
 |
 |
 |
 |
 |
Artigo
|
|
| Polímero de impressão molecular para o tiacloprido: uma investigação teórica Molecularly imprinted polymer for thiacloprid: a theoretical investigation |
|
Camilla Fonseca Silva; Clebio Soares Nascimento Jr.* Departamento de Ciências Naturais, Universidade Federal de São João del-Rei, Campus Dom Bosco, 36301-160 São João del-Rei - MG, Brasil Recebido em 02/06/2020 *e-mail: liurise@139.com Thiacloprid (TCP) is a synthetic insecticide, belonging to the class of neonicotinoids (NNCs), which present potential damages to the environment, biodiversity and human health, being necessary its constant monitoring. In this sense, molecularly imprinted polymers (MIPs) have been shown as an excellent alternative for the separation science, since they can recognize molecularly and selectively the analyte in various types of complex sample, such as food, soil, etc., thus contributing to its efficient extraction. In the present work, we have investigated, through DFT calculations, the mechanism of formation of a MIP for the TCP. As main result we obtained the following theoretical protocol for the MIP synthesis: 2-(trifluoromethyl) aryl acid (TFMAA) as FM (functional monomers), in 1:3 stoichiometry, and toluene as the solvent. This theoretical protocol showed the most favorable stabilization energies for the pre-polymerization complexes formed. In addition, it has been shown that the establishment of hydrogen bonds between the TCP and FM play a fundamental role in the stability of the complexes formed. We consider this work quite relevant since it can be used by experimentalists in order to reach an efficient MIP synthesis for TCP. INTRODUÇÃO Nas últimas décadas, tem-se observado um crescimento substancial da produção agrícola mundial, justificado pela modernização dos equipamentos utilizados no campo e pelo desenvolvimento de técnicas de cultivo, aliados ao uso de fertilizantes e agroquímicos.1 Contudo, verifica-se que, no processo de modernização, os agroquímicos naturais obtidos pela extração de substâncias orgânicas de plantas, a exemplo do piretro, da nicotina e da rotenona, extraídos de flores secas de crisântemo (Chrysanthemum cinerariaefolium), de folhas fumo (Nicotiana tabacum) e de raízes de leguminosas (Derris elliptica), respectivamente,2 deram espaço, após a 2ª guerra mundial, a diversos compostos orgânicos sintéticos, com cadeias carbônicas substituídas por elementos como fósforo (P), cloro (Cl), oxigênio (O), enxofre (S) e nitrogênio (N), atualmente conhecidos como organofosforados, organoclorados, carbamatos, piretróides e neonicotinóides.3 Os neonicotinóides (NNCs) são uma classe de inseticidas sintéticos, derivados da nicotina, que surgiram por volta da década de 1980 e que hoje já representam mais de 25% dos inseticidas totais utilizados no mundo.3 Embora largamente utilizados, os NNCs têm sido relatados na literatura associados à desequilíbrios ambientais e biológicos, devido a sua longa permanência no meio ambiente, bem como sua lixiviação em solos e corpos d'água com consequentes prejuízos à biodiversidade. Além disso, sabe-se que o uso contínuo dos NNCs pode gerar envenenamento da fauna e da flora e trazer danos à saúde humana, dados esses confirmados por eventuais casos intoxicação de trabalhadores rurais e consumidores de produtos alimentícios, devido às suas toxicidades associadas.4 No Brasil, o programa do governo federal para acompanhar a presença de agrotóxicos em alimentos no país é o PARA (Programa de Análise de Resíduos de Agrotóxicos), regulado pela Anvisa (Agência Nacional de Vigilância Sanitária). O relatório mais recente do PARA analisou amostras de 9.680 alimentos, como fubá, farinha de trigo, mel, abacaxi, banana, laranja, entre outros, nos quais foram encontrados traços de NNCs em 2.401 amostras, sendo que em 339 delas haviam níveis irregulares da substância. Isso significa que foram identificadas quantidades dessa classe de inseticidas acima do limite máximo permitido.5 Uma reportagem de 2015, da agência de notícias Reuters, levantou alguns dos inseticidas banidos em outros países do mundo, mas que seguem sendo vendidos no Brasil. Intitulado "Porque o Brasil tem um apetite por pesticidas perigosos", o artigo afirma que o país se transformou em um "mercado tentador para pesticidas banidos de outras nações por causa de riscos ambientais ou à saúde". Há produtos permitidos no país, mas proibidos em países da União Europeia, Estados Unidos e China.6 Dentro desse contexto apresentado, nota-se que o uso de NCCs pode acarretar problemas