
 |
 |
 |
 |
 |
Artigo
| Asarone-derived phenylpropanoids and isoquinoline-derived alkaloids from the bark of Duguetia pycnastera (Annonaceae) and their cytotoxicities |
|
César A. S. de SouzaI; Victória B. NardelliI; Weider H. P. PazI; Maria Lúcia B. PinheiroI; Ana Carolina B. da C. RodriguesII; Larissa M. BomfimII; Milena B. P. SoaresII; Daniel P. BezerraII; Jamal da S. ChaarI; Hector H. F. KoolenIII; Felipe M. A. da SilvaIV; Emmanoel Vilaça CostaI,*
I. Departamento de Química, Instituto de Ciências Exatas, Universidade Federal do Amazonas, 69080-900 Manaus - AM, Brasil Recebido em 23/04/2020 *e-mail: emmanoelvc@gmail.com The phytochemical investigation of the hexane and methanol extracts from the bark of Duguetia pycnastera Sandwith (Annonaceae) afforded seven known compounds, two asarone-derived phenylpropanoids and five isoquinoline-derived alkaloids. The asarones, γ-asarone (1-allyl-2,4,5-trimethoxybenzene) and 2,4,5-trimethoxy-styrene were isolated of the hexane extract while the aporphine alkaloids, O-methylmoschatoline, lysicamine, nornuciferidine, and guatterine N-oxide, and the benzyltetrahydroisoquinoline alkaloid, (S)-reticuline were isolated of the alkaloid fraction of the methanol extract. This is the first report of these compounds in D. pycnastera. γ-Asarone is being reported for the first time in the Annonaceae. Nornuciferidine is described for the second time in the Annonaceae while guatterine N-oxide is the third register. The structures of the isolated compounds were established by extensive analyses using 1D and 2D NMR spectroscopy in combination with MS. The cytotoxic activity of the isolated compounds (except for nornuciferidine) was evaluated against cancer and non-cancerous cell lines, in which lysicamine was the most active compound, mainly against HL-60, HepG2, and K562 with IC50 values of 24.40, 28.86 and 38.75 µmol L-1, respectively. INTRODUCTION The genus Duguetia A. St.-Hil. (Annonaceae) belongs to the Duguetieae tribe and comprises close to 100 species of shrubs and trees considering the recent inclusion of African taxa known as Pachypodanthium Engler & Diels.1 The vast majority of Duguetia occurs from Nicaragua to southeastern Brazil and northern Paraguay, while only four occur on the west coast of Africa. In Brazil, 66 species occur (29 of them endemic) with the greatest diversity focuses on the Amazon region.2 Some studies have been conducted on the secondary metabolites present in different parts of Duguetia plants, from which monoterpenes, diterpenes, triterpenes, lignans, flavonoids, and mostly typical isoquinoline-derived alkaloids have been isolated and characterized.1,3-8 Some of this secondary metabolites that were isolated from different tissues of Duguetia species exhibited important biological activities, such as cytotoxicity against cancer cell lines,5,7-10 anti-inflammatory and urease inhibitory properties,6,11 trypanocidal,7 leishmanicidal,7 antimalarial,9,12 antifungal,9 antioxidant, and anti-rheumatic activities.11 Despite the relevant literature indicates several species of this genus,1 only approximately 25% (~25 species) had previous chemical/pharmacological studies, fact that reinforces the importance of new studies regarding Duguetia. Duguetia pycnastera Sandwith is a small tree popularly known as 'ata', 'envira', 'envira-preta', and 'envira-surucucu' distributed in the Guyanas, Venezuela, and Brazil. In Brazil, it is commonly found in the Amapá, Amazonas and Pará states.13 In the folk medicine this species has several popular uses. The inner and the outer bark are used as a poultice for muscular aches and pains, and to treat coughs and colds while the leaves are used for treating fevers and cold sweat.14 Phytochemical investigations on D. pycnastera described the isolation and identification by GC−MS of essential oils from the leaves and stem.3 Spathulenol, allo-aromadendreno, germacrene D and elemol were the main compounds identified in both parts.3 In our continuous search for bioactive natural products of species of the Annonaceae from Amazonas with cytotoxic activities, the present work describes the phytochemical and pharmacological properties of the chemical constituents isolated from the bark of D. pycnastera. In this, seven compounds (Figure 1) were isolated by classical chromatography techniques and identified by 1D/2D nuclear magnetic resonance (1D/2D NMR) in combination with mass spectrometry (MS), been two asarone-derived phenylpropanoids (1 and 2) and five isoquinoline-derived alkaloids (3-7). In addition, the cytotoxicity of these isolated compounds (except for 7) was assessed against different cancer cell lines by the Alamar blue assay.
