
 |
 |
 |
 |
 |
Artigo
| Theoretical studies on diacetonediperoxide derivatives and comparisons with other multi-peroxidic compounds |
|
Zongcheng MiaoI; Junmei LiII; Guoliang Fanb and Yi LuanIII,*
I. Key Laboratory of Organic Polymer Photoelectric Materials, School of Science, Xijing University, 710123 Xi'an - PR, China Recebido em 22/03/2020 *e-mail: yiluan@ustb.edu.cn Diacetonediperoxide (DADP) derivatives were investigated with density functional theory (DFT) methods at the DFT-B3LYP and M06-2X/6-311++G (d, p) levels and were also compared with other multi-peroxidic compounds. The investigated derivatives were 3,6-dimethyl-3,6-diamine-1,2,4,5-tetraoxane (DADPNH2), 3,6-dimethyl-3,6-dinitro-1,2,4,5-tetraoxane (DADPNO2), and 3,6-dimethyl-3,6-bis(trifluoromethyl)-1,2,4,5-tetraoxane (DADP3F). The investigations were focused on detonation performance and stability which were determined according to geometrical and electronic structure. The results of the simulation revealed that DADP derivatives are less sensitive than some other organic peroxidic compounds. In addition, detonation performance, including explosion heat (Q), detonation velocity (D), and detonation pressure (P), was estimated according to density and heat of formation using EXPLO 5 software. The results revealed that DADPNO2 has better detonation performance (Q = 5543 kJ kg-1, P = 23.7 GPa, D = 7631 m s-1) compared with traditional explosive 2,4,6-trinitrotoluene (TNT; Q = 5418 kJ kg-1, P = 19.0 GPa, D = 6950 m s-1). Therefore, it is a novel candidate for use as an energetic material. INTRODUCTION Energetic materials can be used as explosives, propellants, pyrotechnic agents and so on.1-3 Traditional energetic materials include 2,4,6-trinitrotoluene (TNT),4 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX),5 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX),6 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20),7 1,3,5-triamino-2,4,6-trinitrobenzene (TATB)8 and other poly-nitrogen oxygen-rich compounds. 9,10 TATP (triacetonetriperoxide) and its derivatives have been reported to be novel energetic materials,11 while DADP (diacetonediperoxide), whose structure is shown in Figure 1, is another promising energetic material in the multi-peroxidic compound family.12,13 TATP has been extensively used by terrorists in terrorist attacks because of its low cost and simple production process. However, DADP, a by-product of TATP, has lower sensitivity, more affluent oxygen content, higher detonation velocity and pressure than TATP.14,15 When used as an explosive, DADP is also much more sensitive and has much lower energy than TNT. Therefore, further study and modification are required for DADP to achieve a superior performance. Landenberger et al. have reported that the introduction of cocrystals to DADP could be an effective means to this end.16 In addition, it could also be effective in introducing other energetic substituents.
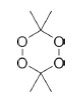 Figure 1. Chemical structure of DADP
Many fused rings, such as azasydnone17 and oxadiazole,18 can become the building blocks of energetic compounds, but they have poor oxygen balance. The skeleton of DADP is a relatively stable six-membered ring containing four oxygen atoms, and there have been few investigations on its novel oxygen-rich backbone. Nitro (-NO2), amino (-NH2), and trifluoromethyl (-CF3) are common functional groups. For example, TNT consists of three -NO2 groups, while TATB consists of three -NO2 groups and three -NH2 groups.4,8 Trifluoromethyl is an inert group widely used in aluminum-containing propellants.19 Therefore, these three energetic and stable substituents could be introduced to DADP to great effect. In this paper, we propose adding -CF3, -NO2, and -NH2 individually to the rich oxygen skeleton of DADP in order to lower its sensitivity and to enhance its detonation performance. The resulting compounds are 3,6-dimethyl-3,6-diamine-1,2,4,5-tetraoxane (DADPNH2), 3,6-dimethyl-3,6-dinitro-1,2,4,5-tetraoxane (DADPNO2), 3,6-dimethyl-3,6-bis(trifluoromethyl)-1,2,4,5-tetraoxane (DADP3F), respectively. The three new compounds were used to compare DADP and other organic peroxides (shown in Figure 2) to screen out the compounds with the best comprehensive performance.
