
 |
 |
 |
 |
 |
Artigo
| Speciation analysis of arsenic in rice using high performance liquid chromatography coupled with hydride generation atomic fluorescence spectrometry (HPLC-HG-AFS) |
|
Gabriella A. BorgesI; Guilhermina de O. SouzaII; Patrícia S. F. LopesII; Virginia S. T. CiminelliII,III; Claudia L. CaldeiraII,III; Guilherme D. RodriguesI,*
I. Departamento de Química, Universidade Federal de Minas Gerais, Av. Antônio Carlos 6627, Campus Pampulha, 31270-901 Belo Horizonte - MG, Brasil Recebido em 13/11/2019 *e-mail: guilhermedr@ufmg.br The aim of this work was to develop a method using high performance liquid chromatography coupled with hydride generation atomic fluorescence spectrometry (HPLC-HG-AFS) for the determination and speciation analysis of the inorganic and organic species of arsenic found most often in rice samples: As(V), As(III), monomethylarsonic acid (MMA), and dimethylarsinic acid (DMA). The best chromatographic resolution was obtained using pH 6.2, a phosphate buffer mobile phase concentration of 20 mmol L-1 and flow rate of 0.57 mL min-1, HCl concentration of 5.55% (v/v), and NaBH4 concentration of 0.90% (w/v). The accuracy of the method was confirmed by the low relative error values obtained for analysis of a certified reference material (NIST 1568b): 6.63% (total inorganic As), 3.44% (DMA), 0% (MMA), and 0.53% (total As). In recovery tests, the proposed method achieved satisfactory recoveries in the range 86-110%. INTRODUCTION Rice is a cereal obtained from Oryza sativa L., a monocotyledonous plant of the family Poaceae (grasses).1 It is an important component of the diet in many countries and is therefore of considerable economic and nutritional importance. According to data of the Brazilian Agricultural Research Corporation (EMBRAPA),2 rice is grown and consumed in all continents of the world. The crop is mainly produced on irrigated and flooded soils, which can be problematic in regions affected by soil contamination, since elements such as inorganic arsenic (As(III) and As(V)) are both highly toxic, but As(III) are 60 times more toxic than As(V), and very soluble in water.3,4 The presence of water increases the mobility of these species in the soil, as a result of which they can be absorbed by the roots of the plants and reach the grains.5 Arsenic is widely distributed in the Earth's crust, where it is generally present in low concentrations.6 It is a component of over 200 mineral species,7 of which 60% are arsenates, 20% are arsenosulfides also containing metals such as Fe, Pb, Cu and Ag and the remainder include arsenites, oxides, arsenides, and elemental arsenic. The most common mineral is arsenopyrite (FeAsS). The mobilization of arsenic in the environment occurs due to natural processes (climatic variations, biological activity, and volcanic activity),6 as well as anthropogenic activities involving industrial emissions (mining, non-ferrous metal smelting, and combustion of fossil fuels) and the manufacture and use of fertilizers, wood preservatives, insecticides, and herbicides.8,9 Arsenic is found naturally in the environment in four oxidation states: As(V), As(III), As(0), and As(-III).10 The methylated acid forms of arsenic, MMA and DMA, are only slightly toxic,11 while arsenobetaine (AsB) and arsenocholine (AsC) are relatively nontoxic.12 However, arsenic is most commonly found as an oxyanion in inorganic compounds, in the species As(V) (AsO43-), highly toxic, and As(III) (AsO33-).10,13 Human exposure to arsenic can occur in several ways. Contamination can occur by skin contact or inhalation,7 but the main routes are by the ingestion of contaminated drinking water or food.14 Studies have shown that the greatest human exposure to arsenic by the food route is due to the consumption of cereals, with rice and rice-based foods being at the top of the list.15 Long-term exposures to high levels of arsenic are associated with higher rates of skin, bladder, and lung cancers, as well as heart disease.7,11,16 Due to their high toxicity to humans, inorganic arsenic compounds are classified as Group I carcinogens by the International Agency for Research on Cancer.10,17 The United States Environmental Protection Agency (USEPA) has