
 |
 |
 |
 |
 |
Nota Técnica
| A simple and sensitive LC-MS/MS method for the determination of S-phenylmercapturic acid in human urine |
|
Andressa Priscila GomesI; Eduardo BarbosaI; Ana Laura Anibaletto dos SantosI; Lilian Feltraco LizotI; Elisa SauerII; Solange Cristina GarciaII; Rafael LindenI; Marina Venzon AntunesI; Mariele Feiffer CharaoI,*
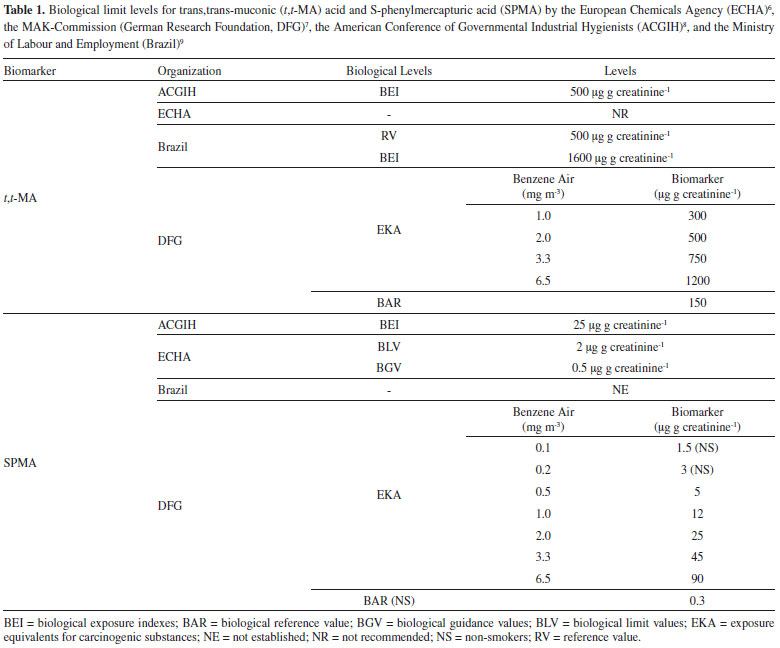
I. Instituto de Ciências da Saúde, Universidade Feevale, 93525-075 Novo Hamburgo - RS, Brasil Recebido em 15/04/2020 *e-mail: marielecharao@feevale.br Benzene is an important environmental and occupational pollutant, and recognized as human carcinogen. S-phenylmercapturic acid (SPMA) is a highly specific biomarker of exposure to benzene, applied in occupational toxicology to assess low levels of benzene exposure. The objective of this study was to develop and validate a one-step liquid extraction bioanalytical method for the quantification of SPMA in urine by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The method was applied to the evaluation of benzene exposure in an occupational setting. The assay was linear from 0.5 to 500 ng mL-1 (r>0.99), accurate (91.4-105.2%) and precise, with CV% between 4.73 and 9.96%. SPMA was stable in urine for 90 days at -20 °C. Airborne benzene concentrations, urinary levels of SPMA and t,t-MA were significantly higher in gas station workers (n=30) in comparison to outdoor workers (n=14) and individuals non-occupationally exposed to benzene (n=34). Benzene airborne levels had higher correlation with urinary SPMA (r=0.532, p<0.001) than with t,t-MA (r=0.338, p=0.04), demonstrating the applicability of this biomarker for monitoring low levels of benzene exposure. INTRODUCTION Benzene is an important environmental and occupational contaminant, which has been proven carcinogenic to humans.1 Its emission to the environment is mainly related to its high volatility from large-scale fossil fuels consumption in factories and the increasing fleet of automotive vehicles. In occupational toxicology benzene is a fairly important substance once it is present in the petrochemical industries, steel mills, gas stations, and many others. Therefore, there is an imperative necessity of constant monitoring of workers exposed to benzene.2,3 Biological monitoring is a useful tool to assess the human exposure to benzene. Exposure to benzene is assessed mainly by the measurement of the urinary levels of its metabolites, such as trans,trans-muconic acid (t,t-MA) and S-phenylmercapturic acid (SPMA).4,5 Different international agencies recommended biological limit values and reference values for t,t-MA and SPMA. The Risk Assessment Committee of the European Chemicals Agency (ECHA),6 the MAK-Commission (German Research Foundation, DFG),7 and the American Conference of Governmental Industrial Hygienists (ACGIH)8 are international committees responsible for the establishment of those limits. In Brazil, the Ministery of Labour and Employment9 established the limits recommended to occupational exposure, according to the technological Reference Value10 of 3.3 mg m-3 and 8.2 mg m-3 of benzene in air, for companies that transport or produce benzene and for steel mills, respectively. However, the biomonitoring of SPMA in Brazil is not mandatory (Table 1).