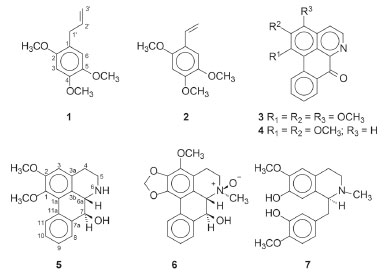 Figure 1. Isolated compounds from the bark of Duguetia pycnastera
EXPERIMENTAL General experimental procedures Optical rotations in methanol (MeOH) were recorded with a Jasco P-2000 polarimeter at 589 nm. 1D and 2D NMR experiments were acquired in CDCl3 at 293 K on a Bruker AVANCE III HD NMR spectrometer operating at 11.75 T (1H and 13C at 500 and 125 MHz, respectively). All 1H and 13C NMR chemical shifts (δ) are presented in ppm relative to the tetramethylsilane signal at 0.00 ppm as an internal reference, and the coupling constants (J) are given in Hz. The NMR spectrometer was equipped with a 5-mm multinuclear inverse detection probe (1D and 2D NMR experiments) with z-gradient. One-bond (HSQC) and long-range (HMBC) 1H-13C NMR correlation experiments were optimized for average coupling constant 1J(C,H) and LRJ(C,H) of 140 and 8 Hz, respectively. LR-ESI-MS spectra were obtained in positive ion detection mode on a Thermo Scientific LTQ XL Linear Ion Trap Mass Spectrometer, equipped with an ESI source. Silica gel 60 (Sigma-Aldrich, 70-230 mesh) was used for the column chromatography (CC), while silica gel 60 F254 (Macherey-Nagel, 0.25 mm, aluminum) was used for analytical and preparative with thin layer chromatography (PTLC) (Macherey-Nagel, 1.00 mm, glass). Compounds were visualized by exposure under UV254/365 light, by spraying with p-anisaldehyde reagent followed by heating on a hot plate, and by spraying Dragendorff's reagent. Plant material The bark of D. pycnastera was collected in August 2017 on the Adolpho Ducke Reserve (geographic coordinates: 02º 55' 37.4" S and 059º 58' 36.0" W), Manaus, Amazonas State, Brazil, and identified by the botanical of the Department of Biology of the Universidade Federal do Amazonas (DB/UFAM). A voucher specimen number 10812 was deposited at the Herbarium of DB/UFAM. The access (specimen) was registered in the 'Sistema Nacional de Gestão do Patrimônio Genético e do Conhecimento Tradicional Associado (SISGEN)' with the record A70EDCD. Extraction and isolation The bark of D. pycnastera was dried in an air circulating oven at approximately 45 °C for 72 h, and subsequently pulverized in a four-knife mill grinder (Marconi) to obtain the powdered material (635.8 g). Then, an exhaustive maceration with hexane (5 × 3 L, 25 °C) followed by MeOH (5 × 3 L, 25 °C) was performed. The extractive solutions obtained were concentrated in a rotary evaporator (Fisatom) at reduced pressure (40-50 ºC) in order to give the hexane (7.67 g) and MeOH (21.73 g) extracts. The hexane extract presented a crystalline solid after being dried. Subsequently, part of the hexane extract (3.52 g) was subjected to silica gel column chromatography (CC) which was eluted with increasing concentrations of CH2Cl2 in hexane (100:0 to 10:90, v/v), followed by EtOAc in CH2Cl2 (100:0 to 10:90, v/v), and MeOH in EtOAc (100:0 to 50:50, v/v), giving 346 fractions (25 mL each). After TLC evaluation using a mixture of hexane-EtOAc in the proportions of 95:05, 90:10, 80:20, and 70:30 as the eluent system, the similar samples were pooled to afforded 14 groups (G1 to G14). Group G7 (752.7 mg) from CC eluted with hexane-CH2Cl2 (60:40, 50:50 and 40:60 v/v) was submitted to a preparative TLC eluted with hexane-EtOAc (90:10, v/v, five elutions) yielding 1 (94.9 mg) and 2 (318.0 mg). TLC analysis revealed with Dragendorff's reagent indicated a high presence of alkaloids in the MeOH extract. Therefore, an aliquot of the MeOH extract (20.0 g) was submitted to an acid-base extraction15 to give an alkaloid (0.591 g) and a neutral (6.990 g) fractions. Subsequently, part of the alkaloid fraction (0.56 g) was subjected to silica gel column chromatography (CC) which was also eluted with increasing concentrations of CH2Cl2 in hexane (100:0 to 10:90, v/v), followed by EtOAc in CH2Cl2 (100:0 to 10:90, v/v), and MeOH in EtOAc (100:0 to 50:50, v/v), affording 151 fractions (20 mL each). After TLC evaluation using a mixture of CH2Cl2-MeOH in the proportions of 95:05, 90:10, 85:05, and 80:20 as the eluent system, the similar samples were pooled to give 7 groups (G1 to G7). Group G2 (45.0 mg) from CC eluted with hexane-CH2Cl2 (70:30, 60:40, and 50:50, v/v, respectively) was submitted to a preparative TLC eluted with CH2Cl2-MeOH (95:05, v/v, one elution) affording 3 (5.8 mg). Group G3 (89.0 mg) from CC eluted with hexane-CH2Cl2 (40:60, 30:70, and 