
 |
 |
 |
 |
 |
Revisão
|
|
| Desenvolvimento nos métodos de glicosilação: uma chave para acessar suas aplicações na síntese de moléculas bioativas Development in glycosylation methods: a key to access its applications in the synthesis of bioactive molecules |
|
Pedro R. de Souza Neto; Bruna M. Guimarães; Jucleiton J. R. de Freitas; Ronaldo N. Oliveira#; João R. de Freitas Filho*
Departamento de Química, Universidade Federal Rural de Pernambuco, 52171-900 Recife - PE, Brasil Recebido em 22/08/2020 *e-mail: joaoveronice@yahoo.com.br Glycosylation reaction is an important class of reactions in organic chemistry, and the development of the method contributes to the synthesis of many biologically active compounds containing various glycoside bonds. Is arguably the most important, albeit challenging, reaction in the field of carbohydrate chemistry. Examples of the products of glycosylation reactions are glycoproteins, glycolipids, glycosaminoglycans, oligosaccharides, and polysaccharides. Glycosylation types are classified according to the identity of the atom which binds the carbohydrate chain, i.e. C-linked, N-linked, O-linked or S-linked. In this short review, recent reports of the main glycosylation methods, basic mechanisms, factors influencing the stereoselectivity and their applications in the synthesis of bioactive molecules are described. CONSIDERAÇÕES INICIAIS Os carboidratos são a classe de biomoléculas mais diversificada e abundante da Terra e desempenham papéis importantes em todas as fases da vida, entre outros no reconhecimento de células e ativação do sistema imunológico.1,2 A obtenção de carboidratos de fontes naturais, se disponível, é um processo tedioso e caro devido à mistura complexa de compostos semelhantes presentes. Por outro lado, a síntese orgânica é um dos fornecedores mais importantes de carboidratos e glicoconjugados bem definidos, em quantidades suficientes e livres de contaminantes que podem interferir ou prejudicar a atividade. No entanto, a obtenção de oligossacarídeos continua sendo uma tarefa complexa e o controle total da estereosseletividade na reação de glicosilação ainda é um grande desafio na química de carboidratos sintéticos. Em uma reação de glicosilação, um doador de glicosila é ativado para fornecer uma espécie eletrofílica capaz reagir com uma molécula aceptora nucleofílica (Figura 1), que pode ser uma molécula simples ou estruturalmente mais complexas, tais como uma proteína.3-8 Em uma reação de glicosilação química, o doador de glicosila possui grupos protetores para desativar temporariamente os grupos hidroxila dos carboidratos reativos e evitar reações colaterais, e um grupo de saída na posição anomérica (C-1).9,10 Esse grupo de saída pode ser ativado por um promotor para torná-lo suficientemente reativo para ser substituído pelo aceptor nucleofílico, que também possui grupos de proteção. As etapas sintéticas adicionais para construir blocos de construção protegidos e trocá-los ou removê-los podem, por si só, ser uma tarefa monumental, mas muitas vezes inevitável devido às similaridades inerentes entre os grupos hidroxila dos carboidratos.11-15
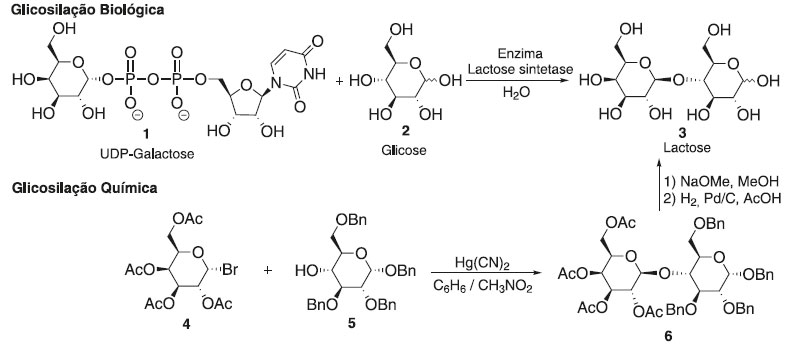 Figura 1. Descrição de uma reação de glicosilação através de uma rota biossintética e uma rota química
As reações de glicosilação podem ser elucidadas pelo tipo de arranjo espacial de seus átomos, diferindo na posição da ligação glicosídica no carbono 1 (C-1) em relação à configuração da cadeia lateral do carbono 5 (C-5), sendo conhecidos por anômeros alfa (α) ou beta (β), sendo a sua formação favorecida por fatores termodinâmicos, cinéticos e estereoeletrônicos (Figura 2).
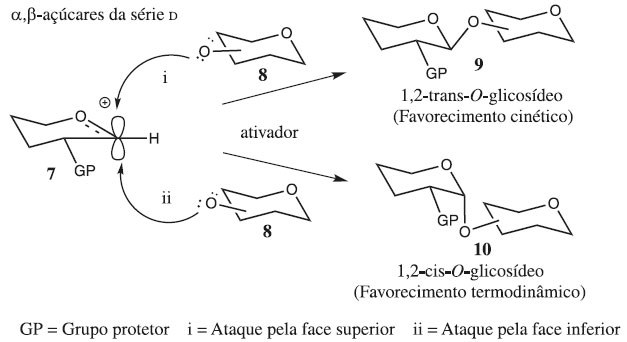 Figura 2. Estereosseletividade sem assistência anquimérica via mecanismo SN1
O favorecimento de estereoisômeros está relacionado a diversos fatores como o orbitais HOMO/LUMO, efeito do solvente, temperatura, pressão, tipo de catalisador, grupos protetores (assistência anquimérica), o grupo de saída na posição C-1, concentração, dentre outros fatores. A glicosilação sucede pela substituição nucleofílica podendo ocorrer de maneira unimolecular (SN1) ou bimolecular (SN2) dependendo dos fatores citados anteriormente.16 A pesquisa e seleção dos artigos e textos relacionados aos métodos de O-glicosilação e as suas aplicações na síntese de moléculas bioativas focalizou-se no período dos anos de 2010 a 2020 e nas publicações indexadas na base de dados Web of Science. Os dados obtidos sofreram uma análise rigorosa para a construção deste trabalho que facilitou a obtenção de textos de qualidade e que visa o desenvolvimento científico, obtendo após seleção 2137 artigos. Os resultados estão ilustrados na Figura 3.
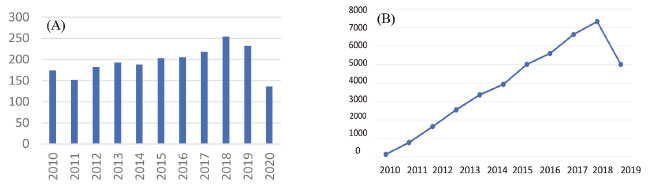 Figura 3. (A) Número de artigos publicados e (B) número de citações sobre métodos de glicosilação entre 2010-2020
Mediante o exposto, esta revisão descreve os principais métodos de glicosilação, mecanismos básicos, fatores que influenciam a estereosselectividade e suas aplicações na síntese de moléculas bioativas.
ABORDAGENS DOS ASPECTOS HISTÓRICOS DOS PRINCIPAIS MÉTODOS DE GLICOSILAÇÃO Definição de glicosilação e características gerais A reação de glicosilação é um processo que ocorre a partir de um doador de glicosila e um aceptor (em geral, ROH), podendo sofrer ativação por um catalisador para tornar possível a formação de uma ligação glicosídica. Os compostos glicosídicos, vastamente encontrados na natureza são baseados na ligação glicosídeo-aglicona ou glicosídeo-glicona, do qual a aglicona pode ser um composto não sacarídico que atua como nucleófilo conectado à porção, açúcar por intermédio de uma ligação glicosídica, do tipo: O-, C-, N- e S-glicosídeos.16 A glicosilação é sem dúvida a reação mais importante, embora desafiadora, no campo da química de carboidratos. A estereosseletividade possui dependência devido a tais fatores: do catalisador utilizado, da densidade eletrônica na região anomérica; do grupo de saída e de grupos anquiméricos; o meio reacional relacionado a solvente, temperatura, pressão e volume; o mecanismo reacional, ocorrendo por substituição nucleofílica unimolecular (SN1) ou bimolecular (SN2), dependendo da estabilidade termodinâmica e cinética do composto; definindo assim a configuração anomérica (Esquema 1).17
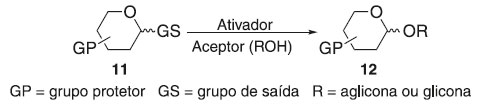 Esquema 1. Reação geral de glicosilação
O estudo desse tipo de reação se torna cada vez mais importante e desafiador, uma vez que as reações de glicosilação geralmente seguem um mecanismo de deslocamento SN1, onde a orientação do grupo de saída no centro anomérico é de pouca importância. No entanto, ocasionalmente as reações de glicosilação prosseguem por meio de um mecanismo semelhante a SN2 com inversão da configuração anomérica.17 Principais métodos de glicosilação As principais metodologias desenvolvidas para reação de glicosilação conduziram a grandes avanços na química orgânica sintética de carboidrato. A facilidade de ativação do grupo de saída, modificação do meio reacional, excelentes rendimentos com régio- e estereosseletividade, agregou valor na obtenção de diversas novas biomoléculas. O Esquema 2 sumariza os principais métodos de glicosilação.
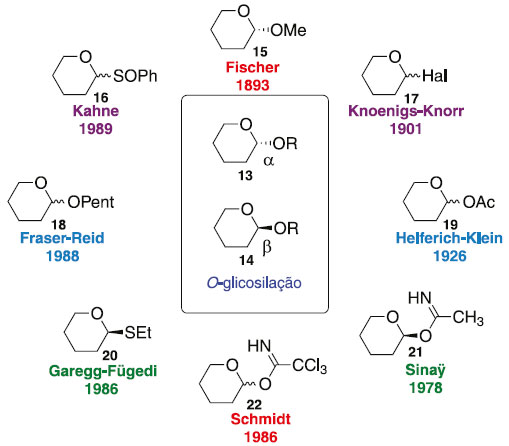 Esquema 2. Diferentes métodos de glicosilação
Em seguida, descrevem-se os principais métodos de glicosilação, conforme mostrado no Esquema 2, para formação de ligações glicosídicas e estratégias para obter determinadas classes de compostos, de importância na química dos carboidratos. Método de Fischer Em 1893, Emil Fischer realizou a síntese de O-glicosídeos, partindo da D-glicose em presença de álcool metílico com adição de HCl até atingir a saturação que em pouco tempo foi capaz de reduzir totalmente a um sólido branco cristalino de α-D-glicopiranosídeo com formação de α- e β-glicopiranosídeos, tendo majoritariamente o α-anômero. Iniciando a reação com o meio catalítico de 0,7% de HCl e com diminuição na temperatura para 20 ºC, obtendo uma mistura dos isômeros α- e β-glicofuranosídeos de metila 26, aumentando a concentração de HCl para 4% em refluxo, originou uma mistura dos glicopiranosídeos 24 e 25 (Esquema 3).18,19
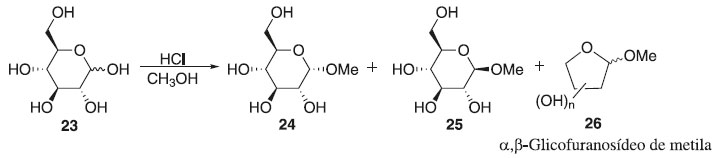 Esquema 3. Método de glicosilação proposto por Fischer
Método de Koenigs-Knorr Em 1901, Koenigs e Knorr modificaram a metodologia de O-glicosilação, utilizando brometo de 2,3,4,6-tetra-O-acetil-α-D-glicopiranosila 27, em presença de um álcool, sendo ativado pelo excesso de carbonato de prata em temperatura ambiente, obteve exclusivamente o β-O-glicosídeo 28, notando-se a quimiosseletividade na substituição nucleofílica, sendo diastereosseletivo na substituição do carbono anomérico. Observou-se também ser possível o emprego catalítico de sais de metais pesados ou ácido de Lewis em baixas temperaturas, obtendo essencialmente β-seletividade anomérica na preparação de O-glicopiranosíedos (Esquema 4). 20,21
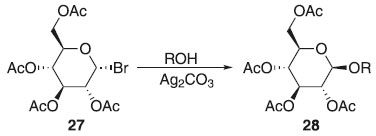 Esquema 4. Método de glicosilação proposto por Koenigs-Knorr
Método de Helferich Helferich e Klein introduziram em 1926 uma eficiente glicosilação a partir do acetato de 2,3,4,6-tetra-O-acetil-β-D-glicopiranosila 29 catalisada por sais de mercúrio II como cianeto de mercúrio II e brometo de mercúrio II, obtendo o composto 28 em rendimentos maiores que 70%, demonstrando ser um método vantajoso e eficiente na síntese de β-O-glicosídeos de alquila e arila pela saída fácil do grupo acetoxila anomérico (Esquema 5).19,22
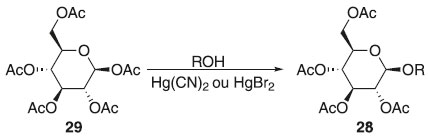 Esquema 5. Método de glicosilação proposto por Helferich
Método de Sinaÿ Em 1978, Sinaÿ publicou a obtenção exclusiva do β-O-imidato de glicosila 30 obtido a partir do brometo de 2,3,4,6-tetra-O-acetil-α-D-glicopiranosila 27 com N-metil-acetamida ativado por óxido de prata e N,N-diisopropiletilamina (DIPEA) com o emprego de um ácido de Brønsted - Lowry na presença de um álcool como acetptor, obtendo em alta seletividade na ligação β-O-imidato de glicosila 30 e sem seguida o composto 31 (Esquema 6). Os estudos revelaram que em presença de uma base orgânica ocorre a formação do α-glicosídeo devido ao produto ser termodinamicamente estável a face β-imidato, no entanto, ao utilizarmos uma base inorgânica forte, obtém essencialmente o β-glicosídeo com interação ao α-tricloroacetamidato, proporcionando excelente estereosseletividade no meio reacional.23
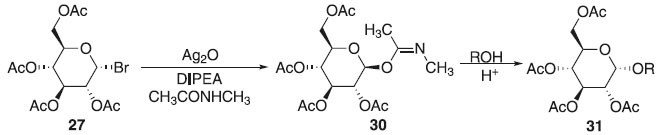 Esquema 6. Método de glicosilação proposto por Sinaÿ
Método de Schmidt Schmidt em 1986 discutiu sobre a obtenção dos diastereoisômeros 34, a partir do doador brometo de 2,3,4,6 tetra-O-benzil-D-glicopiranosila 32 pela ativação com uma base forte di-isopropilamideto de lítio (LDA) e em presença de tricloroacetonitrila à temperatura baixa, promovendo o equilíbrio químico de proporção (1:1) entre os anômeros α- e β-tricloroacetimidato de glicosila 33, sendo a posição alfa termodinamicamente mais estável dependendo da temperatura, pressão e fatores do solvente e a face beta cineticamente mais favorável com o aumento da temperatura. A mistura na presença de um álcool com catálise por um ácido fraco, provoca a saída do grupo imidato de 33, fornecendo o composto 34 como diastereosseletivo (α) e (β) (Esquema 7).24,25
 Esquema 7. Método de glicosilação proposto por Schmidt
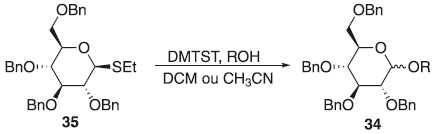
Método de Garegg-Fügedi Fügedi et al. descreveram em 1986 a utilização de derivados do tipo do β-tio-glicopiranosídeo 35 como doadores em presença de um ácido de Lewis como (DMTST) em diclorometano ou acetonitrila, observando a falta de estereosseletividade pela formação da mistura 1,2-trans e 1,2-cis-O-glicosídeos, porém o C-2 protegido com um grupo, acila acarreta a formação exclusiva da ligação 1,2-trans-O-glicosídica, estruturando ser um método β-seletivo. O uso do brometo de 2,3,4,6-O-benzil-α-D-glicopiranosila em excesso de brometo de tetrabutilamônio com DMTST, promoveu apenas a ligação cis-glicosídica, demonstrando α-seletividade (Esquema 8).26
 Esquema 8. Método de glicosilação proposto por Garegg-Fügedi
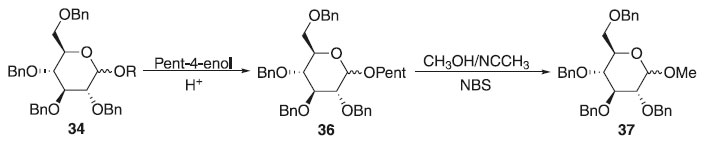
Método de Fraser-Reid Em 1988, Fraser-Reid fez exploração do método de glicosilação de Fischer a partir do 2,3,4,6-tetra-O-benzil-D-glicose 34 em meio ácido com pent-4-enol produzindo essencialmente o α e β-D-glicopiranosídeo de pent-4-enila. O O-glicosídeo de pent-4-enila foi submetido a presença de metanol com acetonitrila e N-bromosuccinimida (NBS) por 3 horas observando a formação da mistura diastereoisomérica de proporção 1:3 de alfa e beta, respectivamente do 2,3,4,6-tetra-O-benzil-D-glicopiranosídeo de metila 37 com 87% de rendimento (Esquema 9); utilizando o perclorato de dicolidina iodada como catalisador, notou-se a sua eficiência na diminuição do tempo reacional passando para 30 minutos e com 75% de rendimento, mantendo a proporção isomérica anterior. Mudou-se também o solvente para diclorometano em perclorato de dicolidina iodada, aumentando para 85% de rendimento com proporções de (1.2:1) da ligação α- e β-glicosídica. Fez também manipulação de uma mistura (1:4) de DCM e Et2O, durando 24 horas a reação para obter 75% de rendimento da mistura (3:1) dos α- e β-O-glicosídeos. Apresentando doadores glicosilas vantajosos na preparação de O-glicosídeos, provenientes da regiosseletividade obtida com o grupo de saída na posição anomérica.27
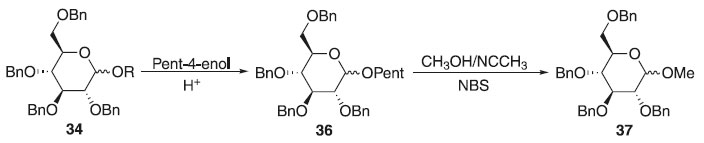 Esquema 9. Método de glicosilação proposto por Fraser-Reid
Método de Kahne Em 1989, os estudos realizados por Kahne descreveram a glicosição do doador fenil sulfóxido de 2,3,4,6-tetra-O-pivaloil-D-glicopiranosila 38 em presença do anidrido trifluorometanossulfônico (Tf2O) a -78 ºC e de um nucleófilo alcoólico, como aceptor, em meio básico utilizando o 2,6-di-tert-butil-4-metilpiridina, então iniciou o processo de aquecimento até -24 ºC, atingindo o final da reação e obtendo 83% de rendimento o composto 39. Em relação ao fenil sulfóxido de 2,3,4,6-tetra-O-benzil-D-glicopiranosila em tolueno originou uma mistura diastereoisomérica de proporção (27:1) de α- e β-glicosídeos, respectivamente e com rendimento de 86%. Fez substituição do solvente por diclorometano ou propanonitrila obtendo majoritarimente a ligação β-glicosídica, apontando ser uma rota sintética eficaz (Esquema 10).28
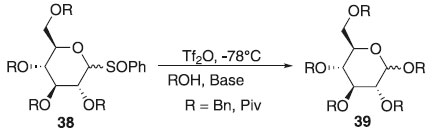 Esquema 10. Método de glicosilação proposto por Kahne
Diante disso, é perceptível mesmo pelas limitações no âmbito científico da época, uma nova abordagem na síntese de carboidratos, mas especificamente nas reações de glicosilação, portanto, é possível analisar o desenvolvimento dos novos protocolos sintéticos empregados atualmente.
