
 |
 |
 |
 |
 |
Educação
|
|
| Uso de softwares livres em aula prática sobre filtros moleculares de biodisponibilidade oral de fármacos The use of free softwares in practical class about the molecular filters of oral bioavailability of drugs |
|
Gabriela dos Santos RodriguesI; Júnior A. AvelinoI; Ariane L. N. SiqueiraI; Luciana F. P. RamosII; Gabriela B. dos SantosI,*
I. Instituto de Saúde Coletiva, Universidade Federal do Oeste do Pará, 68040-070 Santarém - PA, Brasil Recebido em 10/11/2020 *e-mail: gabriela.bds@ufopa.edu.br The study of the physical chemical properties to predict the oral bioavailability of drugs is a widely tool used by researches on the Pharmaceutical and Medicinal Chemistry field. Even though it is a deeply studied subject, it is a complex task to teach to pharmacy students. Lipinski stablished that molecular weight, lipophilicity, hydrogen bond donors and acceptors are of great importance for oral bioavailability. In addition, Veber and Lovering added other properties, such as number of rotatable bonds, fraction of sp3 hybridized carbon and polar surface area that amplified the Lipinski Rule of 5. In this sense, the access to chemical databases and the comprehension of this information is an important task to undergraduate and graduate students. Thus, this work aims a new teaching methodology that values the critical interpretation of data and that favors the students comprehension beyond stablished rules. INTRODUÇÃO Dentre as aplicações práticas da Química Farmacêutica e Medicinal que despertam o interesse da indústria farmacêutica estão aquelas que influenciam a etapa farmacocinética. O estudo das propriedades físico-químicas durante o planejamento de fármacos é uma etapa muito relevante para a predição da biodisponibilidade oral de moléculas promissoras.1 A administração por via oral é preferencial para aplicação de fármacos devido a sua praticidade, maior adesão do paciente, economia e, geralmente, maior segurança na utilização.2 Considerando que a biodisponibilidade de fármacos administrados pela via oral depende da permeabilidade pelas membranas celulares, solubilização nos fluidos estomacais e absorção pela mucosa intestinal, é necessário compreender os descritores moleculares capazes de prever as propriedades farmacocinéticas ideais e fornecer informações relevantes para propor modificações químicas racionais.1,3 Na tentativa de facilitar o processo de tomada de decisão e aumentar a probabilidade de encontrar e desenvolver compostos viáveis farmacologicamente em seres humanos, foram desenvolvidos vários métodos multiparamétricos chamados de regras ou filtros.4 O método mais popular empregado para a predição da absorção oral de novos candidatos a fármacos é a regra de Lipinski (RO5, do inglês, Rule of Five).5 Essa regra prevê que a probabilidade de um fármaco ser biodisponível oralmente está relacionada a propriedades físico-químicas, como peso molecular inferior a 500 Da, coeficiente de partição calculado (cLogP, do inglês, Calculated 1-octanol/water partition coefficients) menor que 5, grupos doadores de ligação hidrogênio (HBD, do inglês, hydrogen bond donors) menor ou igual a 5 e grupos aceptores de ligação de hidrogênio (HBA, do inglês, hydrogen bond acceptors) menor ou igual a 10.6 A RO5 parte da observação de que a maioria dos medicamentos administrados por via oral no mercado são moléculas relativamente pequenas e moderadamente lipofílicas, o que garante a boa solubilidade e permeação, especialmente, daquelas absorvidas por difusão passiva.4 Além das propriedades abordadas por Lipinski, novos descritores foram estudados e auxiliaram na melhor compreensão dos fatores limitantes do processo de permeação celular de forma a complementar a RO5. Entre esses descritores são exemplos a área de superfície polar (TPSA, do inglês, Topological Polar Surface Area), o número de ligações rotacionáveis (NRB, do inglês, Number Rotatable Bonds) e a fração de carbono sp3 (Fsp3).7,8 Veber et al.,8 ao avaliarem mais de 1.100 candidatos a fármacos com ampla diversidade estrutural, demonstraram que a flexibilidade da molécula e a distribuição de grupamentos polares são bons preditores da biodisponibilidade oral. Desse modo, os pesquisadores concluíram que o tamanho da área de superfície polar influencia a absorção intestinal e são considerados ideais os valores de TPSA menores ou iguais a 140 Å2. Ainda, definiu-se o número de ligações rotacionáveis igual ou menor que 10 para uma melhor permeabilidade do fármaco.8 Após isso, Lovering et al.9 correlacionaram o efeito da planaridade na solubilidade de compostos orgânicos. Com base nesse estudo, a planaridade é inversamente proporcional ao número de carbonos sp3. Essa fração pode ser descrita como a razão de carbonos com hibridização sp3 pelo número de carbonos totais do composto. Fármacos com fração de carbono sp3 (Fsp3) igual ou maior que 0,47 apresentam melhor solubilidade devido à capacidade de solvatação pela água.9 Nas últimas décadas, o emprego de novas metodologias didáticas para o ensino de Química Farmacêutica e Medicinal vem resultando em melhoras no processo de ensino-aprendizagem dos cursos de farmácia. Essa evolução tem acompanhado as crescentes publicações de livros e artigos na área de Química Farmacêutica e Medicinal por pesquisadores brasileiros, que têm ampliado e incorporado às aulas experimentais novas abordagens, com conteúdo programático que variam desde práticas envolvendo a síntese, análise de pureza e quantificação de fármacos até o uso de ferramentas computacionais.1,10 Carvalho et al.11 descreveram atividades práticas envolvendo o uso de softwares para Modelagem Molecular, como ChemDraw, Chem3D, Molecular Modeling Pro e ChemSite, para demonstrar a correlação entre a estrutura dos fármacos e atividade biológica.11 Tavares et al.12 propuseram a aplicação de técnicas computacionais para avaliar a evolução dos anti-hipertensivos da classe dos inibidores da enzima conversora de angiotensina (iECA): captopril, enalapril e ramipril. A estratégia objetivou demonstrar aos alunos a análise das interações