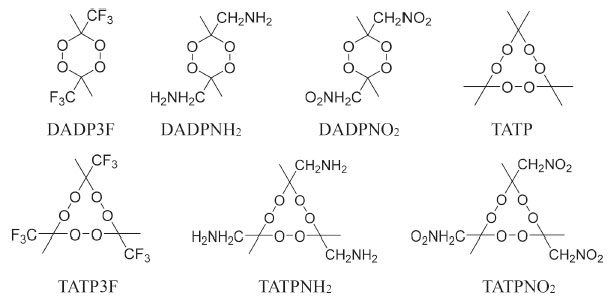 Figure 2. Chemical structures of some multi-peroxidic compounds
The properties of these three new compounds were studied through theoretical calculation. This paper presents crystal density (ρcry) and heat of formation (ΔHf) to calculate detonation performance of the compounds, including explosion heat (Q), detonation velocity (D), and detonation pressure (P). Furthermore, this paper also presents their molecular structures, electrostatic potential analysis (ESP), and energy gap (ΔELUMO-HOMO) to calculate their sensitivity. These results will provide theoretical support for screening out high energy organic peroxides.
COMPUTATIONAL METHODS The calculations were performed with the Gaussian 09 package.20 The DFT-B3LYP and M06-2X methods with the 6-311G++ (d, p) basis set21-23 were used to optimize the molecular structures and to predict the heats of formation. Previous studies have shown that the 6-311G++ (d, p) basis set is an effective tool for precisely predicting the molecular structures and energies of energetic materials.24,25 All of the structures were identified to be local energy minima on potential energy surfaces without imaginary frequencies. Three parameters were used to predict the stability of the compounds. The first, bond length was one of the major focuses of the calculations, especially for O-O bonds. These bond lengths were used for comparison with those of some organic peroxides. In general, bond length is closely related to bond stability; the smaller a bond, the more stable it is.26 The energy gap ΔELUMO-HOMO between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO) was also used as a parameter of energetic materials to predict kinetic stability. However, it is worth noting that this stability refers only to the stability of the compound during its photoinitiated electron-transfer processes, as the gap indicates that the intramolecular charges transfer from the electron donor to the electron acceptor.27 In most cases, the higher the magnitude of the energy gap, the more stable the compound is. Electrostatic potential (ESP) is another important parameter of energetic materials that can be used to predict stability. This parameter can even be used to predict nucleophilic and electrophilic sites. The theoretical basis of these predictions is that molecules always tend to approach one another.28,29 ESP will be shown on a map in which blue and red spheres correspond to electrophilic and nucleophilic ESP surfaces, respectively. With the help of the Multiwfn (multifunctional wave analyzer) program,30 the ESP surface areas can be plotted in a map. The Multiwfn program founded by Tian Lu is an extremely powerful wave function analysis program, supporting almost all of the most important wave function analysis methods. More importantly, results generated by the program can be visualized directly. Most of the plotting parameters are controllable in an interactive interface, so the wave function analysis procedure is remarkably simplified, especially for studying distribution of real space functions.30 Their detonation performances depends on crystal density and heat of formation. In this study, crystal density was predicted using Equation (1), which includes consideration of the intermolecular interactions in the crystals. In Equation (1), M is the molecular weight, and Vm is the molecular volume defined as inside a contour of 0.001 au density that was evaluated using the Monte Carlo integration. What's more, σ2tot and ν are the total variance on the molecular surface and the electrostatic balance, respectively. These parameters were obtained using the Multiwfn program.30 Traditional density was calculated using the formula M/Vm, but these results showed a slight deviation from the measured values. Politzer et al.31 propose introducing vσ2tot into the formula M/Vm to reflect the features of the electrostatic potentials on the molecules' surface. This includes the strengths, the variabilities and the degrees of balance of the positive and negative electrostatic potentials computed on the surfaces of the isolated molecules. They also suggest using the correction parameters α, β, and γ.31 They compared experimental and calculated densities of 36 compounds, revealing that α, β, and γ are 0.9183, 0.0028, and 0.0443, respectively. 