
 |
 |
 |
 |
 |
Artigo
| Insight into the conformational space of N-benzyl-N-(furan-2-ylmethyl)acetamide by nmr spectroscopy and dft calculations |
|
Jeisson D. Corredor MontañaI; Alix E. LoaizaII; Gustavo P. RomanelliIII; Isabelle De WaeleIV; Yeny A. TobónIV; Jovanny A. Gómez CastañoI,*
I. Escuela de Ciencias Químicas, Facultad de Ciencias, Universidad Pedagógica y Tecnológica de Colombia, 050030 Tunja -Boyacá, Colombia Recebido em 27/02/2020 *e-mail: jovanny.gomez@uptc.edu.co In this study, the conformational behavior of N-benzyl-N-(furan-2-ylmethyl) acetamide in chloroform was addressed by using a combined experimental/theoretical strategy using NMR spectroscopy and quantum chemical calculations. The 1H and 13C one-dimensional NMR spectra, as well as the two-dimensional HSQC-DEPT and HMBC-DEPT NMR spectra, evinced the presence of a hindered cis(E)-trans(Z) rotational equilibrium in solution. DFT calculations were performed at different theoretical levels using the polarizable continuum model (PCM) and predicted nine (four Z and five E structures) stable conformations. The interconversion dynamics among the different confirmations were established in terms of four different rotational equilibria in CDCl3. The chemical shifts in the 1H and 13C NMR spectra of the compound are similar to the values calculated for the two most abundant conformational equilibria at room temperature, one caused by two Z rotamers and the other by two E rotamers. The compound was also characterized for the first time by FTIR, Raman spectroscopy, and GC/MS spectrometry. Additionally, several acylation methodologies for synthesizing the title compound from N-benzyl-1-(furan-2-yl)methanamine were tested which resulted in high yields (> 90%) under very convenient conditions (10 min, at room temperature). INTRODUCTION The carboxamide moiety, RC(O)N, is a pervasive building block in naturally occurring systems such as proteins and peptides, as well as in many synthetic polymers (polyamides) and pharmaceuticals. It plays a significant role in constructing the backbone conformation of peptides and proteins by offering structural rigidity and resistance to hydrolysis. This is mainly due to its ability to form hydrogen donor interactions (in secondary amides) and thermodynamically stable cis (E-isomer) or trans (Z-isomer) rotamers in secondary and ternary amides (see Figure 1a).
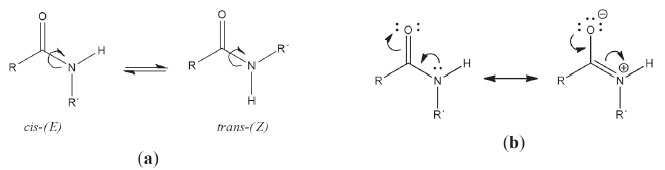 Figure 1. Structural features in amide molecules: (a) Rotational equilibrium between cis-(E) and trans-(Z) rotamers in secondary amides; (b) resonance theory model for the electronic structure of the amide bond
Given its biological relevance and physicochemical interest, the conformational study of secondary and tertiary amides is a subject that has attracted the attention of both experimental and theoretical researchers over several decades.1-13 These systems possess the thermochemical feature of combining a small energy difference between the cis and trans forms (0.5 to 4.0 kcal/mol for both secondary and tertiary amides),1,2 with a relatively high cis-trans internal rotation barrier (ΔG = 15-23 kcal/mol),3-6 leading to a conformational equilibrium characterized by a low interconversion rate. The rotational constraint is attributed to the partial double C-N bond originating from an efficient electron delocalization of the lone pair on the nitrogen atom which can be interpreted using a simple resonance model (Figure 1b). A more sophisticated picture for the pseudo-double bond character in the C-N bond arises alternatively from the molecular orbital (MO) theory in terms of delocalization of the lone pair on nitrogen into the π* orbital of the carbonyl group.1,2 This explains the low ability of amide derivatives to act as Lewis bases.7 As expected, the rotational equilibrium in the amide molecules is largely governed by steric and electronic effects provided by the substituents directly attached to both carbon and nitrogen atoms.8 The effects on secondary amides are also governed by the type of solvent used.3 The hindered cis-trans rotation in amide derivatives is a molecular process that generally falls within the timescale of NMR phenomena, i.e. in the milliseconds-to-seconds range.9 This makes NMR particularly useful for the study of the conformational equilibria of amides in a solution. NMR spectroscopy was used for studying the restricted rotation in amides as early as 1955,10,11 and since then has remained a useful tool for this type of research.7,12 In addition, the interpretation of the NMR spectra of amides can be properly complemented with quantum chemical NMR calculations. This allows for a conformational analysis to be done to determine the preferred molecular orientation of these organic molecules in solution.13 The use of continuum solvation models such as the Polarizable Continuum Model (PCM),14 while utilizing the Density Functional Theory (DFT), may provide an adequate computational strategy for describing the effects of solvent on the conformational analysis of tertiary amides in solution.15 For primary and secondary amides however, continuous solvation models have been associated with poor simulation of the NMR chemical shifts for N-H hydrogens. This is due to the fact that these types of protons are largely influenced by the solvent due to hydrogen bonding and other noncovalent interactions such as van der Waals.15,16 The tertiary amide N-benzyl-N-(furan-2-yl-methyl)acetamide, (C4H3O)CH2N(COCH3)CH2C6H5, has surged as one of our main synthetic equivalents, derived from the application of a synthon approach,17 for attaining new organic structures with antiprotozoal activity against Chagas disease.18 This molecule also has the advantage of synthetic accessibility and structural convenience. Its two aromatic rings can be substituted and it has no proton donor groups. Although this molecule has been around for almost 20 years,19 no study has characterized its molecular structure. Consequently, this study aims to determine the conformational behavior of the title amide in chloroform, through an experimental/theoretical comparative analysis that involves its 1H and 13C NMR spectra and quantum chemical DFT calculations using the Polarizable Continuum Model. The hybrid functional B3LYP, mPW1PW91, and APFD combined with the Pople's basis set 6-311+G(2d,p) or the Dunning's correlation consistent basis set aug-cc-pVDZ, within a gauge-independent atomic orbital (GIAO) methodology have shown to be methods which provide a very good compromise between precision and computational cost for the calculation of NMR chemical shifts on nitrogen molecules in solvated systems.9,20,21 The molecule was also characterized by FTIR, FTRaman spectroscopy, and GC/MS spectrometry, while trying to determine the most efficient synthetic acylation conditions using the corresponding non-acetylated amine.
