
 |
 |
 |
 |
 |
Artigo
|
|
| Método de triagem de microgininas em cianobactérias por LC-MS/MS Microginins screening in cyanobacteria by LC-MS |
|
Kazumi K. TeramotoI,*; Fabiane DorrI; Miriam SanzII; Ernani PintoI
I. Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, 05508-000 São Paulo - SP, Brasil Recebido em 10/03/2020 *e-mail: kazumi.kinoshita@usp.br Cyanobacteria produce a wide variety of bioactive compounds, making them a highly promising group in the search for novel molecules with pharmacological and biotechnological properties. Among these, microginins attract attention for being peptides which inhibit angiotensin-converting enzyme, turning them into potential targets for hypertension and congestive heart failure treatment. This work describes a rapid and sensitive method for untarget screening of microginins in cyanobacteria extracts by LC-QqQ-MS/MS. These compounds are mostly characterized by containing 3-amino-2-hydroxy-decanoic acid at the N-terminus, which often could be chlorinated, dichlorinated or methylated. Based on the fragment ion arising from this decanoic acid derivative, a precursor ion scan (PIS) strategy has been proposed. This approach identified suspect microginins in cyanobacterial strains and environmental samples that were later confirmed by LC-QTOF-MS/MS. Eight new microginins structures were characterized based on the obtained fragmentation spectra from a total of 19 variants detected. This study highlights the applicability of PIS mode acquisition for untarget screening, detecting a wide variety of microginins with amino acids modifications, produced mainly by Microcystis aeruginosa strains. This method is a useful tool for the identification and environmental monitoring of molecules with conserved molecular substructures that possess similar fragmentation pattern. INTRODUÇÃO A eutrofização dos corpos d'água é um dos principais fatores que influenciam a proliferação de cianobactérias nas águas superficiais. Os efeitos das mudanças climáticas (aquecimento global, mudança nos padrões de precipitação, secas mais intensas) intensificaram os episódios tóxicos de florações de cianobactérias, tornando-as mais frequentes e persistentes.1 Essas florações podem afetar negativamente o ecossistema aquático e a qualidade da água e são consideradas um problema de saúde pública.2 Além das cianotoxinas conhecidas, as cianobactérias produzem uma ampla gama de metabólitos secundários biologicamente ativos, muitos dos quais permanecem inexplorados.3,4 A variedade química e estrutural dos metabólitos reflete em atividades biológicas distintas, tais como atividades citotóxica, antitumoral, antiviral, antibacteriana, antimalárica, antifúngica, anti-inflamatória e imunossupressoras, o que as tornam alvos em potencial para aplicações farmacêutica e biotecnológica.5 A maioria desses metabólitos compreende peptídeos cíclicos e lineares, conhecidos como cianopeptídeos, em sua maioria produtos da biossíntese não-ribossomal ou de vias mistas de síntese não-ribossomal e síntese policetídica, mediadas por complexos enzimáticos como NRPS (Nonribossomal Peptide Synthetase) e NRPS/PKS (Polyketide Synthase).3,4,6 Os cianopeptídeos são comumente agrupados de acordo com similaridades estruturais, com vias de biossíntese compartilhadas, como as aeruginosinas, anabaenopeptinas, cianopeptolinas, ciclamidas, microgininas, microviridinas e microcistinas.6,7 Microgininas (MG) (Figura 1) são peptídeos lineares não ribossômicos contendo o ácido 3-amino-2-hidroxi-decanoico (Ahda) na porção N-terminal.8 Em geral, apresentam de 4 a 6 aminoácidos em sua composição,9,10 mas variantes com apenas 3 aminoácidos também foram identificadas.11 Mais de 100 variantes de microgininas foram identificadas em culturas isoladas e florações de cianobactérias, principalmente em espécies do gênero Microcystis, Dolichospermum e Plaktothrix.12 Destas, apenas 46 foram isoladas e estruturalmente caracterizadas por ressonância magnética nuclear (RMN). As sequências peptídicas das demais microgininas foram propostas com base na elucidação estrutural realizada por métodos de espectrometria de massas sequencial (MS/MS). A ocorrência de N- metilação e cloração (mono e dicloração) alifática no Ahda são frequentemente relatadas em microgininas.11,13,14 Variações no resíduo Ahda, como a perda de um grupo hidroxila (-OH) na porção N-terminal, gerando o anidroAhda (Ada) foi reportada para as variantes cloradas MG 99-A e MG 99-B,15 bem como congêneres contendo o ácido 3-amino-2-hidroxi-octanoico (Ahoa)16,17 e seus derivados N-MeAhoa,18 Cl-Ahoa e Cl2-Ahoa.17
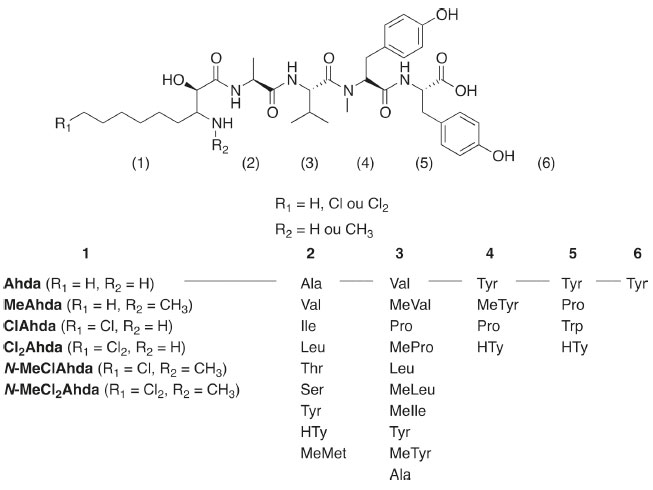 Figura 1. Estrutura geral das microgininas8 e esquema das possíveis combinações de aminoácidos