placed these compounds at the top of the list of priority pollutants.18 According to the Brazilian National Health Surveillance Agency (ANVISA), the legally established maximum tolerable limit (MTL) for this contaminant in rice and its derivatives is 300 μg kg-1.19 In 2014, with the aim of ensuring consumer health, the UN Food and Agriculture Organization (FAO), together with the World Health Organization (WHO), published the FAO-WHO Codex Alimentarius establishing international standards of food safety and quality, according to which the maximum permissible content of arsenic in rice is 200 μg kg-1.20 Since the toxicity of this element depends on the chemical form in which it is present, it is important to develop methods able to determine each species of arsenic in food samples, in order to enable accurate assessment of risks.21-23 Furthermore, since the maximum permissible concentrations are low, such methods must be sufficiently sensitive and accurate for determination of the content of each arsenic species. There are several methods that can be used for arsenic speciation studies. The most sensitive "hyphenated" speciation techniques involve the use of high performance liquid chromatography (HPLC)15,24 coupled with detection using inductively coupled plasma mass spectrometry (ICP-MS),24 hydride generation and atomic absorption spectrometry (HG-AAS), or hydride generation and atomic fluorescence spectrometry (HG-AFS).25 The present work proposes the use of HPLC-HG-AFS, which provides high sensitivity and selectivity for the determination of low concentrations of hydrides of elements of environmental importance, such as those of As, Hg, Sb, Se, Bi, Cd, Sn, and Th.26 The chemical processes involved in this technique are related to the specific chemical reactions for each element. In the case of arsenic, the use of a fairly high concentration of sodium borohydride enables the conversion of DMA, MMA, As(III) and As(V) into volatile species that can be determined by AFS.27,28 As(III) and As(V) form AsH3, MMA forms CH3AsH2, and DMA forms (CH3)2AsH. The main advantage of this technique is that only the volatile hydrides are transferred to the detector in the carrier gas flow, while the sample matrix remains in the liquid, hence eliminating interferences in the detection system.29 AFS provides high sensitivity, comparable to other techniques,30 while offering the advantage of lower cost. Therefore, the main objective of this work was to develop and verify an analytical methodology employing HPLC-HG-AFS for the speciation of arsenic present in different rice samples.
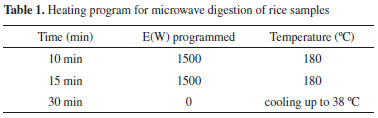
EXPERIMENTAL Apparatus The HPLC-HG-AFS analytical system consisted of a high performance liquid chromatograph equipped with a Dionex pump model UltiMate 3000, a Hamilton PRP-X100 anion exchange column (250 x 4.1 mm, 10 μm particle size) and operated with Chromeleon software, which was coupled to a PS Analytical Excalibur hydride generator with atomic fluorescence spectrometer detector. The optimized operating parameters were as follows: mobile phase consisting of 20 mmol L-1 phosphate buffer (K2HPO4 + KH2PO4) at pH 6.2; mobile phase flow rate of 0.57 mL min-1; HCl concentration of 5.55% (v/v) and flow rate of 2.5 mL min-1; NaBH4 concentration of 0.90% (w/v) and flow rate of 4.5 mL min-1. An inductively coupled plasma mass spectrometer (ICP-MS) (PerkinElmer NexION 300X), equipped with a Meinhard nebulizer and an autosampler, was operated under the following conditions: argon (99.999%) plasma gas flow rate of 15 L min-1; auxiliary gas flow rate of 1.20 L min-1; oxygen (99.99%) flow rate of 0.6 mL min-1; nebulizer flow rate of 0.9 mL min-1; 20 scans with reading time of 75 ms. To eliminate the interference of ArCl in ICP analysis, the equipment has a reaction cell (DRC-ICP-MS). Arsenic was effectively converted to AsO using oxygen as the reaction gas in DRC, and the resulting mass of AsO (91) is determined. Chemicals Purified water was obtained from a Milli-Q system (Elix Technology, Singapore). All reagents were used as received, with no further purification process, as follows: 65% nitric acid (Química Moderna, São Paulo, Brazil), fuming 37% hydrochloric acid (Química Moderna, São Paulo, Brazil), sodium borohydride (Sigma-Aldrich), sodium hydroxide (Sigma-Aldrich), dibasic potassium phosphate (K2HPO4) (Sigma-Aldrich), monobasic potassium phosphate (KH2PO4) (Sigma-Aldrich), sodium arsenite (NaAsO2) (99.0%, Fluka), cacodylic acid (C2H7AsO2) (98%, Sigma-Aldrich), disodium methyl arsenate hexahydrate (CH3AsO(ONa)2.6H2O) (Santa Cruz Biotechnology), dibasic sodium arsenate heptahydrate (HAsNa2O4.7H2O) (98.0%, Sigma-Aldrich), arsenic (1000 mg L-1 in 2% HNO3) (SPEX CertiPrep), 1000 mg L-1 yttrium internal standard (SPEX CertiPrep), and certified reference material NIST SRM 1568b Rice Flour (National Institute of Standards and Technology, USA) containing 180 ± 12 µg kg-1 DMA, 11.6 ± 3.5 µg kg-1 MMA, and 92 ± 10 µg kg-1 inorganic As. For ICP-MS only, double-distilled nitric acid in a quartz distiller is required. Rice samples Rice samples were obtained from national supermarket chains. The samples were rapidly frozen, using liquid nitrogen, in order to facilitate milling in a blender (Cuisinart) until the material passed through a 300 μm sieve (Viatest GmbH). The transformation of the rice into a fine powder was essential in order to ensure the best possible extraction efficiency. The material was then divided into portions of around 10 g. The samples were stored at ambient temperature, avoiding sources of light and heat. Extraction procedure for speciation by HPLC-HG-AFS Approximately 1.00 g portions of milled rice were weighed out into glass tubes, followed by weighing of 10.00 g of 0.28 mol L-1 HNO3. All reagents added to the assays were weighed for the exact final calculation of the arsenic content in the fortified extract. The tubes were placed in a thermostatic bath with water circulation (Model N3, Haake), at 91.7 ºC, for 90 minutes. The tubes were then removed from the bath, noting the color change from white (rice powder) to yellow at the end of the extraction. The tubes were left to cool at ambient temperature, followed by centrifugation (Baby II 206 R, Excelsa) and filtration of the supernatants through 0.22 μm syringe filters. Quantitative determination of the arsenic species was performed using a standard additions procedure, with weighing out of 4.00 g of the filtered extract and 1.00 g of the standard (in nitric acid) containing the four species of arsenic under investigation, totaling 5.00 g of fortified extract. The procedure was based on the previous work by Farías et al.31 and Batista et al.,32 with adaptations according to previous studies in the laboratory. Spiking of the samples with arsenic standards considered the limit of detection of the method for each species studied, the sensitivity, and the estimated initial amounts of each arsenic species in the samples. These estimates were obtained by direct injection of a pure extract of the fortified extract to be studied (with unknown arsenic content) into the HPLC-HG-AFS system, comparing the intensity of the signal obtained with that for a standard containing 10.00 μg kg-1 of each arsenic species, using a directly proportional relation. After spiking with arsenic standards, the fortified extract were homogenized, placed in an ultrasonic bath (Biociclo Instrumentos Científicos) for 20 min, and then analyzed by injection of aliquots into the HPLC-HG-AFS system. Each assay were performed in duplicate. Microwave extraction for determination of total arsenic by ICP-MS The total arsenic content was determined by weighing out 0.2000 g of sample and transferring it to a flask, to which 2.00 mL of double-distilled nitric acid was then added. The samples were submitted to microwave digestion (ETHOS One, Milestone), using the heating program shown in Table 1.
After cooling the microwave to 38 ºC, the samples were removed and left to cool to ambient temperature. The digested material was then quantitatively transferred to 50.0 mL polypropylene Falcon tubes by washing the PTFE digestion flasks with Milli-Q water (18.2 MΩ cm). The final solutions were weighed and the samples were stored in a refrigerator at 4.0 ºC. Prior to analysis, 0.1 mL of yttrium internal standard (50 μg kg-1) was added, with homogenization. The samples were then analyzed using the ICP-MS system.