ambientais e de saúde humana gravíssimos, o que implica na necessidade de constantes determinações de resíduos dessa classe de inseticidas em amostras de interesse analítico, como solo e água, além de produtos alimentícios in natura e/ou processados. O tiacloprido (TCP) (Figura 1) é um inseticida NNC, de fórmula molecular C10H9ClN4S e de massa molar igual a 252,72 g mol-1. Apresenta-se comumente na forma de cristais de coloração branca, com solubilidade em água, à 25 °C, igual a 0,0687 mg mL-1. De acordo com dados da ANVISA, o TCP pertence à classe toxicológica II, considerado um produto altamente tóxico, justificando o apelo analítico para sua determinação.7 Entretanto, tais determinações se mostram como atividades complexas e trabalhosas, devido às diferentes propriedades físico-químicas das substâncias contidas nas amostras, além das baixas concentrações dos analitos (geralmente presentes em concentrações da ordem de µg L-1 ou ng L-1) e das altas concentrações de interferentes,8 exigindo que sejam aplicadas previamente às análises instrumentais, uma etapa denominada preparo de amostra, a qual visa pré-concentrar e isolar o analito em questão.9
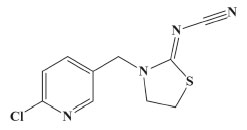 Figura 1. Estrutura química do NNC tiacloprido (TCP)
Nos últimos anos, diversas técnicas de preparo de amostras têm sido utilizadas, com destaque para técnicas clássicas, como a extração com uso de solventes denominada extração líquido-líquido (LLE, do inglês liquid-liquid extraction) e suas diferentes versões e a extração com uso de materiais sorventes, denominada extração em fase sólida (SPE, do inglês solid phase extraction) e suas diferentes versões.10 No que se refere à técnica de extração em fase sólida, SPE, a mesma utiliza de materiais sorventes sólidos e consiste no método mais popular de preparo de amostras, com diversas aplicações que visam o menor consumo de solvente orgânico, a não formação de emulsões, altas porcentagens de recuperação do analito, baixa geração de resíduos tóxicos, além de agregar altas seletividade e recuperação. Em contrapartida, a SPE apresenta como desvantagens o longo tempo de extração, altos custos e em alguns casos, a dificuldade de adequar a natureza do material sorvente para determinadas aplicações.11 Na literatura, há um consenso que os materiais sorventes convencionais, como C8, C18, alumina, sílica gel, silicato de magnésio e alguns polímeros e copolímeros, embora apresentem poder adsorção, têm se mostrado como materiais carentes de seletividade.12 Nesse sentido, estudos das últimas décadas têm relatado o desenvolvimento de novos materiais sorventes, a exemplo dos polímeros de impressão molecular (MIP, do inglês molecularly imprinted polymer), como materiais altamente seletivos a serem empregados em preparo de amostras de diversos tipos de analitos e de matrizes.13,14 Na Figura 2 é mostrado, de forma esquemática, o procedimento de impressão molecular. Na técnica de impressão molecular, monômeros funcionais (MFs) são organizados ao redor da molécula molde (MM), que é o próprio analito ou alguma molécula de estrutura análoga, pelo estabelecimento de ligações covalentes ou mais comumente, de interações intermoleculares, formando um complexo de pré-polimerização, que na presença do agente de ligação cruzada (ALC) e do iniciador radicalar (IR) originam uma matriz polimérica rígida e tridimensional, a qual é submetida a clivagem química ou extração com uso de solventes, a fim de se promover a eluição da MM, de forma que o material obtido passe a exibir sítios seletivos e favoráveis à retenção do analito em posteriores extrações.15
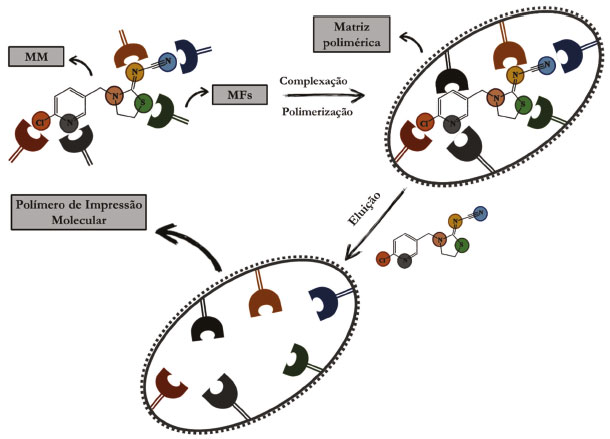 Figura 2. Representação esquemática das etapas envolvidas na síntese de um MIP. Fonte: próprios autores
Embora os MIPs sejam frequentemente relatados na literatura como materiais com grande eficiência e potencialidades, alguns autores têm enfatizado a demanda por mais estudos específicos, que visem o aumento da seletividade dos MIPs em função da formação de sítios mais homogêneos e específicos, além de baixos tempos de extração e maior aplicabilidade, garantidos pela impressão molecular não-covalente.13-16 Nesse contexto, buscando o entendimento a nível molecular do processo de formação de MIPs, além do reconhecimento e seletividade atribuído a eles, a abordagem computacional tem sido relatada na literatura como uma ferramenta útil no planejamento racional de síntese dos MIPs.17,18 Além disso, estudos de modelagem molecular ajudam a evitar consumo excessivo de recursos do laboratório, adequando