20:80, v/v, respectively) was submitted to a preparative TLC eluted with CH2Cl2-MeOH (95:05, v/v, three elutions) giving 4 (40.0 mg). Group G4 (33.6 mg) from CC eluted with hexane-CH2Cl2 (10:90, v/v), CH2Cl2 (100%) and CH2Cl2-EtOAc (90:10, v/v) was submitted to a preparative TLC eluted with CH2Cl2-MeOH (95:05, v/v, three elutions) affording 5 (6.2 mg) and 4 (3.8 mg). Group G7 (70.8 mg) from CC eluted with CH2Cl2-EtOAc (10:90, v/v), EtOAc (100%) and EtOAc-MeOH (90:10, v/v) was submitted to a preparative TLC eluted with CH2Cl2-MeOH (95:05, v/v, three elutions) affording 6 (8.1 mg) and 7 (6.4 mg). γ-Asarone (1): Yellow light amorphous powder; 1H NMR (CDCl3): δ 6.69 (1H, s, H-6), 6.53 (1H, s, H-3), 5.96 (1H, ddt, J = 16.8, 10.4 and 6.5 Hz, H-2'trans/cis), 5.04 (1H, ddt, J = 10.4, 1.4 and 1.4, H-3'cis), 5.02 (1H, ddt, J = 16.8, 1.4 and 1.4 Hz, H-3'trans), 3.87 (3H, s, 4-OCH3), 3.82 (3H, s, 5-OCH3), 3.80 (3H, s, 2-OCH3), and 3.32 (2H, dt, J = 6.5 and 1.4 Hz, H-1'); 13C NMR (CDCl3) δ 151.3 (C-2), 147.9 (C-4), 143.0 (C-5), 137.3 (C-2'), 120.1 (C-1), 115.1 (C-3'), 114.0 (C-6), 98.1 (C-3), 56.6 (2-OCH3 and 5-OCH3), 56.2 (4-OCH3), and 33.6 (C-1'); LR-ESI(+)-MS [M + H]+ m/z 209. 2,4,5-Trimethoxy-styrene (2): White amorphous powder; 1H NMR (CDCl3): δ 7.01 (1H, s, H-6), 6.50 (1H, s, H-3), 5.59 (1H, dd, J = 17.7 and 1.4 Hz, H-2'trans), 6.98 (1H, dd, J = 17.7 and 11.1 Hz, H-1'trans/cis), 5.16 (1H, dd, J = 11.1 and 1.4 Hz, H-2'cis), 3.89 (3H, s, 4-OCH3) 3.86 (3H, s, 5-OCH3), and 3.82 (3H, s, 2-OCH3); 13C NMR (CDCl3) δ 151.4 (C-2), 149.7 (C-4), 143.4 (C-5), 130.9 (C-1'), 112.0 (C-2'), 109.6 (C-6), 97.8 (C-3), 56.7 (2-OCH3), 56.5 (5-OCH3), and 56.1 (4-OCH3); LR-ESI(+)-MS [M + H]+ m/z 195. O-methylmoschatoline (3): Orange needles (CH2Cl2-MeOH 4:1); 1H NMR and 13C NMR in accordance with literature;16 LR-ESI(+)-MS [M + H]+ m/z 322. Lysicamine (4): Yellow amorphous powder; 1H NMR and 13C NMR in accordance with literature;17 LR-ESI(+)-MS [M + H]+ m/z 292. Nornuciferidine (5): Dark orange amorphous powder; [α]D25 − 75.6º (c 0.15, CH3OH); 1H and 13C NMR data; see Table 1. LR-ESI(+)-MS [M + H]+ m/z 298. Guatterine N-oxide (6): Brown amorphous powder; [α]D25 − 40.2º (c 0.75, CH3OH); 1H and 13C NMR data; see Table 1. LR-ESI(+)-MS [M + H]+ m/z 342. (S)-(+)-Reticuline (7): Brown amorphous powder; [α]D25 + 71.6º (c 0.25, CH3OH) 1H NMR and 13C NMR in accordance with literature;18 LR-ESI(+)-MS [M + H]+ m/z 330. Cytotoxicity assay Cancer cell lines B16-F10 (mouse melanoma), HepG2 (human hepatocellular carcinoma), K562 (human chronic myeloid leukemia) and HL-60 (human promyelocytic leukemia) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured following the instructions in the ATCC animal cell culture guide. All experiments were carried out with cells in the exponential growth phase and all cell lines were tested for mycoplasma using a mycoplasma stain kit (Sigma-Aldrich) to validate the use of mycoplasma-free cells. Heparinized blood (from 20-35-year-old, healthy, non-smokers who had not taken any drugs for at least 15 days prior to sampling) was collected, and peripheral blood mononuclear cells (PBMC) were isolated using a standard protocol using a Ficoll density gradient. Concanavalin A (ConA) was used as a mitogen to trigger cell division in T-lymphocytes. ConA (10 μg mL-1) was added at the beginning of culture, and cells were treated with the compounds after 24 h. The Research Ethics Committee of the Oswaldo Cruz Foundation (Salvador, Bahia, Brazil) approved the experimental protocol (# 031019/2013). Cell viability was measured by the Alamar blue assay19 with minor modifications.20 Briefly, the cells were added in 96-well plates (0.7 x 105 cells mL-1 for adherent cells or 0.3 x 106 cells mL-1 for suspended cells in 100 μL of medium) and, after overnight incubation, the drugs (dissolved in DMSO at concentrations ranging from 0.19 to 25 μg mL-1) were added and incubated for 72 h. Doxorubicin was used as the positive control (0.08 to 5.0 μg mL-1). Four hours (for cancer cell lines) and 24 h (for PBMCs) before the end of incubation period, 20 μL of Alamar blue reagent were added to each well. Absorbance was measured on a Spectra Max 190 multiplate reader at 570 nm and 600 nm. Half-inhibitory concentration (IC50) was obtained by nonlinear regression with 95% confidence intervals (CI 95%) using the software GraphPad Prism (Intuitive Software for Science; San Diego, CA, USA).