RESULTS AND DISCUSSION In any speciation study, it is essential to preserve the organic and inorganic species present. Previous studies showed that extraction using 0.280 mol L-1 HNO3 at 95 ± 3 ºC for around 90 min preserved all the species of arsenic and enabled their quantitative recovery.31-33 Use of the same extraction conditions for a longer period of time (120 min) caused the reduction of As(V) to As(III),31 while temperatures higher than 95 ± 3 ºC resulted in lower extraction efficiency. In addition, the concentration of HNO3 should be in the range 0.2-0.7 mol L-1, in order to avoid the reduction of As(V) by sulfur compounds and the oxidation of As(III) due to interaction of HNO3 with the extract.33 Separation of the arsenic species was achieved using an anion exchange column, since most arsenic species are present as neutral or negative ions in solution, depending on the pH.34 It can be seen from the chromatogram shown in Figure 1 that the species with the lowest charge (As(III)) exited the column first, while the species with the highest charge (As(V)) was retained for a longer time. In addition, the efficiencies of hydride formation differ, hence affecting the sensitivity of the method, depending on the analyte under consideration. In the present case, all the species were present in the sample at concentrations of 10 μg kg-1, while the signals showed different heights and areas, as observed previously by Gómez-Ariza et al.35 and Farías et al.31
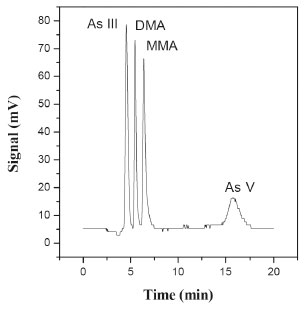 Figure 1. Chromatogram obtained for the arsenic species standard at 10 µg kg-1 in nitric acid, under the optimized conditions: 0.90% (w/v) NaBH4 in 0.1 mol L-1 NaOH; pH 6.2; mobile phase concentration of 20 mmol L-1; mobile phase flow rate of 0.57 mL min-1; HCl concentration of 5.6% (v/v)
Optimization of the method The aim of the optimization procedure was to determine the conditions that provided satisfactory sensitivity and separation of the different species, hence resulting in accurate results. Evaluation was made of the influence on chromatographic resolution of the following parameters and their corresponding levels: pH (5.7, 5.9, 6.2, 6.5, and 6.7); concentration of the K2HPO4/KH2PO4 buffer mobile phase (15, 17.5, 20, 22.5, and 25 mmol L-1); flow rate of the mobile phase (0.44, 0.50, 0.57, 0.64, and 0.70 mL min-1); concentration of HCl (4.6, 5.6, 6.5, 7.4, and 8.3% (v/v)); concentration of NaBH4 (0.70, 0.80, and 0.90% (w/v)). The substances HCl and NaBH4 are used in the hydride formation process for later detection via AFS. After the separation of the species in the chromatographic column, HCl and NaBH4 are the necessary reagents for the formation of volatile hydrides. The kinetics of these chemical reactions and ensuring that they occur completely are key factors in controlling the sensitivity of the technique and the success of the analysis. These chemical reactions occur prior to detection. Although they do not interfere in the separation of arsenic species, they affect the process of hydride formation, thus influencing the quality of the chromatogram. The best conditions were selected by visual analysis of the chromatograms (Figure 2) and the calculation of the resolution according to equation 1: 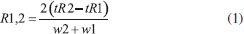