As microgininas são inibidoras de metaloproteinases dependentes de zinco tais como a enzima conversora de angiotensina (ECA)8,11,19 e algumas aminoproteinases (AMP), como as aminopeptidase M,14,20 aminopeptidase N16,21 e leucina aminopeptidase.10,22 A inibição da ECA desempenha um papel importante no tratamento da hipertensão e insuficiência cardíaca congestiva, transformando as microgininas em candidatos de interesse para o desenvolvimento de novos inibidores da ECA para o tratamento de doenças cardiovasculares.19 Elas também são consideradas alvos potenciais para terapia do câncer devido ao papel que as AMP desempenham na promoção da angiogênese, crescimento tumoral e metástase.20 Do ponto de vista toxicológico, evidências de atividade biológica contra crustáceos de água doce (Thamnocephalus platyurus),23 nematódeos (Caenorhabditis elegans)24 e peixes em fase larval (Astyanax altipaeanae)25 foram observadas para substâncias puras e extrato bruto contendo microgininas. Recentemente, estudos in vitro empregando uma linhagem celular de hepatocarcinoma humano (HepG2), demonstraram o potencial genotóxico das microgininas.26 A espectrometria de massas (Mass spectrometry - MS) baseada na ionização por eletrocspray (ESI) ou na dessorção/ionização a laser assistida por matriz (Matrix Assisted Laser Desorption/Ionization - MALDI) tem sido comumente empregada para detectar e caracterizar cianopeptídeos.12,27-29 As microcistinas são de longe a classe de cianopeptídeos mais amplamente estudada e muitas abordagens que descrevem o uso de cromatografia líquida em combinação com espectrometria de massas (LC-MS ou LC-MS/MS) foram relatadas.30 Por outro lado, poucos estudos têm sido empregados para o monitoramento das concentrações ambientais de outros cianopeptídeos, além das microcistinas, em águas superficiais. Até o presente momento, apenas dois trabalhos quantificaram concentrações absolutas de uma única variante de microginina (MG 690) em amostras ambientais.31,32 Entre os diferentes modos de aquisição em que um sistema LC-MS/MS pode operar, o modo de varredura por íons precursores (precursor ion scan - PIS) é especialmente útil para a triagem de classes de compostos que geram o mesmo íon fragmento, permitindo a detecção de compostos conhecidos e desconhecidos com o mesmo perfil de fragmentação característico.33 Após a triagem por PIS, os íons precursores candidatos devem ser submetidos a experimentos de fragmentação (ou seja, varredura de íons do produto) para confirmar os fragmentos e gerar informações adicionais para caracterização estrutural. Experimentos de PIS podem ser realizados em equipamentos com analisadores híbridos do tipo triplo quadrupolo (QqQ), quadrupolo-armadilha de ions (Quadrupole Ion-Trap - Q-IT) ou ainda quadrupolo-armadilha linear de ions (Quadrupole Linear Ion-Trap - Q-LIT).33 Além dos modos de varredura de íon produto, íon precursor e perda neutra, os analisadores do tipo QqQ podem operar no modo de monitoramento de reações múltiplas (MRM), que consiste em selecionar uma ou mais reações de fragmentação, permitindo a quantificação de múltiplos analitos ao mesmo tempo.34 Equipamentos do tipo QqQ são amplamente utilizados na detecção e quantificação de substâncias em matrizes complexas como amostras biológicas35 e ambientais,36 oferecendo alta seletividade, sensibilidade e reprodutibilidade. São inclusive empregados pelas agências de monitoramento de potabilidade de água para a detecção e quantificação das mais diversas substâncias (como cianotoxinas, por exemplo), cujos limites máximos permitidos estão definidos em Portarias estabelecidas pelo Ministério da Saúde.37 O desenvolvimento de um método de PIS por LC-QqQ-MS/MS para detecção de MG trata-se de uma ferramenta bastante útil do ponto de vista toxicológico e de saúde pública, dado que é capaz de detectar de forma simples e rápida esses compostos potencialmente tóxicos em amostras ambientais. As MG, assim como outras substâncias bioativas produzidas por cianobactérias, estão envolvidas na inibição de enzimas que participam de vias metabólicas importantes. No entanto, pouco se sabe sobre o seu potencial risco ecotoxicológico, sendo poucos os trabalhos que avaliaram os efeitos nocivos dessa classe de cianopeptídeos em modelos animais e culturas celulares. O trabalho teve como objetivo estabelecer um método rápido e sensível para o rastreamento não direcionado de microgininas em extratos de cianobactérias por LC-QqQ-MS/MS através da estratégia de PIS. Até onde sabemos, ainda não foram relatados métodos analíticos utilizando essa abordagem para a detecção dessa classe de cianopeptídeos.