a síntese do MIP aos parâmetros estabelecidos pela Química Verde e otimizando as condições a serem empregadas na síntese destes materiais.17 Nos últimos anos, simulações clássicas de dinâmica molecular (DM), bem como cálculos quanto-mecânicos (QM) têm sido usados de forma rotineira no planejamento racional de MIPs, no que diz respeito à obtenção de complexos de pré-polimerização, formados pela organização de MFs ao redor da MM em um meio com solvente.17 Cabe destacar que, atualmente, a Teoria do Funcional de Densidade (DFT) é a metodologia teórica mais empregada em estudos envolvendo MIPs, devido ao fato da DFT ser um método quanto-mecânico rápido, confiável, preciso e eficiente em descrever a energia e estrutura eletrônica destes sistemas químicos.19-29 Pardeshi e colaboradores, em 2012, propuseram o uso da metodologia DFT, na busca de um MIP para o ácido gálico com alta capacidade de reconhecimento molecular. Para tanto, foram otimizados complexos MM:MFs em nível B3LYP/6-31G(d) utilizando 18 diferentes monômeros em estequiometria 1:1, tendo sido o ácido metacrílico (MAA), o MF mais adequado. A validação do estudo computacional foi realizada experimentalmente.26 Em 2013, Tadi e colaboradores, estudaram computacionalmente a interação da molécula molde cis/trans ácido oxálico com o monômero funcional acrilamida (ACR) em razões molares 1:1, 1:2, 1:3 e 1:4, por meio de cálculos DFT em nível B3LYP/6-31G*, tendo obtido a proporção 1:4 como ótima. O MIP foi sintetizado por meio da polimerização em massa e a capacidade de adsorção seletiva verificada.27 Já em 2019, Sajini e colaboradores investigaram a influência da natureza do monômero funcional e do agente de ligação cruzada, bem como da melhor razão molar para síntese de um MIP para o éster benzílico de L-fenilamina (L-PABE). Foram realizados cálculos DFT, em nível B3LYP/6-31G. Os resultados apontaram que o MIP mais apropriado é obtido com o uso do monômero funcional ácido metacrílico (MAA), na proporção 1:2 (L-PABE:MAA) e com etilenoglicol dimetacrilato como agente de ligação cruzada. Em seguida, os autores sintetizaram e caracterizaram o MIP obtido racionalmente e evidenciaram o poder sortivo do material em estudos de adsorção.28 Em relação aos estudos para avaliação da influência do solvente, Riahi e colaboradores avaliaram a influência de acetonitrila e clorofórmio, na estabilidade dos complexos de pré-polimerização da molécula molde clorfenamina em razões molares variadas de 1:1 a 1:4 com o monômero funcional ácido metacrílico, usando a DFT em nível B3LYP/6-311+(d,p) e a DFTB para melhoria da precisão e confiabilidade.29 Nos últimos anos, nosso grupo de pesquisa, o Laboratório de Química Teórica e Computacional (LQTC/UFSJ), tem relatado estudos teóricos, via cálculos DFT, com o objetivo de se planejar racionalmente a síntese de MIPs.30-33 Em 2016, Fonseca e colaboradores realizaram um estudo DFT do processo de interação do tramadol (um opiáceo analgésico) com distintos MFs, em diferentes estequiometrias, na formação de um MIP para esse fármaco. Os resultados possibilitaram estabelecer que as melhores condições na síntese do MIP foram obtidas com ácido acrílico (AA) como MF (na proporção molar MM:MF de 1:3) e com clorofórmio como solvente. Esse protocolo teórico foi utilizado, posteriormente, na síntese eficiente do MIP para o tramadol.30 Mais recentemente, em 2018 e 2019, Silva e colaboradores relataram a otimização das condições de síntese de dois MIPs para os inseticidas neonicotinóides dinotefurano (DNF) e acetamiprido (ACT). Foram avaliados a influência da natureza química dos monômeros funcionais, em diversas estequiometrias e solventes através de cálculos realizados em nível DFT, com o funcional B3LYP e o conjunto de funções de base 6-31G(d,p).31,33 Os resultados da simulação para o DNF foi usado para a síntese de um MIP posteriormente aplicado para recuperação do analito em amostras de saliva artificial e os resultados para o ACT, estão sendo usados como referência para síntese eficiente do MIP. Nesse contexto, dada a relevância atual desse tema nas políticas nacionais envolvendo questões ambientais, além de dar continuidade aos trabalhos sobre MIPs para compostos da classe dos NCCs, o objetivo geral deste trabalho consistiu na investigação teórica das melhores condições de síntese para um MIP para o tiacloprido, um NCC de 2ª geração. Buscou-se realizar a otimização da natureza do monômero funcional a ser complexado à molécula molde, na estequiometria mais adequada, além do solvente a ser empregado na síntese.