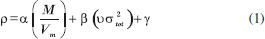 The heat of formation was predicted by the isodesmic reactions, as shown in Scheme 1. Equation (2) was used to calculate the gas phase heats of formation of compounds where ΔfHP and ΔfHR are the heat of formation of the products and the reactants in the isodesmic reactions, respectively. ΔE0 is the change in total energy between the products and the reactants at 0 K, ΔEZPE is the difference between the zero-point energy (EZPE) of the products and the reactants, and ΔHT is the thermal correction from 0 to 298 K. Finally, the value of ΔnRT here is zero.32 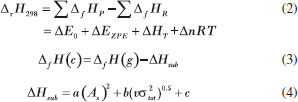
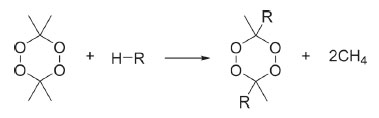 Scheme 1. Isodesmic reactions of DADP derivatives
The condensed phase heat of formation was calculated by Equation (3) and (4), in which ΔfH(c), ΔfH(g), and ΔHsub are the condensed phase heat of formation, the gas phase heat of formation, and the enthalpy of sublimation, respectively. Here the meaning and value of ν and σ2tot are the same as in Equation (1), and As is the molecular surface area. Politzer has reported that a, b, and c in Equation (4) are 0.000267, 1.650087, and 2.966078, respectively.33 The program EXPLO 5 (v6.01) has been widely applied to computing detonation performance, including explosion heat (Q), detonation velocity (D) and detonation pressure (P).34,35 The built-in theoretical models consist of the Becker-Kistiakowsky-Wilson (BKW) gaseous detonation products, the Jacobs-Cowperthwaite-Zwisler (JCZ3) equations of state, the ideal gas equation of state, the virial equation of state and the Murnaghan equation of state. Kamlet and Jacobs36 also used empirical Equations (5) and (6) to predict detonation pressure and detonation velocity. Both methods have a variety of advantages and disadvantages. The Kamlet-Jacobs equation is relatively simple; however, the EXPLO program makes more accurate calculations even in fluorine-containing explosives. Therefore, the latter was used in the present study. 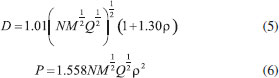
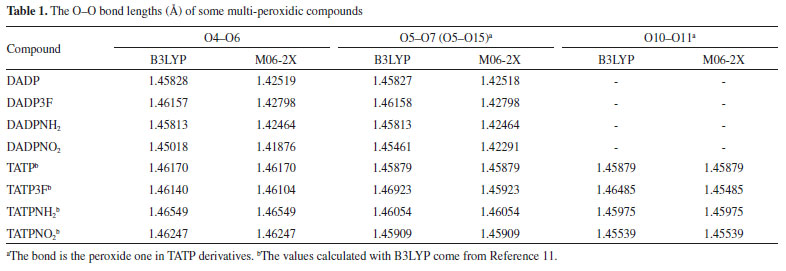
RESULTS AND DISCUSSION Geometrical structures The optimized geometries of the most stable conformers were calculated using the B3LYP and M06-2X by the 6-311G++ (d, p) method. The optimized structures of DADP and its derivatives compounds were shown in Figure 3. Tables 1S−3S show all their harmonic frequencies as well as a summary of their output parameters. It is well known that peroxides can be easily decomposed from weak O-O bonds.37 The bond length is closely related to the bond stability; therefore, we focused on the O--O bond lengths of the title compounds and then compared these with the TATP counterparts, as shown in Table 1. Generally speaking, the shorter the bond, the more stable it was. From Figure 3 and Table 1, it can be seen that the two O-O bonds in the same molecular structure were almost equal. The sequence of the bond lengths was DADP3F > DADP > DADPNH2 > DADPNO2, which is the reverse order compared with that of the compound stability. Since the electron-withdrawing -NO2 groups can decrease electron cloud densities of O-O bonds and then reduce the distance, the bond lengths in DADPNO2 were all lower than the minimum of the bond lengths in the TATP derivatives.11 Tables 4S-7S summarize the detailed information on the standard orientation of each atom.