PARTE EXPERIMENTAL Solventes e reagentes Todos os solventes (metanol, acetonitrila e isopropanol) e demais reagentes (formiato de amônio, formiato de sódio, ácido fórmico e hidróxido de amônio) utilizados para as análises cromatográficas e de espectrometria de massas apresentavam pureza mínima de grau HPLC. Os reagentes utilizados para o preparo do meio de cultura líquido ASM-138 eram de grau analítico. A água ultrapura foi obtida em sistema Milli-Q® Direct 8 (Millipore Corporation, EUA). Seleção e cultivo das linhagens de cianobactérias O Laboratório de Toxinas e Produtos Naturais de Algas (LTPNA) mantém um banco de culturas de cianobactérias, com linhagens isoladas por pesquisadores do grupo e cedidas por colaboradores ou ainda adquiridas comercialmente. As culturas foram cultivadas em tubos de ensaio com meio ASM-1 líquido, sob intensidade luminosa de 20 µmol fótons m-2 s-1, temperatura de 23 ± 2 °C, pH 8,0 e fotoperíodo de 12:12 horas (claro/escuro). Para a obtenção de biomassa para isolamento e purificação das microgininas, as linhagens LTPNA 08 e NIES-299 foram cultivadas em sistema de cultivo em batelada, sob aeração constante e intensidade luminosa de 70 µmol fótons m-2 s-1. Após 30 dias de cultivo, o material celular das culturas foi concentrado por centrifugação (10.000 x g, 10 min, 4 ºC, Eppendorf 5804R), liofilizado e mantido à -20 °C até o momento da extração. Isolamento e purificação das microgininas Os padrões gravimétricos de microgininas utilizados para o desenvolvimento e otimização do método analítico de varredura de íon precursor foram obtidos conforme metodologia proposta em trabalhos anteriores do grupo de pesquisa, com pequenas modificações.19,39 O material liofilizado de LTPNA 08 e NIES-299 foi extraído três vezes com metanol 70% (1 g/40 mL) e submetido à probe de ultrassom (intensidade 30%, pulso de 10, por 10 min - Sonic Omni Disruptor). O material foi centrifugado (8500 x g, 4 °C, 10 min, Eppendorf 5804R) e o sobrenadante coletado e submetido ao sistema rotaevaporador (Modelo B-491 - Büchi) para a remoção do solvente. O extrato reconstituído em 100 mL de metanol 5% foi aplicado em cartucho de extração em fase sólida (Waters Sep Pak C18 Vac 6 cc, 500 mg) e eluído com gradiente de 20 a 100% de metanol acidificado contendo 0,1% de ácido fórmico, para a obtenção de 5 frações (60 mL). Os eluatos foram concentrados sob fluxo de nitrogênio e reconstituídos em acetonitrila:água (30:70, v/v). As amostras foram filtradas em filtros de seringa de PVDF de 0,22 µm e submetidas à análise por LC-HR-QTOF-MS/MS. As frações de 60 e 80% foram submetidas à purificação em escala semi-preparativa por HPLC-DAD, utilizando uma coluna semi-preparativa de fase reversa (Luna C18, 250 x 10 mm, 5 μm - Phenomenex) e fases móveis (A) água e (B) acetonitrila ambos contendo 0,1% de ácido fórmico, a uma vazão de 3,0 mL min-1, empregando um gradiente linear de eluição de 30 - 60% de B em 20 min. Extração e preparo de amostras 66 linhagens de cianobactérias mantidas no banco de culturas do LTPNA e 7 amostras ambientais provenientes de reservatórios e lagos brasileiros foram utilizadas para a busca de variantes conhecidas e inéditas de microgininas. O material liofilizado das linhagens e das amostras ambientais foi pesado (10 mg), extraído com metanol:água (50:50, v/v) e submetido à probe de ultrassom (intensidade de 30%, pulso de 10, por 2 min - Omni Sonic Disruptor). Em seguida, o material foi centrifugado (8500 x g, 4 °C, 15 min, Eppendorf 5415R) e o sobrenadante coletado e filtrado em filtros de seringa de PVDF de 0,22 µm para a análise cromatográfica. Triagem de microgininas por LC-QqQ-MS/MS em modo de varredura de íon precursor (Precursor ion scan) A busca por espécies produtoras de microgininas e a triagem rápida desses compostos em amostras ambientais, foi realizada por LC-MS/MS através do método de precursor ion scan. O método foi desenvolvido em sistema cromatográfico Agilent 1260 Infinity acoplado a espectrômetro de massas do tipo triplo quadrupolo (QqQ - 6460 Triple Quad LC/MS, Agilent Technologies, EUA), com ionização por electrospray (ESI). O espectrômetro foi operado no modo positivo de ionização, com potencial capilar de 3500 V, pressão do gás nebulizador (nitrogênio) de 35 psi, temperatura e vazão do gás secante (N2) de 9 L. min-1 a 300 °C, temperatura e vazão do gás de bainha (N2) de 11 L. min-1 a 325 °C e voltagem do cone de 500 V. Os padrões gravimétricos analíticos das microgininas MG 299-A, MG 299-B, MG 770, MG 756 e MG 299-C (Tabela 1S) foram pesados utilizando uma balança analítica de legibilidade 0,01 mg (Cubis®, Modelo MSU, Sartorius, EUA) e reconstituídos em metanol (1 mg mL-1). Os padrões analíticos foram utilizados para definir e otimizar os parâmetros de ionização, colisão e separação. Uma varredura (full scan) foi realizada para cada variante de microginina, separadamente, na faixa de m/z 100-2000 antes da escolha do íon precursor para experimentos de dissociação induzida por colisão (CID). Parâmetros como voltagem do acelerador de células (cell accelerator voltage - CAV) e energias de fragmentação (fragmentor voltage - FV) e colisão (Collision Energy - CE) foram otimizados injetando soluções individuais dos padrões de microgininas, em que foram selecionadas as condições mais favoráveis para a formação dos íons produtos de interesse (Tabela 1).
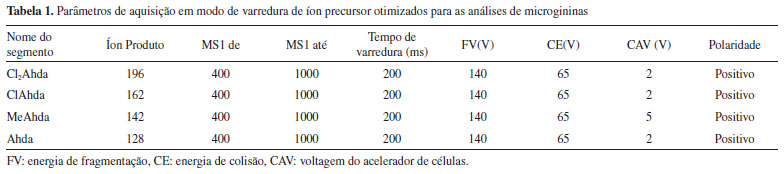