RESULTS AND DISCUSSION The hexane and methanol extracts were subjected to successive chromatographic procedures allowing the isolation of the asarone-derived phenylpropanoids, γ-asarone21 (1-allyl-2,4,5-trimethoxybenzene) (1) and 2,4,5-trimethoxy-styrene4 (2) of the hexane extract, and the isoquinoline-derived alkaloids, O-methylmoschatoline16 (3), lysicamine17 (4), nornuciferidine22 (5), guatterine N-oxide23 (6), and (S)-reticuline18 (7) of the alkaloid fraction of the methanol extract (Figure 1). Although the isolated alkaloids have been described in some species of Duguetia, this is the first reported in D. pycnastera. Since the structures of the alkaloids 5 and 6 have been described a long time ago, their 1H and 13C data are incomplete or scalar coupling constants values have not been assigned or obtained. In this work, the unambiguous assignments of the 1H and 13C NMR data for these alkaloids were described according to 1D and 2D NMR experiments Compound 5 was obtained as a dark orange amorphous powder with the molecular formula C18H19NO3 as determined by LR-ESI(+)MS (m/z 298 [M + H]+) and NMR data. The MSn spectra of the ion at m/z 298 presented an initial loss of 18 Da (− H2O) (m/z 298 → 280), followed by competitive losses of 15 Da (− ∙CH3) (m/z 280 → 265) and 31 Da (− ∙OCH3) (m/z 280 → 249) (Figure 2). This fragmentation pattern is consistent with a 7-hydroxyaporphine derivative containing adjacent methoxyl groups at the A ring.24
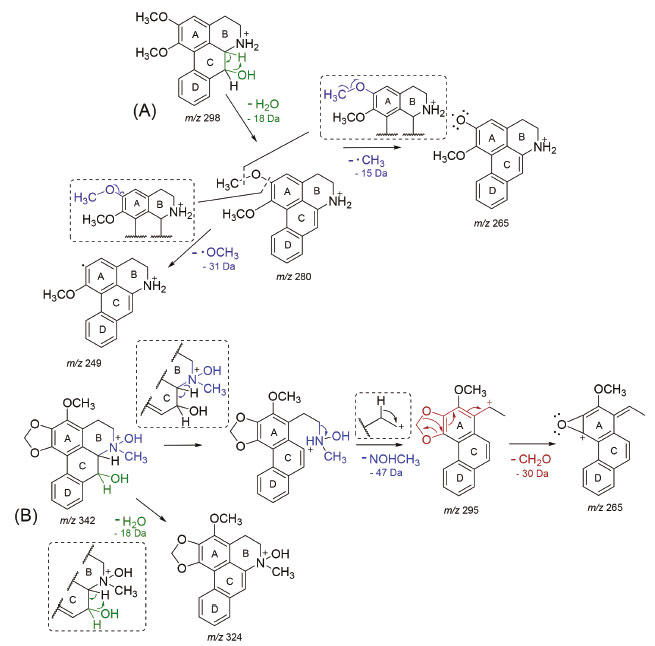 Figure 2.Proposed fragmentation pathways for the aporphine alkaloids nornuciferidine (A, m/z 298) and guatterine N-oxide (B, m/z 342)
The 1H NMR spectrum of compound 5 along with 1H-1H correlation map from COSY NMR experiment revealed the presence of two spin systems; one consisting of the signals at δH 3.06 (1H, ddd, J = 15.7, 11.5, and 5.6 Hz, H-4 pseudoaxial) and δH 2.73 (1H, br d, J = 15.7 Hz, H-4 pseudoequatorial) as well as δH 3.40 (1H, ddd, J = 11.5, 5.6 and 2.6 Hz, H-5 pseudoequatorial) and δH 2.98 (1H, td, J = 11.5 and 3.9 Hz, H-5 pseudoaxial); and the other comprising the signals at δH 3.69 (1H, d, J = 11.7 Hz, H-6a pseudoaxial) and δH 4.58 (1H, d, J = 11.7 Hz, H-7 pseudoaxial). The signal at δH 4.58 also indicate the presence of a β-hydroxyl group in the molecule located at C-7,23 being in accordance with that observed in the mass spectrum. Furthermore, it was observed a singlet at δH 6.64 (1H, H-3), indicative of an pentasubstituted benzene ring, as well as a spin system at δH 8.35 (1H, m, H-11), δH 7.71 (1H, m, H-8), and δH 7.34 (2H, m, H-9 and H-10), confirming the presence a disubstituted benzene ring. These signals observed in the aliphatic and aromatic regions are characteristic of aporphine alkaloids.8,20,25 In addition, the presence of signals at δH 3.88 and δH 3.65 (each, 3H, s) were assigned to two methoxyl groups. The location of the methoxyl groups were established based on the long-range 1H-13C correlation map from HMBC NMR experiment (Table 1 and Figure 3). This analysis revealed that the hydrogen at δH 6.64 (H-3) showed long-range 1H-13C correlation to 3J with the carbons at δC 145.2 (C-1), δC 124.0 (3b), and δC 28.7 (C-4), and to 2J with the carbon at δC 152.6 (C-2). The same analysis was performed for the presence of the hydroxyl group located in the C ring. The signal of the hydrogen at δH 4.58 (H-7) showed long-range 1H-13C correlation to 3J with the signals of the carbons at δC 123.1 (C-8), δC 124.0 (C-3b), and δC 130.2 (11a), and to 2J with the carbons at δC 59.0 (C-6a) and δC 138.9 (C-7a). This substitution was also confirmed by correlation of the signal of the hydrogen at δH 3.69 (H-6a) with the signals of the carbons at δC 42.0 (C-5), δC 71.8 (C-7), δC 124.0 (C-3b), δC 125.9 (C-1a), δC 129.2 (C-3a), and δC 138.9 (C-7a) (Figure 3). Therefore, based on these NMR data, compound 5 was established as the aporphine alkaloid known as nornuciferidine. The complete assignments for all 1H and 13C NMR chemical shifts were established by one-bond (HSQC) and long-range (HMBC) 1H-13C NMR correlation experiments and were described in the Table 1. This alkaloid is described for the second time in the Annonaceae family, only in the Duguetia genus that was reported for the first time in the Duguetia spixiana Mart.1,22,25 After conducting of the 1D/2D NMR experiments for nornuciferidine, it was observed in the spectra signals of its decomposition, probably due to the long wait for analysis in acidic solvent (CDCl3). This observation indicates that the substance is unstable, which may explain its low occurrence in the literature. However, no support for this report was found.