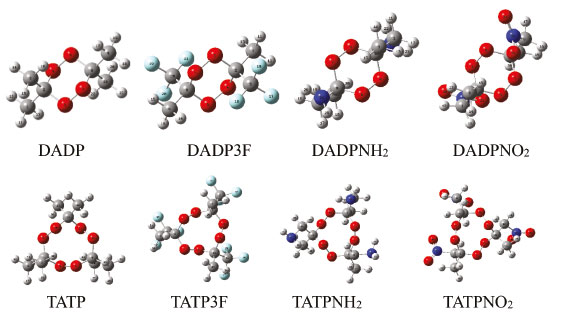 Figure 3. The optimized structures of some multi-peroxidic compounds
Electronic structure The electronic structure, which mainly consists of energy gap ΔELUMO-HOMO and ESP in the paper, reflects photochemistry stability. Explosives with high photochemical stability will not be decomposed under bright light. The ESP maps, the surface areas in each ESP range, and the HOMOs and LUMOs (which were plotted by the DFT method) were depicted in Figures 4, 5, and 6, respectively. In Figure 4, red represents positive phase, and blue represents negative phase. It can be seen from Figure 5 that a large portion of the compounds had a small ESP value ranging from -20 to +30 kcal mol-1. The electron densities of DADP3F and DADPNO2 seem to be distributed more evenly over the surface of the molecules, and then have lower impact sensitivity than other two molecules. The six-membered rings of DADP and DADPNH2 tend to be red, indicating electrophilic sites. O--O bonds in DADP and DADPNH2 are easily attracted by electrophilic reagents, which mean that their rings are fragile enough to be easily broken.
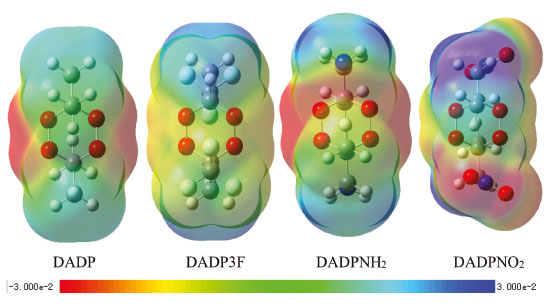 Figure 4. ESP maps of title compounds
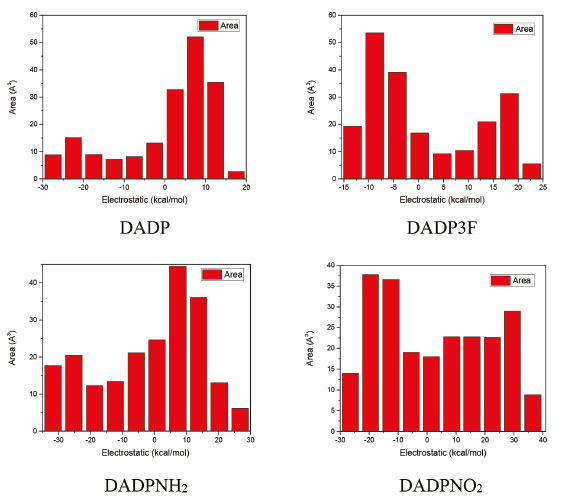 Figure 5. The surface areas in each ESP range of title compounds
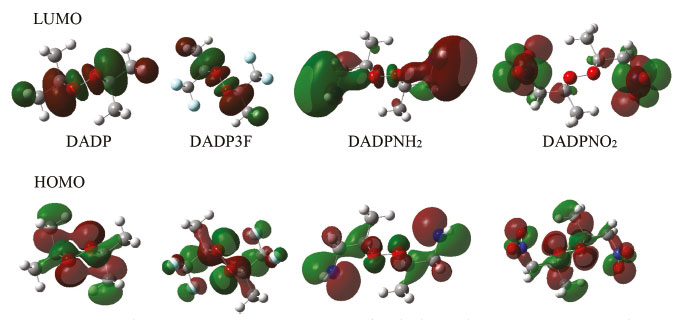 Figure 6. HOMO and LUMO orbital of DADP and its derivatives. (Red and green of the molecular orbitals means positive and negative value of wave function, respectively)
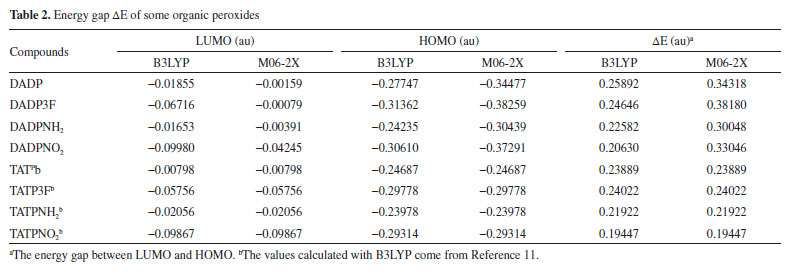
The frontier molecular orbitals, particularly the HOMO and LUMO, are very important because they relate not only to the charge transfer properties but also to their photophysical properties of these complexes. Figure 6 shows that the LUMOs were mainly located in the substituted groups, which is consistent with other multi-peroxidic compounds.11 The HOMOs were almost distributed evenly throughout the atoms. The intramolecular charges transfer from the electron donor to the electron acceptor, substituted groups. Additionally, compared with DADP, the nitro and trifluoromethyl (which is the electron-withdrawing group) makes the orbital energy levels LUMO all down significantly. Whereas nitro group makes the orbital energy levels of HOMO down