
 |
 |
 |
 |
 |
Revisão
|
|
| Redução de CO2 em hidrocarbonetos e oxigenados: fundamentos, estratégias e desafios CO2 reduction to hydrocarbons and oxygenates: fundamentals, strategies and challenges |
|
Gelson T. S. T. da SilvaI; Osmando F. LopesII; Eduardo H. DiasI,III; Juliana A. TorresI; André E. NogueiraIV; Leandro A. FaustinoII; Fernando S. PradoII; Antônio O. T. PatrocínioII,*; Caue RibeiroI,#
I. Laboratório Nacional de Nanotecnologia para o Agronegócio, Embrapa Instrumentação, 13561-206 São Carlos - SP, Brasil Recebido em 24/10/2020 *e-mail: otaviopatrocinio@ufu.br The development of renewable energy sources (e.g., solar and wind) moves foward, the tendance for replacing fossil fuels increases. However, these technologies have as primary barriers to industrial processes' efficiency and especially storage. Thus, CO2 reduction routes using these energy sources could chemically store part of the energy as fuels or chemicals, offering alternatives to current oil and gas industry. This process is inspired by photosynthesis, e.g., photochemical or electrochemical processes, using homogeneous or heterogeneous catalysts. Nevertheless, this reaction is thermodynamically unfavorable and has very slow kinetics, given the high stability of the CO2 molecule and the complexity of the redox reactions involved. Therefore, this review addresses this process's kinetic and thermodynamic challenges, and the fundamental concepts of the photo(electro)chemical processes for CO2 reduction, besides presenting and discussing the materials with the potential to act as catalysts. The main reaction mechanisms and advances in the understanding of such processes are discussed, as well as future perspectives INTRODUÇÃO O elevado crescimento populacional tem resultado em um aumento exponencial pela demanda energética mundial. A utilização de fontes energéticas não-renováveis como os combustíveis fósseis tem levado a um aumento drástico na emissão de CO2 nas últimas décadas, o qual vem se acumulando na atmosfera, deixando assim o seu ciclo desequilibrado e gerando diversos problemas ambientais.1,2 Embora a presença desse gás seja vital para vida humana, em excesso, dificulta a dissipação do calor induzido pelos raios solares devido à sua alta capacidade de absorção de radiação na região do infravermelho. Na tentativa de minimizar os problemas causados pelo acúmulo de CO2 na atmosfera, organizações governamentais têm imposto leis para o controle da emissão de gases na atmosfera, como por exemplo, o Protocolo de Kyoto e o Acordo de Paris.3,4 Entre 2008-2018 a emissão mundial de CO2 cresceu ~17%, com aumento no Brasil de ~19% com grande proporção relacionada ao consumo de energias fósseis (Figura 1).5 Ainda, no país, um dos principais combustíveis líquidos é o etanol de cana-de-açúcar.6 Embora renovável e com "pegada de carbono" inferior à gasolina, diesel e querosene, a cada 789 kg de etanol produzido gera-se entre 461 a 572 kg de CO2 de alta pureza durante a fermentação.7 Esse setor pode ser um alvo de atuação da conversão de CO2 agregando valor ao subproduto das reações e reduzindo o impacto ambiental desse biocombustível. Isso demonstra a necessidade de se desenvolver alternativas sustentáveis de energia, associando tecnologias capazes de neutralizar o balanço de CO2 e agregar valor aos produtos formados. Energias renováveis, tais como a solar e eólica, têm, no entanto, sérios problemas relacionados à sua intermitência e/ou sazonalidade ao longo de um dia ou ano.8 Assim, utilizá-las na conversão de CO2 em produtos como metano, metanol e outras moléculas orgânicas seria uma forma de estocagem.9,10 Esse processo pode ser conduzido via redução eletroquímica ou fotoquímica, apesar de que sua eficiência seja ainda limitada, inviabilizando sua aplicação atual. Nesse cenário, avanços nos processos catalíticos para promover essas reações são essenciais.
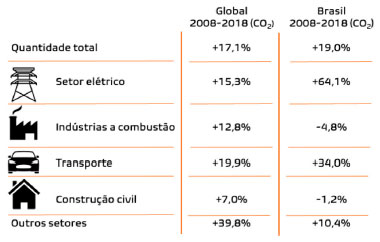 Figura 1. Dados de geração de CO2 no Brasil, por setor, comparado ao restante do mundo.5
Nesse trabalho de revisão serão descritos os principais avanços na conversão fotoquímica e eletroquímica de CO2 em combustíveis e matérias-primas utilizando catalisadores homogêneos e heterogêneos. Serão apresentados os fundamentos da redução de CO2 e os principais desafios termodinâmicos e cinéticos desse processo, seguido pelos princípios da conversão fotoquímica de CO2 utilizando catalisadores moleculares. A seguir, os principais avanços na utilização de semicondutores (óxidos metálicos e materiais metal-free) na conversão fotocatalítica de CO2 serão discutidos. Em seguida, potencial da aplicação do processo de redução eletroquímica de CO2 e os principais avanços relacionados ao desenvolvimento de catalisadores metálicos e arquitetura da célula eletroquímica serão abordados. Por fim, as perspectivas e principais conclusões na área de redução de CO2 pelos diferentes processos serão discutidos de forma crítica.
FUNDAMENTOS DA REDUÇÃO DE CO2 A conversão do CO2 em produtos de maior valor agregado é baseada nos princípios da fotossíntese, mecanismo utilizado por plantas para conversão de CO2 e água em hidrocarbonetos (CO2 + H2O + (h+ + e-) → carboidratos + O2).11 As reações de oxirredução envolvendo o CO2 e H2O para obtenção de diferentes produtos como o CO, CH4 e CH3OH são endotérmicas (ΔrH > 0) e não espontâneas (ΔrG > 0), e.g., DGoCH4 = +818 kJ mol-1.12 O CO2 é quimicamente inerte devido a suas propriedades eletrônicas e geométricas, e apresenta elevada estabilidade termodinâmica devido à alta energia das duplas ligações (750 kJ mol-1).13 Portanto, a redução de CO2 requer energia externa, que pode ser proveniente de uma fonte de radiação (processo fotoquímico), por uma fonte de energia elétrica, i.e., aplicação de uma diferença de potencial elétrico (processo eletroquímico) ou pode ser realizado através do acoplamento desses dois processos num sistema denominado fotoeletroquímico.14-16 Nos processos citados, as reações de redução de CO2 empregando catalisadores heterogêneos em processos eletroquímicos e fotoquímicos ocorrem em três etapas principais: i) adsorção de CO2 (agente oxidante) e/ou de H2O (agente redutor), ii) redução do CO2 (transferência de elétrons) e iii) dessorção dos produtos.13 As etapas de adsorção e ativação de CO2 sobre os sítios ativos geralmente governam a atividade do processo. No entanto, a seletividade depende das três etapas.17-19 O controle do processo de adsorção do CO2 interfere no processo de redução.19,20 Do ponto de vista cinético, a estrutura dos poros e a área superficial específica dos materiais afetam a probabilidade de adsorção. O CO2 pode ser ligado à superfície do material de três maneiras diferentes, (Figura 2).21 Quando a adsorção ocorre pelos átomos de oxigênio (Figura 2a), que doam seus pares de elétrons livres para os centros ácidos de Lewis da superfície do material, a adsorção leva à formação das espécies parcialmente carregadas (CO2•-). O radical CO2•- é um intermediário essencial para a redução, entretanto, exige potencial elevado (-1,85 V vs. EPH).22 No CO2 adsorvido, a transferência de elétron para formação desse radical é facilitada. O CO2 também pode interagir através do átomo de carbono, comportando-se como um ácido de Lewis e adquirindo elétrons dos sítios de base de Lewis do catalisador, resultando em uma estrutura semelhante a um carbonato (Figura 2b). Também pode ocorrer uma interação mista, em que os átomos de oxigênio e de carbono do CO2 atuam como um doador e receptor de elétrons ao mesmo tempo, como mostrado na Figura 2c.21
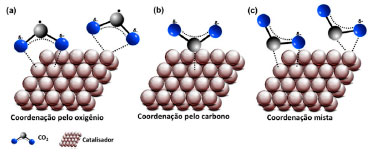 Figura 2. Esquema representativo das possíveis espécies de CO2 adsorvida sobre a superfície de um catalisador. Adaptado de Gattrell et. al.21
Na catalise heterogênea, um aspecto importante da transferência de elétrons para a molécula de CO2 é a posição da banda de condução dos semicondutores, que influencia na seletividade dos produtos gerados. (Tabela 1).22 A redução ocorre em várias etapas, envolvendo a participação de 2, 6, 8 ou 12 elétrons e prótons. Isso torna a cinética do processo extremamente complexa e, além dos desafios termodinâmicos, essa reação tende a apresentar uma cinética lenta. Diversos produtos podem ser gerados, incluindo compostos C1, como moléculas CO, CH4, HCOOH, CH3OH, HCHO e C2, como CH2CH2, C2H5OH e CH3COOH, o que torna o controle da seletividade bastante difícil.23

No processo catalítico homogêneo, como aqueles que utilizam compostos de coordenação, a escolha dos ligantes e centros metálicos exerce papel fundamental. É possível modular as propriedades dos catalisadores utilizando conceitos de engenharia molecular, ou seja, controlar propriedades fundamentais, como por exemplo o tempo de vida de um estado excitado, os potenciais redox ou a labilidade de um dos ligantes.24-26 Nesse contexto, o mecanismo via fotocatálise para redução de CO2 ainda é alvo de intensa discussão na literatura, mas usualmente envolve a supressão redutiva de um estado excitado 3MLCT (Metal-to-ligand charge transfer).27,28 Tal estado pode ser proveniente do emprego de um fotossensibilizador (e.g., [Ru(bpy)3]2+ e seus derivados), ou o catalisador pode também atuar como centro absorvedor. Assim, o fotocatalisador absorve a luz e gera um estado 3MLCT que deve possuir tempo de vida longo o suficiente para que ocorra a supressão redutiva por um agente de sacrifício (D).29 Tipicamente são utilizados supressores como trietanolamina (TEOA), trietilamina (TEA), 1-benzil-1,4-dihidronicotiamida (BNAH) e 1,3-dimetil-2-fenil-2,3-dihidro-1H-benzo[d]imidazol (BIH).30-32 No caso de sistemas com fotossensibilizador, deve ocorrer a transferência eletrônica da espécie fotoreduzida para o catalisador. Após essa etapa, o catalisador reduzido interage com moléculas de CO2 para formação de adutos, seja por meio de alterações na primeira esfera de coordenação ou por meio de interações intermoleculares específicas como ligações de hidrogênio. Posteriormente, esses adutos são novamente reduzidos para geração dos produtos de redução do CO2 e liberados para o meio, permitindo o reinício do ciclo.31 Já o processo eletrocatalítico dispensa o uso de supressores. Isso porque o estado catalítico pode ser alcançado por meio de um potencial eletroquímico externo. Ainda assim, o mecanismo eletrocatalítico possui etapas em comum com o processo fotocatalítico.33 No mecanismo eletrocatalítico ocorre a redução do centro metálico por sucessivas transferências eletrônicas e consequentemente a perda de um ligante ancilar, ponto em comum com o mecanismo fotocatalítico.34-36 Na Figura 3, um esquema geral para os processos foto e eletroquímicos é apresentado para complexos carbonílicos.