MECANISMOS BÁSICOS DA REAÇÃO DE GLICOSILAÇÃO Não existe um mecanismo de reação geral único para descrever todas as reações de glicosilação. Por mais de um século, e especialmente nos últimos 50 anos,29 uma série de novos protocolos de reação de glicosilação foram desenvolvidos e cada um deles requer diferentes reagentes e condições reacionais. O entendimento mecanicista atual, que já foi formulado e proposto nas décadas de 1960 e 1970,30,31 centra-se na estereoquímica de uma reação de glicosilação através de duas vias mecanisticas que pode ser resultado da união entre um doador e aceptor de glicosila (Esquema 11). A ativação de um doador de glicosila leva a uma série de intermediários reativos. As espécies covalentes 46 e 47 com configuração α e β podem ser formadas e estas estão em equilíbrio com os íons 44 e 45, menos estáveis e mais reativas. Por outro lado, os íons 44 (β-CIP) e 45 (α-CIP) podem estar separados de pares de íons separados por solvente (SSIPs) 42 e 43, respecttivamente. Esses intermediários reativos podem ser atacados por um nucleófilo seguindo um mecanismo de reação do tipo SN1 e SN2. As espécies covalentes 46 e 47 são deslocadas em um mecanismo de reação do tipo SN2, enquanto os intermediários 44 e 45 estão envolvidos em uma reação do SN1.
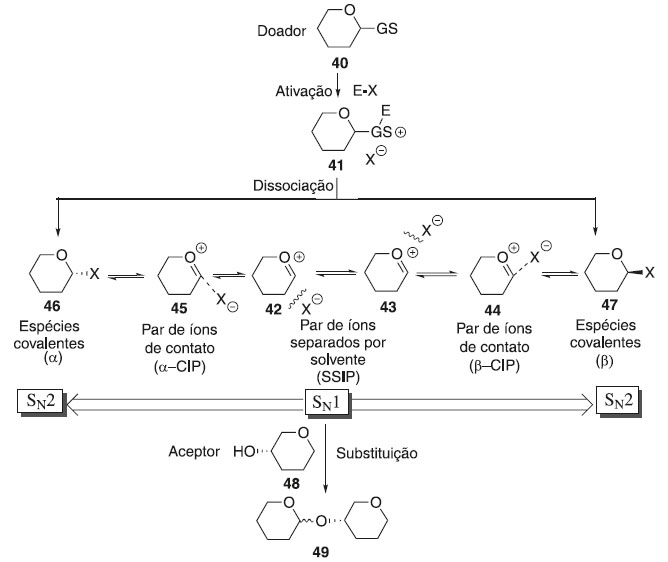 Esquema 11. Mecanismo geral da reação de glicosilação
A substituição pode ocorrer em qualquer um dos estados intermediários, com ligações parcialmente dissociadas ou em uma geometria geralmente não representada em um esquema de reação.32 Embora os doadores possam parecer semelhantes, seu comportamento nas reações de glicosilação pode ser muito diferente. A viabilidade de uma reação é determinada por muitos fatores, entre os quais o potencial de ionização no carbono anomérico, a geometria do doador quando ele sofre glicosilação e a nucleofilicidade do aceptor. De modo geral, a reação de glicosilação é proveniente da interação de um doador de glicosila com um ativador/catalisador (E-Nu), resultando na formação de uma espécie eletropositiva pela dissociação do grupo de saída e então ocorre o ataque do aceptor glicosídico. A classe dos grupos de saída em seu crescente desenvolvimento proporciona o estudo e elucidação reacional dos intermediários e produtos obtidos pelos diversos caminhos de ativação existentes na glicosilação. A essa investigação percebeu o comportamento de vários grupos doadores ao tipo catalítico, podendo ser classificada como:
Além disso, convém destacar que nos últimos vinte anos houve uma evolução nos estudos sobre os mecanismos das reações de glicosilação, há evidências que à intermediação de doadores de glicosila covalentes ativados desempenham um papel importante na reação, mesmo quando usados em quantidades estequiométricas. Outro importante avanço no mecanismo de reação de glicosilação é o estudo da nucleofilicidade do álcool em relação à seletividade. Mais detalhes sobre mecanismo de glicosilação são descritas na literatura.33,34
FATORES QUE INFLUENCIAM NA ESTEREOSSELETIVIDADE DA REAÇÃO DE GLICOSILAÇÃO Muitos fatores podem afetar o rendimento e a estereosseletividade das reações de glicosilação, incluindo, entre outras, estruturas e propriedades do doador e aceptor de glicosila, ativador ou promotor, pressão, concentração, solvente da reação e temperatura. Embora a formação de cada ligação glicosídica específica exija uma condição específica mais adequada, algumas tendências gerais foram observadas ao longo de décadas de investigação. A reação glicosilação 1,3-cis-estereosseletiva é mais robusta do que a dos 1,2-trans. Enquanto a formação dos isômeros 1,2-trans é fortemente favorecida pela participação do grupo vizinho (geração de íon aciloxônio intermediário), a reação de glicosilação 1,2-cis-estereosseletiva frequentemente depende do efeito anomérico.34 Por outro lado, a principal estratégia para a glicosilação é o uso de um substituinte não participante em C-2. O Esquema 15 sumariza princípios de síntese 1,2-cis-glicosilação, ou seja, esta envolve a reação entre um doador de glicosila 62 e aceptor de glicosila 66, em presença de um promotor ou ativador, para formar uma ligação glicosídica de 67. Após a ativação, e saída do grupo de saída de 63 resulta na formação de um cátion glicosila 64, o qual é estabilizado por meio de um intermediário de íon oxacarbênio 65 (Esquema 15a). O nucleófilo, aceitador de glicosila 66, pode então atacar a partir da face superior ou inferior (caminho a e b) do anel de 65. Isto daria origem a 1,2-trans ou 1,2-cis glicosídeos em relação ao substituinte vizinho em C-2, e reações não controladas podem levar a uma mistura dos mesmos.
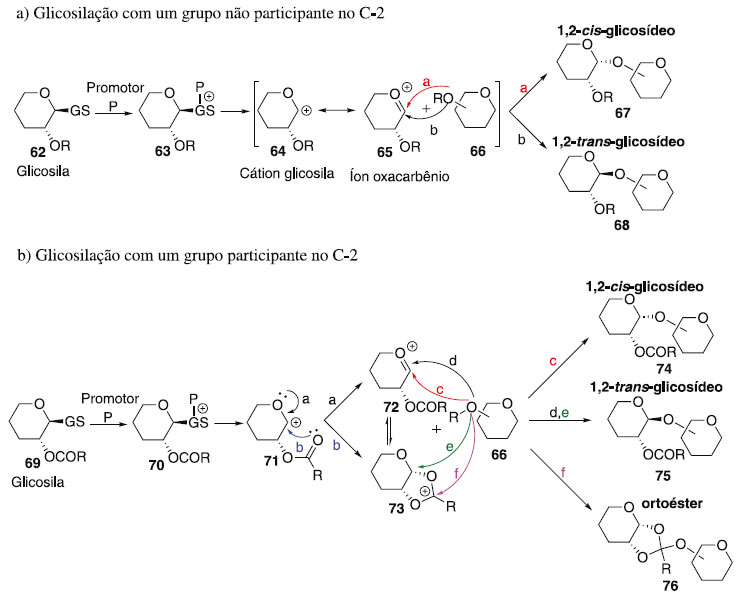 Esquema 15. Esboço geral da reação glicosilação e os principais intermediários envolvidos
A formação de ligações 1,2-trans pode ser realizada usando o efeito participativo do substituinte 2-acila vizinho. Nesse caso, o íon oxacarbênio 71 pode ser estabilizado ainda mais através de um intermediário aciloxônio bicíclico 73, que se torna o principal intermediário no caminho para os produtos de glicosilação (Esquema 15b).34 Uma vez que a face inferior do anel está bloqueada, o ataque nucleofílico do aceptor de glicosila 66 seria direcionado da face superior oposta. Isso normalmente fornece acesso à ligação 1,2-trans com estereosseletividade muito alta ou completa. Ocasionalmente, também são observadas quantidades substanciais de produtos ligados a 1,2-cis ou formação de ortoéster 76. Enquanto a síntese estereosseletiva de ligações 1,2-trans pode ser alcançada de forma confiável com o uso de assistência anquimérica de grupo vizinho,35 a formação de ligações 1,2-cis é tipicamente muito mais desafiadora. A presença de um grupo não participante é necessária para a síntese de 1,2-cis glicosídeos, mas o grupo não participante sozinho não pode garantir a estereosseletividade. Embora o produto α seja favorecido pelo efeito anomérico,36 a estereosseletividade da glicosilação pode ser pobre e requer outros modos de estereocontrole. Embora existam muitos exemplos em que foi alcançada uma excelente estereosseletividade 1,2-cis de determinadas ligações, nenhum método abrangente para a glicosilação da 1,2-cis é disponível.37-39 Sabe-se que vários fatores afetam a estereosseletividade e o rendimento da glicosilação e incluem temperatura, solvente, tipo de doador usado, tipo de aceitador usado, quantidade e tipo de promotor usado, grupos de proteção, etc. (Figura 4). Esses efeitos e métodos especificamente projetados para controlar a estereosseletividade da glicosilação são discutidos em seguida.
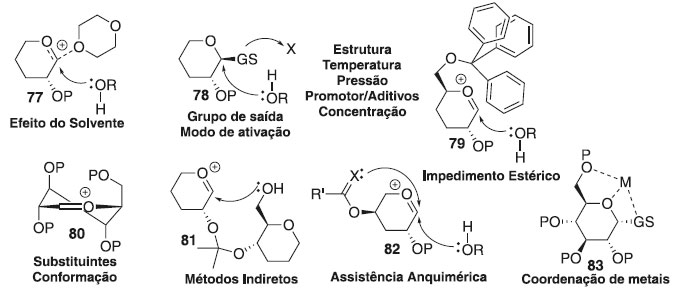 Figura 4. Fatores que afetam a estereosseletividade da reação de glicosilação
Efeito da temperatura As glicosilações são cineticamente controladas a temperaturas mais baixas e geralmente favorecem a formação de β-glicosídeo 90,40 embora também tenham sido relatadas observações inversas.41 Como o α-glicosídeo é favorecido termodinamicamente devido ao efeito anomérico, é predominantemente formado a altas temperaturas. Efeito do solvente O solvente é fator importante que influencia a estereosseletividade no centro anomérico. Assim, foi demonstrado que alguns solventes no meio reacional têm um efeito estereodirecional. Todavia, os solventes de reação polar aumentam a taxa de formação de β-glicosídeo reduzindo o efeito anomérico por meio da separação de carga entre O-5 e β-O1. Se a síntese de α-glicosídeos for desejada, CH2Cl2, ClCH2CH2Cl ou tolueno são candidatos adequados como solventes da reação. No entanto, existem forças mais poderosas que a solvólise que devem ser levadas em consideração quando os chamados solventes participantes são usados. Verificou-se que o acetonitrila e o éter dietílico são os casos limitantes para a formação preferencial dos glicosídeos β-D- e α-D, respectivamente.42 Essas observações foram racionalizadas da seguinte maneira: se as reações são realizadas em acetonitrila, o cátion nitrílico formado in situ adota exclusivamente orientação axial, permitindo, portanto, a glicosilação estereosseletiva para formar glicosídeos equatorialmente substituídos (Esquema 16).