fármaco-receptor a partir de ferramentas livres, como Protein Data Bank, PDBsum e WebLabViewer Lite 4.2 para demonstração dos aspectos bi e tridimensionais relevantes na interação fármaco-receptor.12 Em outra experiência semelhante conduzida por Tavares et al.,13 o objetivo foi analisar os aspectos moleculares envolvidos na interação dos anti-inflamatórios não esteroidais (AINEs), ibuprofeno e celecoxibe, ao sítio ativo da enzima ciclo-oxigenase-2 (COX-2).13 A utilização de plataformas online pode ser realizada conjuntamente a experimentos de síntese, como demonstra a experiência de Araújo et al.,14 que utilizaram a plataforma SciFinder® como recurso auxiliar ao desenvolvimento de protótipos através de hibridação molecular.14 Por outro lado, poucas são as propostas que possuem como tema o ensino das propriedades físico-químicas que influenciam na biodisponibilidade oral de moléculas por meio de uma abordagem computacional.15 No ensino de Química Farmacêutica e Medicinal, a utilização dessas ferramentas em sala de aula é importante para capacitar os discentes a realizarem ensaios preliminares para avaliação de propriedades físico-químicas, estimular a compreensão de alguns filtros desenvolvidos para predição da biodisponibilidade oral de fármacos e até possibilitar que estabeleçam novos limites para os parâmetros já definidos.16 Sabendo a importância das propriedades físico-químicas de moléculas bioativas para a indústria farmacêutica, um dos maiores desafios no ensino da Química Farmacêutica e Medicinal é contemplar todos os aspectos envolvidos na absorção e biodisponibilidade de fármacos no curso de farmácia. Portanto, para um melhor aprendizado, é importante integrar o ensino teórico ao prático para que se desenvolva no estudante um raciocínio crítico a respeito da influência das propriedades estruturais e físico-químicas dos fármacos na absorção pela via oral.17 Para que isso aconteça, são necessárias novas formas de aprendizado, pois já é notório que o ensino teórico expositivo não é capaz de atingir todos os alunos com a mesma intensidade de uma aula experimental.11,12 Portanto, o objetivo deste trabalho é apresentar uma nova estratégia pedagógica para ensino das propriedades físico-químicas importantes no desenvolvimento de fármacos orais, através do uso de plataformas online em aula prática na disciplina de Química Farmacêutica e Medicinal e/ou outras disciplinas com componentes curriculares equivalentes. As disciplinas de Química Farmacêutica I (04 créditos) e Química Farmacêutica II (04 créditos) têm sido ministradas aos estudantes do sétimo e oitavo semestres, respectivamente, do curso de Farmácia da Universidade Federal do Oeste do Pará (UFOPA). Após análise da infraestrutura institucional e do Projeto Pedagógico do Curso, a estratégia pedagógica aqui apresentada foi cuidadosamente planejada e inserida no plano de atividades da disciplina de Química Farmacêutica II da UFOPA e tem sido aplicada como método alternativo à metodologia puramente expositiva. Essa estratégia pode ser abordada em 5 h, sendo melhor dividida em duas aulas práticas de 2 h e uma aula de 1 h para exposição dos resultados. A partir do desenvolvimento desta atividade, realizada em laboratório de informática, espera-se que sejam alcançados os seguintes objetivos pelos discentes:
Ao reconhecer a importância do profissional farmacêutico para a análise de informações contidas em banco de dados de fármacos e medicamentos, torna-se essencial o direcionamento de profissionais com perfil inovador diante das exigências do mercado farmacêutico atual. Portanto, considerando o potencial de geração de conhecimento a partir de um grande volume de informações disponíveis em bases de dados livres, as habilidades e competências supracitadas são de grande relevância para o cenário farmacêutico atual. Dessa forma, com a presente aula experimental, espera-se que os alunos reconheçam as vantagens e limitações da aplicação dos filtros físico-químicos de Lipinski, Veber ou Lovering, pois será possível notar que diversos fármacos não se enquadram nas regras. Além disso, após uma análise detalhada dos dados obtidos e dos cálculos do P90, poderá ser possível criar um novo filtro físico-químico para a classe de fármacos estudada, demonstrando como as métricas atualmente estabelecidas foram desenvolvidas.
ASPECTOS TEÓRICOS Propriedades físico-químicas de fármacos As propriedades físico-químicas dos grupos funcionais são essenciais na fase farmacodinâmica da ação dos fármacos. Tais grupos influenciam diretamente o reconhecimento molecular, pois a afinidade de um fármaco, isso é, "a tendência de um fármaco se ligar ao seu receptor", é dependente do somatório das forças de interação dos grupos farmacofóricos aos sítios complementares da macromolécula.1,18 Essas propriedades também exercem influência no comportamento farmacocinético (absorção, distribuição, metabolismo e excreção), especialmente, nos medicamentos administrados pela via oral, que é a via de administração mais comum. Essa via, em comparação com a parenteral (intravenosa, intramuscular, subcutânea, intradérmica), apresenta como vantagens a facilidade na administração, maior segurança e adesão do paciente, menor custo, além de ser menos invasiva.1,19 Em contrapartida, a fase farmacocinética é uma das principais causas de descontinuidade da pesquisa de candidatos a fármacos orais nos estágios iniciais dos estudos clínicos.20 As propriedades físico-químicas altamente variáveis repercutem diretamente na absorção (passagem do fármaco de seu local de administração para o plasma) e na biodisponibilidade (fração de uma dose oral que chega à circulação sistêmica na forma de fármaco intacto) de fármacos na biofase.1,18 Um fármaco com comportamento farmacocinético inadequado provavelmente deve ser retirado do desenvolvimento e, se necessário, retornado ao laboratório para otimização da estrutura química. Assim, a previsibilidade e estabilidade do perfil farmacocinético são alguns dos aspectos importantes para que um medicamento chegue ao mercado.21 Regra de Lipinski Lipinski et al.6 analisaram 2.245 compostos provenientes do banco de dados do World Drugs Index e identificaram propriedades fármaco-similar (do inglês drug-like) entre os fármacos disponíveis por via oral. Esse trabalho analisou quatro propriedades físico-químicas entre medicamentos orais e candidatos a fármacos, definindo pontos de cortes comuns a 90% das substâncias analisadas. Essas informações foram traduzidas em um conjunto de regras chamado de Regra dos 5 ou RO5 (do inglês Rule of five), pois os fatores descritos envolviam números múltiplos de cinco:22,23