The use of SPMA over t,t-MA as a biomarker of low benzene exposure has been recommended,6,11 due to its longer half-life (~10 hours), and superior selectivity, with urinary concentrations not influenced by the consumption of food additives, as sorbic acid.2,11 Moreover, Jalai et al. reported that t,t-MA may not be a trustworthy biomarker, mainly for biomonitoring low levels of exposure.12 However, the monitoring of SPMA requires highly sensitive analytical methods, capable of quantifying trace levels. In this context, different methodologies were proposed for the biomonitoring of benzene exposure through SPMA in urine.2,13,14 The ELISA (Enzyme-Linked Immunosorbent Assay) immunoassay technique was used, but with limited sensitivity.15-17 The gas chromatography methodology provides advantages related to sensitivity, but sample preparation is laborious, with the need of derivatization of SPMA to a more volatile and thermostable product.18,19 The use of high-performance liquid chromatography with tandem mass spectrometric detection (LC-MS/MS) allows highly sensitive and specific analysis of urinary levels of SPMA, without the need of derivatization. Usually, the use of LC-MS/MS is associated with the use of solid phase extraction,20-23 which makes the procedure more laborious and expensive. Considering this, the objective of this study was to develop and validate a methodology to quantify SPMA in urine by LC-MS/MS employing a simple one-step liquid-liquid extraction and to apply this assay in samples from workers exposed to low levels of benzene, as well as to verify the correlation between personal exposure to benzene in air and the concentration of the t,t-MA and SPMA levels.
MATERIALS AND METHODS Standards, chemicals and materials SPMA was acquired from Sigma Aldrich (Saint Louis, USA), and methanolic solution of the deutered standard SPMA-d5 (1 mg mL-1) was purchased from Toronto Research Chemicals (Toronto, Canada). Methyl-tert-butyl ether (MTBE) was acquired from Honeywell (Morris Plains, USA), and acetic acid, acetonitrile and methanol from Merck (Darmstadt, Germany). A Milli-Q Reference system from Millipore was used to obtain the ultra-pure deionized water (Lane End, Reino Unido). Solutions and mobile phase Stock solution of SPMA (1 mg mL-1) was prepared from the standard by diluting with methanol. An intermediate solution of SPMA was prepared at a concentration of 50 µg mL-1 in methanol, and SPMA-d5 was prepared at a concentration of 1 µg mL-1. Working solutions were obtained from intermediate solution at the concentrations of 5000, 4000, 3000, 2000, 1000, 500, 400, 250, 100, 20, 10 and 5 ng mL-1. Calibration and quality control samples were obtained by diluting the working solutions with blank urine at 1:10 (v/v) proportion. Mobile phase was prepared daily, by adding 2.5 mL of acetic acid 95% plus ultra-pure water to complete 500 mL. The solution was filtered through 0.2 µm Sartorius celulose acetate membranes (Goettingen, Alemanha), resulting a 0.5% acetic acid water solution. Chromatographic and mass spectrometric conditions Analysis was performed using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system composed of an Ultimate 3000 XRS UHPLC system, coupled to a TSQ Quantum Access triple quadrupole mass spectrometer, operating in negative mode, both acquired from Thermo Scientific (San Jose, USA). Chromatography separation was realized with an Ascentis Express C18 (150 x 4.6 mm x 2.7 µm) column, also from Thermo Scientific, maintained at 30 °C. Mobile phase was a mixture of solvent A (0.5% acetic acid in water) and B (acetonitrile), eluted at a flow rate of 0.4 mL min-1. Initial eluent composition was 90% A maintained for 2 min, followed by a linear 3.0 min gradient to 40%, which was maintained for 1.0 min, and returning to the initial condition at 7.5 min with a 5.5 min equilibration time. The total run time was 13 min. The ionization was performed in electrospray negative ion mode (EIS), with capillary voltage 2.5 kV, sheath gas nitrogen at a flow rate of 50 arb, auxiliary gas nitrogen at flow rate of 15 arb, collision gas argon 1.5 mTorr, skimmer offset 12 V, vaporizer temperature of 240 °C, and ion transfer capillary temperature of 204 °C. The scan time was set at 0.3 seconds per transition. The following transitions were used for MRM acquisition: SPMA m/z 238 → 109.1 (quantitation) and m/z 238 → 33.3 (qualification); SPMA-d5 m/z 243 → 114.1 (quantitation) and 243 → 34.5 (qualification). Collision energies were set at 20 and 53 eV for SPMA, and 19 and 52 eV for SPMA-D5. Sample preparation procedure Sample preparation was based on a study of Wang et al., using liquid-liquid extraction.24 The extraction was performed in a 5-mL polypropylene tube with 500 µL of urine, which was added of 50 µL of internal standard (IS) (SPM-d5 1 µg mL-1), 50 µL of 95% acetic acid and 3 mL of MTBE. The tube was homogenized for 10 minutes and then centrifuged for 5 minutes at 3400 rpm. An aliquot from the supernatant (2.6 mL) was transferred to another 5 mL polypropylene tube and evaporated to dryness in a vacuum centrifuge at 45 ºC. The dried extract was reconstituted with 100 µL of mobile phase and mixed in a vortex for 30 seconds. A 25 µL aliquot was injected into a liquid chromatograph coupled with tandem mass detectors (LC-MS/MS). Linearity The linearity of the method was performed by analyzing quintuplicates