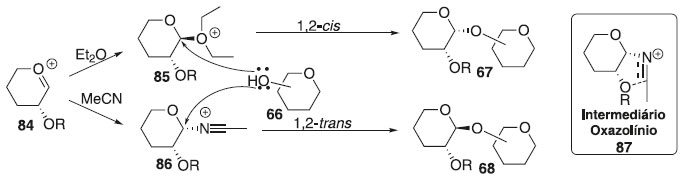 Esquema 16. Efeito estereodirecional do solvente em reação de glicosilação
Por outro lado, solventes de reação do tipo éter, tais como, éter dietílico ou tetra-hidrofurano (THF) também podem participar dos processos de glicosilação, no entanto, nesses casos, o intermediário equatorial é formado preferencialmente, levando, portanto, à formação da ligação glicosídica axial. Efeito da pressão A alta pressão aplicada a reações de glicosilação com doadores de glicosila participantes aumenta ainda mais a seletividade 1,2-trans;43 quando as condições de alta pressão foram aplicadas à glicosilação com um doador de glicosila não participante, um aumento notável no rendimento da reação foi observado apenas com alterações marginais na estereosseletividade.44 As interações estéricas desfavoráveis que ocorrem entre o doador e o aceitador de glicosila, no estado de transição, podem levar a uma dupla diastereodiferenciação45 como fator sem precedentes que governa a razão α/β na reação de glicosilação. Efeito da concentração A concentração da mistura de reação também influencia na seletividade anomérica, aumentando a taxa relativa da formação do produto ligado a glicosilação 1,2-cis-estereosseltivas; por outro lado, a diluição diminui consideravelmente a reação, portanto, há sempre um delicado equilíbrio entre seletividade e eficiência.46 Efeito do aceptor de glicosila Muitos exemplos em que diferentes aceptores de glicosila têm seletividades diferentes podem ser vistos ao longo do texto desta revisão. Uma regra prática é que a reatividade do álcool está inversamente relacionada com a estereosseletividade e as hidroxilas mais reativas apresentam as menores relações α/β: quanto mais forte o nucleófilo, mais rápida a reação e, mais difícil é controlar seu resultado. Como um exemplo, a glicosilação do 4-OH axial da galactose geralmente fornece excelente estereosseletividade do isômero 1,2-cis na reação de glicoislação. Ocasionalmente, a hidroxila primária proporciona maior estereosseletividade em comparação com as hidroxilas secundárias. Isso pode servir como evidência para a reação de glicosilação que ocorre através de um mecanismo bimolecular, pelo menos parcialmente. Os álcoois primários também apresentaram maior estereosseletividade nas reações de glicosilação.47 Efeito do doador de glicosila Glicosilações usando tricloroacetimidatos (TCAI)48 e tioglicosídeos49 como doadores tornaram-se os métodos mais amplamente estudados nas reações de glicosilação. As reações de glicosilação seguem geralmente um mecanismo de deslocamento SN1 unimolecular, onde a orientação do grupo de saída no carbono anomérico é de pouca importância. No entanto, ocasionalmente, as reações de glicosilação ocorrem através de um mecanismo semelhante ao SN2 com inversão da configuração anomérica. Por outro lado, a estereosseletividade da glicosilação pode ser profundamente influenciada por grupos de proteção.50 Os grupos de proteção vizinhos em C-2 tradicionalmente conhecidos como grupos participantes na síntese de 1,2-trans-glicosídeos podem influenciar na formação de 2-cis-glicosídeos ou 1,2-trans-glicosídeos. Grupos de proteção nas posições C-3, 4 e/ou 6 podem afetar a estereosseletividade por meio de participação da aglicona mediada por ligação de hidrogênio, impedimento estérico e/ou retirada de elétrons. Efeito do promotor A maioria das reações de glicosilação é realizada misturando o doador e o aceptor de glicosila, seguido pela adição de um promotor. Os métodos sintéticos para a glicosilação requerem a ativação do doador de glicosila. Devido ao amplo escopo das reações de glicosilação, há uma necessidade contínua de novos promotores para ajustar a reação para doadores e aceptores específicos. Vários promotores são baseados em metais pesados e a desvantagem é o desperdício gerado. Para evitar o desperdício de metais pesados, BF3∙OEt2 e TMSOTf são usados como alternativas comuns. No entanto, na glicosilação mediada por halogenetos, os halogenetos de tetraalquilamônio são os promotores padrões de escolha. As condições de ativação mais brandas são geralmente benéficas para a glicosilação 1,2-cis-estereosseletiva.51 Assim, as reações catalisadas por íons halogenetos fornecem os melhores resultados para a glicosilação utilizando haletos de glicosila; os tioglicosídeos apresentam melhor desempenho quando ativados com o promotor leve, tipo perclorato de dicollidina de iodônio (IDCP); enquanto os tricloroacetimidatos são os mais ativados com catalisadores do tipo ácidos fortes, como o trifluorometanossulfonato de trimetilsilila (TMS-triflato, TMSOTf) ou o ácido triflourometanossulfônico (ácido tríflico, TfOH). Várias adições aos sistemas promotores influenciam frequentemente o resultado estereoquímico da glicosilação: esses exemplos são discutidos ao longo das seções subsequentes que tratam de métodos específicos de glicosilação.
APLICAÇÕES DA REAÇÃO DE GLICOSILAÇÃO NA SÍNTESE DE MOLÉCULAS BIOATIVAS Aplicações sintéticas Na reação de síntese de O-glicosídeos A reação de glicosilação química envolvendo duas espécies para formar uma nova ligação glicosídica depende de um conjunto de variáveis. Por outro lado, desde 1928, a reação de O-glicosilação se tornou um dos métodos mais empregados na síntese de glicosídeos devido a sua obtenção de forma rápida e alta estereosseletividade. Em 2010, Rajaganesh e colaboradores52 reportaram a O-glicosilação de 1,2,3-tri-O-acetil-4,6-O-butilideno-β-D-glicopiranose e 1,2,3-tri-O-acetil-4,6-O-etilideno-β-D-glicopiranose 88a/b, como doador de glicosila catalisada por BF3.Et2O em presença de álcool propargílico e aromáticos, fornecendo os compostos desejados 89a/b com bons rendimentos e β-seletividade (Esquema 17), ou seja, completa retenção da configuração anomérica. Segundo os autores, os compostos apresentaram atividades antioxidantes fracas a moderadas com a atividade inibitória máxima de 77,3%.
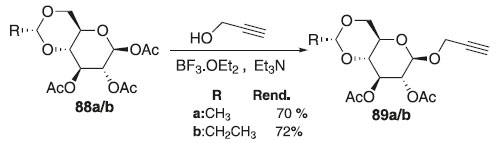 Esquema 17. O-Glicosilação de 1,2,3-tri-O-acetil-4,6-O-butilideno-β-D-glicopiranose e 1,2,3-tri-O-acetil-4,6-O-etilideno-β-D-glicopiranose catalisado por BF3.Et2O
No mesmo ano, Zhang, Wei e Du53 descreveram a primeira síntese total da apigenin-4-il-2-O-(p-coumaroil)-β-D-glicopiranosídeo (Esquema 18), no qual as etapas de síntese consistiram na glicosilação do doador 90 em presença de N,N'-diciclohexilcarbodiimida (DCC) e 4-dimetilaminopiridina (DMAP) e do aceptor ácido 4-O-alilcoumárico, para fonecer o composto desejado 91 com 90% de rendimento e seletividade em favor do anômero β. Segundo os autores, a remoção de todos os cinco grupos alila de 91 após mais três etapa foi realizada com sucesso para produzir o composto alvo apigenin-4-il-2-O-(p-coumaroil)-β-D-glicopiranosídeo em rendimento de 79%. Os autores também relataram que o composto alvo demonstrou boa atividade inibidora contra a xantina oxidase (IC50 = 23,95 ± 0,43 µmol L-1), uma enzima chave envolvida na produção de ácido úrico como metabólito de alimentos contendo purina. Os dados espectrais do composto estão em concordância com o descrito na literatura.53
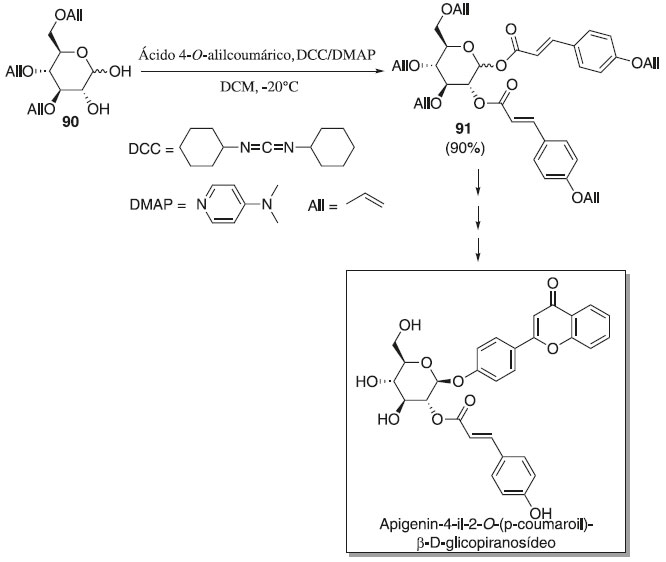 Esquema 18. Glisocilação do doador 90 utilizado na síntese total do apigenin-4-il-2-O-(p-coumaroil)-β-D-glicopiranosídeo
Posteriomente, Mossotti e Panza54 utilizaram o reagente de Hendrickson na reação de O-glicosilação. O agente glicosilante de Hendrickson foi preparado in situ utilizando uma mistura de óxido de trifenilfosfina e anidrido de trifluorometanossulfônico para fornecer o (Ph3P)2O(OTf)2. Em seguida na reação de glicosilação foi utilizado os doadores 92a/b por serem altamente α-seletivo e o aceptor 93 em presença do ativador (Ph3P)2O(OTf)2 para fornecer os compostos desejados 94a/b em bons rendimentos (Esquema 19).
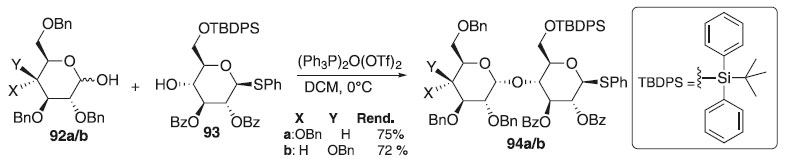 Esquema 19. Glicosilação com doadores de glicosila 92a/b em condições básicas
Outros trabalhos publicados nos anos de 2010 e 2011 descrevem o uso de doadores de glicosila em reações de O-glicosilação.55-58 Em 2012, Luo et al.59 relataram a síntese do 2,3,4,6-tetra-O-benzil-α-D-manopiranosil-2,3,4,6-tetra-O-benzil-α-D-hexopiranosídeo 97a/b utilizando como doador o 2,3,4,6-tetra-O-benzil-α-D-manopiranosídeo de 2-alilfenila 95 (Esquema 20) em presença de uma mistura de monocloreto de iodo (ICl) + AgOTf e como aceptores álcoois benzílico e n-octílico 96a/b. Os compostos foram obtidos com bons rendimentos, alta seletividade em favor do anômero α. Ainda segundo os autores, a utilização do protocolo foi ainda demonstrada com uma síntese eficiente do fragmento do dissacarídeo bleomicinas, fármaco resultante da fermentação de Streptomyces verticillus e indicado normalmente para neoplasias de pele, do trato genitário e câncer do testículo. A molécula de bleomicina é capaz de despolimerizar o DNA.
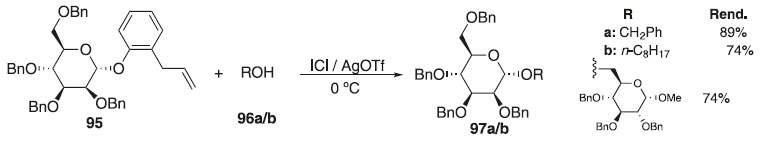 Esquema 20. Glicosilação utilizando como doador 2,3,4,6-tetra-O-benzil-α-D-manopiranosídeo de alilfenila em presença de ICl + AgOTf
Pertel et al.,60 relataram o emprego de 3,4,6-tri-O-acetil-α-D-glicopiranose-2-ozaxolina 98, como doador de glicosila, na reação de glicosilação 1,2-trans estereosseletiva catalisada por 2,4,6-trimetilpiridina (s-colidina) em ácido perclórico sob condições brandas, evitando a anomerização e outras reações colaterais, fornecendo os compostos glicosilados 100a-c em bons rendimentos (Esquema 21).
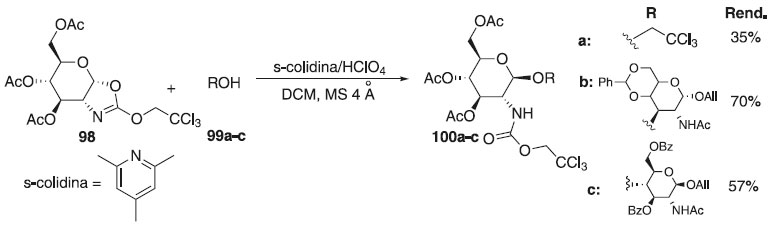 Esquema 21. Glicosilação utilizando como doadores derivados 3,4,6-tri-O-acetil-α-D-glicopiranose-2-ozaxolina em presença de s-colidina/HClO4
Reddy et al.61 descreveram a primeira síntese total enantiosseletiva de novos glicolipídeos, ieodoglicomidas A e B. A metodologia consistiu em uma reação one-pot para obtenção in situ do intermediário imidato de glicosila 103 pelo 2,3,4,6,7,8,9,10-octa-hidropirimidol-[1,2-a]-azepina (DBU), seguido por uma β-glicosilação com o TMSOTf frente aos (R)- e (S)-álcool alifático 102, para fornecer os produtos 104 R e S com bons rendimentos e posteriormente aplicados à síntese de glicopeptídeos (Esquema 22). O presente estudo sintético indicou a ambiguidade na estereoquímica absoluta proposta para o produto natural.
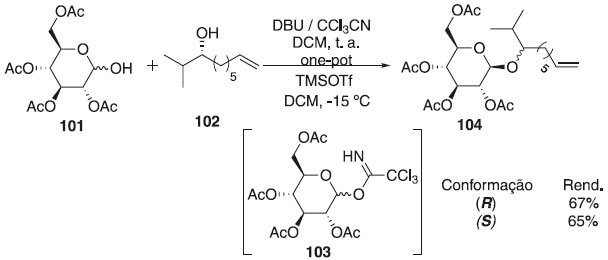 Esquema 22. Síntese enantiosseletivas de novos glicolipídios através de reação de glicosilação
Em 2013, Tatsumi et al.62 desenvolveram uma nova reação de glicosilação, utilizando-se o boronofosfato de glicosila 105 como doador de glicosila e o cátion tritila (Tr+) como ativador (Esquema 23). Os autores descrevem dois tipos de reações que foram estudadas: (1) o triéster de boranofosfato foi ativado com TrNTf2 para reagir com um álcool e (2) éteres de O-tritila funcionaram como aceptores de glicosila e fontes de Tr+. O último tipo de reação forneceu melhores resultados e os produtos de O-glicosilação 106 desejados foram rapidamente gerados e isolados com rendimentos moderados a bons e alta seletividade α e β.
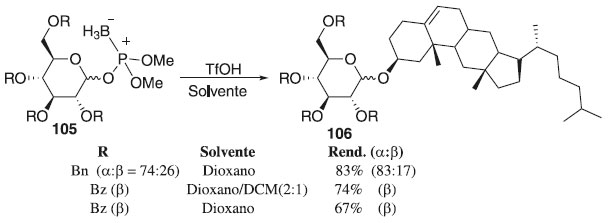 Esquema 23. Glicosilação usando boranofosfatos de glicosila como doadores de glicosila
Uma síntese de α-D-colesterilglicosídeos foi realizada usando uma reação de glicosilação do iodeto de O-trimetilsilil glicosila como doadores 107a/b e relatada por Davis et al.63 Na reação, os autores empregaram derivados de trimetilsilil glicosila e galactosila em uma metodologia one-pot, conseguindo o intermediador iodeto piranosídeo pelo iodeto de trimetilsilila (TMSI), seguido da ativação com DIPEA em presença do colesterol 108, produzindo o glicopiranosídeo de colesterila (αCG) e/ou o α-D-galactopiranosídeo de colesterila 109a/b (Esquema 24). Após reação de glicosidação, os grupos protetores TMS são removidos com facilidade usando a resina ácida Dowex-50WX8-200 em metanol. Convém destacar que tanto o α-D-glicopiranosídeo de colesterila (αCG) quanto o α-D-galactopiranosídeo de colesterila foram preparados em alto rendimento. Os autores também descrevem a reação de esterificação dos compostos obtidos usando acilação enzimática regiosseletiva com éster de vinil tetradecanoila para fornecer 6-O-tetradecanoil-α-D-colesteril glicopiranosídeo (αCAG) e do α-D-galactopiranosídeo de colesterila (αCG) em 78-85% dos rendimentos totais de açúcares livres.