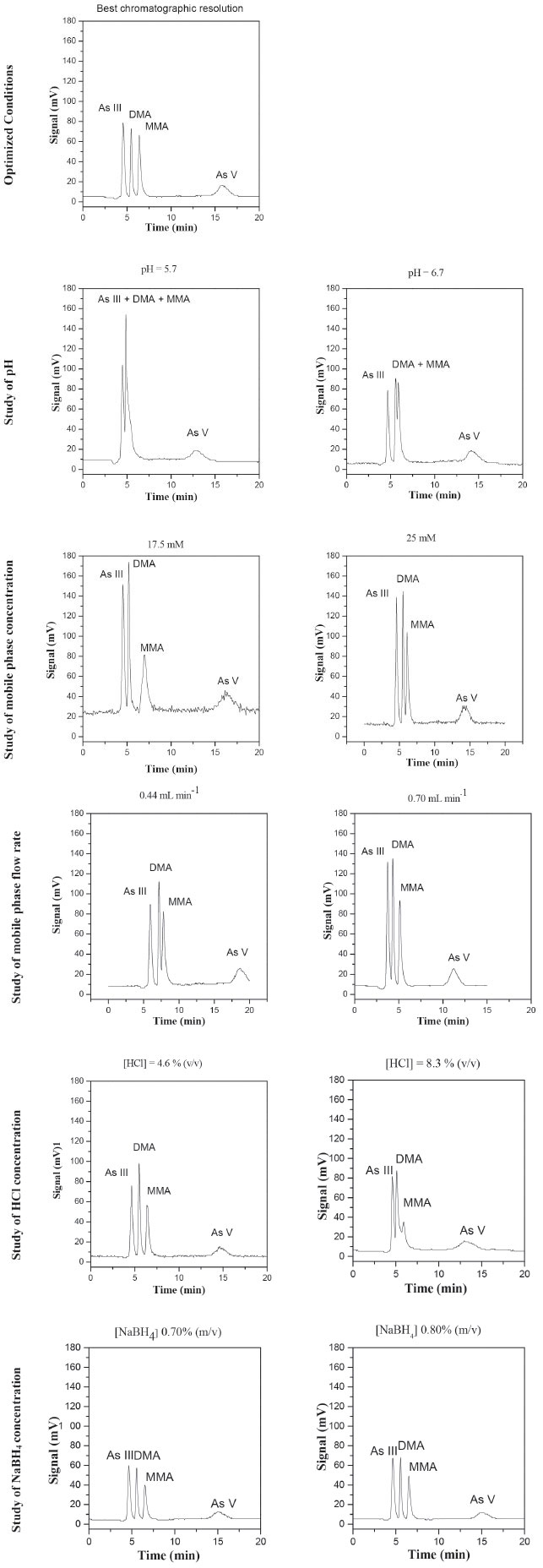 Figure 2. Chromatograms obtained for the arsenic species standards at 10 µg kg-1 in nitric acid. Fixed values of the other experimental parameters in univariate optimization: pH of mobile phase 6.2, concentration of mobile phase K2HPO4/KH2PO4 20 mmol L-1, mobile phase flow rate 0.57 mL min-1, HCl concentration 5.6% (v/v), NaBH4 concentration 0.9% (m/v) in NaOH 0.1 mol L-1
where tR is the retention time and w is the width of the peak base. The optimized experimental conditions, pH of mobile phase 6.2; concentration of mobile phase K2HPO4/KH2PO4 20 mmol L-1, mobile phase flow rate 0.57 mL min-1; concentration of HCl 5.6% (v/v) and concentration of NaBH4 0.9% (m/v) in NaOH 0.1 mol L-1 (Table 2), were used in the experiments to determine the content of each arsenic species in the rice samples.
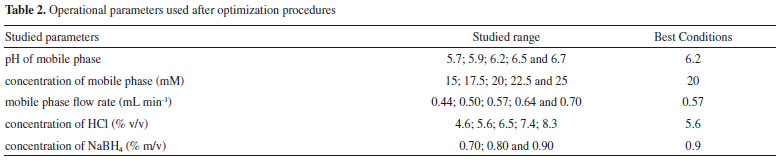
Method verification Verification of the method was performed under the previously optimized conditions, in order to confirm that it was suitable for the speciation of arsenic present in the rice samples. The verification procedure followed the guidelines of the Brazilian Ministry of Agriculture, Livestock, and Supply (MAPA)36 and Instituto Nacional de Metrologia, Qualidade e Tecnologia (INMETRO).37 Firstly, the relationship between the analyte concentration and the chromatographic peak area was evaluated by analyzing 15 standard solutions containing different concentrations of the species of interest, in order to identify the region in which the method provided a linear response. This procedure was performed for each arsenic species. The limits of detection (LOD) and quantification (LOQ) were calculated using 7 replicates of the blank, according to Equations 2 and 3 (INMETRO),37 where SD is the standard deviation.  The LOD and LOQ values obtained (Table 3) showed that the proposed method enabled determination of very low levels of the four arsenic species. The LOQ values of the method complied with the recommended limit established in legislation (200 μg kg-1) for the determination of inorganic arsenic in rice samples, with values of 1.59 μg kg-1 for As(III) and 4.81 μg kg-1 for As(V).
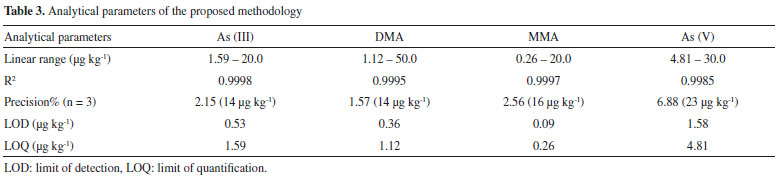
Since a blank matrix without any arsenic was not available, evaluation of the effect of the matrix was performed by comparing quantification of the analyte in an unknown sample using external calibration (employing standards of the analyte in pure solvent) and using a calibration curve constructed using extracts of the unknown sample spiked with standards in nitric acid, as recommended by MAPA.36 This revealed the existence of a matrix effect, so it was necessary to use the standard additions method for analysis of the rice samples. The results obtained for the main parameters considered in the method verification are presented in Table 3. The precision of the method was evaluated in terms of repeatability, with the preparation and analysis of nine standards (at three concentration levels, in triplicate) consisting of solvent (0.28 mol L-1 HNO3) containing the analyte. The concentration levels studied were: 3.75, 8.75 and 13.75 µg kg-1 for As(III) and DMA, 4.5, 10.5 and 16.5 µg kg-1 for MMA and 7.5, 17.5 and 27.5 µg kg-1 for As(V). The calculated coefficients of variation (CV) are shown in Table 4.