of 9 calibration levels in concentrations of 0.5, 2, 10, 25, 50, 100, 200, 300, and 500 ng mL-1. The calibration curves were obtained by plotting the nominal concentration of the calibrator (axis x) and the ratio between the SPMA area and SPMA-d5 area (axis y). It was evaluated the heterocedasticity of the method with F-test (95% confidence level). The calibration curves were adjusted using linear regression using several weighted calibration models (1/x, 1/x0.5, 1/x2, 1/y, 1/y0.5, 1/y2). The calibration models were evaluated via the correlation coefficients (r) and cumulative percentage relative error (∑%ER).25 Precision and accuracy To evaluate the precision and accuracy it was performed the analysis of quality control (QC) at three concentration levels as follow: quality control at low concentration (QCL, 1 ng mL-1), quality control at medium concentration (QCM, 40 ng mL-1), and quality control at high concentration (QCH, 400 ng mL-1). The QCL, QCM, and QCH were analyzed on 5 days in triplicate. The accuracy of the method was calculated through analysis of the percentage of the nominal concentration in relation to the concentration estimated obtained by the calibration curve. Variations below than 15% for precision and values in the range of 85 to 115% for accuracy were considered acceptable criteria for method validation.26 Selectivity The urine samples obtained from six volunteers, not occupationally exposed to benzene, were analyzed as in the previously described procedure. Selectivity of the method was considered adequate in the absence of interference peaks in the monitored transitions at the same retention times of SPMA and SPMA-d5. Sensitivity Sensitivity was performed by analysing of the lowest calibrator (quality control at the lowest limit of quantification, (QCLLOQ) in triplicate on 3 different days. The maximum acceptable intra- and inter-assay coefficient of variation (CV%) was 100 ± 20% of the nominal concentration, and accuracy was between 80% and 120%.26 Extract stability at the autosampler SPMA samples at the QCL and QCH were extracted in triplicate, as described in the sample preparation procedure, and the extracts at each control level were grouped together. The extracts were maintained in the autosampler for 12 h and each pooled control was injected at time intervals of 1 h. The peak area ratios obtained in all injections of the series were compared. A decrease or increase below 15% in peak area ratios of SPMA were considered as acceptable. Stability of SPMA maintained at -20 °C QCL and QCH samples were prepared and stored at -20 °C. Each control was extracted as described above and analyzed at 1, 7, 14, 30, 60, and 90 days. The estimated concentration of QCL and QCH were calculated using calibration curves prepared on the analysis' day. Variations range of 85 - 115% of the nominal concentration was considered acceptable. Matrix effect and extraction yield Matrix effect (ME) and extraction yield (EY) were determined from the analysis of three sets of QC samples. The set A was composed of solutions of SPMA and SPMA-d5 in mobile phase, at concentrations equivalent to a 100% extraction yield of QCL, QCM, and QCH samples. The sample set B was composed of fifteen extracts obtained from pooled blank urine (from five different volunteers), recovered with mobile phase containing SPMA and SPMA-d5, also in concentrations corresponding to a 100% extraction yield of QCL, QCM, QCH. The sample set C was composed of QCL, QCM, and QCH samples, prepared and analyzed as described in section 2.4. The ratios between SPMA and IS were used to evaluate the response in each set of samples. EY and ME were calculated as ME% = [((B/A)*100)-100], and EY%= (C/B)*100, respectively.27 Assay application Urine samples were obtained from 78 volunteers, who were divided into three groups: (G1: gas station attendant, (n=30); G2: outdoor workers, using gasoline-powered lawnmowers and general gardening, or parking guards exposed to combustion of motor vehicles (n=14); and G3: individuals non- occupationally exposed to benzene, (n=34). The urine samples were collected at the end of the work shift, after at least three consecutive days of exposure and stored at -20 °C until analyzes. Individuals with chronic disease were not included in this study. Smokers were excluded of this study to avoid the known confounding factor of cigarette smoking. The individuals who accepted to take part in this research project answered a questionnaire, with questions concerning lifestyle habits, use of medication, and exposure time, among other relevant aspects. This study was approved by the Research Committee of the Feevale University (registration number 1.631.574) and an informed consent was obtained from each volunteer. Quantification of benzene in the workplace Benzene exposure was evaluated using personal passive samplers (SKC 575-002). The samplers were placed near the breath zone and removed after a day's work. After collection, 2.5 mL of dichloromethane was added to each sampler and mixed for 30 min in a shaker. The desorbed benzene was removed to a vial and analyzed using a gas chromatography-flame ionization detector (GC-FID; Varian, Middleburg, The Netherlands) and an OV-1 column (30 m x 0.32 mm x 1 µm). The chromatography conditions are as follow: the carrier gas (helium) flow rate was 4 mL min-1, the initial oven temperature was set at 