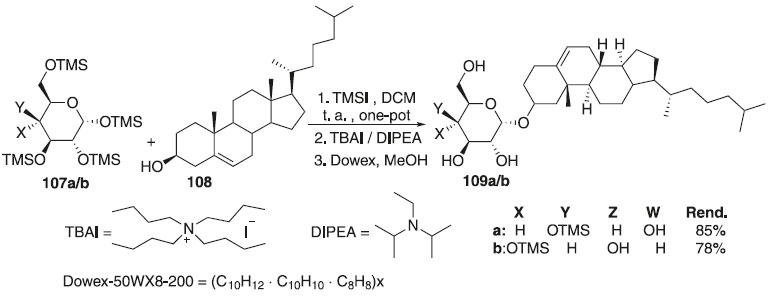 Esquema 24. Glicosilação utilizando iodeto de glicosila para produzir α-D-glicopiranosídeo de colesterila e α-D-galactopiranosídeo de colesterila (αCG)
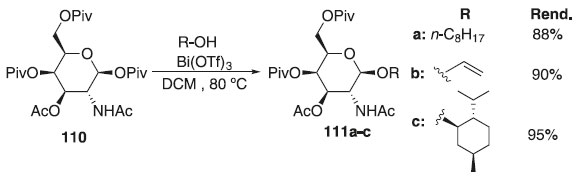
Ainda segundo os autores, pouco se sabe sobre os papéis biológicos dos colesterilglicosídeos, em geral. Embora os análogos da galactose relatados ainda não tenham sido isolados de fontes naturais, sabe-se que o αCG e o αCAG inibem a ativação das células T - célula do sistema imunológico e também um grupo de glóbulos brancos (leucócitos) responsáveis pela defesa do organismo contra agentes desconhecidos (antígenos). Convém destacar também que Rasmussen e colaboradores64 propuseram a reação de glicosilação utilizando o galactopiranosideo de piruvoila 110 como doador de glicosila, diferentes catalisadores, tais como o Bi(OTf)3 e o Fe(OTf)3, os quais foram considerados ácidos de Lewis altamente ativo para essa reação e diferentes álcoois, fornecendo os produtos 111a-c em excelentes rendimentos (Esquema 25). Os autores verificaram também que outros triflatos metálicos menos reativos, como os de Cu (II) e Yb (III), podem ser benéficos em reações de glicosilação em aceptores de glicosila mais exigentes. Ainda foi demonstrado pelos autores que é possível controlar a estereosseletividade anomérica na glicosilação via anomerização in situ pós-glicosilação para obter bons rendimentos dos α-galactopiranosídeos. A metodologia também foi utilizada para preparar importantes carboidratos de ocorrência natural, incluindo um fragmento de tetrassacarídeo denominado clarhamnosídeo, isolado de esponja marinha (Agela clathrodes).
 Esquema 25. Glicosilação estereosseletiva para obtenção α-galactopiranosídeos usando galactopiranosideo de piruvoila como doador de glicosila
Em 2915, um protocolo leve e conveniente para a síntese direta de β-manosídeos foi desenvolvido por Sun et al.65 A glicosilação consistiu em utilizar doadores orto-2-hex-1-in-1-il-D-manopiranosídeos protegidos com 4,6-O-benzilideno 112a-d com vários álcoois como aceptores catalisados pelo complexo ouro (I) de 0 °C até a temperatura ambiente para fornecer o β-O-manopiranosídeos correspondentes 113a-d com altos rendimentos e estereosseletividades satisfatórias (Esquema 26). Ainda conforme os autores, essa reação foi aplicada à síntese total da acremomannolipina A, um glicolipídeo composto por manitol e porção manopiranosídica peracilada através de uma ligação β-glicosídica. A acremomannolipina A foi isolada do Acremonium strictum e é um potencial modulador do sinal de cálcio.
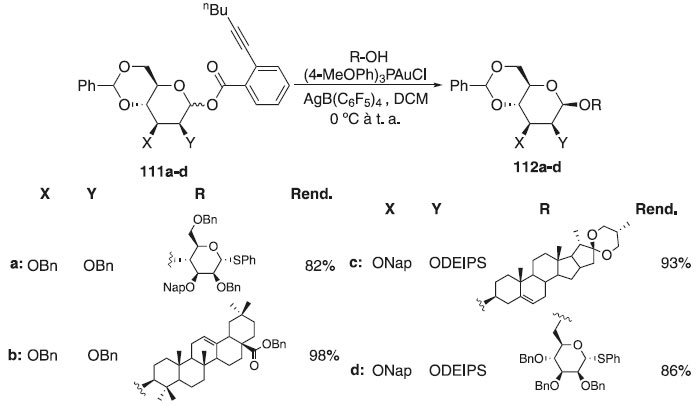 Esquema 26. Glicosilação do doador 4,6-O-Benzilideno-2,3-di-O-benzil-1-(orto-hexinil-benzoato)-D-manopiranosídeo com diferentes aceptores
Metodologia de glicosilação utilizando ouro (III) foi realizada pelo grupo de Shaw, Thakur e Kumar.66 Na reação foi utilizado como doadores o fenilpropriolato de glicosila (PPG) 113 e cloreto de ouro (III) como catalisador e vários aceptores de glicosila à base de carboidratos e sem carboidratos para produzir os correspondentes compostos 114a/b em bons rendimentos e estereosseletividade em favor do anômero α (Esquema 27). Segundo os autores, um estudo mecanístico preliminar revela que a presença da ligação tripla adjacente ao grupo éster é essencial para a ativação e o doador baseado em PPG mostra maior reatividade do que os doadores análogos, tipo acetato e benzoato.
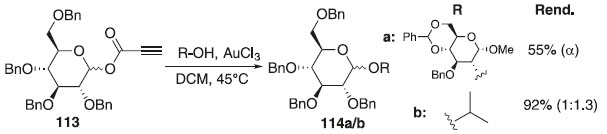 Esquema 27. Glicosilação catalisada por ouro (III) usando doador 113 (PPG)
Recentemente, Liang e colaboradores67 descreveram o desenvolvimento de uma reação de glicosilação utilizando os doadores orto-alilobenzoato de galactosila e manosila 115a/b em presença de TMSOTf em combinação N-iodosuccinimida (NIS) em condições de reação moderadas e aceptores sacarídicos, terpenóides e álcoois alquílicos. Os compostos 116a-c foram obtidos em ótimos rendimentos e β-seletividade (Esquema 28).
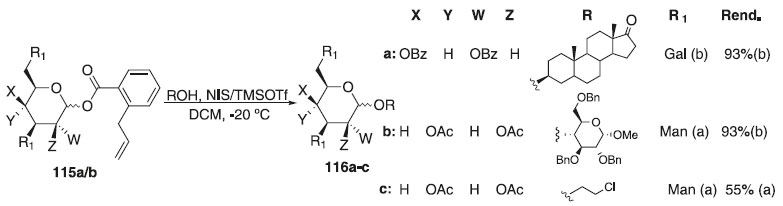 Esquema 28. Glicosilação utilizando como doadores orto-alilobenzoato de galactosila e manosila
Reação de glicosilação utilizando haletos de glicosila A abordagem feita por Koenigs e Knorr em 1901 proporcionaram avanços na síntese de glicopiranosídeos e manopiranosídeos em alta reatividade, regiosseletividade, estereosseletividade e facilidade na obtenção de haletos glicosídeos, impactando diretamente no desenvolvimento metodológico de obtenção desses compostos, como descritos a seguir. Em 2010, Karelin et al.,68 ao sintetizar fragmentos de um oligomanopiranosídeos ramificados, em uma das etapas, promoveram uma reação de glicosilação em que utilizaram o doador α-brometo manosila 118 em presença de AgOTf e o aceptor α-tio-manosila 119 originando o 120 em rendimento de 73%. O composto 120 foi utilizado na síntese de polissacarídeos correspondentes a fragmentos das células que atuam nos antígenos antifúngicos (Esquema 29).
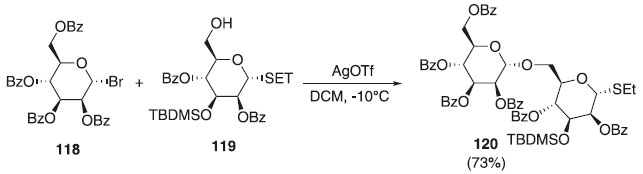 Esquema 29. Glicosilação a partir do doador brometo de manosila em presença de AgOTf
Hou e Kovác69 relataram uma reação de glicosilação, partindo do doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosila 121 e do aceptor 4,6-O-benzilideno-2-desoxi-2-tricloroacetamido-1-tio-β-D-glicopiranosídeo de etila 122 (um hemiacetal) em presença de AgOTf, originando o composto 123 com excelente rendimento (90%) e β-seletividade (Esquema 30). O composto obtido foi utilizado como bloco de construção, após várias etapas reacioanis, para obtenção de uma hexassacaídeo e posteriormente a conjugação deste com transportadores de proteínas usando, por exemplo, a química do ácido esquárico.
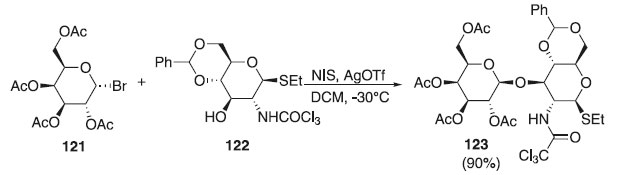 Esquema 30. Glicosilação utilizando o doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosídeo em presença de AgOTf
Em 2011, Ren et al.70 descreveram uma reação de glicosilação utilizando o doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosila 121 em presença de carbonato de potássio (K2CO3) e derivado da quercetina 124, como aceptor, para fonecer o composto 125 em rendimento de 56% (Esquema 31). A quercetina 3-O-[6"-O- (trans-p-coumaroil)]--D-glicopiranosídeo posui atividades anti-hipertensivas, antidiabéticas e inibidoras da tirosinase.
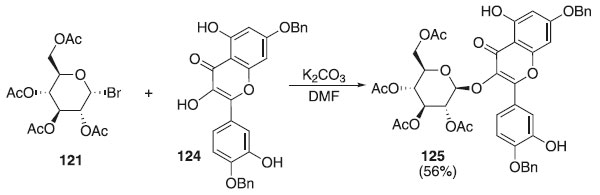 Esquema 31. Glicosilação utilizando o doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosídeo em presença de K2CO3
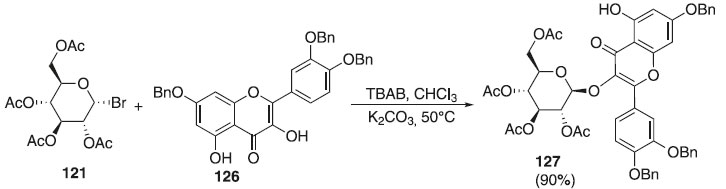
Utilizando derivados da quercentina como aceptor, Zhang et al.71 descreveram a reação de glicosilação regiosseletiva utilizando o doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosila 121 em presença de K2CO3 e 126, como aceptor, para fornecer o composto 127 em excelente rendimento (90%) e β-seletividade (Esquema 32). Esse composto foi submetido a outras etapas de síntese para obtenção de O-glicosídeo de flavonol (trissacarídeo) isolado de sementes de algodão e que demonstrou atividades antidepressivas.
 Esquema 32. Glicosilação utilizando derivados da quercentina como aceptor de glicosila
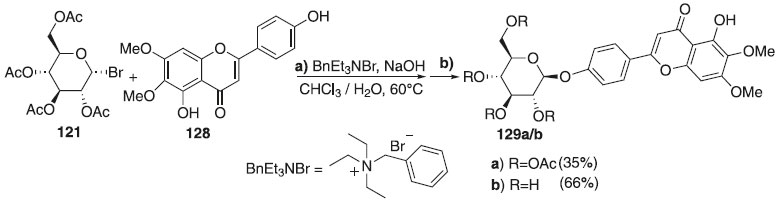
A reação de glicosilação descrita por Soulage et al.72 consiste no emprego do doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosila 121 utilizando o brometo de benziltrietilamônio (BnEt3NBr) como ativador, e a cirsimaritina como aceptor de glicosila 128 para fornecer o composto 129a com rendimento de 35% e β-seletividade (Esquema 33). A cirsimarina (129b), obtida após desacetilação de 129a, em uma mistura metanol/trietilamica/água, apresentou atividades lipolíticas e antilipogênicas. Segundo os autores, embora glicosídeos de flavona sejam compostos amplamente distribuídos na natureza, possuindo muitas atividades biológicas, a glicosilação das flavonas ainda constitui uma tarefa desafiadora.
 Esquema 33. Glicosilação utilizando o doador brometo de 2,3,4,6-tri-O-acetil-α-D-galactopiranosila em presença de BnEt3NBr
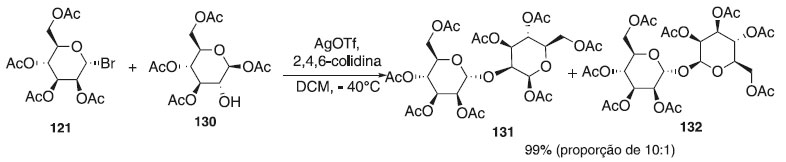
Para síntese dos dissacarídeos 131 e 132, Chen e Tan73 utilizaram na reação de glicosilação o doador brometo de 2,3,4,6-tri-O-acetil-α-D-manopirasosila 121 em presença de AgOTf como catalisador e o aceptor de glicosila, o 1,3,4,6-tetra-O-acetil-β-D-manopiranose 130, fornecendo a mistura dos compostos 131 e 132 em rendimento de 99% na proporção de 10:1. Segundo os autores, o dissacarídeo 131 será utilizado na síntese de blocos de construção de Ser/Thr manosila e oligomanosila (Esquema 34).
 Esquema 34. Glicosilação utilizando o brometo de 2,3,4,6-tri-O-acetil-α-D-manopirasosila em presença de AgOTf
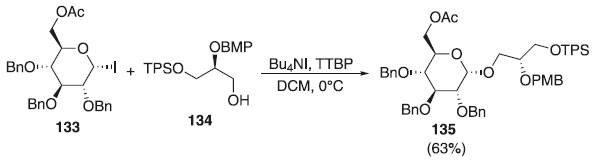
Diferentemente das ligações 1,2-trans-glicosídicas, que podem ser construídas com facilidade, em virtude da participação do grupo vizinho, a formação estereosseletiva das ligações 1,2-cis-glicosídicas é problemática. Por conta disso, a Cao et al.74 realizaram a reação de glicosidação utilizando como doador o iodeto de glicosila 133 e álcool primário 134, como aceptores, em presença de Bu4NI, TTBP e CH2Cl2, para fornecer o α-composto desejado 135 em rendimento de 63% (Esquema 35). O composto 135 foi utilizado na síntese de glicolipídeos e também como bloco de construção para síntese dos isômeros GlcAGroAc2.
 Esquema 35. Reação de glicosilação utilizando como doador o iodeto de glicosila
Dando continuidade, Manabe e Ito75 relataram a reação de glicosilação usando como doador o fluoreto de glicosila 136α:136 (1:4) pela ativação com trifluorometanossulfonato de háfnio (Hf(OTf)4) e como aceptor a glicopiranose protegida 137 fornecendo uma mistura diastereoisomérica 138α:138β (20:80) em 99% rendimentos (Esquema 36). Os autores descreveram a reação de glicosilação sob várias condições e limitações mediadas pelo ativador Hf(Otf)4.
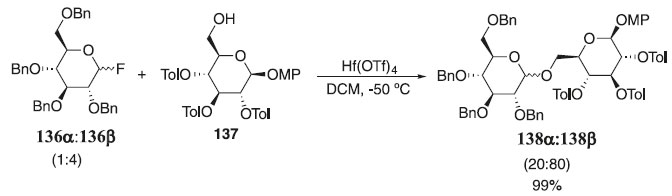 Esquema 36. Reação de glicosilação utilizando como doador o fluoreto de glicosila
Reações de glicosilação utilizando imidatos Os derivados acetimidatos β-glicosídeo descritos por Sinaÿ em 1977 e Schmidt em 1986, como diastereoisômeros, viabilizaram o interesse na área, devido sua facilidade de síntese e sua regioespecíficidade, trazendo modificações e ampliações na obtenção dos glicosídeos, conforme são explanados a seguir. Um método mais conveniente para a preparação e utilização de doadores de glicosila na formação de ligação O-glicosila, foi descrita por Shirahata et al.76 Esses autores reportaram a reação de glicosilação catalítica e estereosseletiva one-pot para obtenção de oligossacarídeos utilizando N-tricloroacetilcarbamato de glicosila 139, como doador de glicosila, com uma quantidade catalítica de ácido de Lewis (TMSClO4) em presença do aceptor de glicosila 140 e peneira molecular de 5 Å para fornecer a mistura de anômeros de um 1→6 dissacarídeos 141 na proporção α:β = 94:6 e com excelente rendimento (Esquema 37).
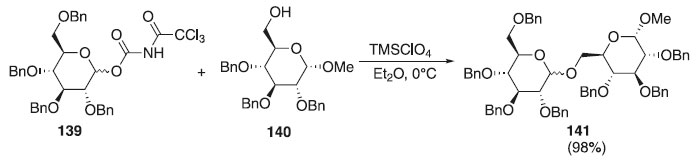 Esquema 37. Reação de glicosilação catalítica e estereosseletiva one-pot usando o N-tricloroacetilcarbamato de glicosila como doador
Outros trabalhos utilizando O-imidatos publicados em 2010 descrevem o uso dele como doadores de glicosila em reações de glicosilação.77-81 Em 2011, Kalikhanda e Li82 reportaram a reação de glicosilação utilizando como doador de glicosila, o 2-azido-2-desoxi-α-D-galactopiranosídeo 142 em presença do ativador TMSOTf e dos aceptores de glicosila 143a/b, em temperatura ambiente, fornecendo os compostos 144a/b em bom e ótimo rendimentos, respectivamente (Esquema 38).