METODOLOGIA TÉORICA Inicialmente, cálculos de otimização de geometria, no nível DFT/B3LYP/6-31G(d,p)34,35 foram realizados para as espécies químicas isoladas, TCP e todos MFs avaliados, tendo sido escolhidos: ácido acrílico (AA), ácido metacrílico (MAA), ácido p-vinilbenzóico (APV) e ácido 2-(trifluorometil)-arílico (TFMAA), devido ao caráter básico do TCP, bem como pelo fato de serem os MFs mais utilizados em estudos de MIPs. Todas as estruturas foram completamente otimizadas e caracterizadas como mínimos verdadeiros na Superfície de Energia Potencial (SEP) através da análise das frequências harmônicas, sendo todas as frequências calculadas reais. A fim de estabelecer o MF mais seletivo e a estequiometria mais adequada para os complexos, calculou-se a variação da energia eletrônica de complexação (ΔE) e a variação da energia livre de Gibbs (ΔG), associadas à formação de cada um dos complexos de pré-polimerização, a partir das Equações (1) e (2), sendo o termo n referente ao coeficiente estequiométrico do MF, o qual teve seu valor variado de 1 a 3, gerando complexos nas razões molares 1:1, 1:2 e 1:3 e os termos G e E referentes respectivamente, a energia de Gibbs e eletrônica absolutas do complexo, da molécula TCP e dos MFs avaliados. 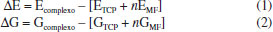 Uma vez definida a estequiometria mais adequada, foi conduzido o estudo da influência de sete solventes na síntese do MIP proposto, por meio do teste de duas metodologias distintas:
Os solventes escolhidos para avaliação foram a água, clorofórmio, metanol, acetonitrila, acetona, dimetilsulfóxido e tolueno, utilizando o modelo SMD (do inglês Solvatation Model based on Density).36 Todos os cálculos DFT foram realizados no pacote computacional Gaussian 0937 e os programas GaussView 5.0 e ArgusLab 4.0.1, utilizados como ferramentas gráficas para construção de modelos moleculares e visualização dos resultados.
RESULTADOS E DISCUSSÃO Seleção do monômero funcional e da estequiometria MM:MF Inicialmente, sabendo que formação de complexos de pré-polimerização se dá pela organização dos MFs ao redor da MM, gerou-se o mapa do potencial eletrostático molecular (MEP, do inglês molecular electrostatic potential)38 para o TCP. O MEP gerado revelou regiões de densidade de carga parcial negativa (δ-), graficamente representada por colorações vermelha e verde e regiões de densidade de carga parcial positiva (δ+), representada por colorações violeta e branca. Cálculos em nível DFT/B3LYP/6-31G(d,p) mostraram a densidade de carga parcial negativa sob os quatro átomos de nitrogênio do TCP, identificados como N-1 (δ = -0,0514) , N-2 (δ = -0,0505), N-3 (δ = -0,0419) e N-4 (δ = -0,0396), que confirmam o caráter doador de elétrons desses átomos (ver Figura 3).
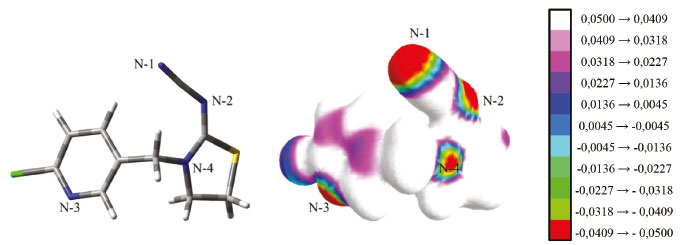 Figura 3. Mapa de potencial eletrostático molecular para o TCP, com a escala de valores de distribuição de carga do tipo Mulliken
Na sequência, levando-se em conta parâmetros quantitativos de distribuição de cargas obtidos via MEP, bem como uma análise do impedimento estérico dos sítios de interação, ou seja, da presença de grupos substituintes volumosos nos átomos de nitrogênio ou próximos espacialmente desses sítios de interação, que em geral podem causar fortes repulsões inter-eletrônicas e uma consequente desestabilização energética, conduziu-se a formação dos complexos de pré-polimerização. Foram consideradas as associações dos monômeros funcionais à estrutura da molécula molde, por meio do estabelecimento de ligações de hidrogênio entre as hidroxilas dos quatro MFs com os átomos de nitrogênio N-1, N-2, N-3 e N-4 do TCP, respectivamente, tendo sido observados a cada nova estequiometria a formação de uma ligação de hidrogênio adicional em relação ao complexo anteriormente formado. As Figuras 4, 5 e 6 apresentam as estruturas otimizadas dos complexos em estequiometria 1:1, 1:2 e 1:3, respectivamente. Deve-se destacar que, ao se considerar a adição de uma quarta molécula de MF aos complexos 1:3, verificou-se que devido ao impedimento estérico desse sítio, não ocorreu a formação de uma ligação de hidrogênio adicional esperada, e sim a condução de uma ligação de hidrogênio entre os próprios MFs adjacentes, o que não é desejável para funcionalidade do material, sendo essa estequiometria desconsiderada.