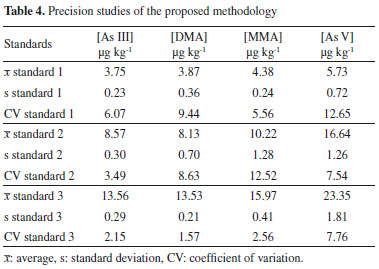
According to the criteria of MAPA36 for acceptable precision, the CV value should be less than 2/3 of the value established according to the concentration. In the case of the present study (with concentrations below 1 ppm), the maximum permitted CV was (2/3 x 35)%, corresponding to 23%. The CV values obtained for standard 1 (with lower arsenic concentration) were 6.07% (As(III)), 9.44% (DMA), 5.56% (MMA), and 12.65% (As(V)). The values for standard 2 (with intermediate arsenic concentration) were 3.49% (As(III)), 8.63% (DMA), 12.52% (MMA), and 7.54% (As(V)), while for standard 3 (with higher arsenic concentration), the values were 2.15% (As(III)), 1.57% (DMA), 2.56% (MMA), and 7.76% (As(V)). It could therefore be concluded that the CV values were well below the maximum permitted value, indicating the good precision of the technique. Accuracy of the method The accuracy of the proposed methodology was evaluated by analysis of the speciation of As in a certified reference material (NIST 1568b). The analytical curves were constructed with six concentration levels. Since each HPLC-HG-AFS analysis lasted about 20 min, the study was done in duplicate for each concentration level. The speciation results obtained for the NIST 1568b certified reference material using the proposed methodology were compared with the labeled values (total inorganic arsenic (As(III) + As(V)) = 92 ± 10 μg kg-1, DMA = 180 ± 12 μg kg-1, and MMA = 11.6 ± 3.5 μg kg-1). The accuracy was obtained by comparing the value obtained experimentally with the certified value, with calculation of the relative error (RE), as recommended by MAPA36 and INMETRO.37 The RE values were 6.63% (total inorganic As), 3.44% (DMA), 0% (MMA), and 0.53% (total As), demonstrating the satisfactory accuracy of the proposed methodology (Table 5).
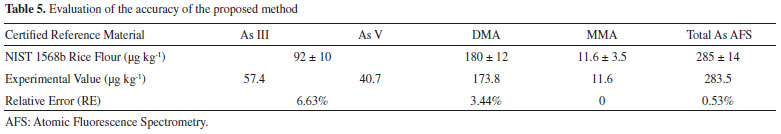
Figure 1S (Supplementary Material) shows the analytical curves for speciation of the NIST 1568b material. The rice extracts were spiked with standards prior to the HPLC-HG-AFS analyses, so the concentration of each arsenic species was obtained from the regression equation of the analytical curve. The concentrations of the arsenic species in the rice grains were then obtained by considering the dilutions performed during the sample preparation procedure. The first dilution was approximately 11-fold (weighing out 1.0000 g of the solid reference material and adding 10.00 g of nitric acid 0.28 mol L-1). The second dilution was 1.25-fold (weighing out 4.0000 g of the filtered extract and completing to 5.0000 g with 0.28 mol L-1 HNO3). The proposed method was applied for the determination and/or speciation of As in nine rice samples. The results obtained for each specie were then summed and compared with the values of total As determined by ICP-MS (Table 6).