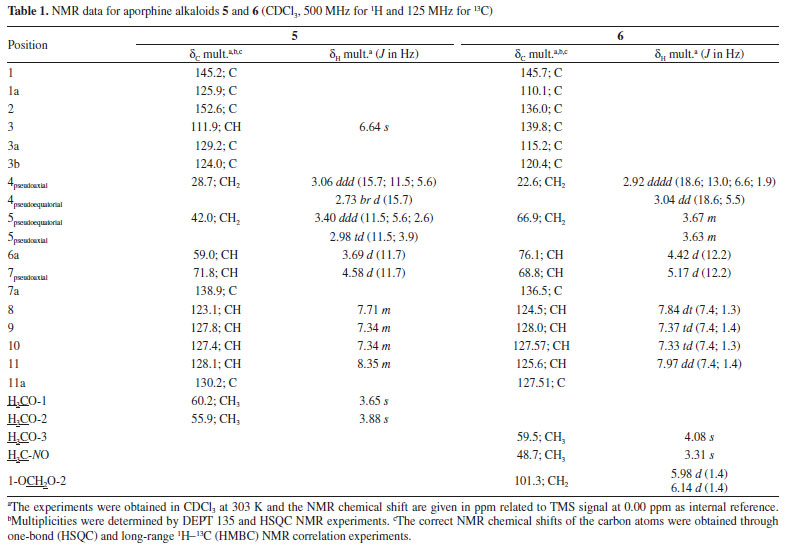
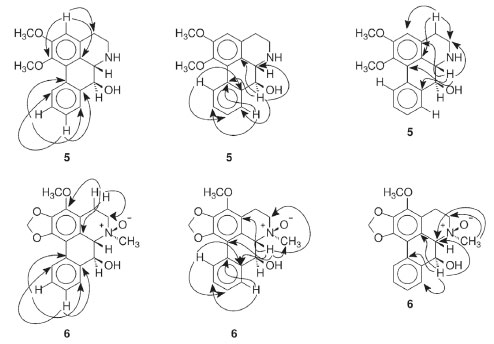 Figure 3. Main key correlations observed in the HMBC (arrows) experiment for the alkaloids 5 and 6
Compound 6 was obtained as a brown amorphous powder with the molecular formula C19H19NO5 as determined by LR-ESI(+)MS (m/z 342 [M + H]+) and NMR data. The MSn spectra of the ion at m/z 342 presented initial competitive losses of 18 Da (− H2O) (m/z 342 → 324) and 47 Da (− NHOCH3) (m/z 342 → 295) (Figure 2). The initial loss of 47 Da is in agreement with an N-methyl N-oxide aporphine structure.8 In addition, the subsequent loss of 30 Da (CH2O) (m/z 295 → 265) is consistent with the presence of a methylenedioxy group at the A ring (Figure 2).24 The 1H NMR spectrum of compound 6 along with 1H-1H correlation map from COSY NMR experiment also revealed the presence of two spin systems; one consisting of the signals at δH 3.04 (1H, dd, J = 18.6 and 5.5 Hz, H-4 pseudoequatorial) and δH 2.92 (1H, dddd, J = 18.6, 13.0, 6.6, and 1.9 Hz, H-4 pseudoaxial) as well as δH 3.67 (1H, m, H-5 pseudoequatorial) and δH 3.63 (1H, m, H-5 pseudoaxial); and other comprising the signals at δH 4.42 (1H, d, J = 12.2 Hz, H-6a pseudoaxial) and δH 5.17 (1H, d, J =12.2 Hz, H-7 pseudoaxial). Similarly, to compound 5, the presence of a β-hydroxyl group in the molecule located at C-7 (ring C) was observed at δH 5.17 (H-7 pseudoaxial).23 The difference between 6 and 5 is the absence of the aromatic hydrogen at C-3 and the methoxyl groups at C-1 and C-2 present in 5 that was replaced with one methoxyl group at C-3 and one methylenedioxy group at C1/C2 in 6 at the A ring. On the other hand, a significant difference between 6 and 5 was observed in δH 3.31 (3H, s) suggesting the presence of methadone N-oxide (CH3-NO) that was established on the basis of one-bond (HSQC) and long-range (HMBC) 1H-13C NMR correlation experiments (Figure 3 and Table 1). This observation is in accordance with that indicated in the mass spectrum (Figure 2). Thus, the signal of the hydrogen at δH 3.31 (CH3-NO) showed long-range 1H-13C correlation to 3J with the signals of the carbons at δC 66.9 (C-5) and δC 76.1 (C-6a), and to 4J with the carbon at δC 22.6 (C-4) (Figure 3). The presence of the methoxyl group at C-3 was also confirmed by HMBC experiment due to correlation of the signals of the hydrogens at δH 3.04 (H-4 pseudoequatorial) and δH 2.92 (H-4 pseudoaxial) with the signal of the carbon at δC 139.8 (C-3) (Figure 3). Therefore, based on these NMR data, compound 6 was established as the aporphine alkaloid known as guatterine N-oxide. The complete assignments for all 1H and 13C NMR chemical shifts were established by one-bond (HSQC) and long-range (HMBC) 1H-13C NMR correlation experiments and were described in the Table 1. The relative configuration of the chiral centers N-6, C-6a and C-7 was determined by NOESY NMR experiments. In this experiment the signal of the hydrogen at δH 3.31 (CH3-NO) showed correlation of the signal of the hydrogen at δH 5.17 (H-7 pseudoaxial) established the relative R-configuration for nitrogen, and relative S-configuration for C-6a. In the same way the configuration of β-hydroxyl group and the relative S-configuration for C-7 was stablished by absence of the correlation in the NOESY