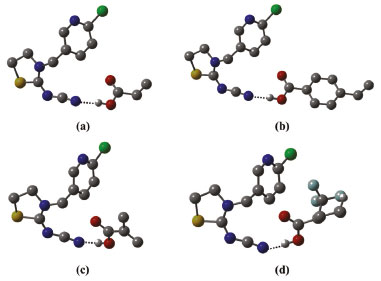 Figura 4. Estruturas otimizadas no nível B3LYP/6-31G(d,p) dos complexos de pré-polimerização, na estequiometria 1:1. (a) TCP-(AA)1; (b) TCP-(APV)1; (c) TCP-(MAA)1 e (d) TCP-(TFMAA)1. As ligações de hidrogênio intermoleculares estabelecidas são mostradas como linhas pontilhadas e os hidrogênios não envolvidos em ligações de hidrogênio omitidos para facilitar a visualização
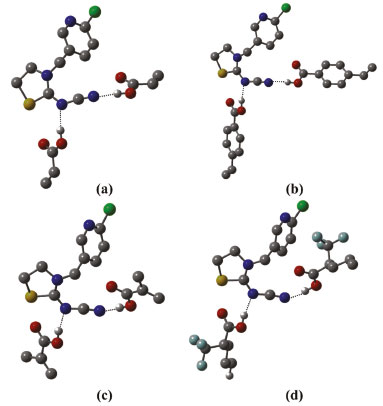 Figura 5. Estruturas otimizadas no nível B3LYP/6-31G(d,p) dos complexos de pré-polimerização, na estequiometria 1:2. (a) TCP-(AA)2; (b) TCP-(APV)2; (c) TCP-(MAA)2 e (d) TCP-(TFMAA)2. As ligações de hidrogênio intermoleculares estabelecidas são mostradas como linhas pontilhadas e os hidrogênios não envolvidos em ligações de hidrogênio omitidos para facilitar a visualização
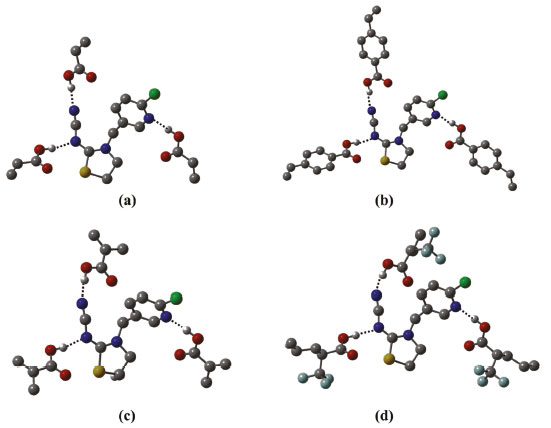 Figura 6. Estruturas otimizadas no nível B3LYP/6-31G(d,p) dos complexos de pré-polimerização, na estequiometria 1:3. (a) TCP-(AA)3; (b) TCP-(APV)3; (c) TCP-(MAA)3 e (d) TCP-(TFMAA)3. As ligações de hidrogênio intermoleculares estabelecidas são mostradas como linhas pontilhadas e os hidrogênios não envolvidos em ligações de hidrogênio omitidos para facilitar a visualização
Frente aos complexos obtidos nas estequiometrias 1:1, 1:2 e 1:3, foram calculadas as variações da energia eletrônica (ΔE) e energia livre de Gibbs (ΔG), no nível B3LYP/6-31G(d,p), em fase gasosa, cujos valores encontram-se apresentados na Tabela 1, pela qual verifica-se que os valores de ΔE e ΔG se tornam mais negativos à medida que mais moléculas de MFs são complexadas à MM, dado o maior número de ligações de hidrogênio estabelecidas.35
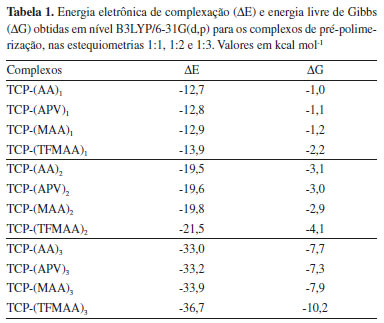
Por meio da análise dos valores apresentados na Tabela 1, nota-se que os complexos mais estáveis são obtidos quando se emprega o ácido 2-(trifluorometil)-arílico (TFMAA) como monômero funcional, sendo em todas as estequiometrias avaliadas a espécie que conduziu a formação de complexos com valores mais estáveis de ΔE e ΔG. A justificativa para essa estabilidade adicional se deve ao fato do TFMAA ser a espécie mais ácida (pKa ≅ 2,8)39 dentre as testadas e, por consequência, atuar como melhor doador de ligações de hidrogênio em comparação as espécies AA, MAA e APV. Já em termos estruturais, a maior estabilidade do complexo com TFMAA pode ser também explicada pelo efeito indutivo negativo do grupo CF3, tendo em vista que o átomo de flúor (F) é o átomo mais eletronegativo da cadeia do TFMAA, de modo que atrai para si maior densidade eletrônica, tornando essa região da molécula mais carregada negativamente.40,41 Em contrapartida, a extremidade que contém o hidrogênio ácido, quando comparada com os demais MFs (Figura 7), apresenta menor densidade eletrônica, ou seja, maior densidade de carga positiva sob o átomo de H ácido (δ+H), com possibilidade de formação de ligações de hidrogênio mais fortes.37
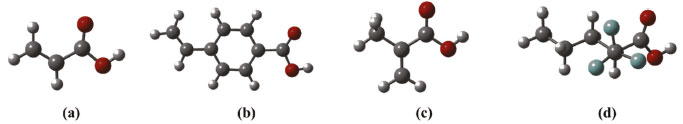 Figura 7. Estruturas otimizadas no nível B3LYP/6-31G(d,p) dos MFs avaliados. Em (a) AA (δ+H = 0,0323), (b) APV (δ+H = 0,0324), (c) MAA (δ+H = 0,0323) e (d) TFMAA (δ+H = 0,0327). O símbolo δ+H se refere à carga de Mulliken sob os átomos de hidrogênio das hidroxilas