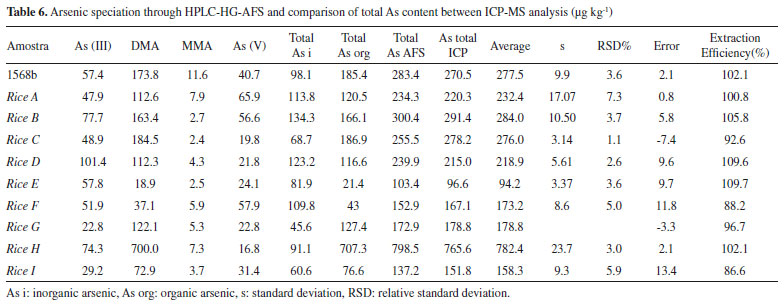
The performance of the method was evaluated using recovery calculations, comparing the total As values obtained by the HPLC-HG-AFS and ICP-MS methods, considering the value obtained by ICP-MS as the reference (expected) value. According to MAPA36 and INMETRO,37 the recovery range considered acceptable depends on the amount of the analyte in the sample. In the case of the present samples with concentrations between 100 and 1000 μg kg-1, recovery values in the range from 80 to 110% were considered acceptable. All the recoveries were within the acceptable range, with values close to 100% (Table 6), confirming the applicability of the proposed HPLC-HG-AFS method for the speciation of arsenic in rice samples. The recoveries obtained here were compatible with those found in previous studies, for the same type of sample, where the values were in the ranges 92-103% (Farías et al.),31 81-117.8% (Batista et al.),32 89-106% (Huang et al.)33 and 83.8-115.6% (Segura et al.).38 Considering the FAO-WHO Codex Alimentarius maximum permissible concentration for arsenic in rice (200 μg kg-1),20 it could be concluded from the results (Table 6) that samples A, B, C, D, and H were unfit for human consumption. The rice samples studied showed heterogeneity in terms of their arsenic contents. For example, sample H had a high total arsenic content (798.5 μg kg-1), although most of this (700 μg kg-1) was in the form of DMA, which is a less toxic organic arsenic species. On the other hand, sample E had the lowest total arsenic content (103.4 μg kg-1), but the highest content of As(III) (57.8 μg kg-1), which is the most toxic inorganic arsenic species, so this sample presented greater risk to human health. A high content of inorganic arsenic (71.9%) was also found for sample F. These results demonstrated that analysis of the total arsenic content alone is insufficient for accurate evaluation of potential risks, highlighting the importance of developing methods capable of arsenic speciation. With the exception of samples E and F, in which there was the predominance of inorganic arsenic, the samples presented higher contents of DMA, compared to the other arsenic species, as previously found by Farías et al.31 for rice from Argentina. Samples A, B, D, and I presented around 50% of inorganic species and 50% of organic species, while samples C, G, and H presented higher contents of organic species.
CONCLUSIONS The methodology applied in this work was effective for the speciation of arsenic present in rice samples. The HPLC-HG-AFS hyphenated technique was selective and provided high sensitivity, enabling the quantification of low levels of arsenic in the samples analyzed. The optimized conditions of the method were pH 6.2, phosphate buffer mobile phase concentration of 20 mmol L-1, mobile phase flow rate of 0.57 mL min-1, HCl concentration of 5.55% (v/v), and NaBH4 concentration of 0.90% (w/v). These conditions enabled accurate speciation of the NIST 1568b certified reference material, as well as determination of the speciated arsenic contents of nine samples of rice. The concentrations of arsenic species found for the rice samples revealed the importance of speciation analyses for accurate evaluation of risks. This was because some samples (such as sample H) presented a high content of total As, although most of the As was in less toxic organic forms, while others (such as sample F) had a low total arsenic content, but most of the As was present in the highly toxic inorganic form.
SUPPLEMENTARY MATERIAL Supplementary Material shows the analytical curves for speciation of the NIST 1568b material. The linear regressions (b / a) applied in the calculation of the respective dilutions and arsenic contents (NIST 1568b) are also presented (Figure 1S).
ACKNOWLEDGEMENTS The authors are grateful to Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, APQ-03210-15 and APQ-03688-18), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Instituto Nacional de Ciência e Tecnologia - Recursos Minerais, Água e Biodiversidade (INCT-Acqua) and Instituto Federal de Minas Gerais Campos Ouro Preto (IFMG - OP) for financial support and scholarships.