NMR experiment of the signal of the hydrogen at δH 5.17 (H-7 pseudoaxial) with the signal of the hydrogen at δH 4.42 (C-6a pseudoaxial). These results are confirmed by comparison of the 13C NMR data (Table 1) of this compound (6) with methadone N-oxide (CH3-NO) aporphine alkaloids models available in the literature such as duguetine and oliveroline β-N-oxide (6a, 7α-H, β-N-oxide),4,25 and dasymaschaline α-N-oxide (6a,7α-H, α-N-oxide).25 This alkaloid is described for the third time in the Annonaceae family, present only in the Duguetia and Guatteria genera.1 In Guatteria it was described only in Guatteria sagotiana23,26 R. E. Fries while in Duguetia was reported in Duguetia confinis23,25 (Engl. & Diels) Chatrou (synonym: Pachypodanthium confine Engl. & Diels).23 All isolated compounds, with the exception of γ-asarone, have been described in several species of the Annonaceae family contributing to the chemotaxonomic knowledge of this family, in addition to providing another alternative source of isolation of these chemical constituents. γ-Asarone is described for the first time in the Annonaceae. So far, only the presence of α-asarone in Duguetia furfuracea (A. St.-Hil.) Benth. & Hook. and β-asarone in Artabotrys hexapetalus (L. f.) Bhandari were previously reported.4,27 2,4,5-Trimethoxy-styrene (2) have been found in D. furfuracea,4 Duguetia panamensis Standley,28 Duguetia eximia Diels,28 and Duguetia staudtii (Engl. & Diels) Chatrou (synonym: Pachypodanthium staudtii Engl. et Diels).28 Among the isolated compounds, the alkaloids are the most representative, particularly O-methylmoschatoline (3), lysicamine (4), and (S)-reticuline (7) that are reported in several genera, with emphasis on Annona, Artabotrys, Cananga, Desmos, Duguetia, Enantia, Guatteria, Miliusa, Monodora, Oxandra, Uvaria, and Xylopia.1,24 Reticuline is considered the biosynthetic precursor of aporphine alkaloids and its presence in this species reinforces this fact. The cytotoxic activity of the isolated compounds, except for compound 5 (due to its decomposition), was evaluated against cancer (B16-F10, HepG2, HL-60, and K562) and non-cancerous (PBMC) cells (Table 2) using the Alamar blue assay after 72 h of incubation. Compound 6 was tested against only HepG2 cells due to the viability of the cells during the experiments. Doxorubicin was used as a positive control and presented IC50 values of 0.71 (HL-60), 1.17 (HepG2), 2.06 (B16-F10), and 2.37 µmol L-1 (K562) for tumor cell lines, respectively, and an IC50 value of 2.61 µmol L-1 for non-cancerous (PBMC) cell line. According to Table 2 only the alkaloids showed cytotoxic activity. Lysicamine (4) was the alkaloid most potent showing IC50 values of 24.24, 28.86 and 38.75 µmol L-1 against HL-60, HepG2 and K562 cells, respectively, and was not active against PBMC at the concentrations tested (IC50 > 85.81 µmol L-1), being moderately active when compared to doxorubicin. The structure-activity relationship established by the alkaloids 3 and 4 (the only difference between them is the presence of an extra methoxyl group that is present in the alkaloid 3) suggests that the presence of a substitution in this position reduces the cytotoxic activity. However, further investigation is needed to confirm this observation.