slightly, suggesting it yield a greatly stabilize, whether on DADP or TATP. Energy gap of title compounds and some organic peroxides were presented in Table 2. In the table, some values coming from previous work were obtained by the same methods and basis set as in this paper. The values of DADP and its derivatives are higher than those of TATP counterparts, respectively. Their values ranged from 0.20 to 0.26 au at the B3LYP level and 0.30 to 0.38 au at the M06-2X level. Six-membered ring is more stable than other rings because of less bond tension in the ring, so the calculation agrees with that theory. The sequence of energy gap is DADP > DADP3F > DADPNH2 > DADPNO2 at the B3LYP level, but DADP3F > DADP > DADPNO2 > DADPNH2 at the M06-2X level, which is the same as that of the stability in order. Nitro and amino introducing the rings enhanced their photochemical activity, so the rings are more vulnerable to decompose in bright light. DADP and DADP3F both have a good stability, but it is not contradictory with the geometrical structures because the energy gap ΔELUMO-HOMO only reflects photochemistry stability.
Detonation performance Detonation performance is proportional to enthalpy of formation and density squared.38 The crystal density parameters of DADP and its derivatives were calculated in Table 3. The density of DADP is 1.30 g cm−3 at the B3LYP level and close to experimental one (1.31 g cm−3),16 which proves that the B3LYP methods are effective. The densities of DADP3F, DADPNH2 and DADPNO2 are 1.90, 1.35 and 1.59 g cm−3 at the B3LYP level respectively. These values are approximately equal to their TATP congeners11 (shown in Table 5), because they all have the same carbon, hydrogen, oxygen, nitrogen and fluorine content. DADP3F and TATP3F both have higher density than others because of the fluorine atom and trifluoromethyl, which is usually used to enhance the density of energetic compounds.39
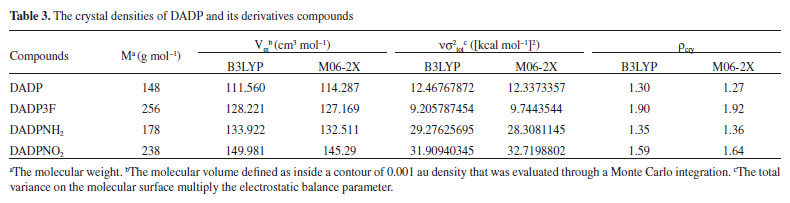
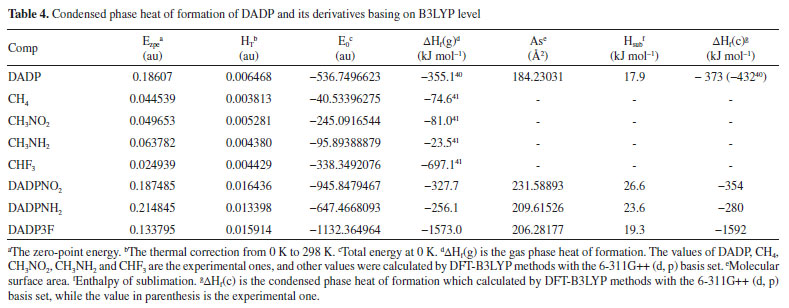
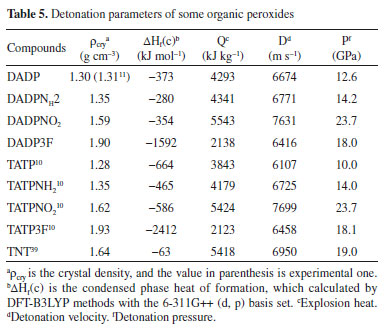