EXPERIMENTAL General information For synthesis procedures, only analytical-grade (purity > 99%) reagents and solvents were used. Both the purity of the products and the composition of the reaction mixtures were verified by thin layer chromatography (TLC) using silica SGF254 TLC plates with a 0.25 mm coating and granularity between 10 and 40 μm. The chromatographic plates were developed under ultraviolet light using iodine and a 1% vanillin-sulfuric solution as revealing agents. For the purification of the products, preparative chromatographic columns packed with silica gel 60 (0.063-0.200 mm) were used. Solvent extraction at different stages of synthesis and purification was performed by rotary evaporation under vacuum. Reaction yields were determined with respect to the isolated product. The boiling points of the compounds were determined by the reflux method at an atmospheric pressure of 553 mmHg. The structures of both the precursor and the product compounds were determined by 1H NMR (300.13 MHz) and 13C NMR (75.47 MHz) through both one- and two-dimensional (COSY, DEPT, HSQC and HMBC) NMR spectra. A Bruker Advance instrument using deuterated chloroform as solvent and tetramethylsilane (TMS) as an internal standard was used. Molecular ions and mass fragmentation patterns of the precursor and amide product were obtained by means of gas chromatography-mass spectrometry (GC-MS). An Agilent 6850 series II chromatograph coupled to an Agilent 5975B VL MSD electron impact (EI) mass spectrometer, equipped with a split/splitless injection port (260 °C, split ratio of 15:1), an automatic injector (Agilent 6850 series), and an Agilent 19091S-433E HP-5MS column was used. The chromatographic method consisted of an initial oven temperature of 80 °C for 3 min then incremented in temperature by 15 °C/min up to 350 °C where it was maintained for 2 min, totaling 28 min. The infrared spectra were obtained on a Shimadzu Prestigie-21 spectrophotometer with Fourier Transform (FTIR), equipped with a Michelson-type interferometer, a KBr/Ge beam-splitter, a ceramic lamp, and DLATGS detector. The FTIR spectra were measured in the range of 4000-500 cm-1 with a resolution of 3.0 cm-1 and 30 scans using the techniques of attenuated total reflectance/reflection (ATR) or KBr windows. The FT-Raman spectra were measured in the range of 3500 to 200 cm-1 with a resolution of 4 cm-1 on a Bruker RFS 100/S device. A Nd:YAG laser excitation line emitting at 1064 nm and a germanium detector cooled with liquid nitrogen were used. Organic synthesis and characterization N-benzyl-1-(2-furanyl)methanamine (1) 10 mmol of furfuraldehyde (99.5%) was mixed with 10 mmol of benzylamine (99%) in 20 ml of absolute ethanol and microwaved (400W) for 30 min at 80 °C. The reaction mixture was then cooled to room temperature, and 10 mmol of sodium borohydride (96%) was added slowly, leaving the reaction under constant stirring for 1 h. Most of the solvent was then removed by rotoevaporation under a vacuum and 30 ml of water was added. The mixture was then extracted with small volumes of dichloromethane and dried over anhydrous sodium sulfate. The solvent was the removed and the compound purified using preparative column chromatography and eluted with a 3:1 hexane-ethyl acetate mixture. The reaction yield was 93%. The compound was obtained as a bright yellow liquid with a boiling point of 268 °C. FTIR/FT-Raman (cm-1) 3328 ν(N-H), 3114 ν(C-H) fur, 3056 ν(C-H) Ph, 2828 ν(CH2), 1600 ν(C=C) Ph, 1504 ν(C=C) fur, 1454 ρ(N-H), 1208 ν(C-C), 1146 ν(C-N), 928 ν(C-O). EI-MS m/z: 188 [M++1], 187 [M+], 91 [100]. RMN 1H (300 MHz, CDCl3) δ 1.79 (s, 1H), 3.77 (s, 4H, J = 9.0 Hz), 6.17 (d, 1H, J = 2.86 Hz), 6.29 (d, 1H, J = 2.86 Hz), 7.24 (m, 1H), 7.35-7.31 (m, 5H). RMN 13C (75 MHz, CDCl3) δ 45.34, 52.77, 107.00, 110.08, 126.99, 128.23, 128.39, 139.86, 141.78, 153.81. N-benzyl-N-(furan-2-ylmethyl)acetamide (2) 8 mmol of (1) was dissolved in 8 mmol of acetic anhydride. The mixture was stirred for 15 min and then neutralized with an aqueous sodium bicarbonate solution. The compound was extracted with small volumes of dichloromethane and dried over anhydrous sodium sulfate. The solvent was then removed and the compound purified using preparative column chromatography eluted with 3:1 hexane-ethyl acetate mixture. The product (2) had a reaction yield of 97%, was a bright yellow-orange liquid, and had a boiling point of 234 °C. IR-Raman (cm-1) 3120 ν(C-H) fur, 3056 ν(C-H) Ph, 3006 ν(CH3), 2933 ν(CH2), 1644 ν(C=O), 1604 ν(C=C) Ph; 1505 ν(C=C) fur, 1420 ρ(C-N), 1250 ρ(CH2), 1077 ρ(C=C-H), 1000 ρ(C-H) Ph, 729 ρ(C-H) fur. EI-MS m/z: 230 [M++1], 229.1 [M+], 96.0 [100]. 1H RMN (300 MHz, CDCl3 (signal duplication): δ 2.18, 2.34 (s, 3H); 4.34, 4.56, 4.59, 4.62 (dd, 1H); 6.19, 6.24 (d, 1H); 6.32, 6.35 (m, 1H); 7.17, 7.19 (s, 1H); 7.25-7.41 (m, 10H). 13C RMN (75 MHz, CDCl3, duplication) δ 21.59, 21.61, 41.0, 44.0, 47.6, 51.1, 108.2, 108.7, 110.2, 110.3, 129-126, 136.3, 137.1, 142.2, 142.6, 149.9, 150.8, 170.8, 170.9. N-Benzyl-1-(2-furanyl)methaminium acetate (3) This compound can be obtained as a by-product of the former reaction or by the addition of acetic acid to (1). The compound was an opaque yellowish solid with a M.P. 84-85 °C. FTIR (ATR, cm-1) 3118 ν(C-H) fur; 3030 ν(C-H) Ph, 2920 ν(C-H), 2821 ν(CH2), 1671 ν(C=O), 1498 ν(C=C), 1450 ρ(N-H), 1391 ρ(CH3), 1147 ν(C-N), 1015 ρ(CH2), 737 ρ(C-H). 1H RMN (300 MHz, CDCl3) δ 2.01 (s, 3H), 3.8 (s, 2H), 3.9 (s, 2H), 6.31 (d, 1H, J = 3.1), 6.36 (dd, 1H, J = 3.1, 1.5 Hz), 7.29-7.37 (m, 5H), 7.42 (d, 1H, J = 1.5 Hz). 13C RMN (75 MHz, CDCl3) δ 22.2, 43.6, 51.2, 108.9, 110.4, 127.7, 128.6, 128.8, 136.6, 142.5, 150.7, 176.4. Computational details All DFT computations were performed using the Gaussian 09 software package (Revision D0.1),22 while molecular models and simulated spectra were visualized through its GaussView 6.0.16 graphical user interface.23 The calculations were performed on an HP Z4-G4 workstation with an Intel(R)-Xeon(R) W-2133 processor and 16 GB of RAM, under the Ubuntu 18.04.2 LTS operating system. The electronic and molecular structure calculations of the stable conformations of compound (2) were performed in both the gas phase and with a PCM implicit solvent model while assuming a temperature of 298.15 K and 1 atmosphere of pressure. Determining the conformational space of compound (2) was initially approached by means of a two-dimensional gas-phase scan of its potential energy surface using the theoretical level B3LYP/6-31+G(d).24,25 During this process the dihedral angles of [C-N-C=O] and [O-C-C-N] were simultaneously modified in consecutive steps of 10° which allowed the relaxation of the rest of the structure. Potential energy paths connecting minima structures in the gas phase were also estimated using Peterson's complete basis set extrapolation model CBS-QB3.26 The conformational search was further complemented with the assistance of the GMMX automated molecular mechanics facility within the Pcmodel program (version 10.075).27 All the energy minima were optimized in both the gas phase and in the condensed phase (implicit solvent) using the exchange-correlation hybrid functional B3LYP24,25 and APFD28 (including dispersion corrections). These were used in combination with the Pople's basis set 6-311+G(2d, p), and the Dunning's correlation consistent basis set cc-pVDZ29 augmented with diffuse functions, i.e., aug-cc-pVDZ. In order to estimate the effects of dispersion forces, condensed phase calculations were also performed using the functional B3LYP augmented with the empirical Grimme's dispersion correction GD3 (B3LYP-D3).30 The equilibrium structures were confirmed as minima in the potential energy surface by the absence of imaginary frequencies. The Boltzmann population distribution (298.15 K) among the equilibrium structures was calculated in terms of Gibbs free energy (ΔG) relative to the global minimum using the same theoretical optimization levels. 1H and 13C magnetic shielding tensors (in ppm) relative to the TMS standard were calculated using the gauge-independent atomic orbital (GIAO) method with the same theoretical optimization levels and by using the high accuracy method for NMR tensor calculations mPW1PW91/6-311+G(2d,p)-SCRF//B3LYP/6-31+G(d,p), as recommended by Tantillo et al.31 Geometric optimizations and NMR calculations were performed in implicit chloroform using the PCM model. All DFT calculations were performed using an ultrafine numerical integration grid (Integral=Ultrafine).