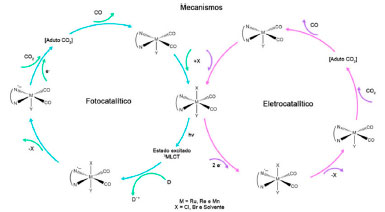 Figura 3. Mecanismos genéricos para redução de CO2 a CO via foto e eletrocatálise. Adaptado de Takeda et al e Manbeck et al.31,34
CATALISADORES MOLECULARES PARA REDUÇÃO DE CO2 Diversos catalisadores moleculares baseados em compostos de coordenação para a redução de CO2 vem sendo estudados. Destacam-se compostos à base de Ru(II), Ir(III), Re(I) e Mn(II) (Tabela 2 e 3), detalhados a seguir. Rutênio
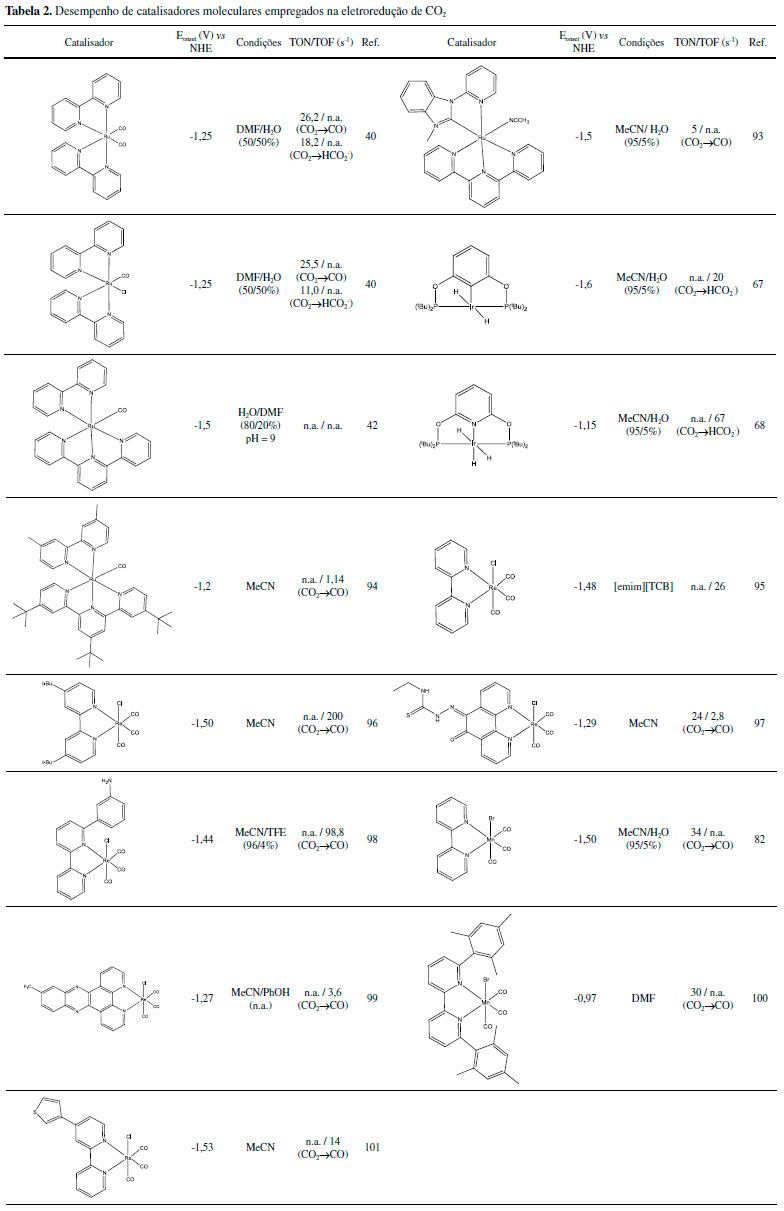
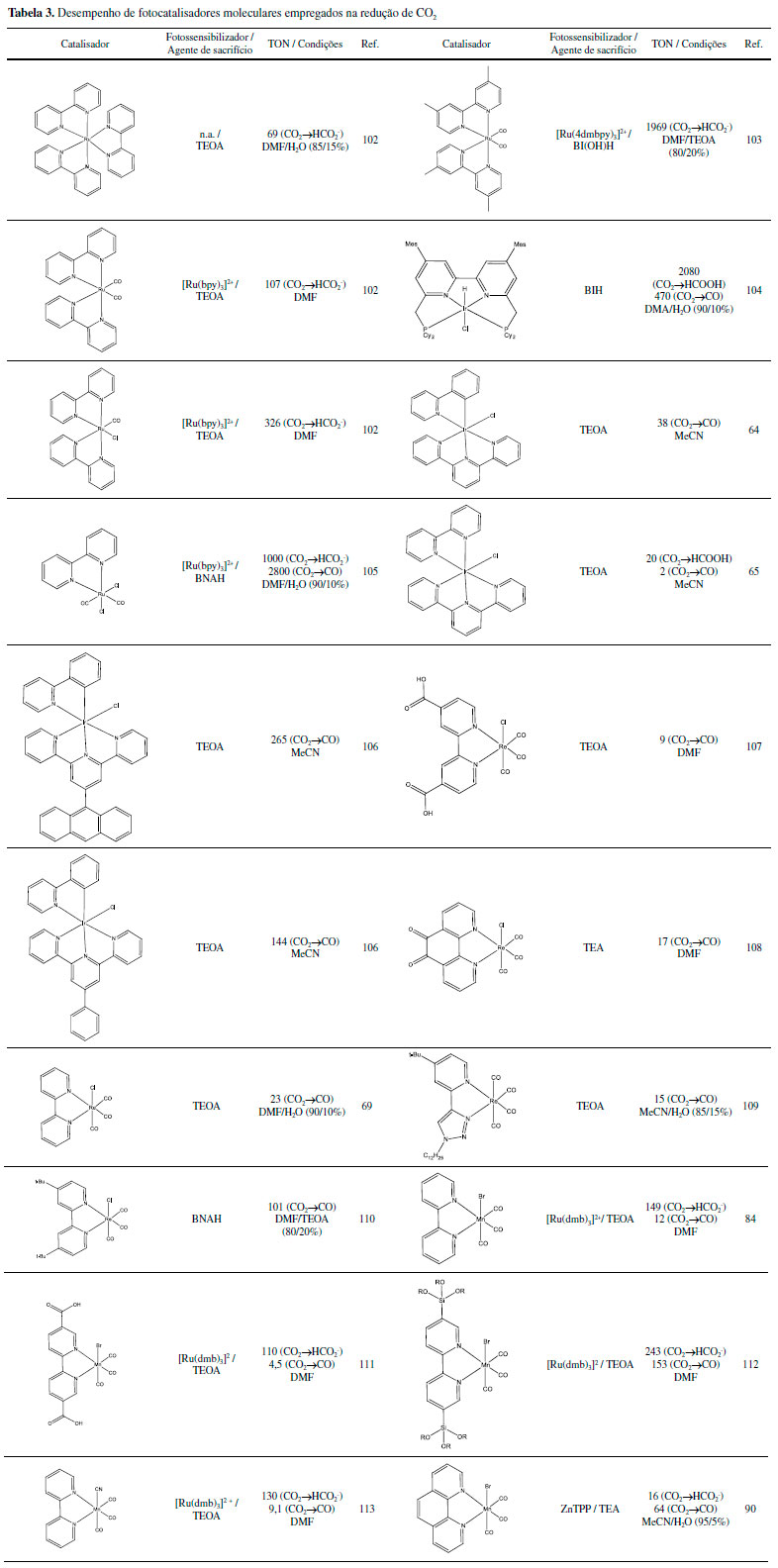
Diversos complexos de Ru(II) são capazes de atuar como foto ou eletrocatalisadores produzindo CO, ácido fórmico, metanol, metilamina e formamida, entre outros.37 Tanaka e colaboradores foram pioneiros no estudo do complexo [Ru(bpy)2(CO)2](PF6)2, bpy=2,2'-bipiridina como eletrocatalisador38,39 assim como na investigação de detalhes mecanísticos, condições e produtos formados utilizando diferentes ligantes.40-44 Desde então, diversos compostos de estrutura [Ru(bpy)2(CO)n(X)m](2-m)+ foram sintetizados e investigados.45 Aspectos da atividade catalítica envolvem o pH do meio e a estabilidade dos compostos. Em pHs ligeiramente ácidos os produtos majoritários são CO e H2. Porém, em condições mais básicas, a espécie HCOO- torna-se o produto majoritário.41,45 A dissociação dos ligantes do tipo bpy (2,2'-bipiridina) e posterior eletropolimerização do centro metálico na superfície do eletrodo foi também reportada como caminhos de desativação do catalisador.46,47 A substituição de alguns ligantes do tipo bpy pelo ligante tpy (2,2';6',2"-terpiridina) auxiliou na prevenção da eletropolimerização.48 Foram propostos mecanismos para a ativação do CO2 utilizando complexos contendo ligantes como bpy (formação de um intermediário hidreto, após a redução do catalisador por dois elétrons) e tpy (CO2 desloca diretamente o solvente coordenado ao catalisador reduzido),45,49,50 porém, o mecanismo ainda não foi totalmente desvendado. Na fotocatálise, os compostos de Ru(II) tem atraído a atenção uma vez que possuem uma intensa absorção de luz na região visível e possuem estados excitados do tipo MLCT de menor energia com longos tempos de vida.50-52 Como o processo, usualmente, envolve a supressão redutiva do estado excitado termicamente equilibrado, os complexos de Ru(II) atuam como fotossensibilizadores e fotocatalisadores.29 Os mecanismos propostos possuem diversas etapas:53 após a excitação do fotossensibilizador, ocorre a supressão redutiva do estado excitado com a fotooxidação do doador de elétrons, tipicamente TEOA ou BNAH. O radical [Ru(bpy)3]•+ formado é oxidado pelo catalisador. Nesse ponto, um dos ligantes ancilares do catalisador é labilizado e o CO2 coordena-se ao metal. A presença de água e a variação do pH do meio determinarão o produto formado, se CO ou HCOO-. Irídio Complexos de irídio têm boa interação com CO2,54-56 formando por exemplo adutos coordenados como reportado para o complexo [Ir(dmpe)2Cl] (dmpe = 1,2-bis(dimetilfosfino)etano).57 Inoue e colaboradores descreveram a hidrogenação catalítica de CO2 gerando ácido fórmico pelo Ir(H)3(PPh3)3 com um turnover (TON) igual a 15.58 Já foram reportados catalisadores de Ir(III) com TON = 222 000 para formação de HCOOH.59 Novas arquiteturas de ligantes já alcançaram TON de até 3,5x106.60 Na hidrogenação catalítica a partir de misturas de CO2 e H2, os catalisadores são solubilizados no solvente apropriado, usualmente água ou THF, e inseridos em um reator. Este é pressurizado com CO2 e H2 em razões conhecidas e aquecido por um determinado tempo.59-62 Estruturas de catalisadores reconhecidamente eficientes para redução de CO2 são apresentadas na Figura 4, cujos mecanismos envolvem a formação de um metal hidreto e posterior redução da molécula de CO2.60-63
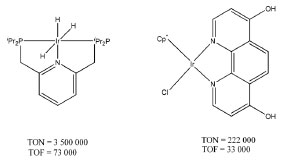 Figura 4. Estrutura dos complexos de Ir(III) para hidrogenação catalítica de CO2.59,60
Apesar desses catalisadores possuírem alto desempenho, as condições de catálise (temperaturas e pressão) são extremas, um agravante em termos práticos.61 Assim, busca-se também empregar complexos de Ir(III) como fotocatalisadores. Apesar desses possuírem ampla faixa de absorção de luz visível e tempo de vida do estado excitado relativamente longo e centrado em estados 3MLCT, apenas recentemente um fotocatalisador baseado em complexos de Ir(III) foi reportado.29,64 O mecanismo de ação é similar aos complexos de Ru(II), envolvendo a supressão redutiva do estado 3MLCT, seguido pela formação de um metal hidreto responsável pela ativação do CO2.65 Os complexos de Ir(III) também podem ser ativados eletroquimicamente,66 com sobrepotenciais que variam entre -0,87 a -1,6V vs NHE e TOF de até 20s-1, sendo formato (HCOO-) o principal produto gerado.65,67-69 Rênio Complexos do tipo fac-[Re(N,N)(CO)3X] (N,N = 2,2'-bipiridina ou 1,10-fenantrolina; X = Cl ou Br) foram propostos como substitutos para [Ru(bpy)3]2+ na fotorredução de CO2.69 Esses complexos apresentam maior seletividade frente a outras reações como evolução de H2, mas possuem menor absorção na região visível. Houve maior investigação das propriedades desses compostos, principalmente, variando os ligantes polipiridínicos,70-77 como por exemplo, a inserção de um grupo aquo como ligante, que permite a solubilização em água mantendo boa atividade eletrocatalítica.75,78,79 Nesses casos o mecanismo de redução de CO2 segue as mesmas premissas descritas para os complexos de Ru(II): o composto é fotorreduzido por um doador de elétrons (D), levando a labilização do ligante ancilar e a formação de um aduto com o CO2. A natureza desse aduto tem sido alvo de intensas investigações mecanísticas75 e depende tanto do meio de reação, bem como do agente de sacrifício empregado. Os complexos de Re(I) também podem atuam como eletrocatalisadores.80 Nesse caso, a aplicação de potenciais catódicos leva tipicamente à redução do ligante N,N e em seguida a redução do centro metálico com consequente dissociação do ligante ancilar. O CO2 associa-se ao centro metálico sendo reduzido a CO ou HCOOH, dependendo do meio e do complexo utilizado. Manganês Catalisadores polipiridínicos de Mn(I) são desejáveis, em comparação a seus análogos de Re(I), devido à maior abundância do manganês e menor custo. Johnson e colaboradores estudaram o complexo fac-[Mn(bpy)(CO)3X] na conversão de CO2, relatando que em THF anidro e em MeCN anidro os mesmos não possuíam atividade, devido a formação de radicais fac-[Mn(bpy)(CO)3], que posteriormente se reduz a fac-[Mn(bpy)(CO)3]-, formando o dímero fac-[Mn(bpy)(CO)3]2.81 Deronzier e colaboradores, relataram o emprego de complexos polipiridínicos de Mn(I), de forma molecular fac-[MnBr(bpy)(CO)3], na eletrocatálise de CO2 em CO em MeCN/H2O (95/5%).82 Nessa vertente, não podemos