35 °C, maintained for 7 min, and then increased at 10 °C min-1 up to 90 °C, which was maintained for 4 min, and after that increased at 30 °C up to 150 °C, which was maintained until the end of the analysis (18.5 min total run). The retention time was 4.6 minutes. The limits of detection (LOD) and quantification (LOQ) were 0.05 and 0.19 µg mL-1, respectively.28 Trans, trans-muconic acid determination The quantification of urinary t,t-MA was carried out using high performance liquid chromatography with diode-array detection (HPLC-DAD) (Acquity®, Waters, Milford, USA), after solid-phase extraction (SPE), according to Lee et al., with modifications.29 The limit of quantification of the method was 0.062 µg mL-1, linear range of 0.062 - 2 µg mL-1, and precision and accuracy were 1.08 - 15% and 95 - 112%, respectively. Briefly, 500 µL of urine was diluted 1:1 (v/v) with phosphate buffer pH 7.2 and centrifuged at 10,000 rpm for 10 min. The diluted sample was applied in SPE Sax cartridge (100 mg) and 10 µL of eluted sample was injected into the HPLC-DAD. Mobile phase consisted of 5 mmol L-1 triethylammonium buffer pH 3.0 (A) and acetronile (B). The gradient of mobile phase started at 98% A, which was maintained for 9 min, followed by another gradient at 88% at 12 min, returning to the initial condition at 15 min, holding until the end of the run time (20 min). Chromatography separation was performed in a Hypersil Gold® C18 column (150 × 4.6 mm, 3 µm), at 25 °C and the chromatography monitoring was realized at 264 nm. Values measured for t,t-MA were normalized by urinary creatinine, which is determined according to Jaffe´s colorimetric method. Statistical Analysis Statistical analysis was carried out using the SPSS program version 22 (Chicago, USA). For groups G1, G2 and G3, analysis of variance (ANOVA) with Bonferroni post hoc test was used to verify the difference in age, and the Kruskal-Wallis test was performed to check the difference for benzene airborne, t,t-MA and SPMA levels. Spearman correlation was utilized to verify the correlation between benzene airborne and urinary biomarkers of exposure. The value of p < 0.05 was considered statistically significant for all analysis.
RESULTS AND DISCUSSION Chromatography and sample preparation The total run time was 13 minutes, with retention times for SPMA and SPMA-d5 of 8.82 minutes and 8.85 minutes, respectively. The run time was similar to other LC-MS methods for the quantification of SPMA in urine samples, in the range of 7.0 to 14 minutes.22,30-34 Figure 1 presents typical chromatograms obtained from a blank urine, first calibration level and a sample from an exposed worker. The method was selective for SPMA, with no interference peaks in the analysis of the blank sample.
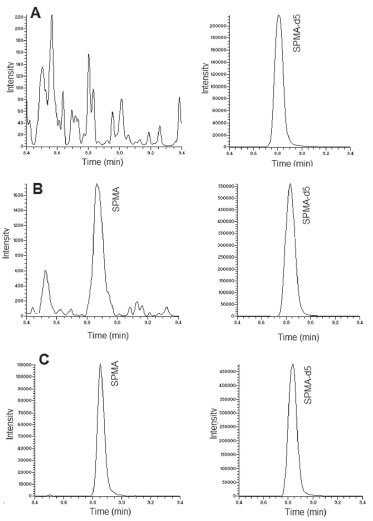 Figure 1. Typical chromatograms obtained from a blank urine (A), first calibration level of curve (5 ng/mL) containing SPMA and SPMA-d5 (B) and urine sample from an exposed worker (C). Monitored transitions were SPMA m/z 238 → 109.1 and SPMA-d5 m/z 243 → 114.1
In this study, the sample preparation consisted of a one-step liquid-liquid extraction (LLE) after acid hydrolysis. The hydrolysis process is an important step, once the pre-SPMA will be converted to SPMA after this step, reducing the variability in the results due to pH differences among the urine samples.35 Sterz et al. evaluated the complete conversion of pre-SPMA to SPMA prior to determination of SPMA by LC/MS-MS. The authors used hydrochloric acid (37%) to the acid hydrolysis step and reported a complete conversion at pH 1.36 In our study, we checked the pH values of the samples after addition of acetic acid, and pH values are approximately near 1 (data not shown). Different organic solvents were analyzed to optimize the efficiency of the extraction, including a mixture of ethyl:isopropanol acetate (3:1, v/v), ethyl acetate, and MTBE. MTBE was chosen due to a higher analytical signal post-SPMA extraction, as well as proving extracts free from impurities. This simple sample extraction is more low cost-effective in comparison to solid phase extraction (SPE), as previously reported for the urinary SPMA quantification by LC-MS/MS.14,21,22,24,34-41 Moreover, the extraction yield in this study was higher than the reported by Wang et al., which used ethyl acetate as solvent of extraction. Recently, Chang et al. developed an analytical method using LLE for biomonitoring of biomarkers of exposure for arsenic (monomethylarsonic acid and dimethylarsonic acida), benzene (SPMA) and polycyclic aromatic hydrocarbons (1-hydroxypirene) by LC-MS/MS.42 The SPMA was extracted using a solution containing chloroform/ ammonium bicarbonate, however, it was necessary multiple steps of extraction due to the chemical characteristics of the compounds, and it is not possible to perform a single one-step extraction.42 General method validation The parameters of validation of the method are presented in Table 2. The calibration data presented significant heteroscedasticity (F=51714, Fcrit=5.05). The lowest relative error (∑%RE) (1.66 x 10-15) was obtained using the weighting factor 1/x, and so it was used for all the quantitative measures. The method developed presented adequate linearity for the 0.5 to 500 ng mL-1 range, presenting a correlation coefficient (r) greater than 0.99, inclination of 0.00851, and intercept of 0.00051.