Considerando que a maioria dos fármacos administrados por via oral são transportados passivamente,1 o aumento do PM pode impedir a difusão passiva através das cadeias laterais alifáticas compactadas na bicamada lipídica das membranas biológicas. Além disso, à medida que o PM aumenta, uma cavidade maior deve ser formada em água para solubilizar o composto, o que pode diminuir a solubilidade. O aumento do peso molecular também reduz a concentração de composto na superfície do epitélio intestinal, em consequência, a absorção também pode ser reduzida.24 A lipofilicidade de um composto é avaliada pelo coeficiente de partição (LogP) que, por sua vez, é a razão entre a concentração da substância na fase orgânica e sua concentração na fase aquosa em um sistema de dois compartimentos em equilíbrio. Experimentalmente, essa propriedade é determinada através do método de shake flask com o emprego de n-octanol na fase orgânica, devido à sua similaridade físico-química com a bicamada de lipídeos das membranas celulares (Equação 1). Fármacos mais lipossolúveis são melhores absorvidos, entretanto, um incremento na lipossolubilidade pode reduzir a absorção, pois a redução da hidrossolubilidade dificulta etapas anteriores, como dissolução e solubilização do fármaco na fase aquosa.1,24 Desse modo, o perfil de lipossolubilidade é descrito como um modelo em parábola bilinear, o qual compreende valores ótimos de LogP na faixa de 1 a 3, capazes de expressar propriedades farmacocinéticas ideais, com prejuízo se houver o aumento progressivo do LogP, uma vez que o balanço hidrofílico-lipofílico é necessário para a absorção.1,25  Uma ligação de hidrogênio ocorre entre um heteroátomo rico em elétrons e um hidrogênio deficiente em elétrons. O hidrogênio é ligado por uma ligação covalente a um átomo eletronegativo (O, N, F) com um ou mais pares de elétrons disponíveis. O grupo funcional que contém um átomo eletronegativo ligado a um H é denominado doador de ligação de hidrogênio, pois fornece o hidrogênio com carga parcial positiva para a ligação. Nessa configuração, o heteroátomo exerce maior atração dos elétrons, ficando com carga negativa em relação ao átomo de hidrogênio que participa da ligação com sua carga parcial positiva (Figura 1A). Já o grupo funcional que fornece o átomo rico em elétrons para receber a ligação de hidrogênio, é chamado de aceptor de ligação de hidrogênio (Figura 1B). Grupos funcionais, como OH e NH2 podem atuar tanto como doadores como aceptores de ligação de hidrogênio. Por apresentar apenas um par de elétrons livres, o nitrogênio atua como aceptor de apenas uma ligação de hidrogênio, enquanto que o oxigênio pode receber duas.22 Apesar de C-H não apresentar polarização suficiente para formar uma ligação de hidrogênio, o grupo diflurometila (F2C-H) é considerado um bom doador de ligação de hidrogênio, devido à introdução de átomos eletronegativos de flúor.1 Lipinski et al.,6 discutiram as implicações dessas propriedades, pois um número excessivo de grupos doadores de ligação de hidrogênio prejudica a permeabilidade através das membranas biológicas.6
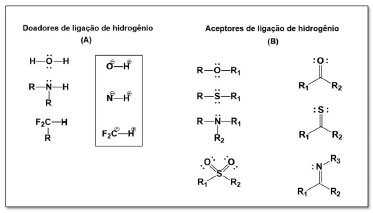 Figura 1. Grupos doadores e aceptores de ligação de hidrogênio. Fonte: Adaptado de Barreiro e Fraga1
A regra de Lipinski foi proposta em resposta ao crescente número de compostos provenientes de síntese química e busca pela potência de fármacos realizadas pela química combinatória. Essa regra visa racionalizar o planejamento de fármacos, especialmente, a procura por compostos líderes em bases de dados, a fim de otimizar a obtenção de moléculas que apresentem características farmacêuticas indesejáveis e reduzida biodisponibilidade oral. Além disso, permitiu correlacionar propriedades físico-químicas a boas propriedades ADME e antecipar a triagem do perfil farmacocinético.24 Entretanto, ressalvas são feitas a esse filtro, uma vez que existem exemplos de fármacos disponíveis no mercado que não obedecem inteiramente à regra, em especial, produtos naturais e semi-sintéticos, moléculas peptídicas e antibióticos também podem não se encaixar nos pontos de corte.7 Assim, a RO5 deve ser empregada com cautela no desenho de moléculas promissoras.26 Regra de Veber Em adição às propriedades discutidas por Lipinski, outros parâmetros são considerados para a avaliação da biodisponibilidade oral. Veber et al.,8 conduziram a análise de banco de dados com mais de 1.100 candidatos a fármacos. Esse estudo demonstrou que a flexibilidade molecular, medida através das ligações rotacionáveis, pode influenciar na biodisponibilidade oral, isso é, quanto maior a flexibilidade da molécula, menor a probabilidade de ser oralmente ativa.8,22 O estudo também demonstrou que a área de superfície polar topológica da molécula (TPSA), relacionada à formação de ligação de hidrogênio, pode ser utilizada como um fator para estimar a biodisponibilidade. Assim os pontos de corte para a regra de Veber são os seguintes:22
Ligações rotacionáveis são definidas como todas as ligações simples acíclicas em que dois átomos da ligação estão conectados a pelo menos dois átomos que não são hidrogênio e não apresentem hibridização sp2 (Figura 2).5,8 A presença de ligações rotacionáveis resulta em conformações significativamente diferentes. Essa definição, portanto, exclui as ligações a substituintes simples, como metil (CH3) ou álcool (O-H), pois a sua rotação não resulta em conformações significativamente diferentes.22 Além disso, ligações amida (C-N) não são consideradas devido à alta barreira de energia rotacional.27 Assim, segundo Veber et al.,8 um menor número de ligações rotacionáveis é ideal para garantir a estabilidade conformacional da molécula e sua passagem através das membranas.8