desconsiderar a semelhança dos complexos fac-[MnBr(bpy)(CO)3] e fac-[ReCl(bpy)(CO)3]. Kubiak e colaboradores elucidaram a eletrocatálise de CO2 comparando os dois complexos.83 Observou-se que o complexo fac-[MnBr(bpy)(CO)3] não atua como a espécie catalítica ativa na redução do CO2 em CO; em vez disso, há a redução de um elétron e a perda do ligante brometo, formando o dímero [Mn2(bpy)2(CO)6], seguido por uma redução subsequente de um elétron para produzir o catalisador ativo, [Mn0(bpy)(CO)3]-. Foi observado também que os catalisadores de manganês reduzem o CO2 em um sobrepotencial marcadamente menor (um ganho de 0,40 V). Também foi relatada a utilização desses complexos na redução de CO2 em ácido fórmico, empregando TEOA como agente de sacrifício.84 O complexo fac-[MnBr(bpy)(CO)3] e seus derivados continuam a ser intensivamente estudados para a eletrocatálise homogênea de CO2 a produtos de maior valor agregado.85-87 Há ainda estudos utilizando ligantes que apresentam frações de coordenação alternativas, como os carbenos imino-N-heterociclicos (NHC). Os ligantes NHC são versáteis possuindo tanto o nitrogênio quanto o carbono como sítios de coordenação. O carbono confere um caráter σ doador mais forte em comparação à piridina e pode resultar em um aumento do gap, HOMO - LUMO, portanto, um deslocamento catódico dos potenciais de redução de um e dois elétrons.85,88 O uso de complexos polipiridínicos de Mn(I), por exemplo fac-[MnBr(bpy)(CO)3], na fotorredução de CO2 apresentou melhores resultados com a utilização do complexo Ru(bpy)32+ como fotossensibilizador e BNAH como agente de sacrifício em DMF/TEOA (TONHCOOH = 157).84 A partir daí, diversos estudos se deram envolvendo investigações mecanísticas,89 uso de novos sensitilizadores90 ou variação da estrutura do catalisador.91 As Tabelas 2 e 3, listam respectivamente exemplos de eletro e fotocatalisadores baseados em complexos de Ru(II), Ir(III), Re(I) e Mn(I). Em cada tabela são apresentados valores de TON e TOF, bem como as condições experimentais em que os dados foram obtidos. Uma vez que as condições experimentais entre os diferentes trabalhos variam significativamente, comparações diretas dos valores numéricos podem levar a conclusões errôneas a respeito da estabilidade/eficiência de cada um dos catalisadores. De forma geral, pode-se observar que os valores de TON para eletrocatalisadores moleculares são relativamente baixos em relação a sistemas heterogêneos, o que pode ser explicado pela ocorrência de reações secundárias como a dimerização.92 Já para os sistemas fotocatalíticos, Tabela 3, os valores de TON podem ser até duas ordens de grandeza maiores, uma vez que há excesso de agente de sacrifício no meio em relação ao fotocatalisador, o que minimiza a ocorrência de reações secundárias. Ainda assim, a estabilidade do sistema é considerada baixa para aplicações reais. Uma das formas de se contornar tal situação é a heterogeneização dos catalisadores moleculares.
REDUÇÃO DE CO2 - FOTOCATÁLISE HETEROGÊNEA Muitos dos processos de fotocatálise aplicada ao abatimento e conversão de CO2 partem de catalisadores heterogêneos (em geral semicondutores), isoladamente ou acoplados a outros materiais114 (Figura 5). O processo de fotorredução de CO2 foi demonstrado pela primeira vez usando semicondutores como o TiO2 e o ZnO que possuem um valor de band gap na região UV.115,116 Ainda hoje, esses materiais são utilizados como ponto de partida para o desenvolvimento de fotocatalisadores mais complexos.
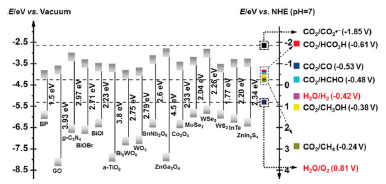 Figura 5. Estrutura eletrônica de diversos semicondutores com os valores das bandas de valência e condução e a comparação com potencial de redução das principais reações de interesse da conversão de CO2.117 (Reproduzida com a permissão da John Wiley and Sons)
Catalisadores heterogêneos como os semicondutores metálicos (CuO, TiO2, SnO2, Nb2O5 entre outros) e não metálicos (grafeno, C3N4, etc),117 apresentam em geral bom custo/benefício, são facilmente recuperados e reativados, e em princípio, podem ser ativados por radiação solar. No entanto, ainda apresentam baixa atividade e seletividade. Adicionalmente, a reação de redução de CO2 compete com a evolução de H2 em meio prótico, portanto, catalisadores eficientes devem exibir alta atividade para conversão de CO2 com atividade baixa ou desprezível para evolução de H2.118 No processo fotocatalítico quando um fóton incide sobre um semicondutor, com uma energia maior ou igual ao seu bandgap, ocorre a absorção do fóton, resultando na transição de um elétron da banda de valência para a banda de condução e geração de um buraco na banda de valência.119 O potencial de redução de um elétron fotoexcitado é determinado pelo nível de energia na parte inferior da banda de condução, e o poder de oxidação do buraco é determinado pelo nível de energia superior da banda de valência (Figura 5). Em uma reação fotoquímica típica, os elétrons e os buracos fotogerados são separados e migram para a superfície do fotocatalisador, na qual ocorrem as reações redox. Em qualquer caso, a posição da banda de condução do semicondutor deve estar acima do potencial redox do CO2. Se a posição da banda de valência estiver abaixo do potencial redox da água, ela pode ser utilizada como fonte protônica. Portanto, materiais com o alinhamento de banda adequado podem favorecer a reação redox antes que os portadores de carga sofram recombinação.120 Outros aspectos importantes incluem o bandgap adequado para absorver a energia solar em uma larga faixa do espectro; uma grande área de superfície ativa para a adsorção máxima de CO2/CO32-; resistência à fotocorrosão; alta eficiência fotocatalítica; e baixa toxicidade. Os óxidos metálicos têm sido amplamente utilizados como fotocatalisadores da redução de CO2 devido à sua estabilidade e resistência à fotocorrosão sob irradiação. As propriedades intrínsecas dos óxidos metálicos desempenham um papel crítico na determinação de sua viabilidade. Dois grupos principais têm sido estudados: óxidos de metais de transição em configuração d0 (Ti4+, Zr4+, Nb5+, Ta5+, V5+ e W6+); e óxidos de metal do grupo principal em configuração d10, com fórmula geral MyOz (M = Ga, Ge, In, Sn ou Sb).121 Na Tabela 4 são apresentados alguns dos principais semicondutores com metais de transição aplicados na fotoredução de CO2 para produção de CO, CH4, HCOOH e CH3OH, bem como suas respectivas taxas de conversão. As reações reportadas foram realizadas sob condições reacionais e/ou geometrias de reatores diferentes, o que pode influenciar diretamente no rendimento e na seletividade dos materiais, de modo que comparações diretas dos valores numéricos podem levar a conclusões errôneas a respeito da estabilidade/eficiência de cada um dos catalisadores.