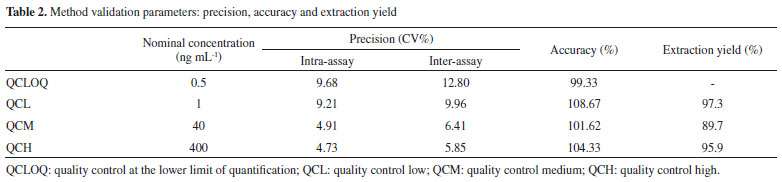
The precision and accuracy assay results were within the acceptance limits, presenting a variation in accuracy of 91.4-105.2% of the nominal SPMA concentration. The precision intra-assay presented values ranging from 4.73 to 9.21% and the inter-assay ranged between 5.85 and 9.96%, in accordance with the acceptance criteria as well. The quality control at the lower limit of quantification (QCLOQ) presented 96.8% accuracy, 9.68% intra-assay precision, and 12.80% inter-assay precision. The LOQ of the present study (0.5 ng mL-1) presented an adequate sensibility for the biomonitoring of workers occupationally exposed to low levels of benzene. The findings for the extraction yield were an advantage of the method, with recoveries in the range of 89.7% to 97.3%, in contrast with other more complex and onerous preparation techniques, such as the use of SPE.31,43 In addition, a higher extraction efficiency was observed when compared to the study of Wang et al., which also used the one-step LLE technique with ethyl acetate, obtaining recovery within the 60% range, thus reinforcing the use of MTBE as an effective organic extraction solvent.24 Wang et al. reported the use a multiple step LLE, however, there is no information about the extraction yield obtained in the study.41 The processed extracts were stable for 12 hours in the automatic sampler, with variations in the peak area ratio for the QCL ranging from +10.3 to -6.1% and for the QCH ranging from +6.9 to +1.3%, respectively. The QCL and QCH samples were stable in 3 freeze-thaw cycles, with variations from +8.02 to +10.9% for the QCL and from -3.9 to 3.26% for the QCH. In addition, the samples were stable for 90 days when stored at -20 °C (Table 3). Thus, these results indicate that reliable SPMA measurements can be carried out for 90 days in the conditions evaluated, with adequate time for processing the sample.
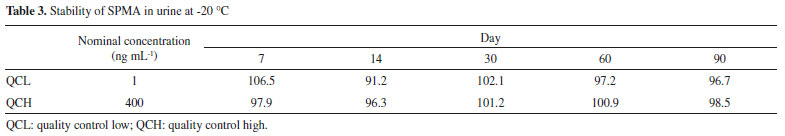
The stability results are consistent with other previous studies. Most of the studies indicate that SPMA in urine samples are stable in storage conditions at temperatures between -20 ºC and -80 ºC, for a period of time between 30 and 90 days.43,44 In addition, the study conducted by Sabatini et al. showed the stability up to 2 months at -20 ºC, and after 3 freeze-thaw cycles.22 SPMA stability at -18 ºC has been shown for up to six months.44 The matrix effect (ME) evaluated at low, medium and high concentrations was compensated with the use of the deuterated internal standard, varying from -7.8% to 3.5%. The use IS is essential to compensate random and systematic errors, from the matrix effect and sample preparation, improving the precision and accuracy of the measurements. In the experiments conducted by Zhang et al., in which a LLE was used, and Sabatini et al., high ion suppression was observed.22,37 Moreover, Chang et al. observed significant matrix effect for SPMA, with a CV% of 20%.42 Method application Senventy-eight subjects were divided into three groups (G1: gas station attendant, n=30; G2: outdoor workers, n=14; and G3: individuals non-occupationally exposed to benzene, n=34). The demographic characteristics of the study groups are presented in Table 4. The age did not present any significant difference (p=0.347) among the groups. Regarding the period in the job for the exposed groups, in group 1, this varied between 7 and 37 months and in group 2 it ranged between 8 and 94 months. There was no significant difference among the groups in relation to alcohol consumption (p=0.546).