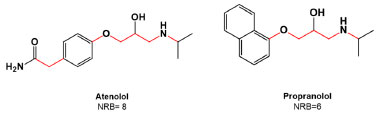 Figura 2. Comparação do número de ligações rotacionáveis (em vermelho) dos anti-hipertensivos atenolol e propranolol
A TPSA é definida como a área de superfície (Å2) ocupada por átomos de nitrogênio e oxigênio ligados ou não a hidrogênio, estando diretamente associada à capacidade de formação das ligações de hidrogênio e à polaridade. Esses dois fatores estão envolvidos na interação da molécula com a superfície polar de membranas celulares e a sua migração para a porção lipídica.28,29 A TPSA é um dos parâmetros utilizados para identificar candidatos a fármacos que podem ser absorvidos em estágios iniciais do processo de descoberta de fármacos.29 Uma TPSA reduzida se correlaciona melhor com o aumento da taxa de permeação do que o coeficiente de partição (LogP), uma vez que a dessolvatação de grupos polares é necessária para a permeação. Por outro lado, a lipofilicidade é necessária para conduzir os solutos até a região interfacial de uma membrana.8 Desse modo, essa dicotomia exige que o fármaco ou candidato a fármaco apresente um balanço hidrofílico-lipofílico, para que se ajuste às características farmacológicas na biofase.1 Regra de Lovering Lovering et al.,9 ao analisar o banco de dados GVK BIO com mais de 2 milhões de compostos em vários estágios do desenvolvimento de medicamentos, verificaram que a complexidade da molécula é um fator significante na transição de candidatos a fármacos da etapa clínica até a aprovação do medicamento. As medidas que refletem essa complexidade molecular são o grau de saturação - definidos pela fração de carbonos com hibridização (Fsp3) - e o número de centros quirais, ou seja, átomos ligados a quatro átomos ou grupos funcionais distintos.9,30 A complexidade da molécula pode ser calculada a partir de ligações arila (grupos derivados de anéis aromáticos), duplas, simples ou número de anéis aromáticos. No entanto, Lovering et al.9 calcularam com base na saturação, a partir da razão entre o número de carbonos hibridizados em sp3 e o número total de carbonos na molécula, demonstrado na equação abaixo: 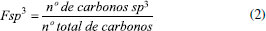 Segundo Lovering et al.,9 níveis aumentados de saturação tornam o composto estruturalmente mais complexo, permitindo a exploração de espaços químicos diversos. Tal efeito pode permitir maior complementaridade com alvos moleculares mais complexos, que não são acessíveis com moléculas planas, por exemplo. Assim, a maior complexidade pode melhorar a potência e/ou especificidade do candidato a fármaco ao sítio ativo, sem aumentar significativamente o peso molecular. Ainda nesse estudo, foi verificado um aumento de 31% da fração saturada de acordo com a progressão clínica, em que a Fsp3 média dos candidatos a fármacos foi de 0,36 enquanto que para os medicamentos aumentou para 0,47. Portanto, uma Fsp3 acima de 0,47 está relacionada à melhora de vários atributos moleculares que contribuem para o sucesso clínico.9
PARTE EXPERIMENTAL Recomenda-se que o presente experimento seja aplicado em duas aulas práticas de 2 h/aula cada e uma aula expositiva de 1 h para apresentação dos resultados obtidos. De forma mais específica, o discente deverá relacionar os objetivos a serem alcançados com a aplicação desta estratégia. O tutorial da aula prática está dividido em cinco etapas, onde cada passo está devidamente descrito e ilustrado no tutorial disponível no material suplementar. Aquisição de estruturas químicas As estruturas dos fármacos a serem analisados serão obtidas a partir da Plataforma Integrity, criada pela editora Thomson Reuters. Essa ferramenta foi desenvolvida para auxiliar setores que investem em pesquisas nas áreas de ciências da saúde e desenvolvimento de fármacos. Cerca de 360 instituições brasileiras têm acesso através da plataforma a uma variedade de informações, como dados biológicos, químicos e farmacológicos de mais de 450 mil compostos com atividade biológica e mais de 2 milhões de citações.31,16 O acesso à plataforma Integrity é realizado a partir de qualquer terminal ligado à internet localizado nas instituições de ensino superior ou conveniadas à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES, através do site www.periodicos.capes.gov.br (Figura 3).32
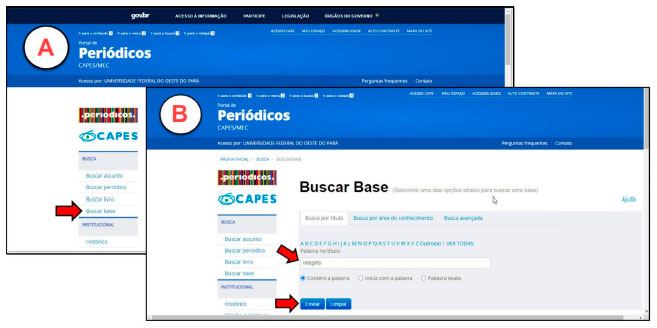 Figura 3. Figura A: página inicial do periódicos capes, seta vermelha indica a Busca na Base. Figura B: Página da busca na base, seta vermelha indica barra de busca
Após acesso ao Portal de Periódicos CAPES, o discente deverá acessar a plataforma Integrity (Figura 4A), onde deverão adquirir as estruturas no formato ".mol" de diversos fármacos. Nesse momento os alunos são orientados a realizar a busca inserindo o nome do fármaco no campo "Drug Name Search" e, em seguida, clicar em "Structure features" para baixar o arquivo do composto (Figura 4A).
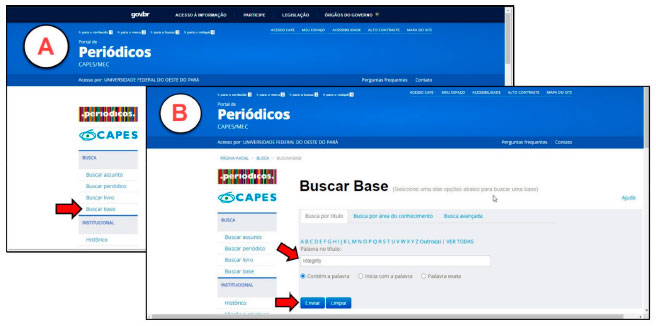 Figura 4. Figura A: Página da plataforma Integrity, setas indicam a aquisição da estrutura em .mol. Figura B: Página da plataforma EMBL-EBI, seta indica a barra de busca
Caso a estrutura química do fármaco em formato ".mol" não esteja disponível na plataforma Integrity, a pesquisa poderá ser realizada também no banco de dados EMBL's European Bioinformatics Institute (EMBL-EBI) (Figura 4B), no qual será possível encontrar informações sobre um grande número de compostos bioativos, fármacos e medicamentos. O acesso está disponível através de uma interface na web em: https://www.ebi.ac.uk/.33 Aquisição da via de administração dos fármacos Após aquisição da estrutura em formato ".mol", o discente é orientado a obter informações sobre a via de administração dos fármacos através da plataforma Integrity com acesso através do periódicos CAPES (Figura 5). Nessa plataforma, os alunos podem acessar uma base de dados que reúne diversas informações sobre os fármacos, como as suas indicações clínicas, grupo terapêutico, alvos, mecanismo celular e molecular.