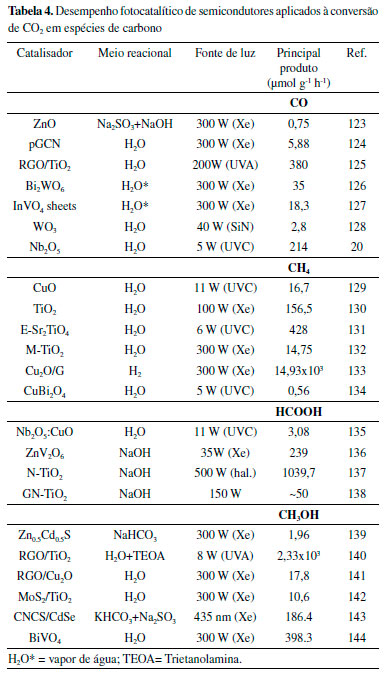
Do ponto de vista cinético, a formação de CO e HCOOH tende a ser mais favorável por ser uma reação mais simples, que envolve apenas 2 elétrons e 2 prótons com apenas um intermediário reacional. Para a formação dos demais produtos, são necessários de 4-18 elétrons e uma série de etapas e intermediários reacionais, o que afeta severamente a cinética do processo. Por outro lado, esses produtos são mais favoráveis energeticamente, ou seja, apresentam um menor potencial de redução, vide Tabela 1. Portanto, o produto favorecido será dependente do balanço cinético/termodinâmico do processo, i.e., se o catalisador facilitar a transferência de uma sequência de elétrons e a formação de intermediários reacionais os produtos mais reduzidos serão favorecidos em relação ao CO e HCOOH. Por exemplo, o Cu2O suportado em grafeno apresenta como principal produto o CH4 (14,93 × 103 μmol g-1 h-1), o qual necessita de 8 elétrons e 8 prótons, pois já foi demonstrado que esse catalisador pode estabilizar os intermediários reacionais específicos e facilitar a transferência de elétrons. Por outro lado, o TiO2 suportado em óxido de grafeno apresentou CH3OH (2,33x103 μmol g-1 h-1) como produto majoritário, no entanto, foi requerido a adição de TEOA que atua como um agente de sacrifício (doador de elétrons) e aumenta a solubilidade do CO2, o que é um indício que esse catalisador em comparação com o Cu2O não apresenta uma transferência de elétrons para os intermediários facilitado.122 Através do processo de fotoredução de CO2 também é possível obter diversas outras espécies C2, tais como C2H4, C2H6, CH3COOH. Entretanto, devido à alta complexidade desses processos, a cinética dessas reações é tão lenta que esses produtos não são favorecidos, mesmo sendo termodinamicamente favoráveis em comparação com os produtos C1. O desempenho desses materiais pode ser melhorado pelo acoplamento com nanopartículas condutoras (tais como: Au, Ag, Cu, Pt), tanto para produzir hidrocarbonetos gasosos, quanto álcoois. Essas nanopartículas melhoram a interação entre a superfície ativa e o CO2, e aumentam o tempo de vida dos portadores de carga fotogerados pela concentração de elétrons.12 Nanopartículas de ouro (AuNPs), cobre (CuNPs) e ligas Au-Cu suportados na superfície do TiO2 aumentam a eficiência e a seletividade do fotocatalisador na conversão de CO2 em hidrocarbonetos. Tanto o TiO2/Cu, quanto o TiO2/Ag apresentam boas performances fotocatalíticas para geração de CH4, a partir do CO2.145,146 O grafeno e seus derivados são materiais compostos majoritariamente por átomos de carbono sp2, porém contém inúmeros grupamentos oxigenados, tais como -COOH e/ou -OH em sua superfície ou borda. Esses grupamentos modificam a capacidade de adsorção do CO2147 e favorecem o acoplamento e a ancoragem de óxidos e nanopartículas metálicas em sua superfície, produzindo assim heterojunções efetivas entre os materiais,148 levando a um aumento considerável na atividade fotocatalítica.149 Fotocatalisadores não metálicos (óxido de grafeno, nitretos150-152 e selenetos) são investigados devido a sua abundância, estabilidade, custo-benefício e boa condutividade elétrica.23 Por exemplo, g-C3N4 tem baixo valor de bandgap (2,7 eV), permitindo sua ativação pela radiação visível. Também possui grupamentos funcionais com características de bases de Lewis e Brønsted, demonstrando potencial aplicação tanto na adsorção quanto na ativação do CO2.153 Entretanto, o posicionamento de suas bandas de valência e condução favorecem uma rápida recombinação dos portadores de cargas fotogerados, e isso pode estar associado a baixa área superficial específica obtida por esses materiais geralmente sintetizados via calcinação. A área superficial do g-C3N4 pode ser aumentada pela formação de compósitos, como Ti-SN/CN.154 Nanopartículas de TiO2 de alta área superficial específica, quando combinadas com o g-C3N4, compartilham sua textura aumentando a capacidade de adsorção de CO2. Com isso, além de melhorar as propriedades eletrônicas suprimindo a recombinação dos portadores de cargas e aumentando o espectro de absorção da luz incidente, há maior interação entre a superfície do compósito e o CO2. Desafios da aplicação de fotocatalisadores heterogêneos O processo de adsorção do CO2 nos sítios ativos do fotocatalisador é um fator crítico para a eficiência da fotorredução. Ela é subsequentemente acompanhada pela ativação do CO2, afetando significativamente a supressão da reação de evolução de hidrogênio, que é a principal reação concorrente da fotorredução de CO2. A interação da molécula de CO2 com a superfície do fotocatalisador pode se dar de diferentes formas, seja através dos átomos de oxigênio, carbono ou por ambos (Figura 2). Quando a interação se dá através do átomo de carbono (ácido de Lewis), a estrutura gerada é semelhante a um carbonato,23,155 que pode ser dessorvido da superfície do fotocatalisador em função da sua fraca interação, sendo consumido durante o processo de fotoredução.134,155 Se essa espécie de carbonato não for dessorvida dos sítios ativos do fotocatalisador, elas podem levar ao envenenamento de seus sítios ativos e consequentemente o seu tempo de vida útil, interferindo dessa forma em sua performance catalítica.129,156 Além da carbonatação, a presença de espécies orgânicas superficiais, que são facilmente adsorvidas principalmente por óxidos metálicos, também pode levar a uma desativação do catalisador.157,158 Para contornar esse inconveniente, alterações morfológicas, estruturais e eletrônicas dos catalisadores podem melhorar a interação da molécula de CO2 com a superfície.13,155,159 Além da estabilidade, há uma busca incessante por catalisadores que sejam altamente ativos sob radiação solar e seletivos para conversão do CO2, e embora os métodos de síntese e modificação dos semicondutores tenham avançado nos últimos anos, sua eficiência ainda está limitada à fraca capacidade de absorção da luz visível pede vários fotocatalisadores.160,161 Soluções em estudo incluem dopagem, controle da morfologia, introdução de defeitos, modificações superficiais e formação de heterojunções (heteroestruturas e Z-Schemes) para aumentar a eficiência dessa etapa.23,135 Compósitos contendo nanopartículas de ouro e microbastões de CuBi2O4 apresentaram excelente performance fotocatalítica atribuída à melhor captação da luz e ao aumento da área superficial específica.161 Estruturas de triazina covalente com pontes de enxofre (SnS2/S-CTFs) formam uma estrutura Z-scheme, melhorando a eficiência de separação dos pares elétron-buraco sob irradiação visível, exibindo excelente performance para redução de CO2 a CO e CH4.162 A dopagem é uma das alternativas para adequar a estrutura eletrônica de semicondutores, visto que forma estados eletrônicos adicionais na estrutura de bandas, aumentando a absorção da luz visível.23,160 A dopagem é a adição de um elemento diferente na rede cristalina de um fotocatalisador, metálico ou não metálico. Na dopagem com metais, por exemplo, pode-se criar estados eletrônicos, como níveis doadores acima da banda de valência e níveis aceptores abaixo da banda de condução de fotocatalisadores. Dessa maneira, a adição desses estados adicionais pode levar a redução da energia de band gap melhorando consequentemente a absorção de luz pelo fotocatalisador.114,160 Catalisadores de TiO2 dopados com diferentes quantidades de níquel (Ni) e bismuto (Bi) apresentaram band gaps mais estreitos, além de uma menor taxa de recombinação dos pares elétron-buraco comparado ao TiO2 puro, o que aumentou consideravelmente a absorção de luz na região do visível. A performance desses materiais na fotoredução de CO2 sob irradiação visível foi cerca de 6,5 vezes maior para produção de CH4 na amostra co-dopada com 1% de Ni e 3% de Bi.163 Nanocompósitos de óxido de grafeno com dióxido de titânio dopado com nitrogênio (TiO2/NrGO) tiveram comportamento superior ao TiO2 e TiO2/rGO puro sob irradiação ultravioleta, aumentando a adsorção do CO2 na superfície do fotocatalisador e facilitando a separação dos elétrons/buraco, impulsionando assim o desempenho fotocatalítico.164 Apesar de os catalisadores na forma de pó serem eficientes pela alta área superficial,165 esses têm forte tendência à aglomeração e requerem operações mais complexas para recuperação e reativação. Os catalisadores perdidos durante o processo de reativação podem levar à uma poluição secundária e entrar no meio aquático produzindo efeitos tóxicos em diferentes seres vivos.166 Portanto, a imobilização em diferentes tipos de suportes como monólitos cerâmicos,167 placas de vidros,168 fibras ópticas,169 argilas,167 entre outros, é desejável, ainda que o suporte possa afetar a seletividade e propriedades eletrônicas do fotocatalisador. Um suporte ideal deve ser resistente à degradação proporcionando adesão firme com o catalisador. Limitações de transferência de massa e baixa eficiência de utilização da radiação devido a pouca ou nenhuma absorção de luz nos poros ou canais dos suportes revestidos com os fotocatalisadores devem ser consideradas.170,171 Materiais 3D, como os aerogéis monolíticos, são suportes ideais por suas redes e canais de difusão interconectados, que permitem a multirreflexão da luz irradiada e ampliam o rendimento quântico, além da sua elevada área superficial.172 Aerogéis de óxido de grafeno reduzido (RGO) heteroestruturados (BiOBr/RGO, TiO2/RGO e Cu2O/RGO) foram facilmente separados dos sistemas de reação aquosa. Nesses sistemas, o aerogel de RGO tem um efeito positivo no desempenho fotocatalítico por aumentar a absorção de luz e retardar a recombinação dos pares elétron-buraco.173 Fotocatalisadores podem ser aplicados em fotorreatores em batelada e de fluxo contínuo. Os reatores fotocatalíticos também podem ser classificados de acordo com as fases envolvidas na reação em i) sistema bifásico e ii) sistema trifásico. Os sistemas de duas fases podem utilizar catalisadores homogêneos ou heterogêneos, enquanto em três fases o catalisador está na fase sólida e os reagentes e produtos estão nas fases líquida ou gasosa, como representado na Figura 6.174
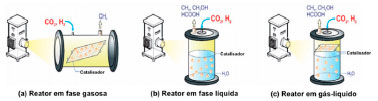 Figura 6. Três tipos de reatores: (a) reator em fase gasosa, (b) reator em fase líquida e (c) reator gás-líquido. Adaptado de Chen et al.174