O espectrômetro de massas foi operado no modo varredura de íon precursor para o monitoramento dos íons precursores que originam os íons produtos de m/z 128, 142, 162 e 196, referentes aos fragmentos não-metilado, metilado, clorado e diclorado, respectivamente, resultantes da clivagem da cadeia lateral do resíduo Ahda. Esses fragmentos são considerados íons diagnósticos da classe das microgininas. A separação cromatográfica foi obtida em uma coluna Luna C18 (2) (150 x 2,0 mm, 4 μ - Phenomenex), protegida com uma pré-coluna de mesma fase estacionária, e mantida à 40 °C. Os eluentes utilizados como fase móvel foram: (A) H2O contendo 5 mmol. L-1 de formiato de amônio e 0,1% de ácido fórmico e (B) acetonitrila, a uma vazão de 0,25 mL min-1. O gradiente de eluição foi de 25 a 55% de B (0-10 min), de 55 a 90% de B (10-10,5 min), mantido a 90% de B (10,5 - 13,5 min) para a limpeza da coluna, e de 90 a 25% de B (13,5-20 min) para reequilíbrio da coluna. Antes de cada injeção de amostra (5 µL), a agulha do injetor foi lavada com isopropanol:metanol:água (80:10:10, v/v). As amostras consideradas positivas foram analisadas posteriormente por LC-QTOF-MS/MS para a identificação e caracterização das microgininas suspeitas. Identificação das microgininas por LC-QTOF-MS/MS A detecção, identificação e caracterização estrutural das microgininas suspeitas foram realizadas por cromatografia líquida de alta eficiência acoplada à espectrometria de massas de alta resolução.28 As análises cromatográficas foram realizadas em sistema cromatográfico Shimadzu Prominence (Shimadzu, Kyoto, Japão) com detector de arranjos de diodos (SPD-M20A) acoplado a um espectrômetro de massas com analisador do tipo quadrupolo-tempo-de-vôo (QTOF) (MicroTOF-QII, Bruker Daltonics, MA, EUA), equipado com fonte electrospray (ESI). O equipamento foi operado no modo positivo de ionização, com potencial capilar de 3500 V, pressão do gás nebulizador (nitrogênio) de 35 psi e temperatura e vazão do gás secante (nitrogênio) de 5 mL min-1 a 300 °C. Uma solução de formiato de sódio 10 mmol L-1 em isopropanol:água (1:1, v/v) foi utilizada como calibrante externo. O instrumento foi operado em modo de varredura (full scan) e em modo Auto MS/MS, na faixa de 50 a 2000 m/z, realizando experimentos de MS/MS sobre os 3 íons mais intensos obtidos no modo MS scan. Os parâmetros de separação cromatográfica foram os mesmos utilizados no método de precursor ion scan por LC-QqQ-MS/MS. O processamento dos dados foi realizado no software Data Analyses 4.0 (Bruker Daltonics). A composição elementar, o desvio entre a massa medida e a massa teórica em [ppm] (err) e a comparação do valor do padrão isotópico teórico e medido do pico de interesse, por meio de score (mSigma), foram calculados usando Smart Formula Editor. A confirmação da fórmula molecular foi baseada nos limites amplamente aceitos de 5 ppm e 20 mSigma. Os resíduos de aminoácidos foram determinados através do cálculo da diferença de massa entre uma série de íons fragmentos sucessivos, identificando íons das séries -a, -b e -y. Além da busca por perdas neutras de CO (-28 u) por parte de um íon -b (íon -a) e perdas neutras de moléculas de H2O (-18 u) e NH3 (-17 u) a partir de íons -b e -y, a busca por íons imônio na região de baixas massas do espectro também pôde fornecer informações inerentes à composição dos aminoácidos dos peptídeos.40
RESULTADOS E DISCUSSÃO Triagem de microgininas pelo método de varredura do íon precursor (Precursor ion scan) O método de precursor ion scan por LC-MS/MS (QqQ) foi testado em linhagens de cianobactérias mantidas em laboratório e amostras ambientais coletadas de lagos e reservatórios brasileiros (Tabela 2S). As amostras positivas para a triagem de microgininas estão compiladas na Tabela 2 e identificadas com os íons precursores que deram origem aos respectivos íons produto selecionados (íons diagnósticos).
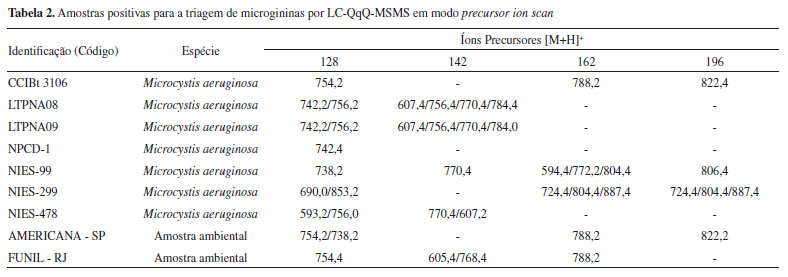
A triagem suspeita detectou possíveis variantes de MG em 7 amostras de linhagens de cianobactérias e em 2 amostras ambientais, sendo alguns íons detectados compatíveis com íons de MG já identificados na literatura (Tabela 3S). Os analisadores de massas do tipo QqQ permitem análises rápidas e sensíveis de compostos em misturas complexas, no entanto, os experimentos realizados nesse tipo de analisador geram dados de baixa resolução e acurácia. Por essa razão, para obtenção de espectros de massas de alta resolução, que pudessem fornecer dados acerca da composição elementar exata dos analitos e, assim, auxiliar na identificação dos compostos suspeitos, as amostras consideradas positivas foram também analisadas por LC-QTOF-MS/MS. A elucidação estrutural por técnicas de espectrometria de massas são baseadas em comparações com perfis de fragmentação de compostos similares já identificados e pelo estabelecimento da fórmula molecular, por meio da precisão de massas (m/z) e do padrão isotópico obtido por técnicas espectrométricas de alta resolução.7,41 A identificação das microgininas foi baseada nos fragmentos diagnósticos de m/z 128 Da (fragmento Ahda), m/z 142 Da (fragmento MeAhda), m/z 162 Da (fragmento Cl-Ahda) e m/z 196 Da (fragmento Cl2-Ahda). Como a maioria das MG descritas apresenta resíduos de tirosina (Tyr), homotirosina (HTy), N-metil tirosina (N-MeTyr) ou prolina (Pro) na porção C-terminal, os fragmentos gerados a partir das perdas neutras correspondentes a esses aminoácidos auxiliaram na identificação da sequência peptídica, assim como a busca por íons imônio, considerados íons diagnóstico, podendo indicar a presença ou ausência de determinados aminoácidos na sequência peptídica. A busca pelos íons detectados no método de PIS permitiu a identificação de 19 variantes de MG. Dessas, 8 apresentaram massa molecular e perfil de fragmentação diferentes daquelas já reportadas na literatura, indicando a possibilidade de ser um novo congênere de MG. Algumas apresentaram perfil de fragmentação semelhante àquelas já descritas, mas com diferença de unidade de massas de 14 Da (-CH2), 34 Da (-Cl) ou 16 Da (-O), referente à metilação, cloração ou desidroxilação na molécula, respectivamente. As MG detectadas que não apresentaram correspondência com nenhuma das listadas na Tabela 3S, através das informações de massa exata e fórmula molecular e perfil de fragmentação, foram consideradas moléculas inéditas e sua estrutura proposta. Na Tabela 3 estão listados os congêneres de MG identificados nas amostras após confirmação por espectrometria de massas de alta resolução e na Tabela 4 a sequência de aminoácidos proposta para as variantes consideradas inéditas. Os espectros MS obtidos por LC-ESI-QqQ-MS/MS no modo PIS e os espectros obtidos via LC-HR-ESI-QTOF-MS/MS das microgininas identificadas nas amostras estão representados nas Figuras 1S - 8S.