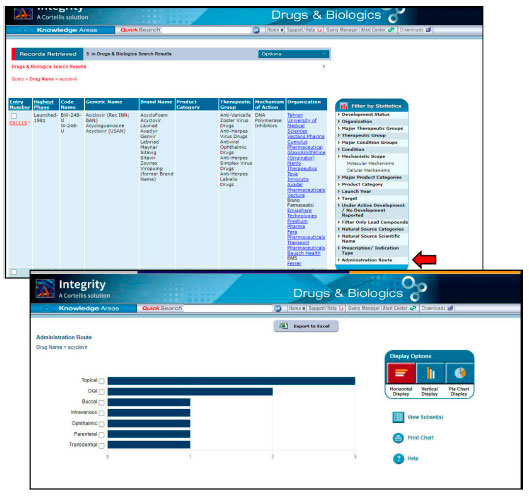 Figura 5. Página para consulta da via de administração na plataforma Integrity
A partir dessas informações, o aluno será capaz de distinguir os fármacos em "oral", quando o medicamento apresentar a via de administração oral, e "não oral", quando o fármaco não apresentar via oral como via de administração. Cabe ao professor orientar que mesmo se o medicamento apresentar inúmeras vias de administração se houver a via oral, esta deve ser a classificação prioritária. Cálculo das propriedades físico-químicas Nesta etapa, os estudantes utilizarão a plataforma online SwissADME (http://www.swissadme.ch/index.php)34 para calcular as propriedades físico-químicas dos fármacos que deverão ser estudados. A ferramenta web SwissADME é gratuita e oferece acesso a uma variedade de modelos preditivos rápidos e robustos para propriedades físico-químicas, farmacocinéticas, semelhança de medicamentos e radar de biodisponibilidade. Além disso, apresenta como vantagem diferentes métodos de entrada (.sdf, .mol, SMILES, por exemplo), cálculo de várias moléculas simultaneamente e possibilidade de exibir, salvar e compartilhar resultados por cada molécula ou através de gráficos que demonstram se os fármacos são capazes de atravessar a barreira hematoencefálica, possuem boa absorção oral e se são substratos ou inibidores da glicoproteína-P (Figura 6).35
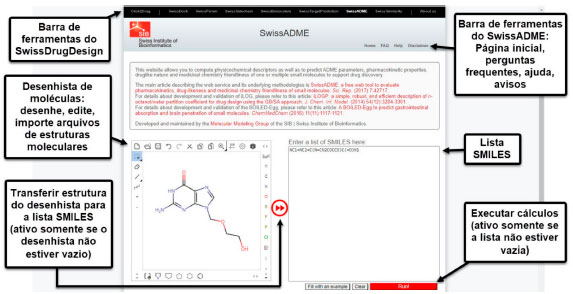 Figura 6. Página principal do SwissADME. Fonte: Adaptado de Daina, Michielin e Zoete35
Portanto, através da ferramenta podem ser calculados os parâmetros físico-químicos a serem considerados na aula: coeficiente de partição (LogP), peso molecular (PM), grupos aceptores de ligação de hidrogênio (HBA), grupos doadores de ligação de hidrogênio (HBD), área de superfície polar topológica (TPSA), número de ligações rotacionáveis (NRB) e fração de carbonos sp3 (Fsp3). Propriedades estruturais e químicas versus rota de administração Além de estabelecer as condições estruturais ideais para predizer a absorção oral de fármacos, Lipinski, Veber e Lovering analisaram as relações entre as propriedades físico-químicas, a fim de verificar possíveis correlações entre estes descritores, como exemplo o aumento da lipofilicidade composta (mais positiva) em detrimento do aumento do peso molecular.5-9 Em vista disso, os estudantes deverão utilizar o LibreOffice Calc (https://pt-br.libreoffice.org/) para gerar gráficos a partir da correlação entre duas das propriedades físico-químicas calculadas, por exemplo PM vs LogP ou HBA vs HBD. Após plotar os gráficos, os estudantes deverão realizar uma avaliação crítica de cada informação gerada, discutindo se cada grupo de moléculas segue ou viola as regras de Lipinski, Veber e Lovering. A RO5 e as demais regras foram baseadas a partir de uma distribuição de propriedade calculadas entre milhares de fármacos. Entretanto, alguns medicamentos de uso oral, como os fármacos peptídicos, estarão fora dos limites da regra.5-9 Sendo assim, o estudante deverá avaliar os resultados e tentar desenvolver ou propor novos filtros para absorção oral da classe de fármacos em estudo. Cálculo do percentil 90º A estatística descritiva é um método científico que auxilia na interpretação de dados coletados. Para uso de dados em literatura científica, é essencial identificar o método estatístico mais apropriado para os diferentes níveis de mensuração, de maneira que os dados brutos sejam descritos por meio de números ou medidas que possam melhor representar as variáveis.36,37 Neste estudo, foram escolhidas as medidas de tendência central (média e mediana) e o cálculo do percentil. As medidas de tendência central são conhecidas por revelarem o valor mais frequente da variável coletada. A média utiliza todos os valores presentes na pesquisa. A análise da média inicia com a contagem do número total de casos (n); em seguida, soma-se os valores obtidos por cada participante da pesquisa e divide-se pelo número total de casos.36 Os percentis (P) são medidas da estatística descritiva que têm como objetivo dividir uma distribuição de frequência, ordenada de forma crescente, em cem partes iguais (Figura 7A).38 Trata-se de uma medida estatística importante, pois representa o comportamento da maior parte da amostra. Lipinski utilizou a medida de percentil 90º para descrever as propriedades importantes que compõe a regra dos cinco.6 Desse modo, 90% dos fármacos orais analisados por Lipinski obedecem às propriedades estabelecidas na RO5.