Diversos grupos de pesquisa estão utilizando diferentes estratégias de modificação dos semicondutores, tais como a utilização de compostos orgânicos e inorgânicos como fotossensibilizadores,175 o acoplamento de semicondutores com diferentes níveis de energia19,135 ou a dopagem com metais ou não metais para suprimir a taxa de recombinação e, assim, aumentar o rendimento quântico.176,177 Além disso, uma nova abordagem no desenvolvimento de fotocatalisadores heterogêneos para aplicação no processo de fotorredução de CO2 tem como objetivo combinar os princípios de desenvolvimento de fotocatalisadores heterogêneos e homogêneos, com a finalidade de aumentar a seletividade dos fotocatalisadores heterogêneos no processo de fotorredução de CO2.175 Muitas variáveis vêm sendo elucidadas no processo fotocatalítico, o que exigirá um esforço sustentado ao longo de muitos anos. No entanto, existe um potencial estimulante para melhorias significativas no campo de fotorredução de CO2, principalmente no desenvolvimento de novos fotocatalisadores heterogêneos. Heterogenização As limitações dos catalisadores heterogêneos podem ser contornadas pela integração com catalisadores homogêneos, em geral mais seletivos, que no entanto sofrem com decomposição e polimerização (na ativação fotoquímica ou eletroquímica)178 além da dificuldade de recuperação, reuso e separação dos produtos de interesse. A heterogenização desses catalisadores moleculares em semicondutores179 confere melhor estabilidade e manutenção da seletividade - por exemplo, a imobilização de complexos de Ru(II) na superfície de nitreto de carbono (C3N4). Esse semicondutor vem sendo estudado como superfície de ancoramento dado seu bom desempenho na oxidação e redução fotocatalítica da água.180-183 Na presença de luz, os buracos na banda de valência oxidam o agente de sacrifício enquanto os elétrons excitados na banda de condução reduzem o complexo de Ru(II) na superfície do semicondutor. Esse centro metálico atua como um mediador redox, transferindo carga para a unidade catalítica, responsável pela redução do CO2. O maior TON obtido nesse sistema foi de 586 em solução aquosa contendo oxalato de potássio, mostrando que esse tipo de sistema é promissor para redução de CO2 em HCOOH.182 Outros exemplos envolvem complexos de Re(I)107,184 que não necessita de um mediador redox, uma vez que o complexo de Re(I) atua como absorvedor de radiação, gerando um estado excitado capaz de transferir elétrons para a banda de condução do semicondutor. O fotossensibilizador fotooxidado é reduzido pelo agente de sacrifício, tipicamente TEOA, levando a formação de adutos entre o centro metálico, o CO2 e o TEOA na superfície do óxido. Esses adutos são reduzidos pelos elétrons fotoinjetados na banda de condução, liberando CO e evitando reações secundárias como a dimerização.107,184 Há ainda exemplos similares empregando complexos de tricarbonílicos de Mn(I).185 É possível estabelecer comparações a respeito da eficiência catalítica após a heterogeneização. Por exemplo, Jain e colaboradores reportam que o processo de fotorredução de CO2 a metanol utilizando apenas óxido de grafeno produz 2201 µmol gcat-1 de metanol. Ao imobilizar um complexo de Ru(II) na superfície do óxido, a formação de metanol chega a 3977 µmol gcat-1, um aumento de cerca de 80%.186 Para os complexos de Re(I), Patrocinio e colaboradores observaram que em solução os complexos complexos [ReCl(CO)3(dcbH2)] e [ReCl(CO)3(phdo)] (dcbH2 = ácido 4,4'-dicarboxílico-2,2'-bipiridina; phdo = 1,10-fenantrolina-5,6-diona) apresentavam TON de 9 e 1, respectivamente, para o processo de redução fotocatalítica de CO2 em CO. Após a imobilização desses complexos na superfície de niobatos lamelares os valores de TON passaram a ser de 58 para o complexo contendo o ligante dcbH2 e 38 contendo phdo. Os autores ainda avaliaram o processo catalítico dos complexos na superfície de outro óxido semicondutor, TiO2, em que os valores de TON também aumentaram, 47 e 40 respectivamente.107 Reisner e colaboradores utilizaram a mesma estratégia para complexos de Mn(I). Em solução o complexo de Mn(I) apresentou TON de 34, enquanto após a imobilização na superfície de TiO2 o valor obtido foi de 112, demonstrando um aumento de eficiência no processo de heterogenização.185 Para os complexos de Ir(III) a heterogenização baseia-se na síntese de polímeros que possam atuar como ligantes (piridinas, carbenos e aminas) do centro metálico. Esses polímeros apresentaram resultados para a redução de CO2 a formato com TONs e TOFs da ordem de 104 e 103 h-1, respectivamente.187-189 O polímero baseado em Y(NO3)3 e o complexo [Ir(ppy)2(Hdcbpy)] (ppy = fenilpiridina; Hdcbpy = ácido 2,2'-bipiridina-4,4'-dicarboxilico) foi eficiente para redução de CO2 sob luz visível, gerando HCOO-. Em solução, os autores reportam que a produção máxima obtida de HCOO- foi de 25 µmol, após imobilização do complexo na matriz polimétrica a produção de HCOO- subiu para 40 µmol com TOF de 118 h-1.190 Outros exemplos de heterogenização consistem na imobilização dos catalisadores para promoção da eletroredução de CO2. Complexos de Ru(II) têm sido imobilizados em superfícies como TiO2 contendo nanotubos de carbono (produzindo CO e H2 na eletrocatálise com TONs de 308 e 597, respectivamente)191 ou carbono vítreo contendo óxido de grafeno.178 Complexos de Ir(III) foram imobilizados em eletrodos de nanotubos de carbono através de interações do tipo π-π, apresentando TONs de 2 x 106 e 5 x 105, e TOFs de 7 e 15,1 s-1 respectivamente.192 Para os complexos de Re(I), observou-se que ao promover a eletrodeposição de complexos derivados do tiofeno, os números de turnover aumentaram de 1,7 para 489.101
REDUÇÃO ELETROQUÍMICA DE CO2 Fundamentos, vantagens e desafios A redução eletroquímica de CO2 em hidrocarbonetos apresenta vantagens como controle do desempenho e seletividade de acordo com o potencial aplicado;193 possibilidade de reciclagem dos eletrólitos;194 dispositivos eletroquímicos são compactos, modulares e podem ser construídos sob demanda para diferentes formas de aplicação o que facilita o escalonamento do processo.195,196 O processo pode utilizar eletricidade oriunda de energias renováveis (i.e energia solar, eólica, hidrelétrica) sem a geração de CO2, o que pode zerar a emissão líquida de CO2.197 Até o momento, a maior parte dos estudos de redução eletroquímica de CO2 foram realizados em células eletroquímicas tipo H196 (Figura 7), que consistem num ânodo que oxida a água por meio da reação de evolução de oxigênio (que consome OH- ou gera H+) e fornece elétrons transportados para o cátodo para redução do CO2 a produtos (CO e CH4) e consome H+ ou gera OH-; um eletrólito para conduzir íons e dissolver e transportar CO2 para a superfície do cátodo; uma membrana de troca iônica para separar o cátodo e ânodo e evitar a oxidação dos produtos; e uma fonte de voltagem para aplicar o potencial necessário para promover a reação.194,196,198,199
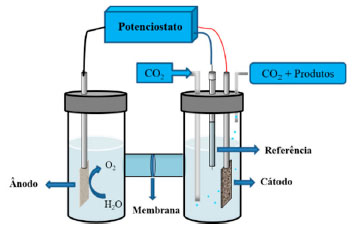 Figura 7. Representação esquemática de uma célula eletroquímica em H para redução do CO2 em diferentes produtos no cátodo e oxidação da água no ânodo
O processo de redução eletroquímica do CO2 pode ser avaliado e comparado com outros trabalhos por meio de 4 parâmetros: eficiência faradaica (EF), densidade de corrente (j), eficiência energética e sobrepotencial (η). A eficiência faradaica é calculada pela razão da quantidade de carga usada para formar um produto em específico e a carga total fornecida ao sistema (Q), e representa a seletividade do processo, i.e. a capacidade de aproveitamento da corrente para o produto de interesse:194,200,201 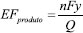 em que n é o número de mols de elétrons envolvidos por mol de produto, F é a constante de Faraday e y é o número de mols do produto formado. A densidade de corrente é a corrente total (I, Ampère) por unidade de área do cátodo (A, cm2) e descreve a taxa total de reação. A densidade de corrente parcial (jproduto) para um produto específico pode ser obtido pela multiplicação da densidade de corrente total e eficiência faradaica.194,200 A eficiência energética (EE) é uma medida do consumo de energia líquida na formação de um produto específico. A EE é obtida pela razão entre a quantidade de energia usada para produzir o produto específico e a energia elétrica líquida fornecida ao sistema: 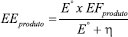 em que Eº é o potencial de equilíbrio da célula para o produto desejado, e η é a soma dos sobrepotenciais do cátodo e ânodo.194,200 Apesar do primeiro trabalho a respeito da redução eletroquímica de CO2 ser de 1946,202 somente na década de 80 foram realizados estudos sistemáticos a respeito do desempenho e seletividade de diversos metais na redução eletroquímica de CO2.203-205 No entanto, os estudos nessa área foram aprofundados apenas recentemente.206 Vários desafios precisam ser superados para tornar esse processo viável para uma aplicação prática:201 i) baixo desempenho catalítico (densidade de corrente menor que 200 mA cm-2); ii) baixa seletividade devido ao grande número de produtos formados: apenas CO e HCOOH apresentam seletividade maior que 90%, no entanto, utilizando metais preciosos como Ag, Au ou tóxicos, como Pb e Cd; e iii) baixa estabilidade dos catalisadores e/ou dos componentes do eletrolisador.206-208 Esses desafios estão relacionados a dois pontos principais, a natureza e as propriedades do catalisador e a arquitetura da célula eletroquímica.209,210 Dessa forma, novos catalisadores ou com diferentes propriedades e células eletroquímicas com diferentes arquiteturas precisam ser desenvolvidos para superar esses desafios.209,210 Catalisadores: seletividade, estabilidade e avanços Um bom catalisador para reação de redução de CO2 deve apresentar uma alta densidade de corrente durante um longo tempo, associado uma alta seletividade para o produto de interesse. Eletrocatalisadores podem ser metais/óxidos metálicos, complexos metálicos e enzimas.206 Os eletrocatalisadores metálicos são os mais intensamente investigados em função do seu relativo baixo custo, simplicidade de obtenção e preparo do eletrodo, e alta condutividade elétrica em eletrólitos líquidos.206 Os eletrocatalisadores metálicos são divididos em 4 grupos em função da sua seletividade. O cobre metálico (Cu) é o único membro do primeiro grupo capaz de converter o CO2 em diferentes hidrocarbonetos (C1 e C2) a depender das suas propriedades físico-químicas e das condições reacionais.205 O segundo grupo consiste em Au, Ag e Zn, que produzem principalmente CO. O terceiro grupo inclui In, Pb, Sn, Bi e Cd, e é caracterizado pela formação de ácido fórmico como principal produto, enquanto o quarto grupo composto por Ni, Fe, Pt e Ti apresenta quase exclusivamente H2 como produto.204,205,211 A seletividade desses metais é explicada pelo sobrepotencial de cada reação e pela força da interação dos intermediários dessa reação com a sua superfície.212 Foi observado ainda que os seguintes eletrodos foram inativos na redução de CO2, mesmo em condições experimentais altamente favoráveis: C, Al, Si, V, Cr, Mn, Fe, Co, Zr, Nb, Mo, Ru, Rh, Hf, Ta, W, Re e Ir.204-206,211 Compreender o mecanismo reacional de redução de CO2 é extremamente importante para racionalizar o desenvolvimento de novos materiais que apresentem propriedades requeridas para aumentar o desempenho do processo e controlar a seletividade na formação de um produto em específico. No entanto, já foi demonstrado que a redução de CO2 pode resultar em mais de 15 produtos diferentes a depender das condições reacionais e da natureza e propriedades do catalisador empregado o que torna o mecanismo desse processo extremamente complexo. Até o momento, foram descritos em detalhes apenas o mecanismo de formação dos produtos C1 e C2 (Figura 8).212