RESULTS AND DISCUSSION Synthesis of N-benzyl-N-(furan-2-ylmethyl)acetamide Compound (2) was obtained by the acylation of N-benzyl-1-(furan-2-yl)methanamine (1), see Figure 2, while testing various experimental conditions as described below. Three acylation methodologies (M1, M2, and M3) were tested against seven different acylation agents to obtain the tertiary amide (2), whose yields and main experimental conditions are listed in Table 1.
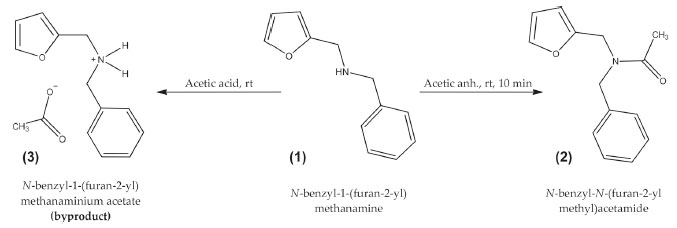 Figure 2. Synthesis of amide (2) by acylation of amine (1)
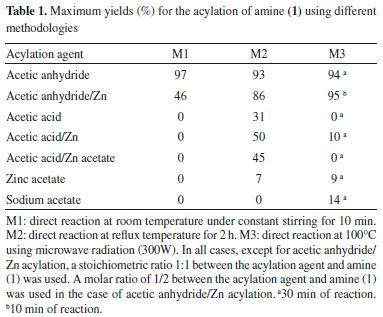
The best yields (≥ 93%) in the synthesis of amide (2) were obtained by reacting directly the amine (1) and acetic anhydride using the three methodologies (M1, M2, and M3). M1 was the most convenient since it had the highest yield (97%), used room temperature, and only took 10 min. The high yield obtained using M1 was comparable to results reported for the production of secondary and tertiary acetamides from amine hydrochloride salts using microwave reactions in solution,32 however, the current study resulted in a faster reaction time. A direct reaction between (2) and acetic anhydride was also tested in the presence of zinc (catalyst), which had yields of 46% forM1, 86% for M2, and 95% for M3. Although these yields are below those reached using only acetic anhydride, the use of a catalyst allows each anhydride molecule to react with two molecules of amine (1), thus improving the atomic efficiency of this acylation reaction. M3 using acetic anhydride in the presence of Zn was shown to be both a very effective and a very efficient synthesis strategy with a yield of 95% after a 10 min reaction. The lower yield of 46% using M1 in the presence of zinc was attributed to the formation of a white crystalline by-product that was subsequently identified as N-benzyl-1-(2-furanyl)methanaminium acetate (3) as seen in Figure 2. This was confirmed by the measurement of its 1H and 13C NMR spectra (Figure 1S in Supplementary Material). In the M1 and M3 procedures using acetic acid as an acylating agent, either pure or in mixture with zinc or zinc acetate; a salt (3) was the dominant by-product, whereas in the M2 procedure moderate yields of (2) were obtained. Formation of (3) instead of (2) using acetic acid as acylating agent was attributed to the protonation of the amine group, which hindered the nucleophilic attack of the nitrogen atom on the carbonyl carbon. Poor or null yields for (2) were obtained using either sodium or zinc acetate as acylating agents given the lack of homogeneity of the reaction mixture. Structural characterization of (2) Mass spectrometry The molecular ion of compound (2) was detected at m/z = 229.1 (C14H15O2N, 3.6%), as seen in Figure 3. The main fragment in the mass spectrum of (2) was observed at m/z = 96 (100%), which corresponds to the furfuryl-amine moiety with a deprotonated nitrogen atom (C5H6NO). Most of the other fragments come from the rupture of the C-N bonds. Thus, fragments containing either furan or phenyl rings are detected at m/z = 138 (C7H8NO2, 43%), m/z = 148 (C9H10NO, 16%), m/z = 106 (C7H8N, 37%), m/z = 81 (C5H5O, 20%), and m/z = 91 (C7H7, 30%). A fragment corresponding to the loss of the acetyl moiety was also observed at m/z = 43 (C2H3O, 29%).
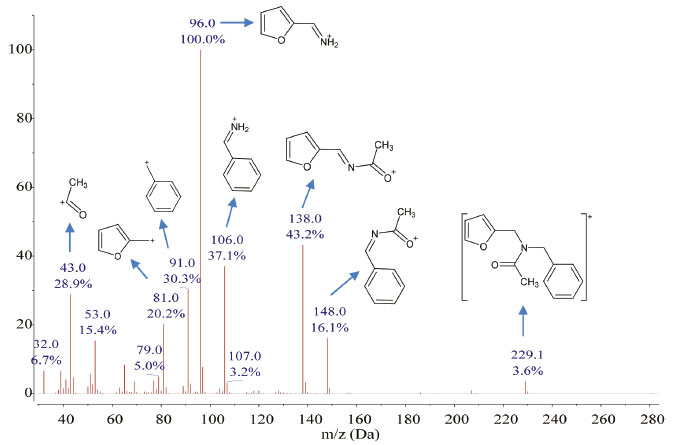 Figure 3. Mass spectrum of amide (2)
FTIR and FTRaman spectra The vibrational spectra (FTIR and FTRaman) of amide (2), and the main vibrational assignments, are shown in Figure 4. The full assignments of the vibrational modes of (2) were determined with the aid of thermodynamic quantum chemical calculations computed at the theoretical level APFD/aug-cc-pVdZ, and are listed in detail in Table 1S in supporting information.