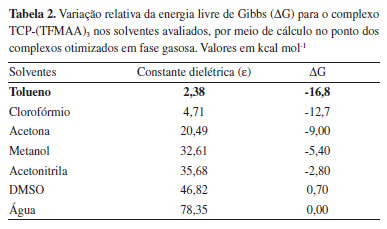
Assim, frente aos dados apresentados na Tabela 1 e obtidos via cálculos de otimização em fase gasosa, pode-se concluir que o complexo de pré-polimerização formado na presença de TFMAA como MF, na estequiometria 1:3, leva à condução do melhor MIP para o TCP, quanto à natureza química e à razão molar da complexação. Seleção do solvente Com a finalidade de definir o solvente mais apropriado para a formação do complexo de pré-polimerização, foram avaliados sete solventes comumente utilizados na síntese de MIPs, de diferentes polaridades e, portanto, de diferentes constantes dielétricas (ε), considerando que a morfologia e porosidade dos MIPs obtidos dependem fundamentalmente das condições sintéticas empregadas, e que solventes porogênicos, apolares e de baixas constantes dielétricas, fornecem um melhor meio reacional para a síntese, por não atuarem como competidores na formação de ligações de hidrogênio, bem como por atribuírem alta porosidade e difusão facilitada do analito em direção aos sítios seletivos do material.16 Utilizando-se do modelo de solvatação SMD,36,42 no qual há a descrição do solvente como um dielétrico contínuo e homogêneo, caracterizado pela sua ε, foram obtidos os valores de ΔG nos sete solventes avaliados por meio de duas metodologias distintas (OPTgas/CPsolv e OPTsolv) a fim de comparar precisão dos resultados e apontar a metodologia mais adequada para o estudo computacional dos complexos de pré-polimerização. Discussão metodológica Frente aos resultados obtidos (Tabelas 2 e 3), pode-se constatar, por meio de ambas as metodologias (OPTgas/CPsolv e OPTsolv), a influência direta dos valores da constante dielétrica dos solventes, gerando valores de ΔG maiores quanto maior ε, tendo sido obtidos os valores mais estáveis de ΔG ao se empregar o tolueno como solvente em todos os casos. Tal comportamento pode ser justificado pelo campo de reação desse solvente ser menor, o que reflete em efeitos de polarização menos pronunciados no complexo de pré-polimerização, que garantem também maior estabilidade, seja para o complexo TCP-(TFMAA)3 otimizado em fase gasosa, seja para os complexos TCP-(AA)3, TCP-(APV)3, TCP-(MAA)3 e TCP-(TFMAA)3, otimizados na presença de solvente.
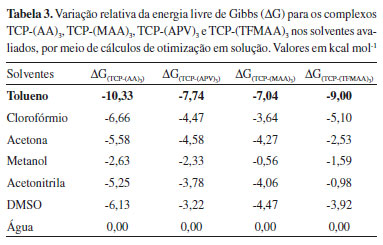
Avaliando os valores das variações de energia (ΔG) para a formação dos complexos em estequiometria 1:3 (Tabela 4), com otimização em fase gasosa e otimização solvente tolueno, identifica-se que os resultados se confluem à medida que os valores de ΔG indicam a maior estabilidade para o complexo formado a partir da espécie TFMAA em solvente tolueno utilizando as duas metodologias de cálculo, porém com menor custo computacional ao se utilizar a Metodologia I (OPTgas/CPsolv). Além disso, de acordo com as estruturas apresentadas na Figura 8, do complexo TCP-(TFMAA)3 formado, verifica-se que ambas metodologias fornecem resultados estruturais semelhantes, com os comprimentos das ligações de hidrogênio estabelecidas apenas ligeiramente diferentes.