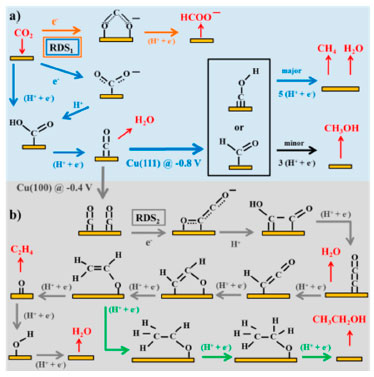 Figura 8. Possíveis caminhos reacionais para a redução eletrocatalítica de CO2 em diferentes produtos por catalisadores metálicos: (a) redução de CO2 em CO, CH4 (setas azuis), CH3OH (seta preta) e HCOO− (setas laranja); (b) redução de CO2 em etileno (setas cinza) e etanol (setas verdes). As espécies em preto são adsorbatos, enquanto àquelas em vermelho são reagentes ou produtos em solução. Os potenciais são relatados em relação a EPH, RDS significa etapa determinante de velocidade, enquanto (H+ + e−) indica etapa na qual ocorre a transferência de próton-elétron combinada ou separada. Reprinted with permission from ref.212 Copyright 2015 American Chemical Society
A formação do HCOOH e CO envolve a transferência de apenas 2 elétrons, dessa forma há apenas um intermediário reacional para cada produto e a descrição do mecanismo se torna bastante simplificado. Os primeiros estudos mecanísticos descobriram que o ácido fórmico não pode ser reduzido a outros produtos, sugerindo que o ácido fórmico não é intermediário dos demais produtos, portanto, a formação dos demais hidrocarbonetos e oxigenados segue um caminho reacional diferente, no qual o monóxido de carbono é proposto como o intermediário.213 Alguns estudos demonstram que a formação preferencial do CO ou HCOOH depende do modo de ligação/interação do CO2 no sítio ativo do catalisador na primeira etapa da reação.214 Feaster et al. demonstraram que os metais do terceiro grupo (In, Sn, Pb) adsorvem o CO2 pelos átomos de oxigênio e na sequência ocorre a hidrogenação do carbono formando um intermediário bidentado (*OCHO), por fim o HCOO- é dessorvido da superfície do catalisador (Figura 8a). Uma curva "Volcano" foi calculada usando a energia de ligação do intermediário *OCHO como descritor para formação do HCOO- com o potencial de -0,9 V vs RHE, essa demonstrou que os metais do terceiro grupo apresenta uma interação otimizada com esse intermediário.215 O mesmo grupo demonstrou que os metais do segundo grupo (Ag, Ag, Zn) adsorvem o CO2 pelo átomo de carbono (Figura 8a), na sequência ocorre o processo de transferência próton-elétron formando o intermediário *COOH (no qual esse grupo de metais apresenta uma interação otimizada com esse intermediário), uma segunda transferência de próton-elétron resulta na liberação de uma molécula de água e dessorve o CO.212 Por fim, o Cu é o único metal capaz de formar hidrocarbonetos e oxigenados (C1 a C3) a depender da condição reacional (pH, potencial aplicado, tipo de eletrólito e outros). Isso ocorre pois o Cu também adsorve o CO2 pelo átomo de carbono (Figura 8a). Porém, enquanto os metais do segundo grupo dessorvem o CO, o Cu apresenta uma maior força de adsorção desse intermediário, o que permite a hidrogenação do CO para formar a espécie *CHO ou *COH. Posteriormente, após uma sequência de transferência de próton-elétron, os intermediários podem ser reduzidos a metanol ou metano, a depender do intermediário, como pode ser visto na Figura 8a. Por outro lado, em altos potenciais de redução pode ocorrer a dimerização do intermediário C1 ou a inserção do CO ao intermediário em uma etapa semelhante a Fischer-Trospch, e formar como produto o etileno (Figura 8b).212 Dentre os catalisadores citados, os metais do grupo IB, Cu, Ag e Au, demonstram o maior potencial de aplicação em processos de redução eletroquímica de CO2.204,205,212,216-218 O Au policristalino é capaz de reduzir CO2 a CO com uma FE de quase 90% em -0,74 V vs. EPH.218 Embora o Au seja atualmente a superfície eletrocatalítica mais eficiente para a redução de CO2 em CO, a baixa abundância e o alto custo do Au podem impedir suas aplicações em grande escala.219,220 Nesse sentido, o Ag atraiu considerável atenção devido ao seu custo significativamente menor em comparação com Au e alta seletividade para a conversão de CO2 em CO.219,220 No entanto, requere-se um alto sobrepotencial (> 0,9 V), atribuído ao obstáculo para a transferência inicial de elétrons para molécula de CO2.221,222 Catalisadores nanoporosos de Ag foram capazes de promover uma rápida transferência de elétrons e reduzindo o sobrepotencial.223 Eletrodos de Ag nanoestruturado são capazes de reduzir eletroquimicamente CO2 a CO com alta seletividade e com sobrepotencial muito menor em comparação com o Ag policristalino, provavelmente em função da estabilização do intermediário COOH•.220 Como descrito acima, o Cu é o eletrodo mais viável para ser aplicado na formação de hidrocarbonetos mais reduzidos ou com 2 e 3 carbonos,209 ainda que com baixa seletividade e alto sobrepotencial.224,225 Há ainda um sério problema de competitividade com a reação de evolução de H2.219 Estratégias de modificação das propriedades físico-químicas do Cu podem aumentar o seu desempenho na redução de CO2.209,217,226 O tamanho de partícula de Cu tem importância crítica no processo: eletrodos de Cu nanoestruturado apresentaram menor sobrepotencial e densidade de corrente aproximadamente 10 vezes maior quando comparado a eletrodos convencionais, e maior eficiência faradaica na conversão do CO2 a C2H4 e CO.227 O aumento do desempenho catalítico do eletrodo de Cu recoberto com nanopartículas foi atribuída ao aumento da sua rugosidade, que apresentou mais arestas e defeitos na superfície provendo mais sítios ativos. Estudos utilizando a teoria do funcional da densidade (do inglês, Density Functional Theory, DFT) previram que partículas menores devem melhorar a atividade e seletividade.227 O mesmo comportamento foi observado em outros trabalhos, atribuído ao aumento da rugosidade e da quantidade de defeitos que podem alterar a seletividade da reação.228,229 Nanoestruturas derivadas de óxido de cobre apresentaram aumento significativo do desempenho catalítico, sendo possível favorecer a produção de C2H4.216,217 No entanto, a razão do aumento da atividade ainda está em aberto.230-233 O aumento pode ser atribuído à modificação nos contornos de grão, que podem atuar como sítios ativos superficiais.217 Outro mecanismo é o aumento do pH local devido às altas densidades de corrente nas superfícies altamente rugosas, o que pode alterar a via de reação em favor do etileno.231 Alternativamente, a nanoestruturação da superfície do catalisador durante a redução do óxido também pode fornecer átomos de baixa coordenação mais reativos.231 Experimentos de absorção atômica de raios-X in operando indicaram que os sítios de Cud+ permaneceram estáveis mesmo sob condições redutoras, sugerindo que esses os sítios ativos dessa reação.232 Ligas metálicas ou catalisadores bimetálicos podem ajustar a força de adsorção do CO2 e seus intermediários na superfície catalítica ou mesmo promover um mecanismo em cascata,234-238 i.e. um catalisador como Ag promove a redução de CO2 em CO e um outro catalisador como o Cu promove a redução do CO em produtos mais reduzidos.239,240 No entanto, esses apresentam um desempenho muito aquém do esperado para uma aplicação prática. Muitos trabalhos afirmam que o desempenho pode ser severamente limitado devido à baixa concentração de CO2 em eletrólito aquoso por conta da sua baixa solubilidade.199,207,241 Isso indica que a limitação do processo possa estar relacionada à arquitetura da célula eletroquímica e não somente à natureza do catalisador.210 Arquitetura de células eletroquímicas A reação em meio aquoso depende da difusão do CO2 do bulk do eletrólito para a superfície do eletrodo, que é bastante limitada pela baixa solubilidade (34 mmol L-1) de CO2 em condições padrão. Essa baixa solubilidade limita a densidade de corrente a apenas algumas dezenas de mA cm-2.207 A maioria das análises técnicas e econômicas do ponto de vista industrial, no entanto, sugerem que a densidade de corrente precisa ser superior a 200 mA cm-2 para que seja viável comercialmente.201 Portanto, alguns estudos têm proposto o uso de eletrolisadores em alta pressão e/ou baixa temperatura,196,242 ou a utilização de líquidos iônicos como eletrólitos que aumentam a solubilidade de CO2.243-245 Essas opções adicionam no entanto custos e/ou complexidade ao processo. A estratégia mais promissora para superar o problema de transferência de massa do CO2 é a utilização de uma célula eletroquímica de fluxo de difusão de gás, baseada no fornecimento do CO2 por trás de um eletrodo de difusão gasosa diretamente na interface entre o eletrodo e eletrólito.199,207,246 O eletrodo de difusão gasosa é um material poroso, hidrofóbico e condutor com um catalisador depositado na sua superfície. Burdyny e colaboradores demonstraram que o caminho de difusão do CO2 pode reduzir de 50 µm (para uma célula eletroquímica em H) que permite o aumento da densidade de corrente para valores de até 1 A.cm-2.193,207-209,247-250 Além das vantagens já apresentadas, esse tipo de célula permite a realização do processo de redução do CO2 em condições mais favoráveis como em eletrólito alcalino (pH maior que 12), que suprime a formação de hidrogênio e reduz o sobrepotencial da redução de CO2.207,246 A utilização desse tipo de eletrólito é inviável em células eletroquímicas típicas pois o CO2 seria convertido em espécies de carbonato antes de atingir a superfície do eletrocatalisador. A utilização de KOH 12 mol L-1 nesse tipo de célula permitiu alcançar uma densidade de corrente maior que 300 mA cm-2 utilizando cobre como catalisador. Além disso, o sobrepotencial da formação do CO foi diminuído em 140 mV, o que auxiliou na cobertura de CO sobre o catalisador em baixos potenciais e aumentou a seletividade para formação de C2H4 para 70%.251 Eletrodos de difusão gasosa de Ag depositada sobre PTFE em meio alcalino apresentaram densidade de corrente superior a 150 mA cm-2 com eficiência faradaica superior a 90% para produção de CO.252 O aumento na concentração de eletrólitos (KOH, KCl e KHCO3), utilizando eletrodos de difusão gasosa à base de Ag, resultou no aumento da densidade de corrente parciais para o CO (jCO). O KOH 3.0 mol L-1 apresentou os melhores resultados, i.e. 440 mA cm-2 com eficiência energética de 42%, atribuídos à diminuição da resistência à transferência de carga e da resistência da célula.208 Efeito similar na formação de etileno e etanol é visto utilizando eletrodos de cobre em condições alcalinas.248 Porém, esses eletrodos apresentam baixa estabilidade quando aplicados em condições extremas (altas concentrações de KOH e altos potenciais).199,201,207 Isso é devido a mudanças nas características dos eletrodos de difusão gasosa, que se tornam hidrofílicos durante o processo de redução de CO2 e permitem que a água entre nos poros do eletrodo (flooding). Esse processo impede o fornecimento do CO2 para interface eletrocatalítica e diminui a eficiência do processo. Foi proposta a utilização de uma membrana de PTFE como camada hidrofóbica e porosa, com uma fina camada de catalisador depositado sobre a membrana, e por fim uma camada condutora depositada sobre o catalisador (e.g. grafite ou grafeno). A principal hipótese é que a separação espacial entre a camada difusora de gás, catalisador e camada transportadora de elétron aumenta a estabilidade do eletrodo mantendo a hidrofobicidade do PTFE. Esse efeito foi demonstrado em diferentes trabalhos, que apresentaram um bom desempenho associado a uma estabilidade superior a 100 h de operação.250,252,253 No entanto, a estabilidade observada ainda está distante daquela esperada para uma aplicação prática.