The condensed phase heats of formation of DADP and its derivatives basing on B3LYP level were shown in Table 4. DADP's calculation and measurement are −373 and −432 kJ mol-1 respectively, but this difference is not significant in energetic compounds.40 Just like TATP and its derivatives, energetic substituted groups -NH2 and -NO2 can increase heats of formation of parent, and -CF3 group will decrease it. It can be seen that heats of formation of DADP and its derivatives are all higher than those of TATP counterpart.11 Nitro, amino and trifluoromethyl are important group in energetic materials. Nitro and amino group can improve density and heat of formation, while trifluoromethyl will reduce heat of formation but increase density dramatically.40 It is interesting to compare parent and derivatives with that three groups in detonation performance. This performance includes explosion heat, detonation velocity and detonation pressure, shown in Table 5. The calculation shows that Q, D, and P ranged from 2138 to 5543 kJ kg-1, 6416 to 7631 m s-1, and 12.6 to 23.7 GPa, respectively. Their detonation performance is close to TATP and derivatives because of their small differences in density. DADP3F and TATP3F both have very high density but poor detonation performance since their heat of formation is quite low. TATPNO2 and DADPNO2 both have excellent performance than other organic peroxides and TNT (Q = 5418 kJ kg-1, P = 19.0 GPa, D = 6950 m s-1).42 In terms of the chemistry of explosives, they release energy based on a redox reaction. The -NO2 group is able to offer oxygen atoms to the redox reaction, which means that that DADPNO2 and TATPNO2 had the best detonation performance. Synthetic routes It is well known DADP can be prepared from acetone and hydrogen peroxide.37 Therefore, DADP3F can be easily prepared from 1,1,1-trifluoroacetone and hydrogen peroxide in the same way. Similarly, bromopropanone reacts with hydrogen peroxide to produce an intermediate, and then to produce DADPNH2 and DADPNO2. The possible synthetic routes are shown in Figure 7, and the key raw materials 1,1,1-trifluoroacetone and bromopropanone are both available commercially.
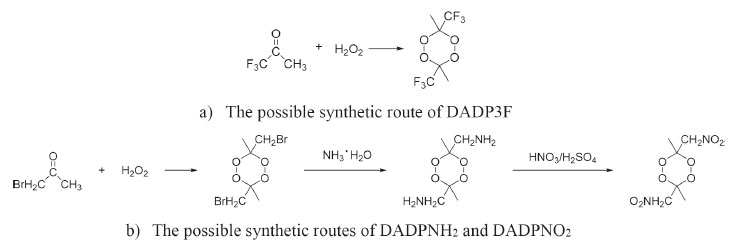 Figure 7. The possible synthetic routes of DADP3F (a) DADPNH2 and DADPNO2 (b)
The above crude products can be recrystallized from acetone by heating acetone solvent with an adjusted pH. Because the DADP crystallize into the P2/c, monoclinic space group and its polymorphism was not observed,43 its title derivatives DADP3F, DADPNH2 and DADPNO2 may form the same state as itself.
