel, M. A.; Greenberg, N. R.; Oblinsky, D. G.; Hyster, T. K.; Nature 2016, 540, 414. 89. Kara, S.; Spickermann, D.; Schrittwieser, J. H.; Weckbecker, A.; Leggewie, C.; Arends, I. W. C. E.; Hollmann, F.; ACS Catal. 2013, 3, 2436. 90. Rothlisberger, D.; Khersonsky, O.; Wollacott, A. M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J. L.; Althoff, E. A.; Zanghellini, A.; Dym, O.; Albeck, S.; Houk, K. N.; Tawfik, D. S.; Baker, D.; Nature 2008, 453, 190. 91. Renata, H.; Wang, Z. J.; Arnold, F. H.; Angew. Chem., Int. Ed. 2015, 54, 3351. 92. Chen, K.; Arnold, F.; Nat. Catal. 2020, 3, 203. 93. Birolli, W. G.; Ferreira, I. M.; Alvarenga, N.; Matos, I. L.; Comasseto, J. V.; Porto, A. L. M.; Biotech. Adv. 2015, 33, 481. 94. Paulo, B. S.; Sigrist, R.; Angolini, C. F. F.; Eberlin, M. N.; de Oliveira, L. G.; J. Braz. Chem. Soc. 2019, 30, 673. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access