REDUÇÃO DE CO2 EM DISPOSITIVOS FOTOELETROQUÍMICOS Uma estratégia promissora para superar os problemas dos processos fotoquímico e eletroquímico é combiná-los num processo fotoeletroquímico.254 De modo geral, esse processo pode reduzir o consumo de energia elétrica em comparação com a redução eletroquímica do CO2, devido à reação espontânea conduzida pela energia solar no ânodo e/ou cátodo. Em comparação com a fotocatálise, pode-se alcançar eficiência muito maior na redução do CO2, já que o potencial externo aplicado conduz à separação do par elétron/buraco fotogerado no semicondutor, i.e., os elétrons são conduzidos para o cátodo, enquanto os buracos são concentrados no ânodo evitando a recombinação desses, de modo que a principal limitação da fotocatálise é superada.255-257 Assim, o processo fotoeletroquímico combina benefícios de ambos os processos e pode superar suas limitações. Os sistemas fotoeletroquímicos utilizados nas reações de redução de CO2, contém três eletrodos: eletrodo de referência, trabalho e contra eletrodo, esses dois últimos separados por membranas de troca iônica, podendo ser catiônica ou aniônica.256 Além disso, a configuração das células fotoeletroquímicas podem ser empregadas em 4 diferentes tipos, representados na Figura 9: (i) sistema utilizando fotoânodo e cátodo metálico; (ii) sistema utilizando fotocátodo e ânodo metálico; (iii) sistema utilizando fotocátodo e fotoânodo; e (iv) sistema utilizando eletrodos metálicos conectados a células fotovoltaicas, como os dispositivos multijunções (tipo Tandem), silício ou perovskita.256,258
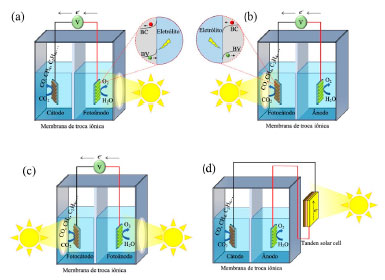 Figura 9. Esquemas representativos de células fotoeletroquímicas utilizadas na redução de CO2 na água usando semicondutor como (a) fotoânodo, (b) fotocátodo e (c) ambos fotoânodo e fotocátodo. (d) Diagrama esquemático do dispositivo que combina uma célula fotovoltaica com eficientes catalisadores eletroquímicos para a redução de CO2 e oxidação de água. Adaptado da Zhang et al.259
Para aplicação das reações fotoeletroquímicas primeiramente deve-se compreender a interface semicondutor/líquido. O contato promove o emparelhamento dos níveis de Fermi do semicondutor e do eletrólito, assim como a transferência das cargas até o equilíbrio. A direção e a magnitude desse fluxo de carga são determinadas pela estrutura da banda do semicondutor, que pode ser tipo n ou tipo p, além do potencial redox das espécies envolvidas na reação presentes no meio líquido.260 Os semicondutores do tipo n, geralmente empregados como fotoânodos, são os que apresentam excesso de elétrons em sua banda de condução, ou seja, o nível de Fermi está localizado próximo a banda de condução. Portanto, ao entrar em contato com o eletrólito (solução eletrolítica contendo CO2) de potencial redox próximo ao do semicondutor, ocorre a drenagem dos elétrons do semicondutor pelo eletrólito, até que haja equilíbrio e emparelhamento dos níveis de Fermi, elevando a curvatura das bandas do semicondutor para cima (Figura 10).261 Com isso, os buracos presentes na banda de valência migram em direção a superfície do material, favorecendo os processos oxidativos, como a quebra da água, gerando oxigênio molecular e íons H+.
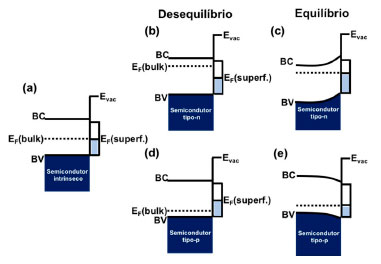 Figura 10. Níveis esquemáticos de energia eletrônica perto da superfície de um semicondutor limpo: (a) semicondutor (intrínseco); (b) antes do equilíbrio e (c) em equilíbrio entre o bulk do tipo n e sua superfície; (d) antes do equilíbrio e (e) equilíbrio entre o bulk do tipo p e sua superfície. Adaptado de Zhang et al.261
BiVO4 e TiO2 foram utilizados como fotoânodo (Figura 9a) nas reações de redução de CO2 para formação de acido fórmico.262 Foi utilizado um novo design de célula fotoeletroquímica, com estanho metálico como cátodo, depositado em um eletrodo de difusão gasosa. Esse modelo de célula, tipo prensa, permite a operação em densidades de corrente mais altas, minimiza a queda ôhmica, aumenta o transporte de massa e permite a alimentação direta de CO2 gasoso na célula, contribuindo significativamente para eficiência da reação. Observou-se que o BiVO4 apresentou maior eficiência em gerar cargas positivas na banda de valência em meio alcalino, no entanto, com baixa estabilidade. O TiO2 resultou em eficiência faradaica de até 65% na produção de ácido fórmico no cátodo de Sn com densidade de corrente de até 5 mA cm-2 num potencial de -1,2 V vs EPH, atingindo eficiências energéticas de até 70%. Esses experimentos demonstraram que a utilização de um fotoânodo, embora não atue diretamente na reação de conversão de CO2 em outros produtos, pode melhorar significativamente a eficiência da reação, demonstrando ser uma opção promissora para essas reações. Semicondutores do tipo p, geralmente aplicados como fotocátodos (Figura 9b), são aqueles que apresentam excesso de vacâncias na banda de condução. Com isso, ao entrar em contato com o eletrólito, com potencial redox menor que a banda de condução, os elétrons migram do eletrólito para o semicondutor, formando um campo elétrico na interface semicondutor/eletrólito. Esse campo elétrico causa uma flexão de banda da estrutura eletrônica do semicondutor. Nesse caso, a banda de condução e valência próxima da interface do semicondutor se curvarão para baixo, como ilustrado na Figura 10.263 A reação de redução de CO2 por fotocátodo ainda é pouco explorada, diferentemente da reação de evolução de H2. Esse apresenta diversos desafios relacionados ao catalisador e ao dispositivo que necessitam ser compreendidos e superados, tais como a absorção de luz limitada, a separação/transferência de carga ineficiente, e a eficiência global insatisfatória da conversão de energia.264 Foi proposto um fotocátodo composto por uma placa de silício num dos lados do eletrodo e no outro o TiO2/Ag e Cu, que atuam como catalisador.257 Com a iluminação diretamente no silício, ele absorve a radiação gerando uma corrente elétrica que passa pelo TiO2/Ag até chegar na superfície do Cu onde o CO2 é reduzido. O sistema foi capaz de reduzir CO2 em -0,4 V vs EPH produzindo predominantemente C2H4, seguido de H2 e CO. Com o aumento do potencial, a formação de hidrogênio foi altamente suprimida, aumentando a formação de C2H4 que alcançou até 40% de eficiência faradaica a -1,0 V vs EPH. O processo sob radiação deslocou até 600 mV no sobrepotencial. Paralelamente, a configuração de célula fotoeletroquímica que integram fotoânodos e fotocátodos também tem sido foco de estudo nos últimos anos (Figura 9c). A utilização desse tipo de sistema possui como principal vantagem a possibilidade de ativar ambos os eletrodos com luz.260 Nesse sentido, propôs-se uma célula com fotocátodo composto por um complexo metálico multinuclear (Ru(II)-Re(I)) suportado na superfície de oxido de níquel, juntamente com um fotoânodo composto por oxido de cobalto (CoOx) suportado na superfície de oxinitreto de tântalo (TaON). Os dois fotoeletrodos são separados por uma membrana de Nafion, pela qual é possível que a radiação incidente possa penetrar e atingir ambos os eletrodos (cátodo e ânodo). Com isso o fotocátodo atuou na redução de CO2 para CO, assim como na geração de H2 com um potencial aplicado de - 0,3 V vs Ag/AgCl, obtendo 37% de eficiência faradaica. Por outro lado, a reação anódica, a formação de O2 a partir da quebra da água, apresentou uma eficiência faradaica de 68%. Essa diferença entre as eficiências faradaicas está relacionada ao consumo energético no cátodo pela redução de íons trivalentes de níquel (Ni3+), que originalmente existiam na forma de NiO. Porém, é valido salientar que os resultados obtidos ao aplicar -0,3 V vs Ag/AgCl só foram possíveis devido à radiação, diminuindo significativamente o potencial necessário para conduzir as reações (de -0,7 V para -0,3V vs Ag/AgCl).265 Deve-se também ressaltar que a utilização de sistemas fotovoltaicos integrados a anodos e catodos também vem sendo estudada, como ilustrado na Figura 9d. Por exemplo, foi reportado um sistema utilizando células fotovoltaicas acopladas a um cátodo de Au (ouro) e um ânodo de IrO2. O sistema obteve um ponto máximo com mais 90% de eficiência faradaica para um potencial aplicado de - 0,4 V, com corrente de 2 mA cm-2 para esse mesmo potencial. A eficiência de conversão do CO2 com relação a quantidade de irradiação recebida pelas placas fotovoltaicas foi maior que 6,5%.266 Células fotoeletroquímicas com modificadores moleculares têm sido também exploradas. Células com diversos tipos de óxidos semicondutores e complexos de Ru(II) apresentaram boa eficiência para redução de CO2.267-270 Alguns dos materiais consistem em semicondutores como CuZnSnS4, p-InP-Zn, Ta2O5 e diferentes complexos de Ru(II), com eficiências faradaicas de até 82%.267-269 A Figura 11 demonstra o funcionamento de uma célula fotoeletroquímica sensibilizada por complexos de Ru(II) polimerizado sobre um fotocátodo.268 O elétron promovido da banda de condução reduz o complexo de Ru(II) polimerizado, que subsequente promove a redução de CO2 em ácido fórmico.