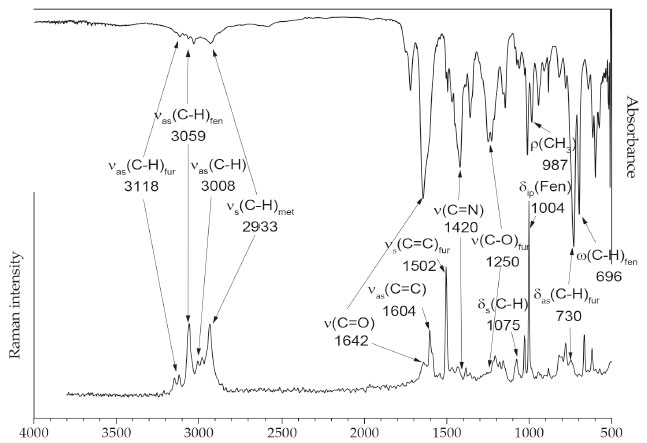 Figure 4. FTIR (top) and FTRaman (bottom) spectra of the title amide (2)
Conformational behavior of (2) through NMR measurements Both the 1H and 13C NMR spectra of the pure compound (2) measured at room temperature in CDCl3, are characterized by two sets of signals in the different chemical shift regions. This reveals a slow molecular interchange at the given NMR measurement temperature (298 K) that was attributed to the presence of stable Z and E configurations of the amide moiety, as shown below. As expected, the chemical shift difference between the Z and E conformations is the highest for signals whose atoms are located close to the amide moiety. In the 1H NMR spectrum of (2) (shown in Figure 5), the protons of the methyl group appear as two main signals at 2.18 and 2.34 ppm, each of which integrated approximately for three hydrogen atoms, and a small signal at 2.08 ppm signifying the presence of different conformations of amide (2). Likewise, the two methylene groups are observed as four singlet signals at 4.34, 4.56, 4.59, and 4.62 ppm (see Figure 5), each integrated for two protons. In this case, the protons closer to the furan ring are more shielded than the methylene protons adjacent to the phenyl ring. Meanwhile, two sets of proton signals at 6.19/6.24 and 6.32/6.35 ppm are observed for the furan ring. The integration of the 1H NMR spectrum signals corresponds to values that double the expected number of protons, which support the presence of two types of isomers.
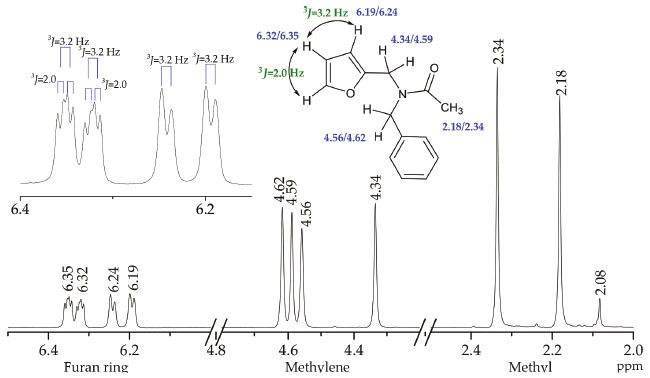 Figure 5. Main signals in the 1H NMR spectrum of amide (2)
Additional evidence for the presence of two main stereoisomers, E and Z, in amide (2) was obtained from the analysis of the two-dimensional NMR spectra. Figure 6 (top) shows the two-dimensional HSQC-DEPT spectrum of (2) along with the assignment of signals in the molecular structure. This two-dimensional correlation reveals the coupling between the different proton signals and the carbon signals observed in the one-dimensional spectra. The signal j11 corresponds to the most unprotected proton of the furan ring. This proton was unassigned in the one-dimensional 1H NMR spectrum because it was hidden with the signals from the protons of the phenyl ring. The signals b5 and c3 correspond to the methylene group attached to the furan ring, while signals e4 and d6 correspond to the methylene group attached to the phenyl ring. Signals a1 and a2 corresponded to two different methyl groups. Figure 6 (bottom) shows the HMBC-DEPT two-dimensional analysis which revealed long-range coupling of up to four bonds. By using this technique, a coupling between the protons of methyl 1 and methyl carbon e (linked to phenyl) was detected. This demonstrated the presence of a structure in Z conformation with respect to the amide function. The same type of coupling occurred between the methyl protons 2 and the methyl carbon c (attached to the furan), which also confirmed the presence of a structure in the E conformation. Orientation Z (together) and E (opposite) in amide (2) were assigned according to the rules of priority proposed by Cahn-Ingold-Prelog.33 This spectrum also detected the two-bond coupling signals l1 and l2, demonstrating that the methyl was attached to acyl carbon. The d-10 and e-10 signals corresponded to the coupling between methylene carbons and the protons of the phenyl ring through three bonds. A coupling signal was also detected in four bonds between carbon k and proton 11. Region A confirmed the coupling through three bonds between the methylene carbons and the opposite methylene protons. Region B revealed the presence of two-, three-, and four-bond couplings in the phenyl ring. Region C confirmed the presence of two- and three-bond couplings in the furan ring. Region D confirmed three-bond coupling between the methylene protons and the carbons of the aromatic rings, as well as with the central carbon of the acyl function.
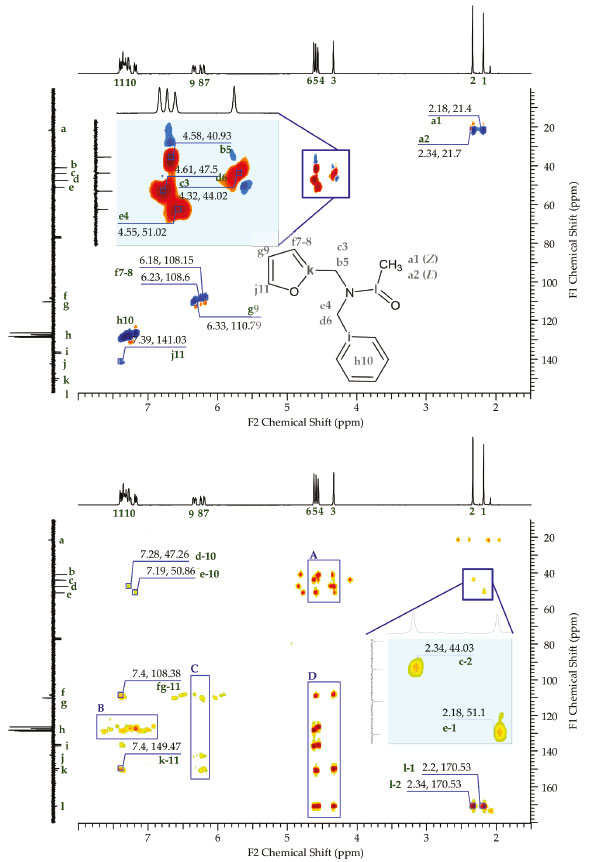 Figure 6. HSQC-DEPT (top) and HMBC-DEPT (bottom) two-dimensional NMR spectra of amide (2)
Computational study of the conformational space of N-benzyl-N-(furan-2-ylmethyl) acetamide Two-dimensional PES scan Further information on the conformational behavior of amide (2) comes from the quantum chemical calculations performed in the framework of the density functional theory. First, an exploration of the potential energy surface (PES) of compound (2) in the gas phase was performed at an approximation level of B3LYP/6-31+G(d) using a relaxed two-dimensional scan (as seen in Figure 7). This was obtained by simultaneously varying the dihedral angles of [C-N-C=O] and [O-C-C-N]. As depicted in Figures 7 and 8, five local energy minima were identified on the PES of (2). Two of the minima had a Z configuration of the amide moiety (structures Za and Zb), while the other three corresponded to the E configuration (structures Ea, Eb, and Ec). Within each set of equilibrium structures (inside the Z or E set) the main structural difference is caused by a slightly restricted rotation (vide infra) of the furan ring.