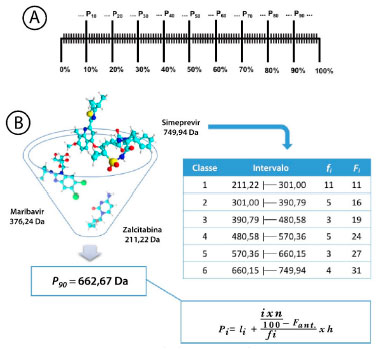 Figura 7. Representação do cálculo de percentil 90º. A: Divisão em cem partes iguais. B: Apresentação da tabela de distribuição de frequência dos pesos moleculares de 35 antivirais (ex. simeprevir, maribavir e zalcitabina) para o cálculo de percentil (P90 = 662,67 Da)
A Figura 7B representa o cálculo de percentil 90º dos pesos moleculares de fármacos antivirais (n=35) (Figuras 18S e 19S) e a tabela de distribuição das frequências simples (fi) e acumulada (Fi). A regra de Sturges (equação 3) é utilizada para determinar o número de classes em função do tamanho da amostra, onde "k" é o número de classes e "n" representa número de amostras:39  A amplitude total da distribuição (h) é obtida a partir da diferença entre o limite superior da última classe (Li) e limite inferior da primeira classe (li) dividido pelo número de classes.39,40 No exemplo representado, os valores de Li e li correspondem ao maior (749,94 Da) e menor peso molecular (211,22 Da) do conjunto de fármacos estudados, respectivamente:  A posição de P90 pode ser encontrada na tabela de distribuição de frequência e substituindo os valores na equação 5, o valor do percentil de interesse pode ser calculado (P90). Onde "li" corresponde ao limite inferior da classe que contém o percentil; "i" o índice de percentil de interesse (ex. 90); "n" é o número de elementos do conjunto de dados; "Fant" é a frequência acumulada da classe anterior à classe que contém o percentil; "fi" é a frequência simples da classe que contém o "Pi"; e "h" corresponde ao valor de amplitude do intervalo de classe encontrado.40 Na representação utilizada neste artigo, 90% dos valores da série de peso molecular dos antivirais analisados são menores ou iguais a 662,67 Da (Figura 7B). 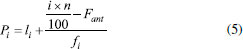 Alternativamente, o discente poderá utilizar um processador de planilha eletrônica e dividir os fármacos em dois grupos, orais e não orais. Em seguida, o estudante deverá calcular o percentil 90º através da fórmula =PERCENTIL(M15:M21;0,90), para cada uma das propriedades. Ao final, esses valores deverão ser comparados com os obtidos por Lipinski, Veber e Lovering.
RESULTADOS E DISCUSSÃO O presente estudo foi aplicado a uma turma de Química Farmacêutica da Universidade Federal do Oeste do Pará. Os alunos foram divididos em grupos, e cada um recebeu uma das seguintes classes de fármaco: antivirais, analgésicos, antineoplásicos, hematológicos, cardioprotetores, antibióticos e antiparasitários. Para a demonstração dos resultados escolhemos a classe dos antivirais, cujos fármacos estão demonstrados nas figuras 18S e 19S do material suplementar. Os fármacos escolhidos foram aprovados pelo Food and Drug Administration (FDA) dos Estados Unidos, destinados ao tratamento das seguintes infecções virais: vírus da imunodeficiência humana (HIV), hepatite B/C, influenza A/B, citomegalovírus, herpes zóster e simplex.41 Dos 35 fármacos selecionados, 88,57% (n=31) apresentam a via oral como forma de administração, enquanto que o restante (11,43%) são administrados unicamente por via parenteral, em específico, a via tópica e inalatória. A Figura 8.1 mostra a relação entre o peso molecular (PM) e a lipofilicidade (LogP) de fármacos classificados como orais e não orais. O PM dos fármacos orais variou de 211,22 Da a 749,94 Da, com média de 419,41 Da; o valor de percentil demonstrou que fármacos com PM até 663,89 Da são bastante favoráveis a absorção oral. Para os fármacos não orais, esse valor não variou significativamente, apresentando média de 314,14 Da e percentil de 347,56 Da (Tabela 1). Todos os fármacos, incluindo aqueles absorvidos por via não oral, apresentaram valores de LogP ≤ 5. Para a via oral, essa propriedade é correlacionada com a atividade biológica, pois acredita-se que fármacos que apresentam maior coeficiente de partição podem se difundir com maior eficiência pelas membranas, aumentando o perfil de biodisponibilidade e, consequentemente, seus efeitos farmacológicos.1 Já compostos com PM muito elevados, com exceção de classes peptídicas, por exemplo, são menos propensos a serem oralmente ativos. Normalmente, à medida que o PM aumenta, o LogP também tende a ser maior.6
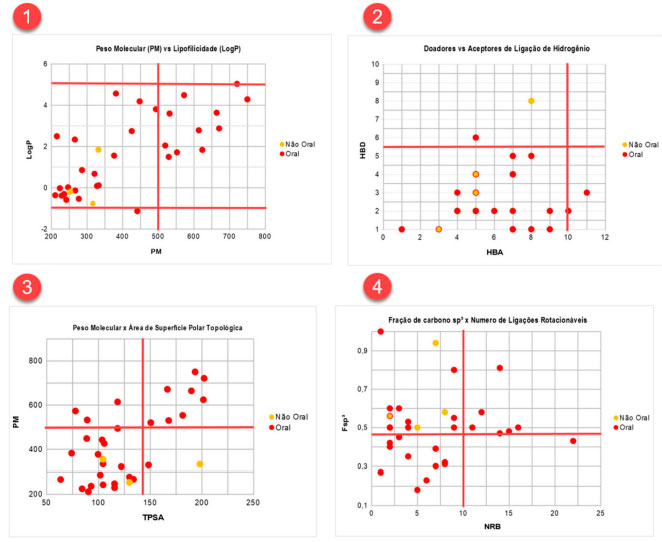 Figura 8. Correlação entre os diferentes descritores físico-químicos. 8.1: Peso molecular versus lipofilicidade; 8.2: Doadores versus aceptores de ligação de hidrogênio; 8.3: Peso molecular versus área de superfície polar topológica; 8.4: Fração de carbono sp3 versus número de ligações rotacionáveis
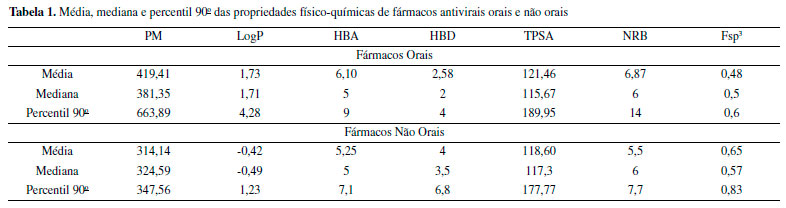