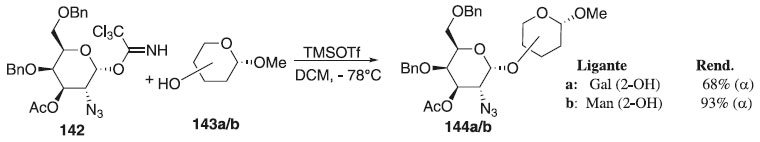 Esquema 38. Reação de glicosilação utilizando o doador de glicosila, 2-azido-2-desoxi-α-D-galactopiranosídeo em presença do ativador TMSOTf
Meses depois, os mesmos autores publicaram um artigo em que utilizaram o 2-azido-2-desoxi-α-galactosídeo 142 como doador de glicosila, em presença do ativador TMSOTf, e como aceptor de glicosila o 2,3,4-tri-O-benzoil-D-glicopiranose 145 para fornecer os compostos 146a/b com α-seletividade e bons rendimentos (Esquema 39).
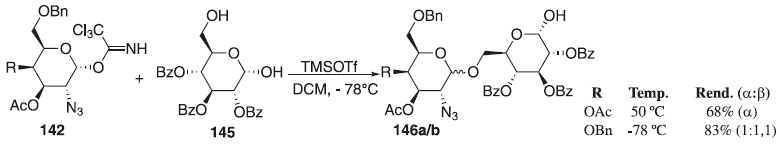 Esquema 39. Glicosilação do 2-azido-2-desoxi-α-galactosídeo com 2,3,4-tri-O-benzoil-D-glicopiranose em TMSOTf
Sob condições anidras, Zhu e Ralph83 reagiram o doador de glicosila 147 em presença do TMSOTf utilizando como aceptores de glicosila, o ácido cloroacetilferúlico e o ácido cloroacetilsinápico 148a/b para fornecer os compostos 149a/b em rendimentos de 72% e 51%, respectivamente (Esquema 40). Segundo os autores, as reações foram β-estereosseletivas e os anômeros α não foram detectados. Após hidrólise de 149a/b, os compostos finais serviram como doadores de acila para a biossíntese de inúmeros metabólitos secundários de plantas.
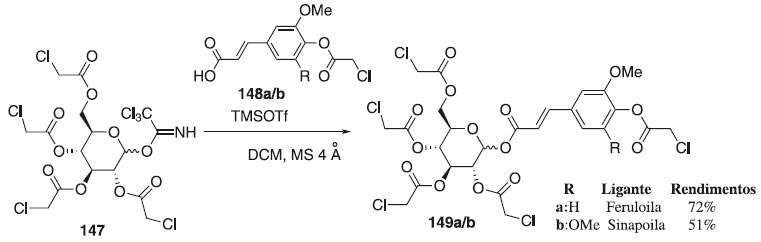 Esquema 40. Glicossilação do tricloroacetimidato de 2,3,4,6-tetra-O-cloroacetilglicosila com diferentes aceptores em presença de TMSOTf
Diferentemente dos autores acima, em 2012 foi relatada por Li et al.84 uma melhor eficiência do catalisador BF3∙OEt2 em relação ao TMSOTf em reação de glicosilação. A reação de glicosilação consistiu em reagir tricloroacetimidatos de glicosila 150 como doador de glicosila e diferentes fenóis 151a-d, como aceptores, para fornecer os glicosídeos desejados 152a-d em excelentes rendimentos e β-estereosseletividade (Esquema 41).
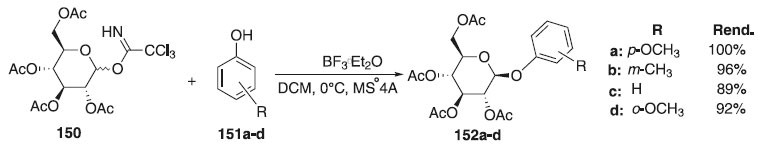 Esquema 41. O-Glicosilação de diferentes fenóis com tricloroacetimidatos de glicosila doador na presença de BF3.Et2O
Outros trabalhos utilizando O-imidatos publicados em 2012 descrevem o uso dele como doadores de glicosila e galactosila em reações de glicosilação e diferentes promotores.85-88 Em 2018, um estudo relatado por Lu et al.89 examinou a utilidade do grupo protetor N-benzilcarbomoílo (BnCar) nas reações de glicosilação, onde utilizou como doador o α-tricloroacetimidato de glicosila 153 em presença do ativador TMSOTf e diferentes álcoois primários ou secundários, como aceptores, fornecendo exclusivamente os produtos 154a/b com β-seletividade, bom e excelente rendimentos (Esquema 42). Um estudo mecanicista revelou que o intermediário ativado é o triflato de glicosila em uma conformação enviesada, o que resulta em glicosilação β-seletiva por uma via do tipo SN2. O grupo BnCar pode ser facilmente clivado usando nitrito de tetrabutilamônio, sem afetar os grupos protetores de éster e éter. Segundo os autores, esses resultados mostram que o BnCar é útil para a síntese de oligossacarídeos complexos.
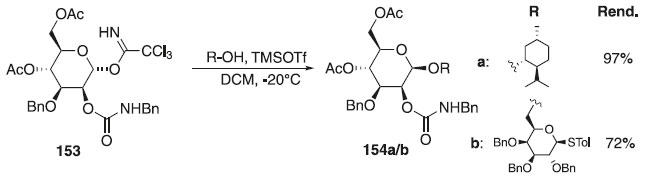 Esquema 42. Glicosilação usando como doador o 4,6-di-O-acetil-3-O-benzil-2-O-(N-benzilcarbamoil)-α-D-glucopiranosídeo de tricloroacetimidila com diferentes aceptores
Ainda em 2018, outros pesquisadores utilizaram diferentes doadores de glicosila em presença de diferentes ativadores e diferentes aceptores, em que os produtos finais foram utilizados como precursores na síntese de oligossacarídeos.90,91 Recentemente, Zhang et al.92 relataram a reação de O-glicosilação utilizando como doador o α-imidato galactopiranosídeo 155 em presença do TMSOTf e como aceptor, o 2,3-O-isopropilideno-β-D-xilopiranosídeo de metila 156, obtendo o produto final 157 com rendimento de 52% (Esquema 43). Segundo os autores, o foco do trabalho de síntese é a análise conformacional de ligação O-glicosídica de fragmentos de oligossacarídeos marcados com 13C de um glicolípido anticongelante.
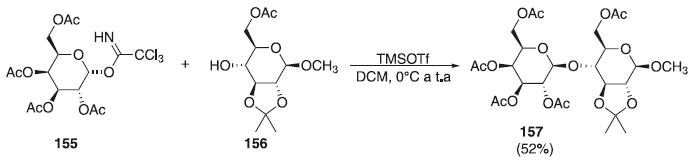 Esquema 43. Glicosilação usando como doador o composto 155 em presença de TMSOTf
Reações de glicosilação utilizando tioglicosídeos Desde a primeira abordagem realizada por Garegg-Fügedi em 1986 com derivados S-glicosila, notou-se o comportamento dos doadores de glicosila na promoção da reação de glicosilação, devido a sua alta reatividade, estabilidade e estereosseletividade frente a aceptores de glicosila. Em seguida descreve-se com detalhes as novas abordagens sintéticas feitas aproximadamente nos últimos 10 anos. Em 2010, Galan et al.,93 relataram a reação de glicosilação utilizando como doador de glicosila o tioglicosídeo 158 em presença de N-iodosuccinimida (NIS) e como aceptor o 1,2:3,4-di-O-isopropilideno-α-D-galactopiranose 159 para fornecer o composto desejado 160 em bons rendimentos dependendo da base imidazólica formada (Esquema 44). Os autores também relataram os efeitos de líquido iônicos na modificação da estrutura do cátion imidazólio e a importância da escolha do contra-íon nas reações de glicosilação dos tioglicosídeos à temperatura ambiente e em presença de N-iodosuccinimida (NIS).
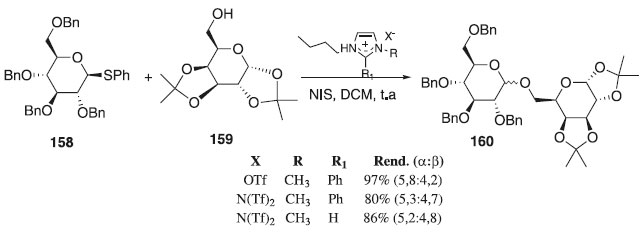 Esquema 44. Líquidos iônicos, doadores de tioglicosídeo e aceptor de glicosila em reação de glicosilação
Ainda em 2010, outros pesquisadores utilizaram tioglicosídeos como doadores de glicosila em presença de diferentes ativadores, tipos AgOTf, NIS e diferentes aceptores, em que os produtos finais foram obtidos com esterosseletividade em favor do anômero α e bons rendimentos.94,95 Adamo et al.96 descreveram uma reação de glicosilação usando como doador de glicosila, o tioglicosídeos 161 em presença de uma mistura de NIS + TfOH e do aceptor de glicosila 162 levando a formação do produto 163 em rendimento de 82% (Esquema 45). O composto 163 foi usado como precursor na síntese de fragmentos de laminarina e avaliação de um hexasaccarídeo β-(1,3) glicano, um conjugado com CRM197 utilizado como candidato a vacina contra Candida albicans.
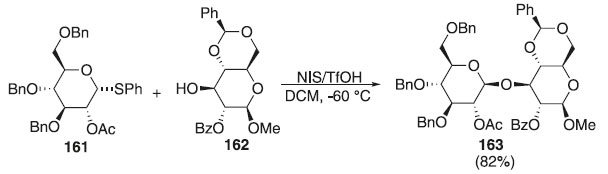 Esquema 45. Reação de glicosilação utilizando como doador tioglicosídeo em presença de NIS + TfOH
Em 2012, Li, Zhu e Kalikanda97 relataram em seu trabalho o uso do tiogalactopiranosídeo de fenila 164, como doador de glicosila em presença da mistura, NIS+TMSOTf e diferentes aceptores para fornecer os produtos de glicosilação finais 165a-c em excelentes rendimentos e α-seletividade (Esquema 46). Segundo esses autores, também foi observada estereosseletividade, quando em comparação com o doador de galactose protegido por tetrabenzila. Por outro lado, essa pesquisa não apenas fornece um novo doador altamente estereosseletivo para a síntese futura de oligossacarídeos complexos, como também demonstra que o design terapêutico do doador de glicosila é uma abordagem poderosa para alcançar reações de glicosilação de alta eficiência e seletividade.
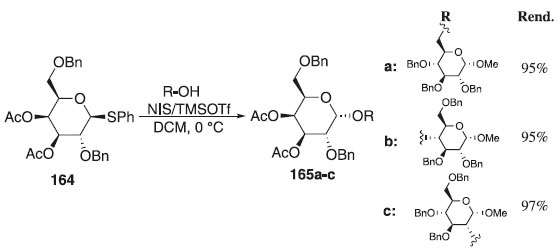 Esquema 46. Glicosilação utilizando o composto 164 como doador de glicosila em presença da mistura NIS+TMSOTf
Segundo estudos anteriores de Lemieux para brometos altamente reativos, Kaeothip, Yasomanee e Demchenko98 relataram a reação de glicosilação, mecanismo, reatividade e estereosseletividade de tioglicosídeos na presença de bromo. Quando o brometo é glicosilado, os α-glicosídeos se formam exclusivamente, no entanto, os rendimentos de tais reações podem ser baixos devido à anomerização competitiva no α-brometo que é totalmente não reativa sob as condições de reação estabelecidas. Outros trabalhos utilizando tioglicosídeos publicados em 2012 descrevem o uso deste como doadores de glicosila em reações de glicosilação.99-102 Muitas propostas de síntese utilizando reação one-pot demonstraram ser essa estratégia capaz de economizar tempo, melhorar a eficiência e eliminar várias etapas de síntese e separação intermediária. Em 2013, Gao e Guo103 relataram três etapas de glicosilação sequencial, com duração de 6 h, para obtenção do composto 168 com rendimento global de 39%, dando uma média de 73% de rendimento para cada etapa de glicosilação (Esquema 47). Todas as reações foram especificadas com estereoquímica α (1JCH anomérico, valores de H de 168 estavam entre 169 e 176 Hz). Em seguida, o composto 168 foi hidrolisado empregando p-TolSCl / AgOTf / TTBP ou NIS / AgOTf / TTBP, DBU, CCl3CN em diclorometano fornecendo o composto 169 com 74%.
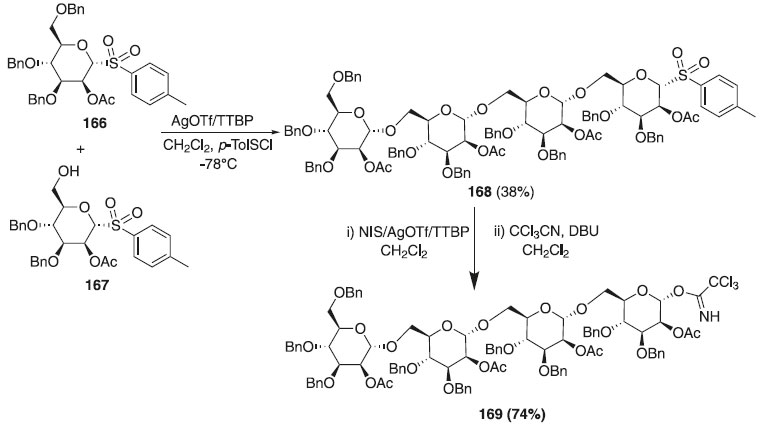 Esquema 47. Glicosilação one-pot utilizando como doador derivados de tioglicosídeos
A síntese e bioatividade de oligomanose com ligação a β-(1→4) e seus derivados parcialmente acetilados foi realizada de maneira eficiente por Ohara et al.,104 utilizando uma glicosilação β-seletiva seguida da epimerização da forma glico para mano. A reação de glicosilação consistiu na reação do doador de glicosila, o tioglicosideos 170, em presença TTBP e diferentes aceptores 171a/b para fornecer os produtos desejados 172a/b em rendimentos variando de 73-79% e β-seletividades (Esquema 48). A bioatividade das oligomanoses sintéticas e derivados parcialmente acetilados foi investigada a fim de identificar o menor oligômero possível para indução de citocinas, como mostrado nos polissacarídeos extraídos de Dendrobium huoshanense.
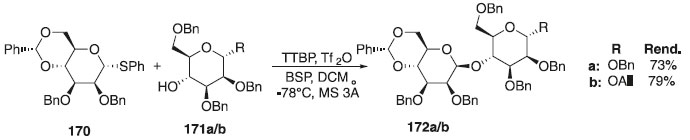 Esquema 48. Glicosilação utilizando doador tioglicosídeo em presença do ativador TTBP
Padungros, Alberch e Wei105 avaliaram os ditiocarbamatos de glicosila (DTCs) com grupos hidroxilas em C2 desprotegidos, como doadores de glicosila, na síntese de oligossacarídeos com β-seletividade. Na reação de glicosilação, esses autores utilizaram os ditiocarbamatos de glicosila 173a/b, como doador de glicosila em presença de sais triflato de cobre e aceptores de glicosila, para fornecer os produtos finial 174a-d, em bons rendimentos (Esquema 49). Segundo os autores, o caráter β-seletivo e regiosseletivo, mesmo pela ausência de assistência anquimérica no C-2 dos aceptores, torna os compostos 174a/b percursor de tetrassacarídeo β-(1→6)-O-glicopiranosídeo com a finalidade de uma possível interação biológica.
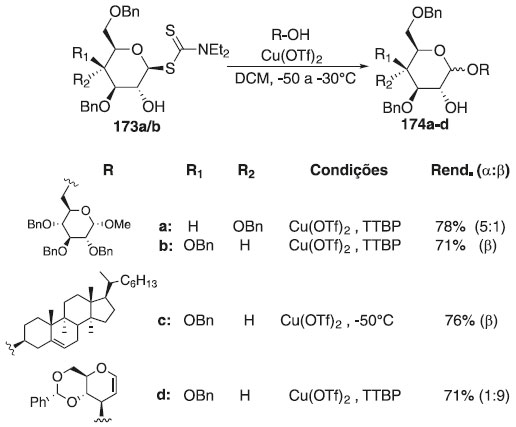 Esquema 49. Glicosilação utilizando ditiocarbamatos de glicosila (DTCs) em presença do sal triflato de cobre
A aplicação de grupos benzila orto-substituídos na reação de glicosilação estereosseletiva foi descrita por Buda et al.106 Segundo os autores o uso dos grupos 2-O-(2-nitrobenzil) e 2-O-(2-cianobenzil) controla a formação estereosseletiva de ligações 1,2-trans-glicosídicas. A estereosseletividade observada provavelmente surge da formação intramolecular do intermediário cíclico entre o substituinte rico em elétrons e o íon oxacarbênio doador, proporcionando a seletividade facial esperada para o ataque do aceptor de glicosila. Na reação de glicosilação foi utilizado como doador de glicosila, o tioglicosídeo 175 em presença Ph2SO/TTBP/Tf2O e diferentes aceptores de glicosila para fornecer os compostos finais 176a-d em bons rendimentos (Esquema 50). Para comprovar o mecanismo postulado com base no efeito da participação de grupos benzila orto-substituídos na estereosseletividade da glicosilação, usou-se a metodologia de cálculo teórico da DFT.