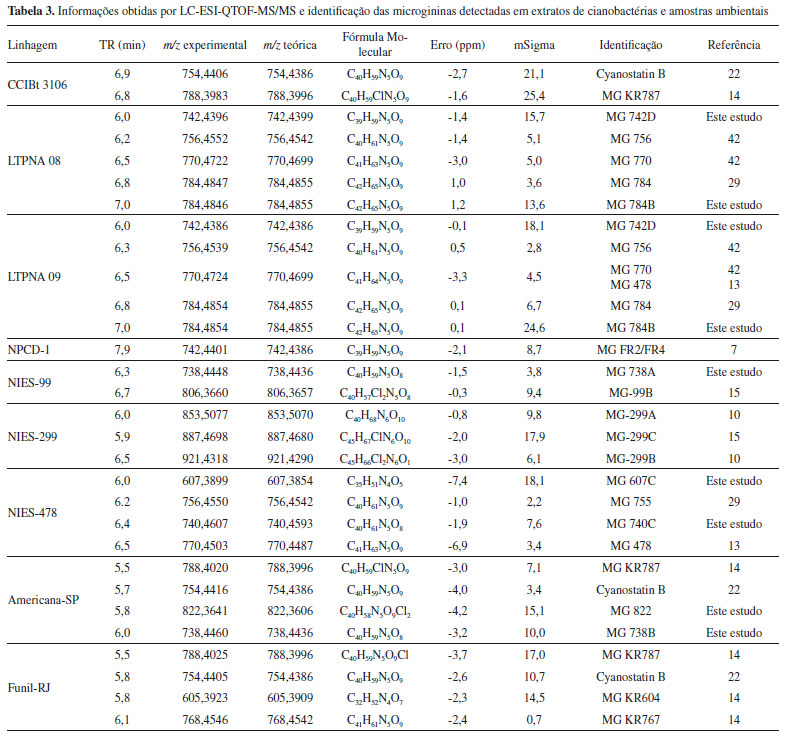
A maioria das sequências peptídicas de MG identificadas continha resíduo de tirosina (Tyr) na porção C-terminal, indicada pela perda neutra de 181 Da, resultando no fragmento de íon [M+H-181]+ ou a presença de prolina (Pro), indicada pela perda neutra de 115 Da do íon precursor, resultando no fragmento de íon [M+H-115]+. Fragmentos com perda neutra de 278 Da e 358 Da e a presença dos íons fragmentos correspondentes de m/z 279 e m/z 359 foram atribuídos à sequência de aminoácidos Pro - Tyr e MeTyr/Hty - Tyr, respectivamente. A detecção dos íons imônio de m/z 100 (N-MeLeu ou N-MeIle), m/z 136 (Tyr) e m/z 150 (Hty ou N-MeTyr) também auxiliaram na determinação dos aminoácidos presentes nas moléculas. A MG 742 (m/z 742,4396), por exemplo, apresentou equivalência com 3 MG da lista de suspeitos, no entanto, os fragmentos gerados nos espectros MS/MS não foram compatíveis com as estruturas propostas. As variantes contendo N-MeAhda, MG FR2/FR4,7,43 quando fragmentadas por CID, geraram íons fragmentos de m/z 142. A MG 742, ao ser fragmentada, gerou um íon intenso de m/z 128, indicando a presença do resíduo Ahda, não metilado. A Nostoginina BN 74116 possui, além do resíduo Ahda (m/z 128), um resíduo de MeTyr na porção C-terminal que, ao ser fragmentada, gera um fragmento com perda neutra de 195 Da do íon precursor. O fragmento gerado na MG 742, por outro lado, indica a presença de Tyr na porção C-terminal, correspondente à perda neutra de 181 Da do íon precursor. Comparando os espectros das outras variantes produzidas pela linhagem LTPNA 08, verificou-se diferença de unidade de massa de 14 Da em relação à MG 755 e de 28 Da em relação à MG 770. Já a MG 784B apresentou um incremento de 14 Da em relação à MG 770, sugerindo uma metilação adicional na molécula. Levando em consideração todos os outros fragmentos formados, como a ausência do íon m/z 86 (íon imônio de Leu, Ile ou MeVal) na MG 742 e do íon m/z 72 (íon imônio de Val) na MG 784B, foram propostas as seguintes sequências de aminoácidos para as MG 742 e MG 784B: Ahda-Val-Val-N-MeTyr-Tyr e N-MeAhda-MeVal-MeVal-N-MeTyr-Tyr. Por meio de comparações dos tempos de retenção e dos espectros de fragmentação das MG 770 e MG 478, detectadas nas amostras LTPNA 08 (LTPNA 09) e NIES-478, respectivamente, e os valores de mSigma e erros experimentais (ppm) em concordância com a fórmula molecular proposta, foi possível concluir que ambas se tratavam da mesma molécula. A elucidação estrutural da MG 478 foi realizada em estudos anteriores, empregando técnicas espectrométricas de RMN e HRFABMS, com determinação da conformação estrutural da molécula e atividade inibitória sobre a ECA.13 Três variantes foram propostas contendo o ácido anidroAhda (Ada) em vez do aminoácido Ahda, as MG 740C, MG 738 e MG 738A. Esse aminoácido é resultado da desidroxilação do aminoácido Ahda e, quando fragmentado, também gera um íon de m/z 128, resultante da quebra entre C2 e C3 do resíduo N-terminal Ahda. O Ada já foi reportado em trabalhos anteriores, sendo identificado nas MG 99-A e MG 99-B, produzidas pela linhagem de Microcystis aeruginosa NIES-99.15 Na amostra Americana - SP foi possível detectar duas variantes cloradas de MG. A presença de cloro na molécula é facilmente identificada devido ao seu perfil isotópico característico. A abundância do isótopo 37Cl (24.23%) na natureza é de mais ou menos 1/3 do isótopo 35Cl (75.77%). Assim, quando o cloro está presente na molécula, o pico M+2 é bastante evidenciado. Na presença de dois cloros, um pico M+4 também vai ser evidenciado. Fragmentos de m/z 128, 142, 162 e 198 foram observados para os íons protonados de m/z 754,4416, m/z 738,4460, m/z 788,4020 e m/z 822,3641 respectivamente. Uma busca na literatura identificou apenas 2 desses compostos, Cyanostatina B (m/z 754,4364) e MG KR 787 (m/z 788,3994). No entanto, todos eles compartilham um mesmo padrão de fragmentação com diferença de unidade de massas associadas a ganho ou perda de cloro e de perda de um grupo hidroxi, no caso do composto MG 738B. A atribuição da sequência de aminoácidos foi dada então a partir das MG previamente descritas.14,22 Apesar de serem coletadas de diferentes reservatórios, inclusive de Estados diferentes, as duas amostras ambientais analisadas (Americana - SP e Funil - RJ) apresentaram perfil de produção de MG semelhantes, com a detecção de variantes cloradas e não cloradas. A maioria dessas variantes foi isolada de florações de Microcystis coletadas de um reservatório em Israel.14 A ampla distribuição geográfica desses compostos pode estar relacionada com o caráter cosmopolita das cianobactérias do gênero Microcystis, principal produtor dessa classe de compostos. É interessante notar que todas as amostras positivas para a triagem de microgininas pertenciam à espécie de cianobactérias M. aeruginosa e que, mesmo dentro de uma mesma espécie, os congêneres produzidos foram variados. Essa capacidade de produzir