CONCLUSIONS Some organic peroxidic compounds are special explosives which are popular among terrorists. TATP is a typical representative as a consequence of its low-cost and simple production process. DADP is a by-product of TATP but has better performance. In spite of this, DADP cannot replace other energetic materials due to its sensitivity and detonation performances. Therefore, three groups, -CF3, -NH2 and -NO2, were linked to DADP to improve it. DADP and its derivatives, DADP3F, DADPNH2, and DADPNO2, were investigated in detail. In this paper, we focused on their geometrical and electronic structures, crystal density, and heat of formation to present their stability and detonation performances. Besides, these parameters were also compared with those of TATP and its derivatives to screen organic peroxidic compounds with the best comprehensive performance. The calculation reveals that DADPNO2 and TATPNO2 both have the highest explosion heat, detonation velocity and detonation pressure. However, the DADP derivatives are all more stable than their TATP counterparts, including DADPNO2. What's more special is that the detonation performance of TATPNO2 (Q = 5543 kJ kg-1, P = 23.7 GPa, D = 7631 m s-1) is higher than that of the traditional explosive TNT (Q = 5418 kJ kg-1, P = 19.0 GPa, D = 6950 m s-1). Considering all of its properties comprehensively, such as detonation performance, stability, availability, and rich-oxygen, DADPNO2 is a promising candidate for use as an energetic materials.
SUPPLEMENTARY MATERIAL Tables 1S to 7S are freely available at http://quimicanova.sbq.org.br, in PDF format.
ACKNOWLEDGEMENTS We thank the National Natural Science Foundation of China (No. 51673157), the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2018JQ5028, No. 2017JM5134 and No. 2018JM5047) and the Science Research Foundation of Xijing University (Grant No. XJ16T02) for financial support.
REFERENCES 1. Zhao, G.; He, C.; Yin, P.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M.; J. Am. Chem. Soc. 2018, 140, 3560. 2. Liu, Y.; Ma, Y.; Yu, T.; Lai, W.; Guo, W.; Ge, Z.; Ma, Z.; J. Phys. Chem. A 2018, 122, 2129. 3. Chen, J.; Yu, Y.; Li, Y.; Pang, S.; J. Fluorine Chem. 2018, 205, 35. 4. Landenberger, K. B.; Matzger, A. J.; Cryst. Growth Des. 2010, 10, 5341. 5. Li, C. F.; Mei, Z.; Zhao, F. Q.; Xu, S. Y.; Ju, X. H.; Phys. Chem. Chem. Phys. 2018, 20, 14192. 6. Landenberger, K. B.; Matzger, A. J.; Cryst. Growth Des. 2012, 12, 3603. 7. Liu, G.; Li, H.; Gou, R.; Zhang, C.; Cryst. Growth Des. 2018, 18, 7065. 8. Richard, H. G.; Szczepan, R.; Krishnan, B.; Laurence, E. F.; J. Chem. Phys. 2004, 15, 7059. 9. Xu, Z.; Cheng, G.; Yang, H.; Zhang, J.; Shreeve, J. M.; Chem. - Eur. J. 2018, 24, 10488. 10. Tang, Y.; Kumar, D.; Shreeve, J. M.; J. Am. Chem. Soc. 2017, 139, 13684. 11. Miao, Z.; Li, F.; Luan, Y.; Quim. Nova 2019, 42, 22. 12. Pyykko, P.; Chem. Commun. 1999, 6, 495. 