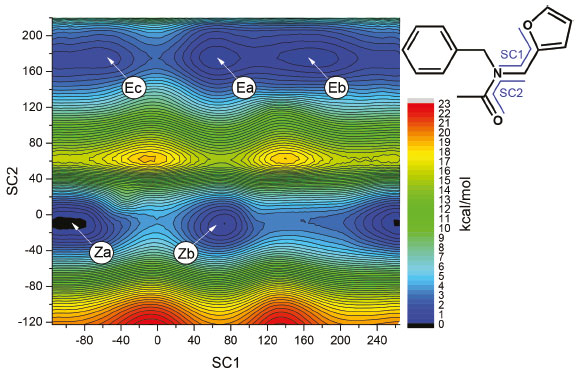 Figure 7. Two-dimensional PES for amide (2) constructed by the variation of [O-C-C-N] and [C-N-C=O] dihedral angles, coordinates SC1 and SC2, respectively, calculated at the B3LYP/6-31+G(d) approximation level
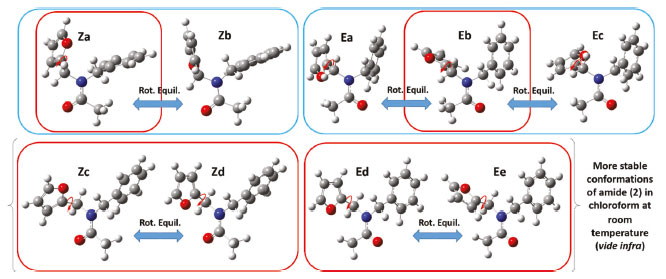 Figure 8. Equilibrium structures for amide (2) optimized with the B3LYP/6-31+G(d) approximation. In the blue frame: energy minima found by a two-dimensional PES scan. In the red frame: energy minima found by an automated conformational search using molecular mechanics
As shown in Figure 9A, rotational interconversion within the Z set is associated with energy barriers (expressed as ΔG+) of 5.2 and 3.4 kcal mol-1 for structures Za and Zb respectively. Figure 9B reveals furan-ring rotations within the E set associated with values of 0.6 and 2.8 (for structure Ec), 3.5 and 2.1 (for structure Ea), and 2.0 and 1.7 (for structure Eb) kcal/mol. Figures 9C and 9D show the two calculated paths through which the Z and E sets are interconnected, i.e., Za ↔ Ec and Zb ↔ Ea respectively. These paths implied overcoming an energy barrier that oscillates between 15.3 and 13.1 kcal mol-1. These barriers were also estimated using the CBS-QB3 method, using a PCM implicit solvent model in chloroform, which resulted in values ranging between 15.6 and 14.2 kcal mol-1 (red values in Figures 9C and 9D). These values coincide with the values reported for this type of rotational exchange.3-6 Consequently, a rapid structural exchange caused by a low rotational impediment of the furan ring is expected at room temperature within each Z or E set, while combined with a slow rotational exchange of the amide-moiety between these two sets.
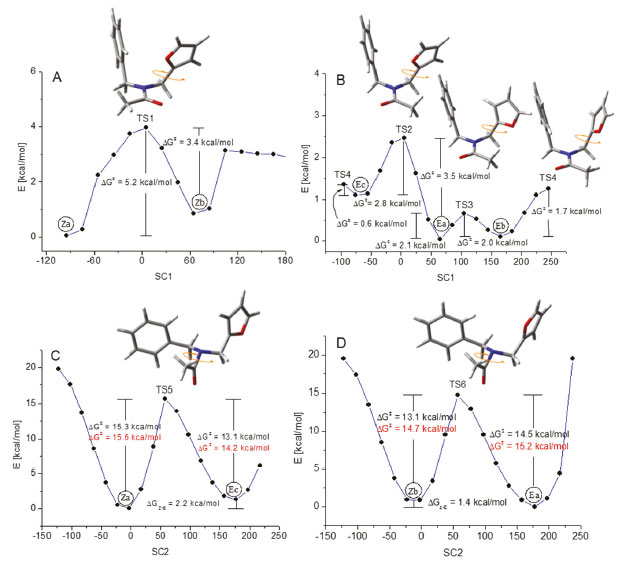 Figure 9. Potential energy paths connecting conformational structures Za, Zb, Ea, Eb and Ec of the amide molecule (2) calculated in gas phase using the B3LYP/6-31+G(d) and the CBS-QB3 approximation levels. Molecular models inserted in this figure correspond to transition structures (TSs)
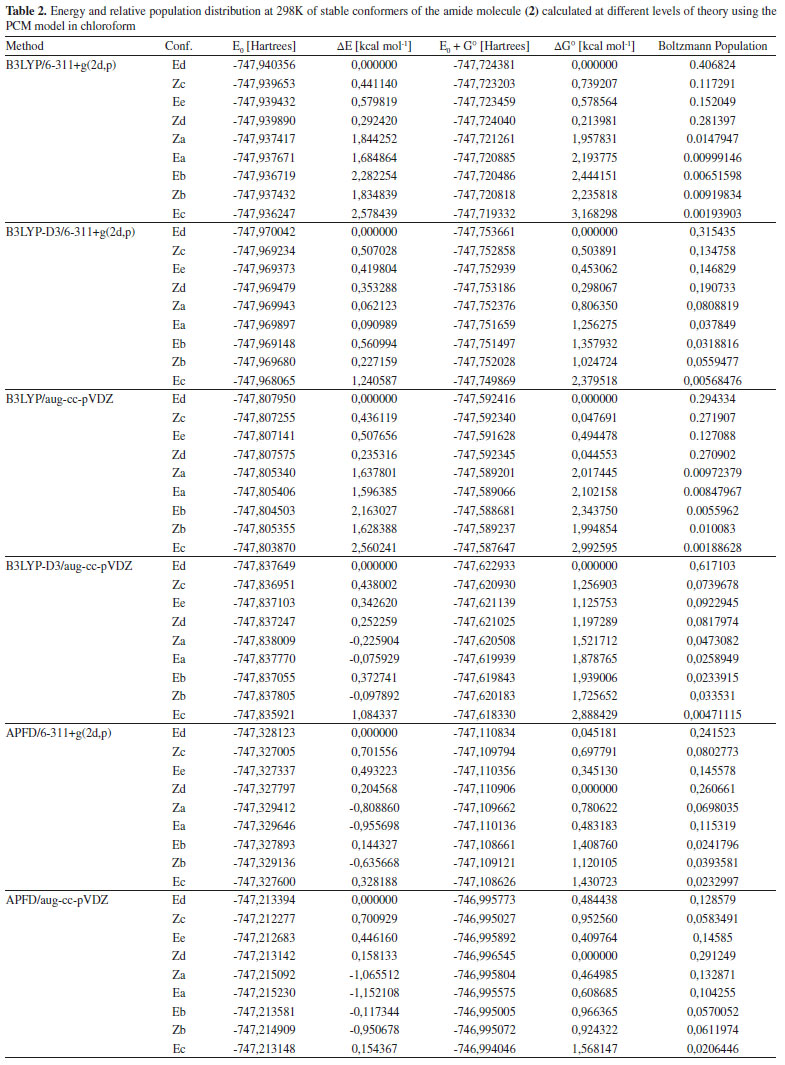
Automated conformational searching using molecular mechanics A second stage in the conformational exploration of amide (2) was carried out in the Pcmodel software, using the GMMX molecular mechanics method. This methodology uses all of the rotatable bonds of the molecule for an automated conformational searching and optimization in gas phase at the approximate GMMX level. As a result, six structures were located as stable conformers, which were then optimized in Gaussian 09 at the B3LYP/6-31+G(d) approximation level. These structures are shown in Figure 8 in the red frame. As shown in Figure 8, two of the energy minima found with the GMMX molecular mechanics approximation coincided with the Za and Eb structures, which were previously found using the two-dimensional PES scan. The other four structures, denoted as Zc, Zd, Ed, and Ee, are new conformers, leading to a total of nine equilibrium structures which were identified in this study for amide (2). Boltzmann averaging in chloroform at room temperature The rotational equilibrium for the nine optimized structures in solution and at room temperature was estimated by introducing the sum of electronic and thermal free energies, obtained from thermochemical calculations at 298 K, into the Boltzmann distribution equation. These calculations were performed using the hybrid functional B3LYP, B3LYP-D3 or APFD in combination with the basis set 6-311+G(2d,p) or aug-cc-pVDZ, and a PCM implicit solvent model in chloroform. As shown in Table 2, all