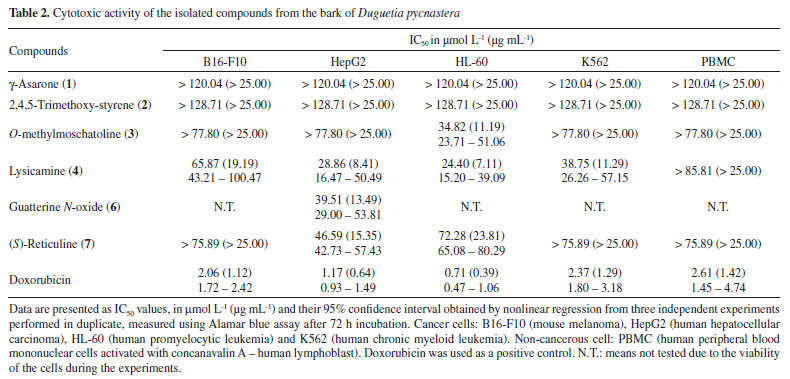
CONCLUSIONS The phytochemical investigation of the bark of D. pycnastera led to the isolation and identification of seven known compounds (1-7); two asarone-derived phenylpropanoids, γ-asarone (1) and 2,4,5-trimethoxy-styrene (2) of the hexane extract; four aporphine alkaloids, O-methylmoschatoline (3), lysicamine (4), nornuciferidine (5), and guatterine N-oxide (6), and one benzyltetrahydroisoquinoline alkaloid, (S)-reticuline (7) of the methanol extract. The isolated compounds are described for the first time in D. pycnastera. γ-Asarone is reported for the first time in the Annonaceae family. Nornuciferidine is described for the second time in the Annonaceae and only in the Duguetia. Guatterine N-oxide has been reported for the third time in the Annonaceae only in the Duguetia and Guatteria genera. These results are especially important for the chemotaxonomic point of view and contributes for the chemotaxonomic knowledge of the species of the Annonaceae family. The cytotoxic activity of the isolated compounds (except for nornuciferidine) were evaluated against cancer and non-cancerous cell lines (except for guatterine N-oxide that was only evaluated against HepG2), in which lysicamine was the most potent one, mainly against HL-60, HepG2, and K562 with IC50 values of 24.40, 28.86 and 38.75 µmol L-1, respectively, being moderately active when compared to doxorubicin.
SUPPLEMENTARY MATERIAL Supplementary information containing 1D and 2D NMR, and MS data for isolated compounds is available free of charge at http://quimicanova.sbq.org.br as a PDF file.
ACKNOWLEDGEMENTS The authors are grateful to Analytical Center of the Multidisciplinary Support Center at the UFAM (CA/CAM/UFAM) for the NMR and MS analysis, and CNPq, CAPES, FINEP, FAPESB, and UFAM for financial support and fellowship.
REFERENCES 1. Pérez, E. G.; Cassels, B. K. In The Alkaloids; Cordell, G. A., ed.; Academic Press: Chennai, pp. 83-156, 2010, v. 68, chapter 3, doi: 10.1016/S1099-4831(10)06803-3. 2. Lopes, J. C.; Mello-Silva, R.; Rev. Bras. Frutic. 2014, 36, 125. 3. Maia, J. G. S.; Andrade, E. H. A.; Carreira, L. M. M.; Oliveira, J.; J. Essent. Oil Res. 2006, 18, 60. 4. Silva, D. B.; Tulli, E. C. O.; Garcez, W. S.; Nascimento, E. A.; Siqueira, J. M.; J. Braz. Chem. Soc. 2007, 18, 1560; Santos, T. C. B.; Oliveira, R. C.; Vasconcelos, L. G.; Souza Jr., P. T.; Silva, V. C. P.; Carvalho, M. G.; Ribeiro, T. A. N.; Biochem. Syst. Ecol. 2019, 87, 103951. 5. Rodrigues, A. C. B. C.; Bomfim, L. M.; Neves, S. P.; Menezes, L. R. A.; Dias, R. B.; Soares, M. B. P.; Prata, A. P. N.; Rocha, C. A. G.; Costa, E. V.; Bezerra, D. P.; Planta Med. 2015, 81, 798. 6. Ngouonpe, A. W.; Mbobda, A. S. W.; Happi, G. M.; Mbiantcha, M.; Tatuedom, O. K.; Ali, M. S.; Lateef, M.; Tchouankeu, J. C.; Kouam, S. F.; Biochem. Syst. Ecol. 2019, 83, 22. 7. Silva, D. B.; Tulli, E. C. O.; Milita, G. C. G.; Costa-Lotufo, L. V.; Pessoa, C.; Moraes, M. O.; Albuquerque, S.; Siqueira, J. M.; Phytomedicine 2009, 16, 1059. 8. Paz, W. H. P.; Oliveira, R. N.; Heerdt, G.; Angolini, C. F. F.; Medeiros, L. S.; Silva, V. R.; Santos, L. S.; Soares, M. B. P.; Bezerra, D. P.; Morgon, N. H.; Almeida, J. R. G. S.; Silva, F. M. A.; Costa, E. V.; Koolen, H. H. F.; J. Nat. Prod. 2019, 82, 2220. 9. Muhammad, I.; Dunbar, C.; Takamatsu, S.; Walker, L. A.; Clark, A. M.; J. Nat. Prod. 2001, 64, 559. 