13. Cazut, S. A. I.; Ramírez Maisuls, E. H.; Delfino, M. R.; Romero, J. M.; Jorge, N. L.; Castro, E. A.; Russ. J. Gen. Chem. 2009, 79, 2187. 14. Oxley, J. C.; Smith, J. L.; Bowden, P. R.; Rettinger, R. C.; Propellants, Explos., Pyrotech. 2013, 38, 244. 15. Härtel, M. A. C.; Klapötke, T. M.; Stiasny, B.; Stierstorfer, J.; Propellants, Explos., Pyrotech. 2017, 42, 623. 16. Landenberger, K. B.; Bolton, O.; Matzger, A. J.; J. Am. Chem. Soc. 2015, 137, 5074. 17. Dalinger, I. L.; Serushkina, O. V.; Muravyev, N. V.; Meerov, D. B.; Miroshnichenko, E. A.; Kon'kova, T. S.; Suponitsky, K. Y.; Vener, M. V.; Sheremetev, A. B.; J. Mater. Chem. 2018, 6, 18669. 18. He, C.; Imler, G. H.; Parrish, D. A.; Shreeve, J. n. M.; J. Mater. Chem. 2018, 6, 16833. 19. Yang, T.; Xu, Z.; Meng, Z.; Zhai, L.; ChemistrySelect 2019, 4, 6338. 20. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Iiskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 09 (Revision A.01); Gaussian Inc., Wallingford CT, 2009. 21. Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A.; J. Chem. Phys. 1980, 72, 650. 22. Calais, J.-L.; Int. J. Quantum Chem. 1993, 47, 101. 23. Abashkin, Y.; Russo, N.; Sicilia, E.; Toscano, M.; Theor. Comput. Chem. 1995, 2, 255. 24. Gao, H.; Ye, C.; Piekarski, C. M.; Shreeve, J. n. M.; J. Phys. Chem. C 2007, 111, 10718. 25. Fan, X. W.; Ju, X. H.; J. Comput. Chem. 2008, 29, 505. 26. Jin, X.; Zhou, J.; Wang, S.; Hu, B.; Quim. Nova 2016, 39, 467. 27. Chi, W. J.; Li, L. L.; Li, B. T.; Wu, H. S.; J. Mol. Model. 2012, 18, 3695. 28. Hammerl, A.; Klapotke, T. M.; Noth, H.; Warchhold, M.; Holl, G.; Propellants, Explos., Pyrotech. 2003, 28, 165. 29. Murray, J. S.; Politzer, P.; Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 153. 30. Lu, T.; Chen, F.; J. Comput. Chem. 2012, 33, 580. 31. Politzer, P.; Martinez, J.; Murray, J. S.; Concha, M. C.; Toro-Labbé, A.; Mol. Phys. 2009, 107, 2095. 32. Zhang, X.; Gong, X.; Cent. Eur. J. Energ. Mater. 2015, 60, 2869. 33. Byrd, E. F.; Rice, B. M.; J. Phys. Chem. A 2006, 110, 1005. 34. Suceska, M.; Propellants, Explos., Pyrotech. 1999, 24, 280. 35. Yu, Y.; Chen, J.; Zhang, R.; Li, Y.; Pang, S.; RSC Adv. 2017, 7, 23709. 36. Kamlet, M. J.; Jacobs, S. J.; J. Chem. Phys. 1968, 48, 23. 37. Oxley, J. C.; Smith, J. L.; Chen, H.; Propellants, Explos., Pyrotech. 2002, 27, 209. 38. He, P.; Zhang, J. G.; Wang, K.; Yin, X.; Zhang, T. L.; J. Org. Chem. 2015, 80, 5643. 39. Dalinger, I. L.; Kormanov, A. V.; Suponitsky, K. Y.; Muravyev, N. V.; Sheremetev, A. B.; Chem. Asian J. 2018, 13, 1165. 40. Sinditskii, V. P.; Kolesov, V. I.; Egorshev, V. Y.; Patrikeev, D. I.; Dorofeeva, O. V.; Thermochim. Acta 2014, 585, 10. 41. https://webbook.nist.gov/chemistry/, accessed in May 2020. 42. Politzer, P.; Murray, J.; Cent. Eur. J. Energ. Mater. 2011, 8, 209. 43. Bowden, P. R.; Tappan, B. C.; Manner, V. W.; Preston, D. N.; Scott, B. L.; Shock Compression Condens. Matter (2017), doi: 10.1063/1.4971504. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access