methods using the functional B3LYP found the Ed structure to be the most stable conformation (29% ≤ %Ed ≤ 62%), while methods using the APFD functional reported the Zd structure as the lowest energy conformer (%Zd = 26 or 29%). The Ec structure was predicted as the least stable conformer, regardless of the method. It had a configuration percentage between 0.2 and 0.6% with all methods using the functional B3LYP, and 2% by the methods using the functional APFD. Methods using the functional B3LYP in combination with the basis set 6-311+g (2d, p) or aug-cc-pVDZ estimated that 96% of the total configurations of (2) in chloroform is concentrated in only four structures: the two Z configurations Zc and Zd and the two E configurations Ed and Ee. As shown in Figure 8, each of these two pairwise combinations (i.e. Zc ↔ Zd or Ed ↔ Ee) results in a rapid conformational interchange by simply rotating of the furan ring. Addition of Grimme's dispersion correction (GD3) to functional B3LYP caused the concentration of (2) in chloroform to be somewhat more distributed among the conformers. The B3LYP-D3/6-311+g(2d, p) method resulted in the structures Zc, Zd, Ed, and Ee representing 79% of the total configurations, while the structures Za, Zb, Ea, and Eb presented percentages between 3 and 8% each. The B3LYP-D3/aug-cc-pVDZ method predicted a percentage of 62% for the Ed structure, while the rest of the structures presented percentages between 2 and 9% each (except Ec, percentage = 0.5%).
Computational modeling of 13C and 1H NMR spectra 1H and 13C magnetic shielding tensors (in ppm) relative to the tetramethylsilane (TMS) standard were calculated for all optimized equilibrium structures of compound (2). This was accomplished using the functional B3LYP, B3LYP-D3 or APFD in combination with the basis sets 6-311+G(2d,p) or aug-cc-pVDZ, as well as the mPW1PW91/6-311+G(2d,p)//B3LYP/6-31+G(d,p) method. Every calculation was performed taking into consideration a PCM implicit solvent model in chloroform. A comparison between the calculated chemical shifts and the experimental shifts for 1H and 13C signals in the one-dimensional NMR spectra of amide (2) in CDCl3 is shown in Tables 3 and 4. As the methyl (CH3) and methylene (CH2) proton signals appear integrated in the experimental 1H NMR spectrum a comparison with the calculated signals was performed taking into consideration the arithmetic average of the theoretical values. Due to the minimal difference in the values calculated for both the 1H and 13C NMR shifts of the phenyl ring, it was not possible to make an appropriate assignment in terms of Z and E structures for these signals.
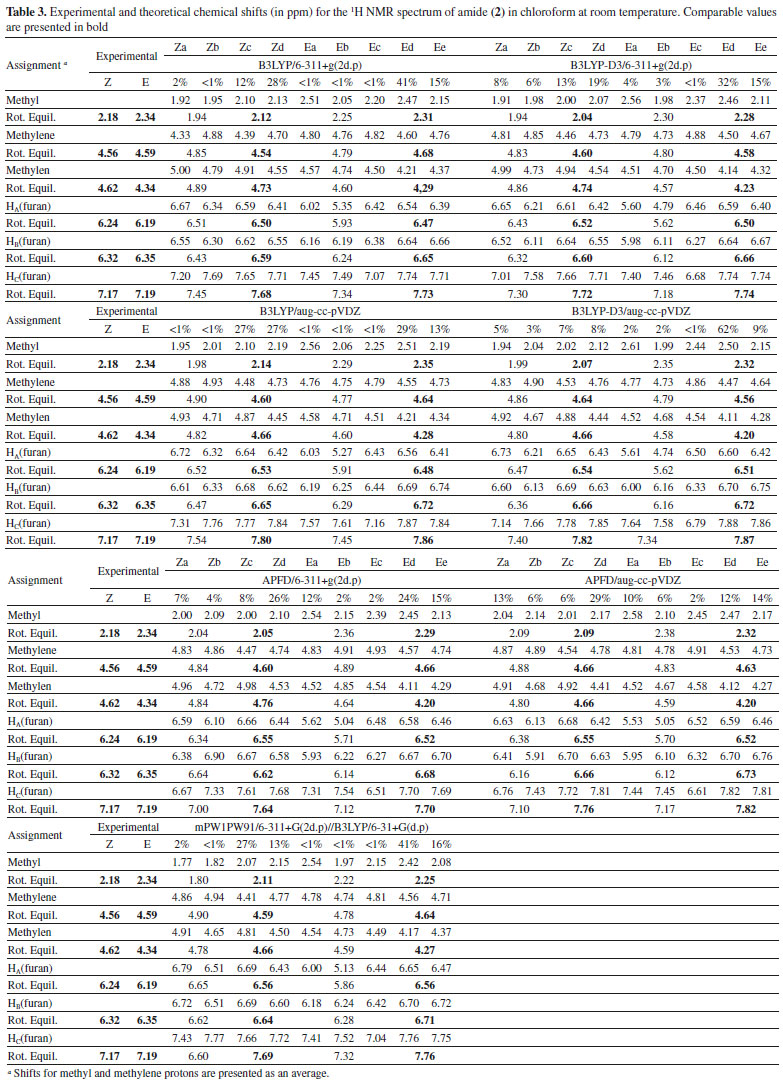
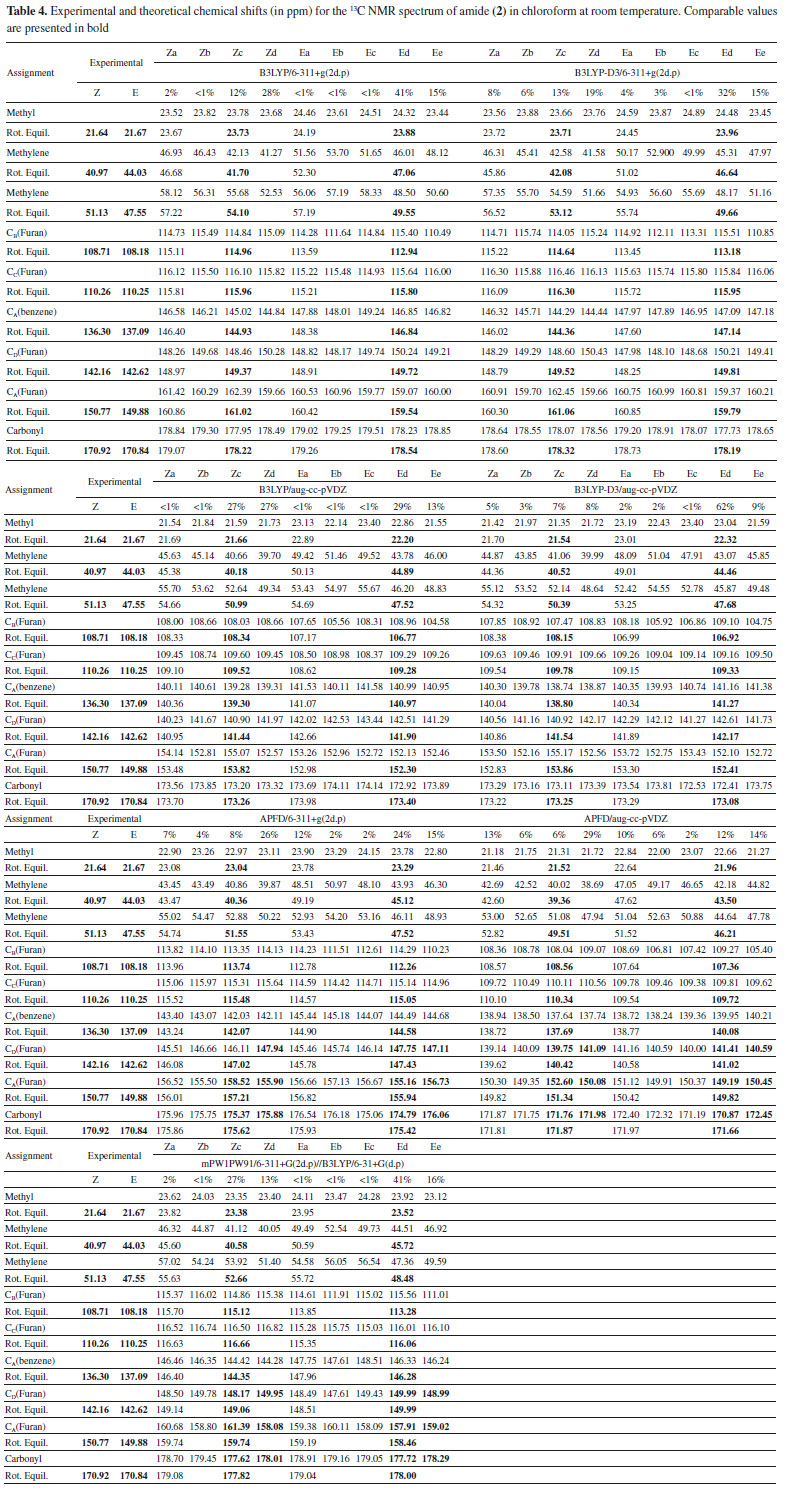
As previously noted (see Figure 9), the conformational space of the molecule (2) is characterized by a rapid interconversion within each set of Z or E rotamers. This is caused by a low-hindered rotation of the furan moiety, along with an exchange between configurations Z and E which is thermochemically restricted. Consequently, the nine stable structures of compound (2) lead to their classification into four different conformational equilibria: i) Za ↔ Zb, ii) Zc ↔ Zd, iii) Ea ↔ Eb ↔ Ec and iv) Ed ↔ Ee (see Figure 8). However, since the calculations performed predict a predominant percentage for the Zc, Zd, Ed, and Ee conformations in chloroform at room temperature (see Table 2), the NMR spectra of compound (2) in CDCl3 are expected to be governed by both Zc ↔ Zd and Ed ↔ Ee rotational equilibria which explains the duplication of signals in the NMR spectra in this study. There is an excellent correlation between the calculated average for the chemical shifts of Zc ↔ Zb or Ed ↔ Ee combination and the experimental chemical shifts in the 1H and 13C NMR spectra of the amide (2), as seen in Tables 3 and 4 (see values in bold) and in Figure 10.