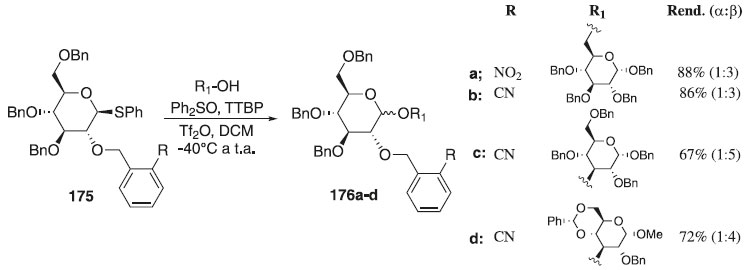 Esquema 50. Glicosilação estereosseletiva usando como doador o tioglicosídeo 175
A síntese do dissacarídeo 179 iniciou a partir da reação de glicosilação do doador de glicosila 177 em presença do p-TolSCl e AgOTf a -78 °C e do aceptor 178 e da base 2,4,5-tritert-butilpirimidina (TTBP), para fornecer o dissacarídeo 178 em rendimento de 85% e como um único anômero isolado (Esquema 51).107 A estereoquímica da ligação glicosídica formada foi confirmada por análise de RMN com 3JH1B-H2B = 3,7 Hz e 1JC1B-H1B = 171 Hz.
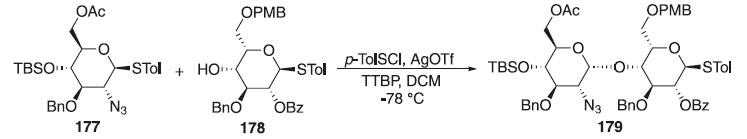 Esquema 51. Reação de glicosilação a partir do doador tioglicosídeo em presença p-TolSCl e AgOTf
Em 2016, Heuckendorff, Poulsen e Jensen108 descreveram os efeitos eletrônicos de grupos de proteção de éter e a reatividade de doadores de glicosila. De posse dos doadores de glicosila, os autores testaram sua reatividade nas reações de glicosilação. As condições padrão de ativação foi realizada usando o doador 180a-d em presença dos catalisadores NIS / TfOH, usando L-mentol 181, como aceptor de glicosila, para fornecer os compostos desejados 182a-d em excelentes rendimentos (Esquema 52). Devido à dificuldade de encontrar uma temperatura de reação apropriada para que ocorre uma dada reação de glicosilação, a mistura reacional foi resfriada a -78 °C antes da adição do catalisador (TfOH). Em seguida a temperatura foi aumentada lentamente para 0 °C durante várias horas. Foi observado que nenhuma reação ao longo deste estudo foi efetivada a -78 °C, mas sim concluídas antes de atingir 0 °C. As proporções anoméricas foram estabelecidas por comparação das intensidades das integrais dos prótons e carbonos anoméricos nos espectros de RMN 1H e RMN 13C da mistura reacional bruta.
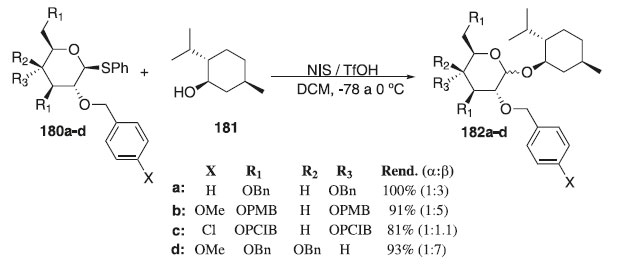 Esquema 52. Glicosilação usando L-mentol como um aceptor de glicosila
Um trissacarídeo cíclico foi sintetizado pela reação de cicloglicosilação de um trissacarídeo linear, modificado com a porção hidroximetil na posição C4 da porção glicopiranose. O trissacarídeo cíclico possui uma simetria trigonal perfeita raramente observada no grupo espacial P3, em forma de cone estreito, e um arranjo do tipo parede de tijolos de moléculas no estado sólido e exibe uma afinidade de ligação significativamente aumentada ao 1-aminoadamantano em solução aquosa.109 Esta síntese foi realizada por Maiti e Jayaraman109 e consistiu na cicloglicosilação utilizando inicialmente o doador de glicosila 183 em presença dos ativadores NIS/TfO e do aceptor 184 para fornecer o dissacarídeo 185, em rendimento de 67%, que após uma sequência de várias etapas reacional forneceu o trissacarídeo cíclico 187 em rendimento de 80% (Esquema 53).
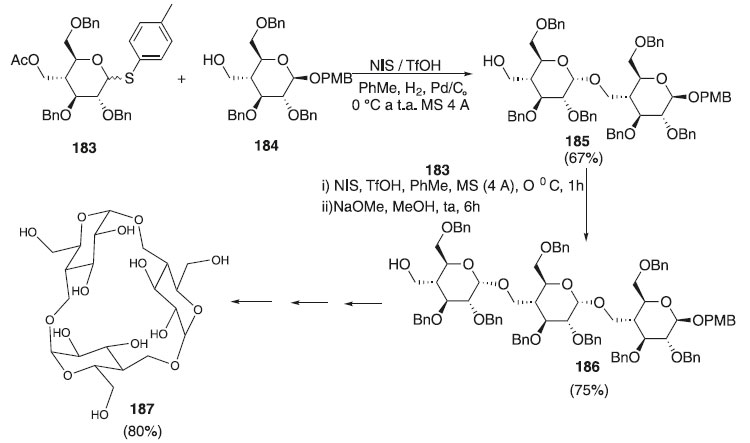 Esquema 53. Cicloglicosilação utilizando como doador o 4-desoxi-4-C-acetoximetil-2,3,6-tri-O-benzil-1-tio-α/β-D-glicopiranosídeo de 4-metilfenila
Recentemente, com os fragmentos de carboidratos individuais sintetizados, Susanto et al.110 realizaram a reação de glicosilação utilizando o doador de glicosila, β-tioglicosídeos 188 em presença da mistura, NIS/TfOH e do aceptor 189 para fornecer o dissacarídeo desejado 190 em boa seletividade e com rendimento de 78% (Esquema 54). Convém destacar que a estereosseletividade completa para cada glicosilação foi alcançada através da ortogonalidade e dos efeitos participantes dos grupos bloqueadores. No decorrer desta síntese, os autores também demonstraram uma nova metodologia para efetuar a piruvilação do trans-diol.
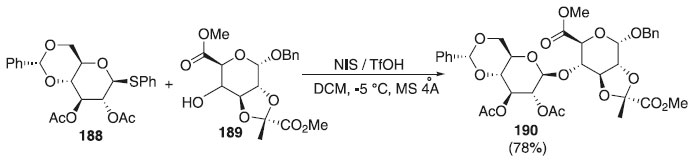 Esquema 54. Utilização de fragmentos de carboidratos para promover reação de glicosilação
Reações de glicosilação utilizando glicais Diante de todos os doadores de glicosila encontrados na literatura, uma nova abordagem de doador vem adquirindo interesse na síntese de carboidratos. Os glicais são compostos altamente importantes na química dos carboidratos que podem ser usados com eficácia para a formação de glicosídeos 2,3‐insaturados, ou seja, como doadores de glicosila. Por outro lado, a presença da ligação insaturada proporciona alta seletividade aos compostos finais. Em seguida, descreve-se a utilização de glicais como doadores de glicosila. A ativação seletiva de doadores de glicais "armado" e "desarmado" permitindo reações de glicosilação estereocontrolada empregando o Cu (II) como promotor foi realizado pelo grupo de Kumar et al.111 Assim, a glicosilação estereosseletiva foi realizada utilizando como doadores de glicosila, galactais protegidos 191a/b, empregando o 6-hidroxi-2,3,4-tri-O-benzil-α-D-glicopiranosídeo de metila, como aceptor em presença do triflato de cobre (II) como catalisador para fornecer os compostos 192a/b, em excelente rendimento e alta estereosseletividade em favor do anômero α (Esquema 55). Segundo os autores, a praticidade sintética do método foi verificada para a síntese estereosseletiva de trissacarídeos. Estudos mecanísticos envolvendo experimentos deuterados validaram a 1,2-adição syn-diastereosseletiva do aceptor a dupla ligação dos doadores armados. A vantagem de "armar" e "desarmar" doadores de glicosila consiste na sua utilização sintética. Por exemplo, ao desarmar um doador de glicosila, um acoplamento seletivo pode ser conseguido.
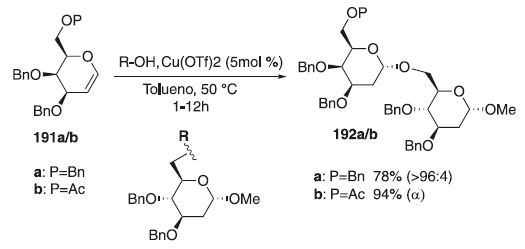 Esquema 55. Glicosilação usando como doador os galactais protegidos 191a/b
Outro método bastante utilizado com a finalidade de obter O-glicopiranosídeo é através da mistura intermediária in situ de epóxidos, permitindo uma alta α-seletividade do produto final, devido ao menor efeito estérico em relação ao C-2, contribuindo para que a mistura glicosídica contenha majoritariamente o α-anômero. De acordo com Marín et al.,112 a reação de glicosilação foi realizada utilizando o doador de glicosila 193 que reage com o ácido m-cloroperbenzóico (MCPBA) para fornecer derivados de manno e alo-1-O-m-clorobenzoato, respectivamente, como resultado da reação de epoxidação syn que após outras etapas reacionais fornecem o dissacarídeo 194 em bom rendimento (49%) e alta estereosseletividade (Esquema 56).
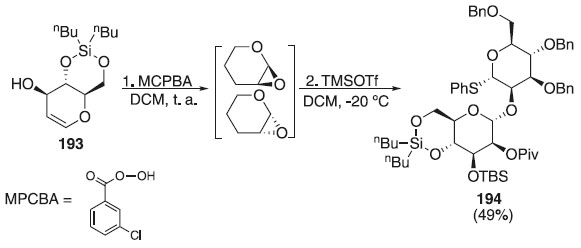 Esquema 56. Glicosilação utilizando doadores de glicosila protegidos através da epoxidação com MCPBA
Por outro lado, Cui et al.113 relataram a síntese exclusiva de 2-deoxy-α-O-D-glicosídeo utilizando doadores de glicosila do tipo 3,4,6-tri-O-benzil-D-glical e galactal 195a/b em presença da mistura de iodeto de trimetilsilano (TMSI) + PPh3 e diferentes aceptores de glicosila para fornecer os compostos finais 196a-h em bons rendimentos (70 - 86%) e alta estereosseletividade em favor do anômero α (> 19:1) (Esquema 57).
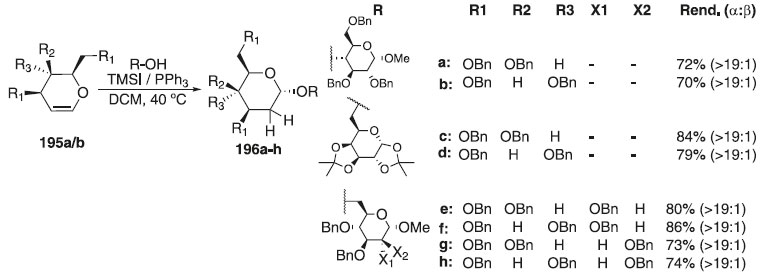 Esquema 57. Glicosilação utilizando doadores 3,4,6-tri-O-benzil-D-glical e galactal em presença de TMSI + PPh3
Em 2015, os pesquisadores Kimura, Takahashi e Toshima114 descreveram reações de glicosilação do doador de glicosila 197 com diferentes álcoois, como aceptores, usando como ativador N-iodosuccinimida (NIS) e uma quantidade catalítica de PPh3 sob condições suaves para fornecer os correspondentes 2-iodo-O-glicopiranosídeo e 2-iodo-O-manopiranosídeo 198a-d, em altos rendimentos (Esquema 58). Convém destacar que a reatividade das iodoglicosilações com PPh3 aumentou significativamente em comparação com a utilização apenas de NIS como ativador.
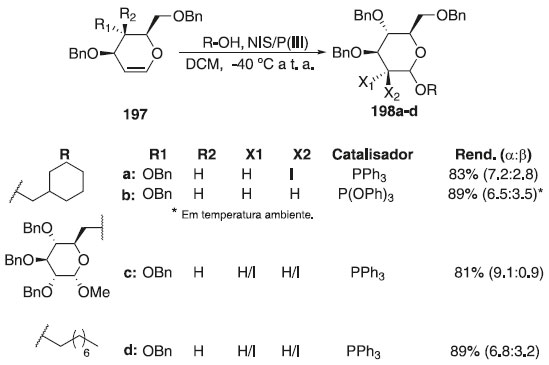 Esquema 58. Glicosilação de glicais com diferentes álcoois usando N-iodosuccinimida (NIS) e PPh3
Por outro lado, Melo e Colaboradores115 relataram que na reação de glicosilação utilizou como doador de glicosila, o tri-O-acetil-D-glical 199, em presença da montmorilonita K-10/5%FeCl3 e dos aceptores alcinóis 200a-j, fornecendo os compostos 201a-j em altos rendimentos e α-estereosseletividades (Esquema 59). Subsequentemente, os glicosídeos obtidos foram acoplados com 2-azido-1,4-naftoquinona para produzir uma nova série de derivados de 1,2,3-1H-triazolil O-glicosídeos através de uma reação "click".
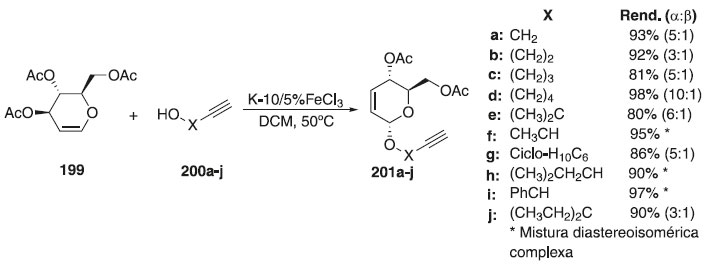 Esquema 59. Glicosilação do doador tri-O-acetil-D-glical em presença da montmorilonita K-10/5%FeCl3
Dando continuidade, Costa et al.116 reagiram tri-O-acetil-D-glical 199, como doador de glicosila, e como aceptor o carbonato de glicerol 202 em presença do BF3∙OEt2 ou montmorilonita K-10 em 5% FeCl3 para fornecer o composto 203 com 84% ou 94%, dependendo do catalisador utilizado (Esquema 60). A cristalização espontânea produziu 28% de um diastereoisômero puro com a configuração (S), conforme determinado por cristalografia de raios-X.
 Esquema 60. Glicosilação do doador tri-O-acetil-D-glical e o carbonato de glicerol como aceptor
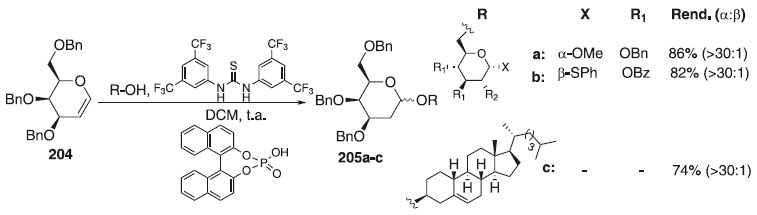
Uma abordagem prática para a síntese α-estereosseletiva de desoxi glicosídeo usando organocatálise cooperativa do tipo ácido de Brønsted foi desenvolvida por Palo-Nieto et al.117 Nessa metodologia, os autores utilizaram como doador glicosila, o glical 204 e como aceptores álcoois primários e secundários, catalisada por tioureia e um ácido de Brønsted-Lowry para fornecer os compostos desejados 205a-c em bons rendimentos e alta esterosseletividades (>30:1) (Esquema 61).