diferentes cianopeptídeos pode estar relacionada com os genes envolvidos na via biossintética desses compostos. Dentro de uma espécie existem cepas não produtoras, que carecem do gene envolvido na biossíntese de um determinado cianopeptídeo, assim como cepas que carregam o gene, mas que nem sempre são expressos.44 Devido à essa regulação genética, a produção de cianopeptídeos é altamente variável entre as cianobactérias e pode ser influenciada por fatores bióticos e abióticos.45 Microgininas foram detectadas em diferentes florações ao redor do mundo,12,14,31,46 inclusive no trato alimentar de carpas29 e isoladas de cianobactérias cultivadas em laboratório11,42 para elucidação estrutural de novos peptídeos. Apesar do grande sucesso da espectrometria de massas para o sequenciamento de peptídeos e pequenas proteínas, aminoácidos isobáricos como a leucina (Leu), isoleucina (Ile), ou metil valina (MeVal), são dificilmente distinguidos por MS porque suas massas moleculares são exatamente as mesmas.47 O mesmo ocorre para os aminoácidos Hty e MeTyr, dificultando a correta caracterização de novas moléculas, sendo necessário o emprego de técnicas como RMN para o correto assinalamento dos aminoácidos e a atribuição das conectividades e orientações entre resíduos de aminoácidos. No entanto, miligramas de material puro são necessários para as análises, o que muitas vezes é impraticável de se obter se tratando de um produto natural.6 As inúmeras possibilidades de combinação e variação de aminoácidos dentro da molécula, além de modificações que incluem metilação, cloração, hidroxilação e sulfatação, refletem a elevada variabilidade estrutural das MG e de outras classes de peptídeos. Isso reflete também no nível ou tipo de atividade biológica exercida por uma determinada molécula. Estudos mostraram que a sequência de aminoácidos que compreende a estrutura de uma microginina e até mesmo a sua conformação estereoquímica estão diretamente ligadas à capacidade de inibição enzimática.13,14 Os grupos amino e hidroxil do Ahda e a porção C-terminal da molécula composta por dois resíduos de Tyr desempenham um papel importante na inibição da ECA,13 assim como os dipeptídeos formados por aminoácidos aromáticos ou Pro na porção C-terminal e os aminoácidos Val ou Ile na porção N-terminal.48 Resultados dos ensaios de inibição da aminopeptidase M revelaram que o ácido carboxílico terminal e a N-metilação do aminoácido Ahda estão diretamente relacionados com a atividade inibitória da AMP.14 Embora se saiba que as MG são biologicamente ativas, dados referentes aos seus efeitos e impactos ambientais ainda são escassos. Os poucos estudos que avaliaram a toxicidade aguda de microgininas em modelos de organismos aquáticos são bastante recentes e reportaram efeitos negativos sobre os animais testados.23,24 A toxicidade aguda da MG FR3 foi avaliada em larvas de T. platyurus (crustáceo) e observou-se atividade biológica com elevada taxa de mortalidade dos animais após 48 h de exposição, obtendo-se uma CL50 de 7,78 µg mL-1.23 Lenz et al.24 avaliaram o efeito tóxico de diferentes classes de cianopeptídeos (anabaenopeptinas, cianopeptolinas, MG 690 e MC-RR), na reprodução, tempo de incubação, taxa de crescimento, expectativa de vida e integridade vulvar relacionada à idade do nematódeo Caenorhabditis elegans. Apesar de ser considerada a menos tóxica entre os compostos testados (na concentração de 10 µg L-1), a MG 690 afetou de forma negativa quase todos os parâmetros avaliados. O mais recente dos estudos avaliou os efeitos tóxicos de 4 congêneres de MG (MG FR3, MG GH787, Cyanostatin B e MGL 402) em linhagem celular humana de carcinoma hepatocelular (HepG2).26 Todas as variantes testadas induziram dano ao DNA após 4 e 24 h de exposição, em concentrações entre 0,01 e 1 µg mL-1. Este foi o primeiro estudo a fornecer evidências do potencial genotóxico das MG e o risco associado à exposição desses compostos. A exposição humana aos compostos genotóxicos está associada com o desenvolvimento de diversas doenças crônicas, incluindo o câncer, e nos organismos aquáticos afeta principalmente a reprodução.26 Apesar de sua ampla ocorrência, a função ecológica de MG ainda permanece incerta. A complexidade estrutural desses metabólitos secundários produzidos por cianobactérias e os dados biológicos disponíveis ainda não são suficientes para afirmar a sua verdadeira função e relevância ecológica. Ainda assim, dados acerca da toxicidade de alguns compostos dessa classe, que podem representar riscos ao meio ambiente ou à saúde humana, reforçam a necessidade de maior investigação desses cianopeptídeos. Com os eventos de florações de cianobactérias cada vez mais frequentes e persistentes, a detecção analítica padronizada e sensível desses compostos em águas destinadas ao abastecimento público se mostra relevante, podendo o seu monitoramento ser incluso futuramente em análises de rotina atendendo aos padrões de potabilidade de água.