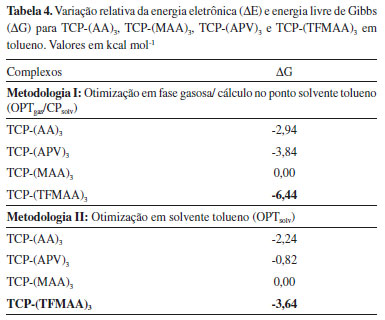
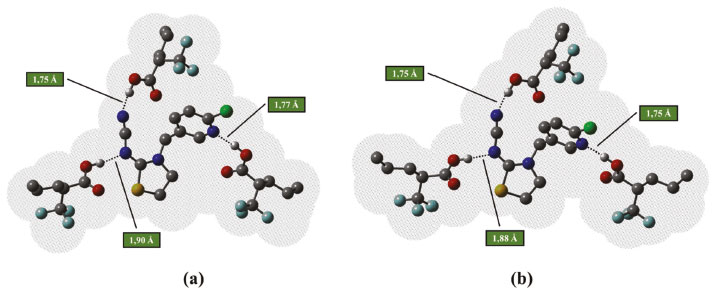 Figura 8. Estruturas otimizadas no nível B3LYP/6-31G(d,p) dos complexos de pré-polimerização na presença de TFMAA, na estequiometria 1:3, mostrando a superfície de acesso ao solvente implícito: tolueno. Em (a) estrutura otimizada em fase gasosa/cálculo no ponto em solvente tolueno e em (b) estrutura otimizada em solvente tolueno. As ligações de hidrogênio intermoleculares estabelecidas são mostradas como linhas pontilhadas e os hidrogênios não envolvidos em ligações de hidrogênio omitidos para facilitar a visualização
Por fim, destaca-se que os resultados apontam que o procedimento computacional comumente adotado nos trabalhos anteriores que envolvem a otimização das condições sintéticas de MIP, via cálculos teóricos, por meio da obtenção de parâmetros termodinâmicos em fase gasosa, seguido de cálculos no ponto para estudo da influência do solvente (OPTgas/CPsolv) se mostram eficientes, à custos computacionais consideravelmente menores. Por essas razões, conclui-se que para a formação do complexo de pré-polimerização envolvendo a molécula de tiacloprido, deve-se utilizar o ácido 2-(trifluorometil)-arílico (TFMAA) como monômero funcional, na razão molar 1:3, e deve-se empregar como solvente o tolueno.
CONCLUSÕES O presente trabalho descreveu uma análise teórica totalmente inédita, via cálculos DFT, do processo de formação de um complexo de pré-polimerização formado entre os monômeros funcionais AA, APV, MAA e TFMAA e a molécula molde TCP, a fim de se obter as melhores condições de síntese para o MIP em questão. Como conclusões gerais do trabalho, pode-se destacar: (i) a definição das melhores condições de síntese do complexo de pré-polimerização para o TCP, quanto à natureza química do MF, a razão molar MM:MF e o solvente, sendo TFMAA, 1:3 e tolueno, respectivamente, sendo o mesmo resultado apontado por meio de duas metodologias distintas (OPTgas/CPsolv e OPTsolv); (ii) a obtenção das estruturas eletrônicas e de propriedades termodinâmicas envolvidas da formação dos complexos de pré-polimerização e a influência significativa das ligações de hidrogênio formadas entre as hidroxilas dos MFs e os átomos de nitrogênio da MM, para a estabilidade dos complexos formados; (iii) a influência do efeito indutivo negativo atribuído ao grupo CF3 presente na estrutura química do TFMAA, que favorece o carácter doador da ligação de hidrogênio dessa espécie, evidenciado pela maior densidade de carga positiva sob o hidrogênio da hidroxila deste monômero funcional; (iv) a eficiência metodológica dos trabalhos comumente realizados nesta área, no que diz respeito a adoção da metodologia sequencial (OPTgas/CPsolv) de precisão considerável à metodologia não-sequencial (OPTsolv). Por fim, destaca-se a importância da Química Computacional como uma ferramenta útil para o planejamento racional de MIPs e na compreensão do reconhecimento molecular atribuído a esses materiais. Além disso, deve-se ressaltar também a necessidade do desenvolvimento de mais trabalhos, que, aliados aos experimentos, conduzem à otimização dos resultados obtidos no desenvolvimento de novos materiais.
AGRADECIMENTOS Os autores agradecem à Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo suporte financeiro.