REFERENCES 1. Gomes, A. S.; Magalhães Júnior, A. M.; Arroz Irrigado no Sul do Brasil, 1ª ed., EMBRAPA Informação Tecnológica: Brasília, 2004. 2. MAPA e EMBRAPA. Projeções do Agronegócio. Brasil 2014/15 a 2024/25. Projeções de Longo Prazo. Brasília, DF. Julho, 2015. 3. Kumarathilaka, P.; Seneweera, S.; Meharg, A.; Bundschuh, J.; Water Res. 2018, 140, 403. 4. Lewchalermvong, K.; Rangkadilok, N.; Nookabkaew, S.; Suriyo, T.; Satayavivad, J.; J. Agric. Food Chem. 2018, 66, 3199. 5. Weiss, D.; Khondoker, R.; J. Braz. Chem. Soc. 2013, 24, 690. 6. Nearing, M. M.; Koch, I.; Reimer, K. J.; Spectrochim. Acta, Part B 2014, 99, 150. 7. European Food Safety Authority. Dietary exposure to inorganic arsenic in the European population. EFSA Journal 2014, 12, 3597. 8. Ribeiro, R. V.; Vieira, J. C.; Lobo, F. A.; Froes-Silva, R. E. S.; J. Braz. Chem. Soc. 2018, 29, 873. 9. Hughes, M. F.; Beck, B. D.; Chen, Y.; Lewis, A. S.; Thomas, D. J.; Fundam. Toxicol. Sci. 2011, 123, 305. 10. Shamsipur, M.; Fattahi, N.; Assadi, Y.; Sadeghi, M.; Sharafi, K.; Talanta 2014, 130, 26. 11. González-Martínez, F.; Sánchez-Rodas, D.; Cáceres, D. D.; Martínez, M. F.; Quiñonese, L. A.; Johnson-Restrepo, B.; Chemosphere 2018, 212, 927. 12. Liu, L. H.; He, B.; Yun, Z. J.; Sun, J.; Jiang, G. B.; J. Chromatogr. A 2013, 1304, 227. 13. Nogueira, R.; Melo, E. A.; Figueiredo, J. L. C.; Santos, J. J.; do Nascimento Neto, A. P.; J. Braz. Chem. Soc. 2018, 29, 1593. 14. Abdolmohammad-Zadeh, H.; Talleb, Z.; Talanta 2014, 128, 147. 15. Cubadda, F.; Jackson, B. P.; Cottingham, K. L.; Van Horne, Y. O.; Kurzius-Spencer, M.; Sci. Total Environ. 2017, 579, 1228. 16. http://www.fda.gov, acessada em Abril 2020. 17. Ma, J.; Sengupta, M. K.; Yuan, D.; Dasgupta, P. K.; Anal. Chim. Acta 2011, 831, 1. 18. Chen, M. L.; Ma, L. Y.; Chen, X. W.; Talanta 2014, 125, 78. 19. http://portal.anvisa.gov.br, acessada em Abril 2020. 20. http://www.fao.org, acessada em Abril 2020. 21. Narukawa, T.; Inagaki, K.; Kuroiwa, T.; Chiba, K.; Talanta 2014, 130, 213. 22. Quináia, S. P.; Rollemberg, M. C. E.; J. Braz. Chem. Soc. 2001, 12, 37. 23. Chen, H.; Tang, Z.; Wang, P.; Zhao, F.; Environ. Pollut. 2018, 238, 482. 24. Gorny, J.; Dumoulin, D.; Lesven, L.; Noiriel, C.; Madé, B.; Billona, G.; J. Anal. At. Spectrom. 2015, 30, 1562. 25. Komorowicz, I.; Barałkiewicz, D.; Talanta 2011, 84, 247. 26. Chen, Y. W.; Belzile, N.; Anal. Chim. Acta 2010, 671, 9. 27. Vilano, M.; Padro, A.; Rubio, R.; Anal. Chim. Acta 2000, 411, 71. 28. Narsito, J.; Agterdenbos, J. S. S.; Anal. Chim. Acta 1990, 237, 189. 29. Barra, C. M.; Santelli, R. E.; Abrão, J. J.; de la Guardia, M.; Quim. Nova 2000, 23, 58. 30. Lindberg, A. L.; Goessler, W.; Grander, M.; Nermell, B.; Vahter, M.; Toxicol. Lett. 2007, 168, 310. 31. Farías, S. S.; Londonio, A.; Quintero, C.; Befani, R.; Soro, M.; Smichowski, P.; Microchem. J. 2015, 120, 34. 32. Batista, B. L.; Souza, J. M. O.; De Souza, S. S.; Barbosa Jr, F.; J. Hazard. Mater. 2011, 191, 342. 33. Huang, J. H.; Ilgen, G.; Fecher, P.; J. Anal. At. Spectrom. 2010, 25, 800. 34. Sanchez-Rodas, D.; Corns, W. T.; Chen, B.; Stockwel, P. B.; J. Anal. At. Spectrom. 2010, 25, 933. 35. Gómez-Ariza, J. L.; Sanchez-Rodas, D.; Giraldez, I.; Morales, E.; Talanta 2000, 51, 257. 36. Ministério da Agricultura, Pecuária e Abastecimento; Guia: Validação e Controle de Qualidade Analítica (Fármacos em Produtos para Alimentação Animal e Medicamentos Veterinários), Brasília, 2011. 37. Instituto Nacional de Metrologia, Qualidade e Tecnologia; Orientação sobre validação de métodos analíticos DOQ-CGCRE-008. Revisão 05 - Agosto 2016. 38. Segura, F. R.; Souza, J. M. de O.; de Paula, E. S.; Martins Jr, A. da C.; Paulelli A. C. C.; Barbosa Jr., F.; Batista, B. L.; Food. Res. Int. 2016, 89, 169. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access