Considerando o número de grupos doadores e aceptores de ligação de hidrogênio (HBD e HBA, respectivamente), a maioria dos fármacos apresentou HBA ≤ 10 e HBD ≤ 5, incluindo os fármacos não orais (Figura 8.2). Os valores médios de HBA e HBD para os fármacos orais foram 6,10 e 2,58, respectivamente, e o valor de percentil corresponde àquele apontado na RO5. Os valores médios de aceptores (HBA = 5,25) e doadores de ligação de hidrogênio (HBD = 4) das moléculas absorvidas por via parenteral também se mostraram dentro da faixa estabelecida no estudo de Lipinski para candidatos orais, exceto o percentil de HBD (P90=6,8). Segundo Lipinski, um número excessivo de grupos doadores ou aceptores de ligação de hidrogênio pode prejudicar a permeabilidade de fármacos através da bicamada lipídica.6 O valor médio e o percentil de TPSA para fármacos orais foi de 121,46 Å2 e 189,95 Å2, respectivamente. Perfil semelhante é observado para os fármacos não orais (média = 118,60 Å2; P90 =177,77 Å2). Na Figura 8.3, nota-se uma correlação positiva entre TPSA e PM, em que os fármacos com PM ≥ 500 Da também demonstram elevada TPSA (≥ 140 Å2). De acordo com Veber et al.,8 um impacto negativo de uma alta superfície polar na absorção intestinal é reconhecido. No que diz respeito ao número de ligações rotacionáveis (NRB), os valores encontrados para os fármacos orais variaram de 1 a 22, com média de 6,87 e percentil de 14. Já para os não orais os valores da média e percentil foram 5,5 e 7,7, respectivamente. Os fármacos orais apresentaram Fsp3 entre 0,18 e 1,00, média de 0,48 e valores próximos de 0,6 são considerados favoráveis para absorção oral (Figura 8.4). Em relação aos fármacos não orais, todos demonstraram valores de Fsp3 superiores a 0,47, variando de 0,5 a 0,94, média de 0,65 e percentil de 0,83. A saturação das moléculas, determinada pela Fsp3, torna as substâncias estruturalmente mais complexas, resultando em maior exploração de espaços químicos.9 Por outro lado, um número alto de ligações rotacionáveis tem um efeito negativo sobre a taxa de permeação.8 Ambas as propriedades são afetadas com aumento do PM.8,9 Apesar de os antivirais não orais avaliados nesse estudo se encaixarem nos limites de propriedades físico-químicas definidos para fármacos orais, alguns pontos devem ser levados em consideração, como toxicidade sistêmica e alta polaridade, que justificam as suas vias de administração. A idoxuridina (Figura 19S), um análogo de timidina indicado para ceratoconjutivite e ceratite provocadas pelo vírus Herpes simplex (HVS), não apresenta toxicidade significativa quando administrada topicamente, porém é altamente tóxica quando administrada por via oral.42,43 O penciclovir (Figura 19S), um análogo de guanina destinado ao tratamento local de herpes labial (HSV-1) e HSV em pacientes imunocomprometidos por via intravenosa, apresenta baixa biodisponibilidade oral (<5%) devido à polaridade. Os dois grupos -OH do penciclovir são mascarados com grupos ésteres que tornam a estrutura menos polar, formando o pró-fármaco famciclovir (Figura 18S), o qual apresenta melhor absorção (~77%).22 Explicação semelhante pode ser dada ao inibidor de neuraminidases de influenza A e B, zanamivir (Figura 19S). Esse fármaco é administrado por via intrapulmonar e sua biodisponibilidade é apenas de 2% (intervalo de 1-5%). A baixa biodisponibilidade oral pode ser explicada pela alta hidrofilicidade resultante de sua natureza polar e zwitteriônica, sendo a última devido à semelhança estrutural com o ácido siálico e presença de grupo guanidina.22,44 A RO5 é melhor aplicada em compostos que são transportados por difusão passiva, desse modo, a violação de dois ou mais parâmetros é considerada como um alerta de baixa absorção ou permeabilidade. No entanto, algumas classes de fármacos, como antibióticos, antifúngicos, cardiotônicos e vitaminas, podem não se encaixar na regra dos cinco. A explicação para isso é que tais fármacos podem apresentar propriedades estruturais que permitem que atuem como substratos de transportadores ativos. Portanto, a regra, apesar de ser uma importante ferramenta de rastreamento no desenvolvimento de fármacos, não impede que químicos medicinais se dediquem a compostos que violem seus parâmetros.6
CONCLUSÕES A proposta foi executada por 20 alunos, todos responderam o questionário. Desses, 65% classificaram seu conhecimento sobre filtros de absorção oral como "bom"; 15% como "muito bom" e 20% como "ruim", antes da realização da atividade. Por outro lado, 85% afirmaram ter melhorado o conhecimento sobre esses filtros após a realização da atividade. 95% dos estudantes relataram não ter dificuldade nas etapas de aquisição das estruturas dos fármacos nas plataformas Integrity e EMBL-EBI; 85% dos alunos não tiveram dificuldades na busca das vias de administração; 70% e 60% não encontraram dificuldades na construção dos gráficos e no cálculo de média, mediana e percentil 90º no LibreOffice Calc, respectivamente. Todos os alunos relataram não ter dificuldades para realizar os cálculos de descritores físico-químicos na plataforma SwissADME e afirmaram ter compreendido o objetivo da proposta didática. Em consonância com as novas tendências do mercado, as diretrizes curriculares atuais estabelecem que o egresso do curso de farmácia deve estar habilitado ao planejamento e desenvolvimento de novos fármacos. Para tal, é necessário estimular o desenvolvimento de habilidades e competências que vão desde a compreensão de propriedades químicas e estruturais até a pesquisa e interpretação de bases de dados, utilizando softwares e análises estatísticas. Assim, é possível a formação de recursos humanos com perfil multidisciplinar e visão abrangente acerca do desenho racional de novas entidades químicas. Portanto, a presente atividade pretende desenvolver um alto valor de competência acerca dos fatores que influenciam na farmacocinética de candidatos a fármacos. Para tal, essa atividade cria a oportunidade de discussão sobre o papel das propriedades físico-químicas e estruturais dos fármacos nos processos de solubilização, absorção e excreção do fármaco no organismo.