REFERÊNCIAS 1. http://www.planalto.gov.br/ccivil_03/Leis/L7802.htm, acessada em agosto 2020. 2. Braibante, M. E. F.; Zappe, J. A. A.; Quim. Nova Esc. 2012, 34, 10. 3. Baird, C.; Cann, M.; Química ambiental, 4ª ed., Bookman: Porto Alegre, 2011. 4. Wood, T. J.; Goulson, D.; Environ. Sci. Pollut. Res. Int. 2017, 24, 17285. 5. https://www.nexojornal.com.br/expresso/2018/03/05/O-lugar-dos-neonicotinoides-na morte-das-abelhas-no-Brasil-e-na-Europa, acessada em agosto 2020. 6. https://www.reuters.com/investigates/special-report/brazil-pesticides, acessada em agosto 2020. 7. Correa, J. C. R.; Salgado, H. R. N.; Rev. Bras. Planta. Med. 2011, 13, 500. 8. Biziuk, M.; Namieśnik, J.; Czerwiński, J.; Gorlo, D.; Makuch, B.; Janicki, W.; Polkowska, Z.; Wolska, L.; J. Chromatogr. A 1996, 733 (1-2), 171. 9. Borges, K. B.; Figueiredo, E. C.; Queiroz, M. E. C.; Preparo de amostras para análise de compostos orgânicos, LTC: Rio de Janeiro, 2015. 10. Jardim, I. C. S. F.; Andrade, J. A.; Queiroz, S. C. N.; Quim. Nova 2009, 32, 996. 11. Martín-Esteban, A.; Fresenius J. Anal. Chem. 2001, 370, 795. 12. Nazario, C. E. D.; Lanças F. M.; Sci. Chromatogr. 2013, 5, 111. 13. Piletsky, S. A.; Turner, N. W.; Laitenberger, P.; Med. Eng. Phys. 2006, 28, 971. 14. Zhou, T.; Ding, L.; Che, G.; Jiang, W.; Sang, L.; TrAC, Trends Anal. Chem. 2019, 114, 11. 15. Mayes, A. G.; Mosbach, K.; TrAC, Trends Anal. Chem. 1997, 16, 321. 16. Tarley, C. R. T.; Sotomayor, M. D. P. T.; Kubota, L. T.; Quim. Nova 2005, 28, 1076. 17. Marc, M.; Kupka, T.; Wieczorek, P. P.; Namieśnik, J.; TrAC, Trends Anal. Chem. 2018, 98, 64. 18. Cowen, T.; Karim, K.; Piletsky, S.; Anal. Chim. Acta 2016, 936, 62. 19. Wang, S.; Yang, B.; Zhu, Q.; J. Chin. Chem. Soc. 2017, 64, 434. 20. Nezhadali, A.; Mojarrab, M.; Sens. Actuators, B 2010, 190, 829. 21. Khodadadian, M.; Ahmadi, F.; Talanta 2010, 81, 1446. 22. de Barros, L. A.; Pereira, L. A.; Custódio, R.; Rath, S.; J. Braz. Chem. Soc. 2014, 25, 619. 23. Fernandes, L. S.; Homem-de-Mello, P.; de Lima, E. C.; Honorio, K. M.; Eur. Polym. J. 2015, 71, 364. 24. Marestoni, L. D.; Wong, A.; Feliciano, G. T.; Marchi, M. R. R.; Tarley, C. R. T.; Sotomayor, M. D. P. T.; J. Braz. Chem. Soc. 2016, 27, 109. 25. Barros, L. A.; Custodio, R.; Rath, S.; J. Braz. Chem. Soc. 2016, 27, 2300. 26. Pardeshi, S.; Patrikar, R.; Dhodapkar, R.; Kumar, A.; J. Mol. Model. 2012, 18, 4797. 27. Tadi, K. K.; Motghare, R. V.; J. Chem. Sci. 2013, 125, 413. 28. Sajini, T.; Thomas, R.; Mathew, B.; Chin. J. Polym. Sci. 2019, 37, 1305. 29. Riahi, S.; Edris-Tabrizi, F.; Javanbakht, M.; Ganjali, M. R.; Norouzi, P.; J. Mol. Model. 2009, 15, 829. 30. Fonseca, M. C.; Nascimento Jr., C. S.; Borges, K. B.; Chem. Phys. Lett. 2016, 645, 174. 31. Silva, C. F.; Borges, K. B.; Nascimento Jr., C. S.; The Analyst 2018, 143, 141. 32. Pereira, T. F. D.; Silva, A. T. M.; Borges, K. B.; Nascimento Jr., C. S.; J. Mol. Struct. 2019, 1177, 101. 33. Silva, C. F.; Borges, K. B.; Nascimento Jr., C. S.; J. Mol. Model. 2019, 25, 1. 34. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. 35. Hehre, W. J.; Ditchfield, R.; Pople, J. A.; J. Chem. Phys. 1972, 56, 2257. 36. Marenich, A. V.; Cramer, C. J.; Truhlar, D. G.; J. Phys. Chem. B 2009, 113, 6378. 37. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.; Gaussian 09, revision A.1, Gaussian Inc., Wallingford CT, 2009. 38. Tasi, G.; Palinko, I.; Nyerges, L.; Fejes, P.; Foerster, H.; J. Am. Chem. Soc. 1993, 33, 296. 39. Zhang, T.; Liu, F.; Chen, W.; Wang, J.; Li, K.; Anal. Chim. Acta 2001, 450, 53. 40. Matsui, J.; Kubo, H.; Takeuchi, T.; Anal. Chem. 2000, 72, 3286. 41. Yilmaz, E.; Mosbach, K.; Haupt, K.; Anal. Commun. 1999, 36, 167. 42. Pliego Jr., J. R.; Quim. Nova 2006, 29, 535. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access