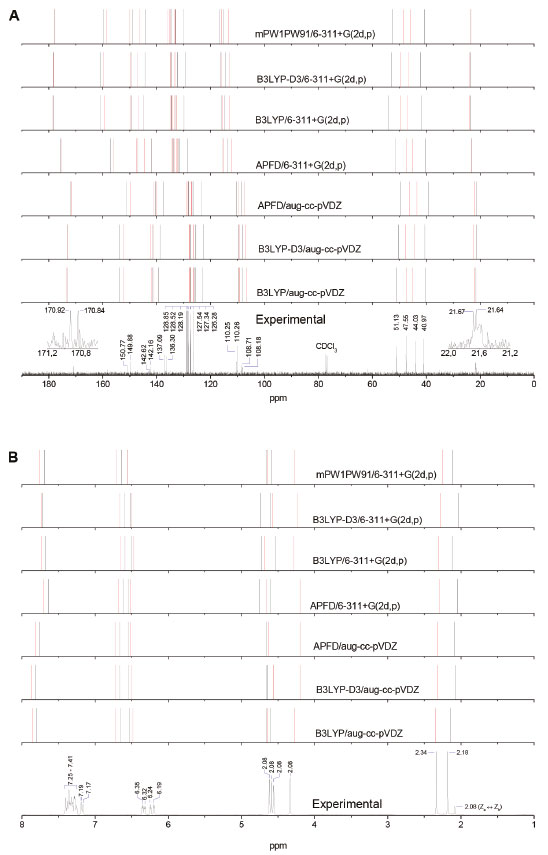 Figure 10. Experimental and theoretical NMR spectra for 13C (A) and 1H (B) nuclei of amide (2) in chloroform. Average chemical shifts for rotational equilibria Zc ↔ Zd and Ed ↔ Ec are presented in black and red, respectively
As can be noted in Figure 10A, the best correlation with the experimental 13C NMR shifts was obtained by using the functional B3LYP, B3LYP-D3 or APFD in combination with the basis set aug-cc-pVDZ. Figure 10B reveals that the chemical shifts for the methyl and methylene protons in the experimental 1H NMR spectrum were best approached by the B3LYP/aug-cc-pVDZ method, followed by the B3LYP/6-311+G (2d,p) and mPW1PW91/6-311+ G(2d, p) methods. Figure 10B also shows that all the approximation levels used in this study overestimated the shifts found experimentally which ranged between 0.23 and 0.68 ppm related to the furan protons.
CONCLUSIONS The best conditions for the synthesis of N-benzyl-N-(2-furanylmethyl)acetamide using acylation of N-benzyl-1-(furan-2-yl)aminomethane were established using a methodology framed in the principles of green chemistry. A direct reaction with acetic anhydride provided the maximum yield of 97%. Mass spectrometry confirmed the molecular formula while the vibrational spectra were consistent with the expected structure; C=O, C=C (furan and phenyl), C-O stretches. The spectroscopic characterization using 1H and 13C NMR of the title compound in chloroform revealed the formation of spatial isomers of type E-Z. They were distinguishable due to the variation in chemical shifts caused by the orientation of the acyl function in the structure. The presence of two distinct isomer types could be inferred from the integration of signal intensities. This suggested that there were two distinct methyl groups (six protons). Two-dimensional NMR characterized long range coupling. Both the vibrational assignments and the analysis of the NMR spectra were assisted by DFT calculations. In the latter, signals characteristic of protons of the furan ring, the methylene links, and the methyl group were evident. The computational calculations allowed the explanation of the distribution of the various conformations present in chloroform. Minimum-energy geometries and their relative energies were determined by DFT calculations. Initial structure optimization using the model B3LYP/6-31+G(d) model was accomplished by a relaxed scan of two dihedral angles. This defined the orientations of the benzyl fragment and the furan ring relative to the amide plane. In total, nine stable rotamers were theoretically identified as a result of the exploration of the amide conformational space by combining a two-dimensional PES scan (B3LYP/6-31+G(d)) with an automated molecular mechanics searching at the GMMX approximation level. These structures were further refined with more advanced DFT models including a polarizable continuum model for chloroform solvent. A comparative quantitative analysis using both Boltzmann population and NMR DFT calculations allowed the explanation of the signal duplication in the experimental NMR spectra of amide in terms of two predominant rotational equilibria in chloroform at room temperature. One of which was caused by two Z-type structures and other by two E-type structures.