Esquema 61. Glicosilação utilizando como doador de glicosila o D-galactal 203 e como aceptores álcoois primários e secundários catalisada por ácido de Brønsted-Lowry
Outros trabalhos utilizando glicais como doadores de glicosila e diferentes aceptores em reações de glicosilação são descritos.118-120 As aplicações das reações de glicosilação enzimaticamente são altamente estereosseletivas.121 As reações de glicosilação intermolecular comum na ausência de um auxiliar participante geralmente prosseguem com baixa estereosseletividade. As primeiras tentativas de alcançar algum estereocontrole das reações de glicosilações foram dedicadas principalmente ao desenvolvimento dos grupos participantes e à otimização das condições da reação. Mais recentemente, a ênfase da pesquisa está a mudar para a compreensão de outros fatores e aspectos mais fundamentais da glicosilação. Estudos extensos dedicados à conformação, configuração, estereoquímica do material de partida e principais intermediários de reação surgiram.122-126 Diante desses métodos glicosídicos, nota-se a constante evolução na síntese de carboidratos, por excelentes rendimentos e a oportunidade da criação de diferentes moléculas bioativas com aplicação farmacêutica, tornando importância para a sociedade o uso desses compostos. Aplicações na farmacologia Abordagens de reação de glicosilação são amplamente úteis para a síntese de oligossacarídeos, tioaçúcares e glicoconjugados de interesse e relevância para a indústria farmacêutica. O controle da reação de glicosilação é de grande importância durante o desenvolvimento desses fármacos, porque as suas cadeias glicosídicas têm efeitos marcantes na estabilidade, atividade, antigenicidade e farmacodinâmica em organismos intactos. Na Figura 5, são mostrados alguns carboidratos isolados de fonte natural, que também foram obtidos através de uma síntese total, tais como o composto 206, com atividade diurética, o 207 que possui atividade anticâncer e o composto 208 que apresenta adividade antibacteriana.127 Os compostos possuem atividades biológicas e algumas rotas de síntese envolvem reação de glicosilação empregando doadores como brometos de glicosila, fluoretos, iodetos, tricloroacetimidatos, trifluoroacetimidatos de N-fenila, tioglicosídeos, sulfóxidos, heteroaril tioglicosídeos, açúcares 1-hidroxil, 1-O-acetatos e orto-alcinilbenzoatos.127
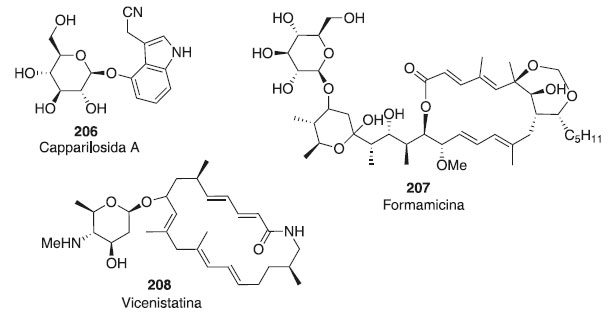 Figura 5. Carboidratos isolados de fontes naturais que também foram obtidos através de reação de glicosilação
Nos últimos anos, houve um grande esforço dedicado à investigação dos papéis dos carboidratos em vários processos biológicos essenciais e no desenvolvimento de carboidratos em medicamentos. Em 2015, também foram incluídas diversas substâncias farmacêuticas já comercializadas a base de carboidratos e com ação terapêutica, antitumoral, anti-hepática, antiviral e dentre outros. A Figura 6 exemplifica dois polissacarídeos, o astrágalo 209 [polissacarídeo com ligação β(1-4) e α(1-6)] e o lentinano 210 [polissacarídeo com ligação β(1-3 e 1-6)], ambos com atividade antitumoral.128
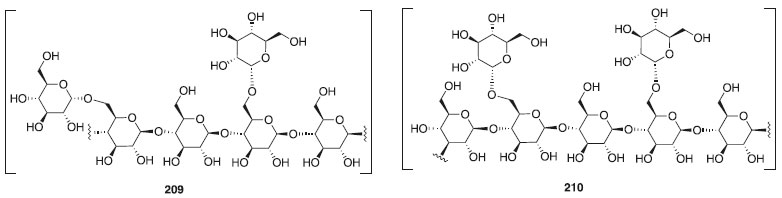 Figura 6. Glicosídeos complexos com atividade antitumoral
A indústria tem demonstrado crescente interesse na utilização de enzimas para várias aplicações nas reações de glicosilação, pois, as mesmas catalisam reações com alta especificidade e são consideradas ambientalmente amigáveis. Existem dois tipos principais de glicosilação: N-glicosilação (ligada a resíduos de asparagina) e O-glicosilação (ligada ao átomo de oxigênio em resíduos de serina ou treonina, geralmente em regiões ricas em prolina).129 Nas vias de síntese de glicanos, suas estruturas variam entre células de mamíferos, leveduras e plantas130 e podem até diferir dentro de um filo.131 A glicosilação pode afetar o dobramento de proteína131-133 estabilidade,134 agregação,135 ligação de substrato,136 dinâmica estrutural137 e atividade catalítica.138 Um grande número de produtos farmacêuticos proteicos atualmente aprovados é glicosilado para exibir uma eficácia terapêutica ideal. Isso se deve ao fato de que a glicosilação pode influenciar uma variedade de processos fisiológicos, tanto nos níveis celulares (por exemplo, direcionamento intracelular) quanto nos níveis de proteínas (por exemplo, ligação proteína-proteína, estabilidade molecular da proteína).139,140 Glicosilação refere-se à ligação covalente de moléculas à base de carboidratos (glicanos) à superfície da proteína. A glicosilação é a mais prevalente e estruturalmente complexa das modificações químicas que ocorrem naturalmente nas proteínas. Nesse contexto, a glicosilação pode exibir heterogeneidade estrutural em relação ao local de ligação do glicano (macro-heterogeneidade) e em relação à estrutura do glicano (micro-heterogeneidade). Além disso, uma vez que todos os potenciais locais de glicosilação não estão ocupados simultaneamente, isso pode levar à formação de glicoformas com diferenças no número de glicanos ligados.141-144Outras aplicações das reações de glicosilação são descritas na literatura.145-147
CONSIDERAÇÕES FINAIS Esta revisão aborda tópicos especiais em química de carboidratos - principais métodos de glicosilação, mecanismos básicos, fatores que influenciam a estereosseletividade e as suas aplicações na síntese de moléculas bioativas. A obtenção de ligações glicosídicas altamente estereosseletivas é uma das tarefas mais desafiadoras da síntese orgânica, pois, é afetada por vários fatores de controle. Conclui-se que nesses 10 anos muitos pesquisadores aprimoraram e se dedicaram a produzir protocolos bastante eficientes, independentes dos doadores e aceptores glicosídicos empregados. A grande parte das metodologias empregadas está interligada a certos grupos de saída, podendo gerar a glicosilação mediante os doadores O-glicosídeo, haleto de glicosila, imidato de glicosila, S-glicosídeo, glicais e intermediários epóxidos, sendo eficientemente consolidados por reagentes específicos, solventes, ativadores e temperatura, conseguindo obter o produto desejado com alta seletividade. A reação de glicosilação não é apenas uma reação de substituição simples, com um perfil de reação SN2 ou SN1. A carga positiva no centro anomérico pode ser estabilizada pelos pares de elétrons não ligantes do átomo de oxigênio do anel, mas é indutivamente desestabilizado pelos substituintes do anel de oxigênio. Em geral, o intrincado equilíbrio de efeitos estereeletrônicos estabilizadores e desestabilizadores no sistema determina quão bem a carga positiva pode ser acomodada durante uma reação de glicosilação. Dependendo da nucleofilicidade do aceptor, espécies eletrofílicas mais fortes ou mais fracas podem ser necessárias para uma glicosilação eficaz. Atualmente, é impossível prever, de antemão, onde no "continuum" da reação SN2-SN1 ocorrerá uma glicosilação e uma visão mais profunda dos fatores que decidem essa posição (reatividade do doador ativado, reatividade do aceitador, papel do solvente) são muito necessários. O progresso na área de glicosilação química melhorou significativamente a nossa capacidade de sintetizar glicosídeos com rendimentos impressionantes e uma melhor estereosseletividade. Logo, esperamos que esta revisão tenha apresentado ao leitor o desafio da glicosilação química, uma variedade de fatores, condições e forças motrizes que influenciam todos os aspectos dessa complexa reação química. Felizmente, o leitor terá informações sobre métodos e estratégias especializados empregados na química moderna dos carboidratos. Essas informações, quando combinadas com o nosso conhecimento existente sobre a reação de glicosilação, os estudos aqui apresentados e os estudos futuros inspirados por esses trabalhos podem servir apenas para aprimorar as nossas capacidades sintéticas no campo desafiador da química de carboidratos.
AGRADECIMENTOS Os autores deste trabalho agradecem a todas as agências de fomento que financiaram a nossa pesquisa: CAPES, CNPq e FACEPE pelas bolsas concedidas aos estudantes de Pós-Graduação.
REFERÊNCIAS 1. Demchenko, A. V. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2008. 2. Mydock, L. K.; Demchenko, A. V.; Org. Biomol. Chem. 2010, 8, 497. 3. Crich, D.; Acc. Chem. Res. 2010, 43, 1144. 4. Paulsen, H.; Angew. Chem., Int. Ed. Engl. 1982, 21, 155. 5. Cid, M. B.; Alfonso, F.; Martín-Lomas, M.; Chem. - Eur. J. 2005, 11, 928. 6. Islam, M.; Gayatri, G.; Hotha, S.; J. Org. Chem. 2015, 80, 7937. 7. Buda, S.; Nawój, M.; Gołębiowska, P.; Dyduch, K.; Michalak, A.; Mlynarski, J.; J. Org. Chem. 2015, 80, 770. 8. Buda, S.; Gołębiowska, P.; Mlynarski, J.; Eur. J. Org. Chem. 2013, 2013, 3988. 9. Komarova, B. S.; Orekhova, M. V.; Tsvetkov, Y. E.; Nifantiev, N. E.; Carbohydr. Res. 2014, 384, 70. 10. Baek, J. Y.; Lee, B.-Y.; Jo, M. G.; Kim, K. S.; J. Am. Chem. Soc. 2009, 131, 17705. 11. Kim, K. S.; Fulse, D. B.; Baek, J. Y.; Lee, B.-Y.; Jeon, H. B.; J. Am. Chem. Soc. 2008, 130, 8537. 12. Lee, Y. J.; Lee, K.; Jung, E. H.; Jeon, H. B.; Kim, K. S.; Org. Lett. 2005, 7, 3263. 13. Ryzhov, I. M.; Korchagina, E. Y.; Popova, I. S.; Tyrtysh, T. V.; Paramonov, A. S.; Bovin, N. V.; Carbohydr. Res. 2016, 430, 59. 14. Lee, J. K.; Bain, A. D.; Berti, P. J.; J. Am. Chem. Soc. 2004, 126, 3769. 15. Schmidt, T. H.; Madsen, R.; Eur. J. Org. Chem. 2007, 3935. 16. Specker, D.; Wittmann, V.; Top. Curr. Chem. 2007, 267, 65. 17. Demchenko, A. V.; Curr. Org. Chem. 2003, 7, 35. 18. Adero, P. O.; Amarasekara, H.; Wen, P.; Bohé, L.; Crich, D.; Chem. Rev. 2018, 118, 17, 8242. 19. Fischer, E.; Ber. Dtsch. Chem. Ges 1893, 26, 2400. 20. Arias, M. B.; Química de los glicósidos, 1st ed., México: Instituto Politécnico Nacional, 2010. 21. Koenigs, W.; Knorr, E.; Chem. Ber. 1901, 34, 957. 22. László, K.; Czakó, B. ; Strategic Applications of Named Reactions in Organic Synthesis, 1st ed., Elsevier : Califórnia, 2005. 23. Helferich, B.; Klein, W.; Liebigs Ann. 1926, 450, 219. 24. Sinaÿ, P.; Pure Appl. Chem. 1978, 50, 1437. 25. Schmidt, R. R.; Angew. Chem., Int. Ed. Engl. 1986, 25, 212. 26. Schmidt, R. R.; Pure Appl. Chem. 1989, 61, 1257. 27. Fugedi, P. ; Garegg, P.J. ; Lonn, H. ; Noberg, T.; Glycoconjugate J. 1987, 4, 97. 28. Fraser-Reid, B.; Konradsson, P.; Mootoo, D. R.; Udodong, U.; J. Chem. Soc., Chem. Commun. 1988, 823. 29. Kahne, D.; Walker, S.; Cheng, Y.; Van Engen, D.; J. Am. Chem. Soc. 1989, 111, 6881. 30. van der Vorm, S.; Hansen, T.; Overkleeft, H. S.; van der Marel, G. A.; Codẽe, J. D. C.; Chem. Sci. 2017, 8, 1867. 31. Crich, D.; Acc. Chem. Res. 2010, 43, 1144. 32. Santana, A, G.; Montalvillo-Jiménez, L.; Díaz-Casado, L; Corzana, F.; Merino, P.; Cañada, F. J.; Jiménez-Osés, G.; Jiménez-Barbero, J.; Gómez, A. M.; Asensio, J. L.; J. Am. Chem. Soc. 2020, 142, 12501. 33. Mydock, L. K.; Demchenko, A. V.; Org. Biomol. Chem. 2010, 8, 497. 34. Nigudkar, S. S.; Demchenko, A. V.; Chem Sci. 2015, 6, 2687. 35. Das, R.; Mukhopadhyay, B.; ChemistryOpen 2016, 5, 401. 