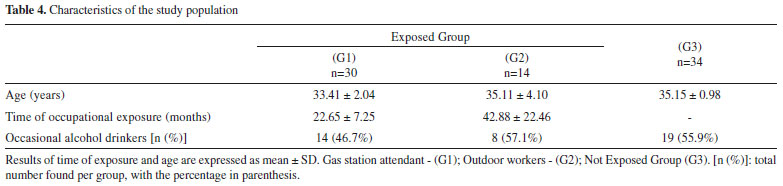
Groups G1 and G2 were exposed to ambient benzene levels significantly higher than group G3 (Table 4, p<0.001). The median benzene levels were below the limit preconized by NIOSH45 (0.3 mg m-3). Only five exposed workers from group G1 presented levels higher than 0.3 mg m-3. Besides, these median levels are far below the recommendation of others agencies, ACGIH8 and OSHA46 (1.6 and 3.3 mg m-3, respectively), and the limits preconized in Brazil (1.6 mg m-3).10 This characterizes the low levels of benzene that workers are exposed during their working day, and they are in accordance with other studies.4,12,47-52 The median t,t-MA levels at the studied groups were below the biological limits.7,8,9 However, the urinary t,t-MA concentrations were significantly higher in the G1 group in comparison to G2 and G3 groups (p<0.002, Table 5). In addition, 33.3% of the gas station attendants (G1 group), had urinary t,t-MA above the ACGIH recommendation.8 Our results are in agreement with previous studies, with similar levels of t,t-MA in groups of gas station attendants.49,52 Jalai et al. reported that t,t-MA presented a great linear correlation (r = 0.904) with high benzene levels in air, above 1.5 ppm (4.5 mg m-3).12 However, for exposure levels below 0.5 ppm (1.6 mg m-3), this correlation was not observed, indicating that at low levels of benzene exposure, t,t-MA cannot be used as a reliable and selective biomarker.12
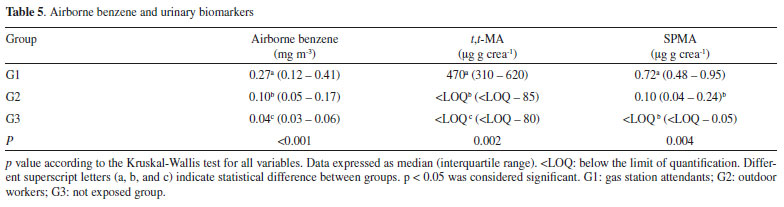
According to the observed results, 72.4% of the samples from group G1 presented quantifiable SPMA levels, while in G2, 34.4% of the samples had quantifiable SPMA values. In addition, in the group of workers non-occupationally exposed to benzene (G3), 91.2% presented values lower than the quantification limit of the method (Table 5). SPMA levels were significantly different among the groups (Table 5, p=0.004). G1 group presented significantly higher levels of SPMA than groups G2 and G3, with median values of 0.72 µg g creatinine-1, below the levels stablished by DFG7 (1.5 µg g creatinine-1, at levels of 0.1 mg m-3 of benzene in air), by ECHA6 (2.0 µg g creatinine-1), and far below the levels preconized by ACGIH8 for this biomarker of exposure (25 µg g-1 creatinine-1). Three of the exposed subjects presented levels of SPMA higher than the levels recommended by DFG7. Furthermore, recent study with coke oven workers found median SPMA levels of 0.31 (0.04 - 2.98) µg g creatinine-1, comparable with the results obtained in this study.4 It was observed a better correlation between benzene levels in air and SPMA (r = 0.532, p < 0.001) than benzene in air and t,t-MA (r = 0.338, p = 0.004), demonstrating that SPMA has a great applicability for monitoring workers exposed to low levels of benzene in the workplace (Figure 2). A study carried out in petrochemical industry operators (non-smoking workers) exposed to low levels of benzene (range < 0.003 - 0.924 mg m-3) found similar results to ours.53
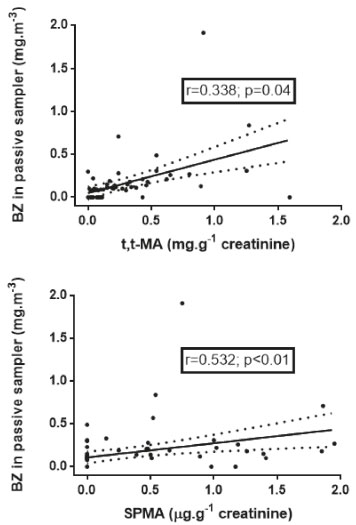 Figure 2. Correlation between personal benzene exposure and the urinary biomarkers (t,t-MA and SPMA)
CONCLUSIONS The validated method presented satisfactory selectivity, precision and accuracy, with adequate sensitivity to measure low levels of SPMA in urine samples. The sample treatment consisted of an efficient one-step liquid-liquid extraction, conferring a cost-effective sample analysis and easy execution, making this an applicable biomarker in the laboratory routine. Regarding the evaluation of environmental monitoring and biomarkers of exposure, our results indicated that workers were occupationally exposed to low levels of airborne benzene. Moreover, SPMA seems to be a more specific and trustworthy biomarker when compared to t,t-MA, demonstrated by the better correlation observed in this study for SPMA and benzene in air.