CONCLUSÕES Foram propostas diversas microgininas com base na elucidação de seus espectros de fragmentação. O método de triagem desenvolvido para a detecção de microgininas por LC-QqQ-MS/MS em modo precursor ion scan foi capaz de detectar compostos suspeitos em 9 amostras de cianobactérias. A confirmação desses compostos foi feita por LC-HR-QTOF-MS/MS, identificando as mesmas microgininas nas 9 amostras analisadas, mostrando a boa correlação entre os métodos. No total foram identificadas 19 variantes de MG, das quais 8 foram consideradas moléculas inéditas por não apresentarem equivalência com microgininas conhecidas descritas na literatura. As estruturas das novas MG foram propostas com base nos espectros de fragmentação obtidos por espectrometria de massas de alta resolução. Algumas apresentaram perfil de fragmentação semelhante ao de MG conhecidas, com diferença de massas referentes às modificações químicas tais como metilação, cloração e desidroxilação. O método de triagem em modo precursor ion scan se mostrou uma análise rápida e sensível na busca por microgininas conhecidas e desconhecidas, demonstrando ser uma ferramenta analítica potencialmente útil na detecção e monitoramento ambiental de compostos que apresentam perdas características de um grupamento químico, como as microcistinas e microgininas.
MATERIAL SUPLEMENTAR A lista de microgininas, os espectros de MS e MS2 dos compostos, assim como outros materiais de apoio, estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem à Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP (Processos Nº 2013/07914-8 e 2014/50420-9), ao Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (Processos Nº 311048/2016-1 e 439065/2018-6), à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES (Processo Nº 23038.001401/2018-92), e à Fundação de Apoio à Universidade de São Paulo - FUSP (Projeto Nº 1979) pelo suporte financeiro.
REFERÊNCIAS 1. Paerl, H. W.; Paul, V. J.; Water Res. 2012, 46, 1349; Glibert, P. M.; Mar. Pollut. Bull. 2017, 124, 591; Paerl, H. W.; Huisman, J.; Environ. Microbiol. Rep. 2009, 1, 27. 2. Codd, G. A.; Ecol. Eng. 2000, 16, 51; Huisman, J.; Codd, G. A.; Paerl, H. W.; Ibelings, B. W.; Verspagen, J. M. H.; Visser, P. M.; Nat. Rev. Microbiol. 2018, 16, 471. 3. Elersek, T.; Blaha, L.; Mazur-Marzec, H.; Schmidt, W.; Carmeli, S. Em Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; Meriluoto, J.; Spoof, L., Codd, A., eds.; Wiley: New Jersey, 2017, cap. 19. 4. Huang, I. S.; Zimba, P. V.; Harmful Algae 2019, 83, 42. 5. Singh, R. K.; Tiwari, S. P.; Rai, A. K.; Mohapatra, T. M.; J. Antibiot. 2011, 64, 401; Swain, S. S.; Paidesetty, S. K.; Padhy, R. N.; Biomed. Pharmacother. 2017, 90, 760; Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B.; Mar. Drugs 2019, 17, 320. 6. Welker, M.; Von Döhren, H.; FEMS Microbiol. Rev. 2006, 30, 530. 7. Welker, M.; Maršálek, B.; Šejnohová, L.; von Döhren, H.; Peptides 2006, 27, 2090. 8. Okino, T.; Matsuda, H.; Murakami, M.; Yamaguchi, K.; Tetrahedron Lett. 1993, 34, 501. 9. Sano, T.; Kaya, K.; Phytochemistry 1997, 44, 1503. 10. Ishida, K.; Matsuda, H.; Murakami, M.; Yamaguchi, K.; Tetrahedron 1997, 53, 10281. 11. Stewart, A. K.; Ravindra, R.; Van Wagoner, R. M.; Wright, J. L. C.; J. Nat. Prod. 2018, 81, 349. 12. Zervou, S. K.; Gkelis, S.; Kaloudis, T.; Hiskia, A.; Mazur-Marzec, H.; Chemosphere 2020, 248, 125961. 13. Ishida, K.; Kato, T.; Murakami, M.; Watanabe, M.; Watanabe, M. F.; Tetrahedron 2000, 56, 8643. 14. Lodin-Friedman, A.; Carmeli, S.; Mar. Drugs 2018, 16, 78. 15. Ishida, K.; Matsuda, H.; Murakami, M.; Tetrahedron 1998, 54, 13475. 16. Ploutno, A.; Carmeli, S.; Tetrahedron 2002, 58, 9949. 17. Strangman, W. K.; Wright, J. L. C.; Tetrahedron Lett. 2016, 57, 1801. 18. Reshef, V.; Carmeli, S.; Tetrahedron 2001, 57, 2885. 19. Paiva, C. R. F.; Ferreira, M. G.; Trossini, H. G. G.; Pinto, E.; Molecules 2017, 22,1884. 20. Ferreira, G. M.; Kronenberger, T.; de Almeida, É. C.; Sampaio, J.; Terra, C. F.; Pinto, E.; Trossini, G. H. G.; Molecules 2019, 24, 4369. 