36. Frihed, T. G.; Bols, M.; Pedersen, C. M.; Chem. Rev. 2015, 115, 4963. 37. Demchenko, A. V.; Synlett 2003, 1225. 38. Christina, A. E.; van der Marel, G. A.; Codee J. D. C. In Modern Synthetic Methods in Carbohydrate Chemistry; Werz, D. B., Vidal, S., ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2014, pp. 97. 39. Crotti, S.; Adamo R.; Curr. Org. Synth. 2013, 10, 501. 40. Dohi, H.; Nishida, Y.; Tanaka, H.; Kobayashi, K.; Synlett 2001, 1446. 41. Ishiwata, A.; Munemura, Y.; Ito, Y.; Tetrahedron 2008, 64, 92. 42. Nigudkar, S. S.; Demchenko, A. V.; Chem Sci. 2015, 6, 2687. 43. Klimov, E. M.; Malysheva, N. N.; Demchenko, A. V.; Makarova, Z. G.; Zhulin, V. M.; Kochetkov, N. K.; Dokl. Akad. Nauk 1989, 309, 110. 44. Kochetkov, N. K.; Zhulin, V. M.; Klimov, E. M.; Malysheva, N. N.; Makarova, Z. G.; Ott, A. Y.; Carbohydr. Res. 1987, 164, 241. 45. Makoto Sasaki, M.; Gama, Y.; Yasumoto, M.; Ishigami, Y.; Tetrahedron Lett. 1990, 31, 6549. 46. Demchenko, A.; Stauch, T.; Boons, G. J.; Synlett 1997, 818820. 47. Yasomanee, J. P.; Demchenko, A. V.; J. Am Chem Soc. 2012, 134, 20097. 48. Zhu, X.; Schmidt, R. R.; Angew. Chem., Int. Ed. Engl. 2009, 48, 1900. 49. Zhong, W.; Boons, G.-J., In Handbook of Chemical Glycosylation; Demchenko, A. V., ed.;, Wiley-VCH: Weinheim, 2008, p. 261. 50. Fraser-Reid, B.; Jayaprakash, K. N.; López, J. C.; Gómez, A. M.; Uriel, C., In ACS Symp. Ser. (Frontiers in Modern Carbohydrate Chemistry); Demchenko A. V., ed.; Oxford Univ. Press: Oxford, 2007, 960, p. 91. 51. Smith, R. S.; Müller-Bunz, H.; Zhu, X.; Org. Lett. 2016, 18, 3578. 52. Rajaganesh, R.; Jayakumar, J.; Sivaraj, C.; Raaman, N.; Das, T. M.; Carbohydr. Res. 2010, 345, 1649. 53. Zhang, L.; Wei, G.; Du, Y.; Carbohydr. Res. 2010, 345, 2714. 54. Mossotti, M.; Panza, L.; J. Org. Chem. 2011, 76, 9122. 55. Yao, N.; Fung, G.; Malekan, H.; Ye, L.; Kurth, M. J.; Lam, K. S.; Carbohydr. Res. 2010, 345, 2277. 56. Wang, Y.; Liang, X.; Wang, P.; Tetrahedron Lett. 2011, 52, 3912. 57. Liao, J.; Sun, J.; Niu, Y.; Yu, B.; Tetrahedron Lett. 2011, 52, 3075. 58. Matsuo, K.; Nishikawa, K.; Shindo, M.; Tetrahedron Lett. 2011, 52, 5688. 59. Luo, S.-Y.; Tripathi, A.; Zulueta, M. M. L.; Hung, S.; Carbohydr. Res. 2012, 352, 197. 60. Pertel, S. S.; Kononov, L. O.; Zinin, A. I.; Chirva, V. J.; Kakayan, E. S.; Carbohydr. Res. 2012, 356, 172. 61. Reddy, C. R.; Jithender, E.; Prasad, K. R.; J. Org. Chem. 2013, 78, 4251. 62. Tatsumi, S.; Matsumura, F.; Oka, N.; Wada, T.; Tetrahedron Lett. 2013, 54, 3731. 63. Davis, R. A.; Fettinger, J. C.; Gervay-Hague, J.; J. Org. Chem. 2014, 79, 8447. 64. Rasmussen, M. R.; Marqvorsen, M. H. S.; Kristensen, S. K.; Jensen, H. H.; J. Org. Chem. 2014, 79, 11011. 65. Sun, P.; Wang, P.; Zhang, Y.; Zhang, X.; Wang, C.; Liu, S.; Lu, J.; Li, M.; J. Org. Chem. 2015, 80, 4164. 66. Shaw, M.; Thakur, R.; Kumar, A.; J. Org. Chem. 2018, 84, 589. 67. Liang, H.; Ma, L.; Li, C.; Peng, Q.; Wang, Z.; Zhang, Z.-X.; Yu, L.; Liu, H.; An, F.; Xue, W.; Tetrahedron Lett. 2019, 60, 84. 68. Karelin, A. A.; Tsvetkov, Y. E.; Paulovicóvá, L.; Bystrický, S.; Paulovicóvá, E.; Nifantiev, N. E.; Carbohydr. Res. 2010, 345, 1283. 69. Hou, S.; Kovác, P.; Carbohydr. Res. 2011, 346, 1394. 70. Ren, X.; Shen, L.; Muraoka, O.; Cheng, M.; J. Carbohydr. Chem. 2011, 30, 119. 71. Zhang, Y.; Wang, K.; Zhan, Z.; Yang, Y.; Zhao, Y.; Tetrahedron Lett. 2011, 52, 3154. 72. Soulage, C. O.; Grand, L.; Géloën, A.; Chambert, S.; Tetrahedron Lett. 2012, 53, 480. 73. Chen, L.; Tan, Z.; Tetrahedron Lett. 2013, 54, 2190. 74. Cao, B.; Chen, X.; Yamaryo-Botte, Y.; Richardson, M. B.; Martin, K. L.; Khairallah, G. N.; Rupasinghe, T. W. T.; O'Flaherty, R. M.; O'Hair, R. A. J.; Ralton, J. E.; Crellin, P. K.; Coppel, R. L.; Mcconville, M. J.; Williams, S. J.; J. Org. Chem. 2013, 78, 2175. 75. Manabe, S.; Ito, Y.; J. Org. Chem. 2013, 78, 4568. 76. Shirahata, T.; Matsuo, J.; Teruya, S.; Hirata, N.; Kurimoto, T.; Akimoto, N.; Sunazuka, T.; Kaji, E.; Omura, S.; Carbohydr. Res. 2010, 345, 740. 77. Wang, A.; Auzanneau, F. I.; Carbohydr. Res. 2010, 345, 1216. 78. Zhang, Z.; Zong, C.; Song, G.; Lv, G.; Chun, Y.; Wang, P.; Ding, N.; Li, Y.; Carbohydr. Res. 2010, 345, 750. 79. Khaja, S. D.; Kumar, V.; Ahmad, M.; Xue, J.; Matta, K. L.; Tetrahedron Lett. 2010, 51, 4411. 80. Pastore, A.; Matteo, A.; Iadonisi, A.; Silvia, V.; Carbohydr. Res. 2010, 345, 1316. 81. Schmidt, R. R.; Huchel, U.; Tiwari, P.; J. Carbohydr. Chem. 2010, 29, 61. 82. Kalikanda, J.; Li, Z.; Carbohydr. Res. 2011, 346, 2380. 83. Zhu, Y.; Ralph, J.; Tetrahedron Lett. 2011, 52, 3729. 84. Li, Y.; Mo, H.; Lian, G.; Yu, B.; Carbohydr. Res. 2012, 363, 14. 85. Mattson, A. L.; Michel, A. K.; Cloninger, M. J.; Carbohydr. Res. 2012, 347, 142. 86. Sarkar, S.; Dutta, S.; Sen, A. K.; Synthesis 2012, 44, 1079. 87. Ren, S.; Chen, Q.; Ding, N.; Zhang, W.; Li, Y.; Guo, Y.; J. Carbohydr. Chem. 2012, 31, 647. 88. Manzo, E.; Ciavatta, M. L.; Pagano, D.; Fontana, A.; Tetrahedron Lett. 2012, 53, 879. 89. Lu, Y.-J.; Lai, Y.-H.; Lin, Y.-Y.; Wang, Y.-C.; Liang, P.; J. Org. Chem. 2018, 83, 3688. 90. Yang, S.; Liu, Q.; Zhang, G.; Zhang, X.; Zhao, Z.; Lei, P.; J. Org. Chem. 2018, 83, 5897. 91. Mishra, K. B.; Singh, A. K.; Kandasamy, J.; J. Org. Chem. 2018, 83, 4204. 92. Zhang, W.; Meredith, R.; Yoon, M.-K.; Wang, X.; Woods, R. J.; Carmichael, I.; Serianni, A. S.; J. Org. Chem. 2019, 84, 1706. 93. Galan, M. C.; Jouvin, K.; Alvarez-Dorta, D.; Carbohydr. Res. 2010, 345, 45. 94. Kaeothip, S.; Akins, S. J.; Demchenko, A. V.; Carbohydr. Res. 2010, 345, 2146. 95. Beaver, M. G.; Woerpel, K. A.; J. Org. Chem. 2010, 75, 1107. 96. Adamo, R.; Tontini, M.; Brogioni, G.; Romano, M. R.; Costantini, G.; Danieli, E.; Proietti, D.; Berti, F.; Costantino, P.; J. Carbohydr. Chem. 2011, 30, 249. 97. Li, Z.; Zhu, L.; Kalikanda, J.; Tetrahedron Lett. 2011, 52, 5629. 98. Kaeothip, S.; Yasomanee, J. P.; Demchenko, A. V.; J. Org. Chem. 2012, 77, 291. 99. Fang, M.; Gu, L.; Gu, G.; Fang, J.; J. Carbohydr. Chem. 2012, 31, 187. 100. Maity, S. K.; Basu, N.; Ghosh, R.; Carbohydr. Res. 2012, 354, 40. 101. Liu, R.; Wei, A.; J. Carbohydr. Chem. 2012, 31, 384. 102. Yang, F.; Wang, Q.; Yu, B.; Tetrahedron Lett. 2012, 53, 5231. 103. Gao, J.; Guo, Z.; J. Org. Chem. 2013, 78, 12717. 104. Ohara, K.; Lin, C.-C.; Yang, P.-J.; Hung, W.-T.; Yang, W.-B.; Cheng, T.-J. R.; Fang, J.-M.; Wong, C.; J. Org. Chem. 2013, 78, 6390. 105. Padungros, P.; Alberch, L.; Wei, A.; J. Org. Chem. 2014, 79, 2611. 106. Buda, S.; Nawoj́, M.; Gołębiowska, P.; Dyduch, K.; Michalak, A.; Mlynarski, J.; J. Org. Chem. 2015, 80, 770. 107. Dulaney, S. B.; Xu, Y.; Wang, P.; Tiruchinapally, G.; Wang, Z.; Kathawa, J.; El-dakdouki, M. H.; Yang, B.; Liu, J.; Huang, X.; J. Org. Chem. 2015, 80, 12265. 108. Heuckendorff, M.; Poulsen, L. T.; Jensen, H. H.; J. Org. Chem. 2016, 81, 4988. 109. Maiti, K.; Jayaraman, N.; J. Org. Chem. 2016, 81, 4616. 110. Susanto, W.; Kong, K.-H.; Hua, K.-F.; Wu, S.-H.; Lam, Y.; Tetrahedron Lett. 2019, 60, 288. 111. Kumar, M.; Reddy, T. R.; Gurawa, A.; Kashyap, S.; Org. Biomol. Chem. 2020, DOI:10.1039/D0OB01042A. 112. Marín, I.; Castilla, J.; Matheu, M. I.; Díaz, Y.; Castillón, S.; J. Org. Chem. 2011, 76, 9622. 113. Cui, X.-K.; Zhong, M.; Meng, X.-B.; Li, Z.-J.; Carbohydr. Res. 2012, 358, 19. 114. Kimura, T.; Takahashi, D.; Toshima, K.; J. Org. Chem. 2015, 80, 9552. 115. Melo, V. N.; Dantas, W. M.; Camara, C. A.; Oliveira, R. N.; Synthesis 2015, 47, 3529. 116. Costa, P. L. F.; Melo, V. N.; Guimarães, B. M.; Schuler, M.; Pimenta, V.; Rollin, P.; Tatibouët, A.; Oliveira, R. N.; Carbohydr. Res. 2016, 436, 1. 117. Palo-Nieto, C.; Sau, A.; Williams, R.; Galan, M. C.; J. Org. Chem. 2017, 82, 407. 118. Freitas, J. C.R.; Couto, T. R.; Paulino, A. A. S.; Freitas Filho, J. R.; Malvestiti, I.; Oliveira, R. A.; Menezes, P. H.; Tetrahedron 2012, 68, 10611. 119. Srivastava, R. M.; Freitas Filho, J. R.; da Silva, M. J.; Souto, S. C. M.; Carpenter, C. B.; Faustino, W. F.; Tetrahedron 2004, 60, 10761. 120. Freitas Filho, J. R.; Freitas, J. J. R.; Cottier, L.; Sinou, D.; Srivastava, R. M.; J. Chil. Chem. Soc. 2015, 60, 2646. 121. Muthana, S.; Cao, H.; Chen, X.; Curr. Opin. Chem. Biol. 2009, 13, 573. 122. Mydock, L. K.; Demchenko, A. V.; Org. Biomol. Chem. 2010, 8, 497. 123. Crich, D.; Acc. Chem. Res. 2010, 43, 1144. 124. Frihed, T. G.; Bols, M.; Pedersen, C. M.; Chem. Rev. 2015, 115, 4963. 125. Bohé, L.; Crich, D.; Carbohydr. Res. 2015, 403, 48. 126. Jia, X. G.; Demchenko, A. V.; Beilstein J. Org. Chem. 2017, 13, 2028. 127. Yang, Y.; Zhang, X.; Yu, B.; Nat. Prod. Rep. 2015, 32, 1331. 128. Zhang, Y.; Wang, F.; Drug Discov. Ther. 2015, 9, 79. 129. Wayman, J. A.; Glasscock, C.; Mansell, T. J.; DeLisa, M. P.; Varner, J. D.; Metab. Eng. Commun. 2019, 9, e00088. 130. Nadeem, T.; Khan, M. A.; Ijaz, B.; Ahmed, N.; Rahman, Z.; Latif, M. S.; Ali. Q.; 131. Rana, M. A.; Cancer Res. 2018, 78, 2787. 132. Gusakov, A. V.; Antonov, A. I.; Ustinov, B. B.; Carbohydr Res. 2008, 343, 48. 133. Benoit, I.; Asther, M.; Sulzenbacher, G.; Record, E.; Marmuse, L.; Parsiegla, G.; 134. Gimbert, I.; Asther, M.; Bignon, C.; FEBS Lett. 2006, 580, 5815. 135. Hanson, S. R.; Culyba, E. K.; Hsu, T-L.; Wong, C-H.; Kelly, J. W.; Powers, E. T.; Proc. Natl. Acad. Sci. 2009, 106, 3131. 136. Bonzom, C.; Hüttner, S.; Mirgorodskaya, E.; Chong, S-L.; Uthoff, S.; Steinbüche, A.; Verhaert, R. M. D.; Olsson, L.; AMB Expr. 2019, 9, 126. 137. Bosques, C. J.; Imperiali, B.; Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7593. 138. Goettig, P.; Int. J. Mol. Sci. 2016, 17, 1969. 139. Bosques, C. J.; Collins, B. E.; Meador, J. W.; Sarvaiya, H.; Murphy, J. L.; Dellorusso, G.; Bulik, D. A.; Hsu, I.-H.; Washburn, N.; Sipsey, S. F.; Myette, J. R.; Raman, R.; Shriver, Z.; Sasisekharan, R.; Venkataraman, G.; Nat. Biotechnol. 2010, 28, 1153. 140. Sola, R. J.; Griebenow, K.; BioDrugs 2010, 24, 9. 141. Clerc, F.; Reiding, K. R.; Jansen, B. C.; Kammeijer, G. S. M.; Bondt, A.; Wuhrer, M.; Glycoconjugate J. 2016, 33, 309. 142. Dalziel, M.; Crispin, M.; Scanlan, C. N.; Zitzmann, N.; Dwek, R. A.; Science 2014, 343, 1235681. 143. Huttenhain, R.; Surinova, S.; Ossola, R.; Sun, Z.; Campbell, D.; Cerciello, F.; Schiess, R.; Bausch-Fluck, D.; Rosenberger, G.; Chen, J.C.; Rinner, O.; Kusebauch, U.; Hajduch, M.; Moritz, R. L.; Wollscheid, B.; Aebersold, R.; Mol. Cell. Proteomics 2013, 12, 1005. 144. Audfray, A.; Varrot, A.; Imberty, A.; C. R. Chim. 2013, 16, 482. 145. Zhang, Y.; Zhang, H.; Zhao, Y.; Guo, Z.; Gao, J.; Org. Lett. 2020, 22, 1520. 146. Sacco, P.; Cok, M.; Scognamiglio, F.; Pizzolitto, C.; Vecchies, F.; Marfoglia, A.; Marsich, E.; Donati, I.; Molecules 2020, 25, 1534. 147. Busold, S.; Nagy, N. A.; Tas, S. W.; van Ree, R.; Jong, E. C.; Geijtenbeek, T. H.; Front. Immunol. 2020, 11, 1. |
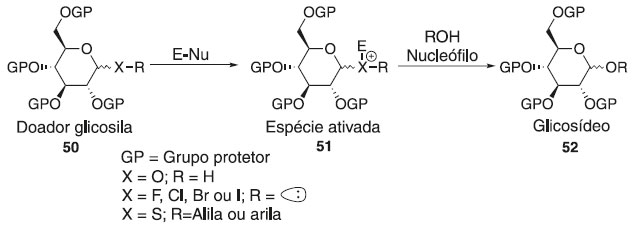
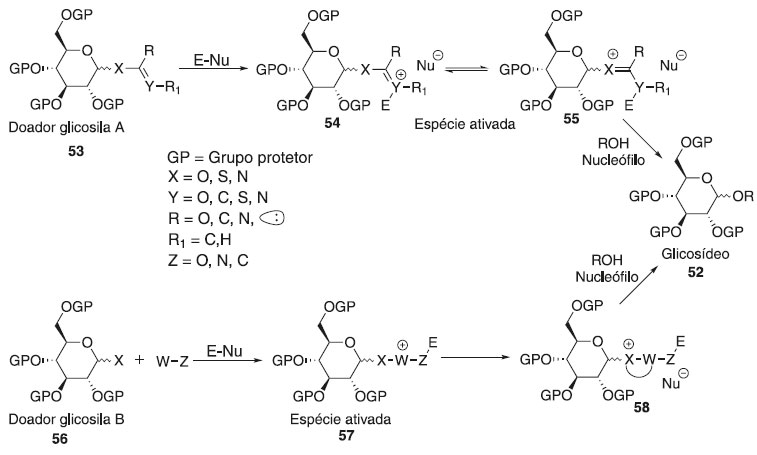
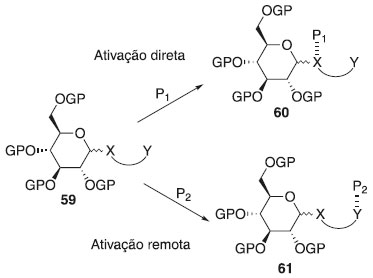
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access