10. Matos, M. F. C.; Leite, L. I. S. P.; Brustolim, D.; Siqueira, J. M.; Carollo, C. A.; Hellmann, A. R.; Pereira, N. F. G.; Silva, D. B.; Fitoterapia 2006, 77, 227. 11. Santos, R. C.; Souza, A. V.; Andrade-Silva, M.; Cruz, K. C. V.; Kassuya, C. A. L.; Cardoso, C. A. L.; Vieira, M. C.; Formagio, A. S. N.; J. Ethnopharmacol. 2018, 211, 9. 12. Pérez, E.; Sáez, J.; Blair, S.; Franck, X.; Figadère, B.; Lett. Org. Chem. 2004, 1, 102. 13. Maas, P. J. M.; Maas, H.; Miralha, J. M. S.; Rodriguésia 2007, 58, 617. 14. DeFilipps, R. A.; Maina, S. L.; Crepin, J.; Medicinal Plants of the Guianas (Guyana, Surinam, French Guiana), Departament of Botany, National Museum of Natural History, Smithsonian Institution, Washington, D.C., 20013-7012, 2004. 15. Costa, E. V.; Pinheiro, M. L. B.; Xavier, C. M.; Silva, J. R. A.; Amaral, A. C. F.; Souza, A. D. L.; Barison, A.; Campos, F. R.; Ferreira, A. G.; Machado, G. M. C.; Leon, L. L. P.; J. Nat. Prod. 2006, 69, 292. 16. Costa, E. V.; Pinheiro, M. L. B.; Souza, A. D. L.; Barison, A.; Campos, F. R.; Valdez, R. H.; Ueda-Nakamura, T.; Dias Filho, B. P.; Nakamura, C. V.; Molecules 2011, 16, 9714. 17. Teles, M. N. O.; Dutra, L. M.; Barison, A.; Costa, E. V.; Biochem. Syst. Ecol. 2015, 61, 465. 18. Cruz, P. E. O.; Costa, E. V.; Moraes, V. R. S.; Nogueira, P. C. L.; Vendramin, M. E.; Barison, A.; Ferreira, A. G.; Prata, A. P. N.; Biochem. Syst. Ecol. 2011, 39, 872. 19. Ahmed, S. A.; Gogal Jr., R. M.; Walsh, J. E.; J. Immunol. Methods 1994, 170, 211. 20. Costa, E. V.; Sampaio, M. F. C.; Menezes, L. R. A.; Dutra, L. M.; Costa, C. O. S.; Pinheiro, M. L. B.; Silva, F. M. A.; Soares, M. B. P.; Bezerra, D. P.; Barison, A.; Koolen, H. H. F.; Quim. Nova 2020, 43, 419, doi: 10.21577/0100-4042.20170504. 21. Sinha, A. K.; Acharya, R.; Joshi, B. P.; J. Nat. Prod. 2002, 65, 764. 22. Rasamizafy, S.; Hocquemiller, R.; Cavé, A.; J. Nat. Prod. 1987, 50, 674. 23. Rasamizafy, S.; Hocquemiller, R.; Cavé, A.; J. Nat. Prod. 1986, 49, 1078; Bévalot, F.; Leboeuf, M.; Bouquet, A.; Cavé, A.; Ann. Pharm. Fr. 1977, 35, 65; Guinaudeau, H.; Leboeuf, M.; Cavé, A.; J. Nat. Prod. 1979, 42, 325; Guinaudeau, H.; Leboeuf, M.; Cavé, A.; J. Nat. Prod. 1988, 51, 389. 24. de Lima, B. R.; da Silva, F. M. A.; Soares, E. R.; de Almeida, R. A.; da Silva-Filho, F. A.; Barison, A.; Costa, E. V.; Koolen, H. H. F.; de Souza, A. D. L.; Pinheiro, M. L. B.; J. Braz. Chem. Soc. 2020, 31, 79; Stévigny, C.; Jiwan, J. L. H.; Rozenberg, R.; de Hoffmann, E.; Quetin-Leclercq, J.; Rapid Commun. Mass Spectrom. 2004, 18, 523; Carnevale Neto, F.; Andréo, M. A.; Raftery, D.; Lopes, J. L. C.; Lopes, N. P.; Castro-Gamboa, I.; Maia, B. H. L. N. S.; Costa, E. V.; Vessecchi, R.; Rapid Commun. Mass Spectrom. (2019), doi: 10.1002/rcm.8533. 25. Guinaudeau, H.; Leboeuf, M.; Cavé, A.; J. Nat. Prod. 1994, 57, 1033; Asaruddin, M. R.; Kiuchi, F.; Honda, G.; Nat. Med. 2001, 55, 149. 26. Lúcio, A. S. S. C.; Almeida, J. R. G. S.; da-Cunha, E. V. L.; Tavares, J. F.; Barbosa-Filho, J. M. In The Alkaloids; Knolker, H.-J., ed.; v. 74, pp. 233-409, 2015, chapter 5, doi: 10.1016/bs.alkal.2014.09.002. 27. Thang, T. D.; Dai, D. N.; Hoi, T. M.; Ogunwande, I. A.; J. Essent. Oil Res. 2013, 25, 85. 28. Wang, Z.-W.; Ma, W.-W.; McLaughlin, J. L.; J. Nat. Prod. 1988, 51, 382; Gottlieb, O. R.; Magalhães, A. F.; Magalhães, E. G.; Maia, J. G. S.; Marsaioli, A. J.; Phytochemistry 1978, 17, 837; Waterman, P. G.; Phytochemistry 1976, 15, 347. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access