21. Lifshits, M.; Carmeli, S.; J. Nat. Prod. 2012, 75, 209. 22. Sano, T.; Takagi, H.; Morrison, L. F.; Metcalf, J. S.; Codd, G. A.; Kaya, K.; Phytochemistry 2005, 66, 543. 23. Bober, B.; Bialczyk, J.; J. Appl. Phycol. 2017, 29, 1355. 24. Lenz, K. A.; Miller, T. R.; Ma, H.; Chemosphere 2019, 214, 60. 25. Fernandes, K.; Gomes, A.; Calado, L.; Yasui, G.; Assis, D.; Henry, T.; Fonseca, A.; Pinto, E.; Toxins 2019, 11, 220. 26. Ujvárosi, A. Z.; Hercog, K.; Riba, M.; Gonda, S.; Filipič, M.; Vasas, G.; Žegura, B.; Chemosphere 2020, 240, 124880. 27. Welker, M.; Brunke, M.; Preussel, K.; Lippert, I.; von Döhren, H.; Microbiology 2004, 150, 1785. 28. Sanz, M.; Andreote, P. A.; Fiore, F. M.; Dörr, A. F.; Pinto, E.; Mar. Drugs 2015, 13, 3892. 29. Riba, M.; Kiss-Szikszai, A.; Gonda, S.; Boros, G.; Vitál, Z.; Borsodi, A. K.; Krett, G.; Borics, G.; Ujvárosi, A. Z.; Vasas, G.; Toxins 2019, 11, 288. 30. Mekebri, A.; Blondina, G. J.; Crane, D. B.; J. Chromatogr. A 2009, 1216, 3147; Dörr, F.; Oliveira-Silva, D.; Lopes, N.; Iglesias, J.; Volmer, D.; Pinto, E.; Rapid Commun Mass Spectrom. 2011, 25, 1981; Miles, C. O.; Sandvik, M.; Nonga, H. E.; Rundberget, T.; Wilkins, A. L.; Rise, F.; Ballot, A.; Toxicon 2013, 70, 21; Zastepa, A.; Pick, F. R.; Blais, J. M.; Saleem, A.; Anal. Chim. Acta 2015, 872, 26; McMullin, D. R.; Hoogstra, S.; McDonald, K. P.; Sumarah, M. W.; Renaud, J. B.; J. Visualized Exp. (2019), doi: 10.3791/59712. 31. Beversdorf, L. J.; Rude, K.; Weirich, C. A.; Bartlett, S. L.; Seaman, M.; Kozik, C.; Biese, P.; Gosz, T.; Suha, M.; Stempa, C.; Shaw, C.; Hedman, C.; Piatt, J. J.; Miller, T. R.; Water Res. 2018, 140, 280. 32. Bartlett, S. L.; Brunner, S. L.; Klump, J. V.; Houghton, E. M.; Miller, T. R.; J. Great Lakes Res. 2018, 44, 924. 33. Domon, B.; Aebersold, R.; Science 2006, 312, 212. 34. Sismotto, M.; Paschoal, J. A. R.; Reyes, F. G. R.; Quim. Nova 2013, 36, 449; Vidova, V.; Spacil, Z.; Anal. Chim. Acta 2017, 964, 7. 35. Sagratini, G.; Fernández-Franzón, M.; De Berardinis, F.; Font, G.; Vittori, S.; Mañes, J.; Food Chem. 2012, 132, 537; Palagama, D. S. W.; Baliu-Rodriguez, D.; Lad, A.; Levison, B. S.; Kennedy, D. J.; Haller, S. T.; Westrick, J.; Hensley, K.; Isailovic, D.; J. Chromatogr. A 2018, 1573, 66. 36. Stolker, A. A. M.; Niesing, W.; Hogendoorn, E. A.; Versteegh, J. F. M.; Fuchs, R.; Brinkman, U. A. T.; Anal. Bioanal. Chem. 2004, 378, 955; Asimakopoulos, A. G.; Kannan, P.; Higgins, S.; Kannan, K.; Anal. Bioanal. Chem. 2017, 409, 6205. 37. https://portalarquivos2.saude.gov.br/images/pdf/2018/marco/29/PRC-5-Portaria-de-Consolida----o-n---5--de-28-de-setembro-de-2017.pdf, acessada em agosto de 2020. 38. Gorham, P. R.; McLachlan, J.; Hammer, U. T.; Kim, W. K.; SIL Proceedings, 1922-2010. Internationale Vereinigung für Theoretische und Angewandte Limnologie: Verhandlungen 1964, 15, 796. 39. Sanz, M.; Salinas, R. K.; Pinto, E.; J. Nat. Prod. 2017, 80, 2492. 40. Paizs, B.; Suhai, S.; Mass Spectrom. Rev. 2005, 24, 508; Cantú, M. D.; Carrilho, E.; Wulff, N. A.; Palma, M. S.; Quim. Nova 2008, 31, 669. 41. Pilon, A. C.; Selegato, D. M.; Fernandes, R. P.; Bueno, P. C. P.; Pinho, D. R.; Carnevale Neto, F.; Freire, R. T.; Castro-Gamboa, I.; Bolzani, V. S.; Lopes, N. P.; Quim. Nova 2020, 43, 329. 42. Carneiro, R. L.; Dörr, F. A.; Dörr, F.; Bortoli, S.; Delherbe, N.; Vásquez, M.; Pinto, E.; FEMS Microbiol. Ecol. 2012, 82, 692. 43. Le Manach, S.; Duval, C.; Marie, A.; Djediat, C.; Catherine, A.; Edery, M.; Bernard, C.; Marie, B.; Frontiers in Microbiology 2019, 10, 791. 44. Janssen, E. M. L.; Water Res. 2019, 151, 488. 45. Benayache, N.; Nguyen-Quang, T.; Hushchyna, K.; McLellan, K.; Afri-Mehennaoui, F.-Z.; Bouaïcha, N. Em Limnology: Some New Aspects of Inland Water Ecology; Gokce, D., ed.; IntechOpen: London, 2019, cap. 2. 46. Gesner-Apter, S.; Carmeli, S.; Tetrahedron 2008, 64, 6628. 47. Xiao, Y.; Vecchi, M. M.; Wen, D.; Anal. Chem. 2016, 88, 10757. 48. Cheung, H. S.; Wang, F. L.; Ondetti, M. A.; Sabo, E. F.; Cushman, D. W.; J. Biol. Chem. 1980, 255, 401. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access