REFERENCES 1. International Agency for Research on Cancer; IARC; Monogr. Eval. Carcinog. Risks Hum. 2018, 120, 1. 2. Coutrim, M. X. ; Carvalho, L. R. F.; Arcuri, A. S.; Quim. Nova 2000, 23, 653. 3. Bulcão, R. P.; Maria, L. S.; Charão, M.; Moro, A.; Roehrs, M.; Garcia, S. C.; Limberger, R.; Quim. Nova 2008, 31, 1343. 4. Frigero, G.; Campo, L.; Mercadante, R.; Miel˙zy´nska-Svach, D.; Pavanello, S.; Fustinoni, S.; Int. J. Environ. Res. Public Health 2020, 17, 1801. 5. Arnold, S. M.; Angerer, J.; Boogaard, P. J.; Hughes, M. F.; O´Lone, R. B.; Robison, S. H.; Schnatter, A. R.; Crit. Rev. Toxicol. 2013, 43, 119. 6. https://echa.europa.eu/documents/10162/13641/benzene_opinion_en.pdf/4fec9aac-9ed5-2aae-7b70-5226705358c7, acessada em setembro 2020. 7. https://onlinelibrary.wiley.com/doi/pdf/10.1002/9783527818402, acessada em setembro 2020. 8. American Conference of Governmental Industrial Hygienists, ACGIH; 2017 TLVs and BEIs, ACGIH Signature Publications, 2017. 9. Brasil, Ministério do Trabalho e Emprego. Portaria. N°. 34, de 20 de dezembro de 2001, disponível em https://www.legisweb.com.br/legislacao/?id=182693, acessada em setembro 2020. 10. Brasil, Ministério do Trabalho. Portaria n°14, De 20 De Dezembro de 1995, disponível em http://portal.mte.gov.br/data/files/FF8080812C12AA70012C12C576E3005C/p_19951220_14.pdf, acessada em setembro 2020. 11. Lv, B.; Song, S.; Zang, Z.; Mei, Y.; Ye, F.; JOEM 2014, 56, 319. 12. Jalai, A.; Ramezani, Z.; Ebrahim, K.; Saf. Health Work 2017, 8, 220. 13. Weisel, C. P.; Chem. Biol. Interact. 2010, 184, 58. 14. Mathias, P. I.; B'hymer, C.; J. Chromatogr. B 2014, 964, 136. 15. Lohse, C.; Jaeger, L. L.; Staimer, N.; Sanborn, J. R.; Jones, A. D.; Langó, J.; Gee, S. J.; Hammock, B. D.; J. Agric. Food Chem. 2000, 48, 5913. 16. Carrieri, M.; Bonfiglio, E.; Scapellato, M. L.; Maccà, I.; Tranfo, G.; Faranda, P.; Paci, E.; Bartolucci, G. B.; Toxicol. Letters 2006, 162, 146. 17. Seow, W. J.; Pesatori, A. C.; Dimont, E.; Farmer, P. B.; Albetti, B.; Ettinger, A. S.; Bolatti, V.; Bolognesi, C.; Panev, T. I.; Georgieva, T.; Merlo D. F.; Bertazzi, P. A.; Baccarelli, A. A.; PLoS One 2012, 7, 12. 18. Angerer, J.; Schildbach, M.; Krämer, A.; J. Anal. Toxicol. 1998, 22, 211. 19. Waidyanatha, S.; Rothman, N.; Li, G.; Smith, M. T.; Rappaport, S. M.; Anal. Biochem. 2004, 327, 184. 20. Manini, P.; De Palma, G.; Andreoli, R.; Poli, D.; Mozzoni, P.; Folesani, G.; Mutti, A.; Apostoli, P.; Toxicol. Letters 2006, 167, 142. 21. Barbieri, A.; Violante, F. S.; Sabatini, L.; Graziosi, F.; Mattioli, S.; Chemosphere 2008, 74, 64. 22. Sabatini, L.; Barbieri, A.; Indiveri, P.; Mattioli, S.; Violante, F. S.; J. Chromatogr. B 2008, 863, 115. 23. Manini, P.; De Palma, G.; Andreoli, R.; Poli, D.; Petyx, M.; Corradi, M.; Mutti, A.; Apostoli, P.; Toxicol. Letters 2008, 181, 25. 24. Wang, Z.; Zhao, B.; Liu, X.; Zheng, Y.; Wang, J.; Zhang, R.; Abliz, Z.; Anal Methods 2013, 5, 6081. 25. Almeida, A. M.; Castel-Branco, M. M.; Falcão, A. C.; J. Chromatogr. B 2002, 774, 215. 26. Food and Drug Administration, FDA; Guidance for Industry: Bioanalytical method validation, 2018, 4. 27. Matuszewski, B. K.; Constanzer, M. L.; Chavez-eng, C. M.; Anal. Chem. 2003, 75, 3019. 28. Occupational Safety and health Agency, OSHA; Method 1501 Hidrocarbons aromatic, 2003. 29. Lee, B. L.; Ong, H. Y.; Ong, Y. B.; Ong, C. N.; Anal. Chem. 2005, 818, 277. 30. Pieri, M.; Miraglia, N.; Acampora, A.; Genovese G.; Soleo, L.; Sannolo N.; J. Chromatogr. B 2003, 795, 347. 