SUPPLEMENTARY MATERIAL The following are available on http://quimicanova.sbq.org.br in the form of a PDF file, with free access. Figure 1S: 13C and 1H NMR spectra of N-benzyl-1-(furan-2-yl)methanaminium acetate, Table 1S: Experimental (Raman and IR) and calculated (APFD/Aug-cc-pVDz; correction factor λF: 0.97044) vibrational wavenumbers (cm-1) of amide (2).
ACKNOWLEDGEMENTS The authors would like to acknowledge the financial support provided by the host institutions, i.e., Universidad Pedagógica y Tecnológica de Colombia (UPTC), Pontificia Universidad Javeriana, Universidad Nacional de La Plata, Argentina, and the Université de Lille, France. J.A.G.C especially like to thank the French Ministère de l'Enseignement Supérieur et de la Recherche for his post-doctoral grant developed at the laboratory LASIR (Laboratoire de Spectrochimie Infrarouge et Raman) in France, as well as the Dirección de Investigaciones (DIN) of the UPTC for financial support provided through the project SGI 2619.
REFERENCES 1. Avalos, M.; Reyes B.; Barneto, J. L.; Bravo, J. L.; Cintas, P.; Jiménez, J. L.; Palacios; J. C.; J. Org. Chem. 2001, 66, 7275. 2. Ramírez, J. Z.; Vargas, R.; Padilla-Martínez, I. I.; Flores-Huerta A. G.; Garza, J.; J. Mex. Chem. Soc. 2012, 56, 275. 3. Young Kee Kang, Y. K.; Park, H. S.; J. Mol. Struct.: THEOCHEM 2004, 676, 171. 4. Stewart, W. E.; Siddall, T. H.; Chem. Rev. 1970, 70, 517. 5. Abraham, R. J.; Griffiths, L.; Perez, M.; Magn. Reson. Chem. 2013, 51, 143. 6. Fontoura, L. A. M.; Rigotti, I. J. da C.; Correia, C. R. D.; J. Mol. Struct. 2002, 609, 73. 7. Ledneczki, I.; Forgo, P.; Kiss, J. T.; Molnár, Á.; Pálinkó, I.; J. Mol. Struct. 2007, 834-836, 349. 8. Coronado, J. L. Gu.; Martín, E.; Montero, L. A.; Fierro, J. L. G.; García de la Vega, J. M.; J. Phys. Chem. A 2007, 111, 9724. 9. Mazzanti, A.; Casarini, D.; WIREs Comput. Mol. Sci. 2012, 2, 613. 10. Phillips, W. D.; J. Chem. Phys. 1955, 23, 1363. 11. Gutowsky, H. S.; Holm, C. H.; J. Chem. Phys. 1956, 25, 1228. 12. Suárez-Castillo, O. R.; Meléndez-Rodríguez, M.; Castelán-Duarte L. E.; Sánchez-Zavala, M.; Rivera-Becerril, E.; Morales-Ríos, M. S.; Joseph-Nathan, P.; Tetrahedron: Asymm. 2009, 20, 2374. 13. Da Silva, H. C.; De Almeida, W. B.; Chem. Phys. 2020, 528, 110479. 14. Mennucci, B. Cances, E.; Tomasi, J.; J. Chem. Phys. 1997, 101, 10506. 15. Abraham, R. J.; Byrne, J. J.; Griffiths, L. G.; Perez, M.; Magn. Reson. Chem. 2006, 44, 491. 16. Benzi, C.; Crescenzi, O.; Pavone, M.; Barone, V.; Magn. Reson. Chem. 2004, 42, S57. 17. Warren, S.; Designing Organic Syntheses: A Programmed Introduction to the Synthon Approach, 1st ed., Wiley: New York, 1991. 18. Nossa, D. L.; Gómez, J. A., Rozo, W. E.; Duchowicz, P. R.; J. Biomed. Sci., submitted. 19. Jacobi, P.A.; Yongkai, L.; J. Am. Chem. Soc. 2001, 123, 9307. 20. Iron, M. A.; J. Chem. Theory Comput. 2017, 13, 5798. 21. Casabianca, L. B.; Magn. Reson. Chem. (2020), doi.org/10.1002/mrc.4994. 22. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.; Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009. 23. Dennington, R.; Keith, T.; Millam, J.; GaussView 6.0.16, Semichem, Inc., Shawnee Mission, 2016. 24. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. 25. Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J.; J. Phys. Chem. 1994, 98, 11623. 26. Peterson, G. A.; Bennett, A.; Tensfeldt, T. G.; Al-Laham, M. A.; Shirley, W. A.; Mantzaris, J.; J. Chem. Phys. 1988, 89, 2193; Montgomery Jr., J. A.; Frisch, M. J.; Ochterski, J. W.; Petterson, G. A.; J. Chem. Phys. 1999, 110, 2822; Montgomery Jr., J. A.; Frisch, M. J.; Ochterski, J. W.; Petterson, G. A.; J. Chem. Phys. 2000, 112, 6532. 27. Gilbert, K. E.; Pcmodel version 10.075, Serena Software, Bloomington, USA, 2020. 28. Austin, A.; Petersson, G. A.; Frisch, M. J.; Dobek, F. J.; Scalmani, G.; Throssell, K.; J. Chem. Theory Comput. 2012, 8, 4989. 29. Dunning Jr., T. H.; J. Chem. Phys. 1989, 90, 1007; Kendall, R. A.; Dunning Jr., T. H.; Harrison, R. J.; J. Chem. Phys. 1992, 96, 6796; Peterson, K. A.; Woon, D. E.; Dunning Jr., T. H.; J. Chem. Phys. 1994, 100, 7410; Woon, D. E.; Dunning Jr., T. H.; J. Chem. Phys. 1993, 98, 1358. 30. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.; J. Chem. Phys. 2010, 132, 154104. 31. Lodewyk, M. W.; Siebert, M. R.; Tantillo, D.; J. Chem. Rev. 2012, 112, 1839; Lodewyk, M. W.; Soldi, C.; Jones, P. B.; Olmstead, M. M.; Larrucea, J. R.; Shaw, J. T.; Tantillo, D. J.; J. Am. Chem. Soc. 2012, 134, 18550; Lodewyk, M. W.; Tantillo, D. J.; J. Nat. Prod. 2011, 74, 1339. 32. Di Grandi, M. J.; Bennett, C.; Cagino, K., Muccini, A.; Suraci, C.; Saba, S.; Synth. Commun. 2015, 45, 2601. 33. Cahn, R. S.; Ingold, C.; Prelog, V.; Angew. Chem., Int. Ed. 1966, 5, 385. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access