MATERIAL SUPLEMENTAR O tutorial completo aplicado aos estudantes referente ao procedimento prático descrito neste trabalho segue amplamente detalhado e ilustrado no material suplementar disponível em: (http://quimicanova.sbq.org.br/) na forma de arquivo PDF, com acesso livre.
REFERÊNCIAS 1. Barreiro, E. J.; Fraga, C. A. M. Em Química Medicinal As Bases Moleculares da Ação dos Fármacos, 3I ed., Artmed: Porto Alegre, 2015, cap. 1. 2. Gilman, A. G.; A. S. Goodman. Em Goodman & Gilman: As bases farmacológicas da terapêutica, 8ª ed., Guanabara Koogan: Rio de Janeiro, 1991, cap. 2. 3. Amaral, A. T.; Montanari, C. A.; Quim. Nova 2002, 25, 39. 4. Mignani, S., Rodrigues, J., Tomas, H., Jalal, R., Singh, P. P., Majoral, J. P.; Vishwakarma, R. A.; Drug Discovery Today 2018, 23, 605. 5. Merlitz, H. B.; Burghardt, B.; Wenzel, W.; Nanotechnology 2003, 1, 44. 6. Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J.; Adv. Drug Delivery Rev. 1997, 3. 7. Santos G. B.; Ganesan A.; Emery F. S.; ChemMedChem 2016, 20, 2245. 8. Veber, D. F.; Johnson, S. R.; Cheng, H. Y.; Smith, B. R.; Ward, K. W.; Kopple, K. D. J.; Med. Chem. 2002, 45, 2615. 9. Lovering, F.; Bikkler J.; Humblet, C.; J. Med. Chem. 2009, 6752. 10. Amaral, A. T.; Andrade, C. H.; Kummerle, A. E.; Guido, R. V. C.; Quim. Nova 2017, 40, 694. 11. Carvalho, I.; Pupo, M. T.; Borges, A. D. L.; Bernardes, L.S.C.; Quim. Nova 2003, 26, 428. 12. Tavares, M. T.; Primi, M. C.; Polli, M. C.; Ferreira, E. I.; Parise-Filho, R.; Quim. Nova 2015, 38, 1117. 13. Tavares, M. T.; Primi, M. C.; Silva, N. A.; Carvalho, C. F.; Cunha, M. R.; Parise-Filho, R.; J. Chem. Educ. 2017, 94, 380. 14. Araújo, C. R. M.; Filho, C. A. L.; Santos, V. L. A.; Maia, G. L.A.; Gonsalves, A. A.; Quim. Nova 2015, 38, 868. 15. Santos, V. L. A.; Gonsalves, A. A.; Araújo, C. R. M.; Quim. Nova 2018, 41, 110. 16. http://www5.usp.br, acessada em Abril de 2018. 17. Lima, L. M.; Quim. Nova 2007, 30, 1456. 18. Rang, R.; Ritter, J. M.; Flower, R. J.; Henderson, G.; Rang & Dale Farmacologia, 8ª ed., Rio de Janeiro: Elsevier, 2015, cap. 8. 19. Allen, J. R.; Loyd, V.; Popovich, N. G.; Ansel, H. C.; Formas Farmacêuticas e Sistemas de Liberação de Fármacos, 9ª ed., Porto Alegre: Artmed Editora, 2013. cap 5. 20. Schuster, D.; Laggner, C.; Langer, T.; Curr. Pharm. Des. 2005, 11, 3545. 21. Goodwin, R.; Giaccone, G.; Calvert, H.; Lobbezzo, M; Eisenhauer, E. A.; Eur. J. Cancer 2012, 48, 170. 22. Patrick, G. L.; An Introduction to Medicinal Chemistry, 5th ed., Oxford: Oxford University Press, 2013. 23. Walters, W. P.; Exp. Opin. Drug Discovery 2012, 7, 99. 24. Di, L.; Kerns, E. H.; Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. Elsevier: Amsterdam, 2015. 25. Kubinyi, H.; J. Med. Chem. 1977, 20, 625. 26. Zhang, M. Q.; Wilkinson, B.; Curr. Opin. Biotechnol. 2007, 18, 478. 27. Verma, A.; Asian Pac. J. Trop. Biomed. 2012, 2, 1735. 28. Pajouhesh, H.; Lenz, G. R.; NeuroRx 2005, 2, 541. 29. Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P.; Pharm. Res. 1997, 14, 568. 30. Kingwell, K.; Nature Rev. Drug Discovery 2009, 8, 931. 31. https://integrity.clarivate.com/integrity/xmlxsl/, acessada em março 2021. 32. https://www-periodicos-capes-gov-br.ezl.periodicos.capes.gov.br/, acessada em março 2021. 33. https://www.ebi.ac.uk/, acessada em março 2021. 34. http://www.swissadme.ch/, acessada em março 2021. 35. Daina, A.; Michielin, O.; Zoete, V.; Sci. Rep. 2017, 7, 42717. 36. Twycross, A.; Shields, L.; Paediatr. Nurs. 2004, 16, 28. 37. McHugh, M. L.; Hudson‐Barr, D.; Journal for Specialists in Pediatric Nursing 2003, 8, 111. 38. Falco, J. G.; Estatística aplicada, Ed. UFMT: Cuiabá, 2008. 39. Silva, J. L. C.; Fernandes, M. W.; Almeida, R. L. F.; Estatística e Probabilidade, 3ª ed., EdUECE: Fortaleza, 2015, cap. 5. 40. Silva, E. M.; Gonçalves, W.; Silva, E. M.; Murolo, A. C.; Estatística 1, 2ª ed., Atlas: São Paulo, 1996, cap. 4. 41. https://go.drugbank.com/, acessada em março 2021. 42. Paintsil, E.; Cheng, Y. C.; Em Encyclopedia of Microbiology, Schaechter, M., ed.; 3I ed., Elsevier Inc.: Amsterdam, 2009, pp. 223-257. 43. Wiltink, E. H. H.; Janknegt, R.; Pharm. Weekbl. 1991, 13, 58. 44. Shanmugam, S.; Ther. Delivery 2015, 6, 403. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access