31. Lin, L. C.; Chiung, Y. M.; Shih, J. F.; Shih, T. S.; Liao, P. C.; Anal. Chim. Acta. 2006, 555, 34. 32. Schettgen, T.; Musiol, A.; Alt, A.; Kraus, T.; J. Chromatogr. B 2008, 863, 283. 33. Ding, Y. S.; Blount, B. C.; Valentin-Blasini, L.; Applewhite, H. S.; Xia, Y.; Watson, C. H.; Ashley, D. L.; Chem. Res. Toxicol. 2009, 22, 1018. 34. Tranfo, G.; Bartolucci, G. B.; Pigini D.; Paci, E.; Scapellato, M. L.; Doria D.; Manno, M.; Carrieri M.; J. Chromatogr. B 2010, 878, 2529. 35. Paci, E.; Pigini, D.; Cialdella A. M.; Faranda P.; Tranfo, G.; Biomarkers 2007, 12, 111. 36. Sterz, K.; Kohler, D.; Schettgen, T.; Scherer, G.; J. Chromatogr. B 2010, 878, 2502. 37. Zhang, X.; Xiong, W.; Shi, L.; Hou, H.; Hu, Q.; J. Chromatogr. B 2014, 967, 102. 38. Lovreglio, P.; Barbieri, A.; Carrieri, M.; Sabatini, L.; Fracasso, M. E.; Doria D.; Drago, I.; Basso, A.; D'Errico, M. N.; Bartolucci, G. B.; Violante, F. S.; Soleo L.; Int. Arch. Occup. Environ. Health 2010, 83, 341. 39. B´Hymer, C.; J. Chromatogr. Sci. 2011,49, 547. 40. Mansi, A.; Bruni, R.; Capone, P.; Paci, E.; Pigini, D.; Simeoni, C.; Gnerre, R.; Papacchini, M.; Tranfo G.; Toxicol. Lett. 2012, 213, 57-62. 41. Carbonari, D.; Proietto, A.; Fioretti, M.; Tranfo, G.; Paci, E.; Papacchini, M.; Mansi, A.; Toxicol. Lett. 2014, 231, 205. 42. Chang, F. C.; Chen, C. Y.; Lin, C. Y.; Sheen, J. F.; Talanta 2019, 198, 137. 43. Melikian, A. A.; O'Connor, R.; Prahalad, A. K.; Hu, P.; Li, H.; Kagan, M.; Thompson S.; Carcinogenesis 1999, 20, 719. 44. Alwis, K.U.; Blount, B. C.; Britt, A. S.; Patel, D.; Ashley, D. L.; Anal. Chim. Acta 2012, J.750, 152. 45. National Institute of Occupational and safety Health, NIOSH; Pocket Guide to Chemical hazards - Benzene, 2007. 46. Occupational Safety and health Agency, OSHA; Occupational Exposure to Benzene Final Rule, Standards-29 CFR Benzene, 2014, 1910. 47. Moro, A. M.; Charão, M. F.; Brucker, N.; Durgante, J.; Baierle, M.; Bubols, G.; Goethel, G.; Fracasso, R.; Nascimento, S.; Bulcão, R.; Gauer, B.; Barth, A.; Bochi, G.; Moresco, R.; Gioda, A.; Salvador, M.; Farsky, S.; Garcia, S. C.; Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2013, 754, 63. 48. Moro, A. M.; Brucker, N.; Charão, M. F.; Sauer, E.; Freitas, F.; Durgante, J.; Bubols, G.; Campanharo, S.; Linden, R.; Souza, A. P.; Bonorino, C.; Moresco, R.; Pilger, D.; Gioda, A.; Farsky, S.; Duschl, A.; Garcia, S. C.; Environ Res. 2015, 137, 349. 49. Moro, A. M.; Brucker, N.; Charão, M. F.; Baierle, M.; Sauer, E.; Goethel, G.; Barth, A.; Nascimento, S. N.; Gauer, B.; Durgante, J.; Amaral, B. S.; Neto, F. R.; Gioda, A.; Garcia, S. C.; Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2017, 813, 1. 50. Li, J.; Zhang, X.; He, Z.; Sun, Q.; Qin, F.; Huang, Z.; Zhang, X.; Sun, X.; Liu, L.; Chen, L.; Gao. C.; Wang, S.; Wang, F.; Li, D.; Zeng, X.; Deng, Q.; Wang, Q.; Zhang, B.; Tang, H.; Chen, W.; Xiao, Y.; Biomarkers 2017, 22, 470. 51. Xiong, F.; Li, Q.; Zhou, B.; Huang, J.; Liang, G.; Zhang, L.; Ma, S.; Qing, L.; Liang, L.; Su, J.; Peng, X.; Li, Q.; Zou, Y.; Research and Public Health 2016, 13, 1212. 52. Sauer, E.; Gauer, B.; Nascimento, S.; Nardi, J.; Goethel, G.; Costa, B.; Correia, D.; Matte, U.; Charão, M.; Arbo, M.; Duschl, A.; Moro, A.; Garcia, S. C.; Environ. Res. 2018, 166, 91. 53. Carrieri, M.; Tranfo, G.; Pigini, D.; Paci, E.; Salamon, F.; Scapellato, M. L.; Fracasso, M. E.; Manno, M.; Bartolucci, G. B.; Toxicol. Lett. 2010, 192, 17. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access