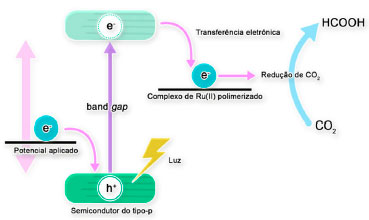 Figura 11. Esquema da célula fotoeletroquímica sensibilizada por complexo de Ru(II) polimerizado. Adaptado de Arai et al.268
Foi também descrita uma célula fotoeletroquímica em meio aquoso, empregando o complexo fac-[Re(bh-bpy)(CO)3(OH2)] (bh-bpy = 4,4'-dihidroximetil-2,2'-bipiridina) no fotocátodo, e IrO2/FTO no anodo. Os autores obtiveram eficiências faradaicas de 95 e 4% para conversão de CO2 em CO e HCOOH, respectivamente a pH = 6,9. Mesmo em pHs ligeiramente ácidos, 4,2, o sistema apresentou uma boa seletividade para CO (84%) gerando apenas 3% de HCOOH. Apesar dos relevantes avanços até o momento, o desempenho energético e faradaico na conversão de CO2 em produtos de interesse é ainda aquém do esperado para uma aplicação pratica. Isso se deve a dois fatos basicamente: i) complexidade do sistema, pois deve ser acoplado e otimizado dois processos diferentes, sendo que nem sempre as condições ótimas de ambos se sobrepõem; ii) a característica eletrônica intrínseca aos semicondutores limita severamente a densidade de corrente que pode ser obtido em um sistema como esse. Portanto, esforços precisam ser investidos nessa área para que ambos os processos sejam otimizados.
PERSPECTIVAS Nesta revisão foram apresentados os principais avanços e desafios na conversão do CO2 em produtos químicos por processos foto-, eletro- e fotoeletroquímicos, aplicando tanto catalisadores homogêneos quanto heterogêneos. A redução de CO2 é uma alternativa promissora e com grande potencial para produzir combustíveis renováveis e commodities químicas em um ciclo com emissão líquida de CO2 zero. Ainda são necessários desenvolvimentos de novos catalisadores e de arquiteturas de células para o uso em larga escala dos processos de redução fotoeletroquímicos para conversão de CO2 em combustíveis e produtos químicos, devido à sua baixa eficiência energética e produtividade. O entendimento dos mecanismos de redução do CO2 por compostos de coordenação tem permitido grandes avanços em termos de seletividade e velocidade dos catalisadores. Há, porém, a necessidade de se desenvolver sistemas mais estáveis e menos sensíveis à variações do meio reacional. A heterogenização se mostra uma alternativa interessante para aumento de escala, mas resta o desafio de se substituir metais raros por espécies mais abundantes. Apesar do progresso significativo nos últimos anos, dos métodos de síntese, caracterização e elucidação dos mecanismos envolvidos no processo de fotorredução de CO2 utilizando fotocatalisadores heterogêneos, ainda existem questões importantes que precisam ser enfrentadas e tratadas para um possível escalonamento do processo, como: i) a forma de utilização do catalisador, ii) maior aproveitamento da radiação solar e iii) e seletividade dos produtos formados. No entanto, do ponto de vista da síntese dos catalisadores heterogêneos, continua sendo um desafio realizar a produção em massa de fotocatalisadores com um bom custo/benefício e que apresente um elevado controle da composição, morfologia e da densidade de defeito dos semicondutores. O desenvolvimento de eletrodos e da arquitetura das células eletroquímicas são os principais desafios para alcançar um desempenho viável na conversão de CO2 em combustíveis. Estratégias como oxidação proposital dos eletrodos metálicos ou formação de ligas metálicas podem ajustar a força de adsorção do CO2 e dos intermediários reacionais que resultam na diminuição do sobrepotencial da reação. No entanto, para se obter densidades de corrente acima de 200 mA cm-2 é necessária a utilização de eletrolisadores na fase líquida/gasosa pelo uso de eletrodo de difusão gasosa. Estratégias para aumentar a estabilidade do catalisador de centenas para milhares de horas, enquanto mantém as densidades de corrente, serão essenciais para a comercialização dessa tecnologia. Também é esperado que a combinação dos processos eletroquímicos e fotoquímicos possam melhorar a economia e até mesmo a produtividade em comparação com as respectivas abordagens individuais.
AGRADECIMENTOS Os autores agradecem à FAPESP (2016/21516-7 e 2018/01258-5), FAPEMIG (PPM-00220-17, APQ-00330-14), ao CNPq (402287/2013-4, 38294/2020-0, 407497/2018-8, 406392/2018-8, 305432/2017-6) e à CAPES-PrInt (88887.374340/2019-00) pelo apoio financeiro e pelas bolsas concedidas. O. F. L., A. O. T. P. e C. R. também agradecem ao acordo Capes/Humboldt pela bolsa de Pós-Doutorado concedida (CAPES - Código de Financiamento 001 - processos: 88881.368050/2019-01, 23117.002652/2015-61 e 88881.145566/2017-1).
REFERÊNCIAS 1. Roe, S.; Streck, C.; Obersteiner, M.; Frank, S.; Griscom, B.; Drouet, L.; Fricko, O.; Gusti, M.; Harris, N.; Hasegawa, T.; Hausfather, Z.; Havlík, P.; House, J.; Nabuurs, G.-J.; Popp, A.; Sánchez, M. J. S.; Sanderman, J.; Smith, P.; Stehfest, E.; Lawrence, D.; Nat. Clim. Change 2019, 9, 817. 2. Peters, G. P.; Andrew, R. M.; Canadell, J. G.; Friedlingstein, P.; Jackson, R. B.; Korsbakken, J. I.; Le Quéré, C.; Peregon, A.; Nat. Clim. Change 2020, 10, 3. 3. Moreira, H. M.; Giometti, R.; Contexto Internacional 2008, 30, 9. 4. Sumaila, U. R.; Tai, T. C.; Lam, V. W. Y.; Cheung, W. W. L.; Bailey, M.; Cisneros-Montemayor, A. M.; Chen, O. L.; Gulati, S. S.; Sci. Adv. 2019, 5, eaau3855. 5. Crippa, M.; Oreggioni, G.; Guizzardi, D.; Muntean, M.; Schaaf, E.; Lo Vullo, E.; Solazzo, E.; Monforti-Ferrario, F.; Olivier, J. G. J.; Vignati, E.; European Commission, Joint Research Centre, Fossil CO2 and GHG emissions of all world countries; Publications Office of the European Union, 2019. 6. Análise de conjuntura dos biocombustíveis - ano 2019 https://www.epe.gov.br/pt/publicacoes-dados-abertos/publicacoes/analise-de-conjuntura-dos-biocombustiveis-2019, acessada em Março 2021. 7. De Oliveira, M. E. D.; Vaughan, B. E.; Rykiel, E. J.; BioScience 2005, 55, 593. 8. Yu, K. M. K.; Curcic, I.; Gabriel, J.; Tsang, S. C. E.; ChemSusChem 2008, 1, 893. 9. Centi, G.; Quadrelli, E. A.; Perathoner, S.; Energy Environ. Sci. 2013, 6, 1711. 10. Gielen, D.; Boshell, F.; Saygin, D.; Nature Mater 2016, 15, 117. 11. Wang, Q.; Domen, K.; Chem. Rev. 2020, 120, 919. 12. Ulmer, U.; Dingle, T.; Duchesne, P. N.; Morris, R. H.; Tavasoli, A.; Wood, T.; Ozin, G. A.; Nature Communications 2019, 10, 1. 13. Chang, X.; Wang, T.; Gong, J.; Energy Environ. Sci. 2016, 9, 2177. 14. Du, C.; Wang, X.; Chen, W.; Feng, S.; Wen, J.; Wu, Y. A.; Mater. Today Adv. 2020, 6. 15. Romero Cuellar, N. S.; Scherer, C.; Kaçkar, B.; Eisenreich, W.; Huber, C.; Wiesner-Fleischer, K.; Fleischer, M.; Hinrichsen, O.; J. CO2 Util. 2020, 36, 263. 16. Ding, P.; Jiang, T.; Han, N.; Li, Y.; Mater. Today Nano 2020, 10. 17. Nguyen, V. H.; Nguyen, B. S.; Jin, Z.; Shokouhimehr, M.; Jang, H. W.; Hu, C.; Singh, P.; Raizada, P.; Peng, W.; Shiung Lam, S.; Xia, C.; Nguyen, C. C.; Kim, S. Y.; Le, Q. Van; Chem. Eng. J. 2020, 402, 126184. 18. Ma, Y.; Wang, Z.; Xu, X.; Wang, J.; Chin. J. Catal. 2017, 38, 1956. 19. Torres, J. A.; Nogueira, A. E.; Da Silva, G. T. S. T.; Lopes, O. F.; Wang, Y.; He, T.; Ribeiro, C.; J. CO2 Util. 2020, 35, 106. 20. da Silva, G. T. S. T.; Nogueira, A. E.; Oliveira, J. A.; Torres, J. A.; Lopes, O. F.; Ribeiro, C.; Appl. Catal., B 2019, 242, 349. 21. Gattrell, M.; Gupta, N.; Co, A.; J. Electroanal. Chem. 2006, 594, 1. 22. Yin, W.-J.; Wen, B.; Ge, Q.; Li, X.-B.; Teobaldi, G.; Liu, L.-M.; Dalton Trans. 2020, 49, 12918. 23. Shen, H.; Peppel, T.; Strunk, J.; Sun, Z.; Sol. RRL 2020, 4, 1900546. 24. Zhang, S.; Fan, Q.; Xia, R.; Meyer, T. J.; Acc. Chem. Res. 2020, 53, 255. 25. Horváth, I.; Kiss, G.; Cook, R.; J. Am. Chem. Soc. 1998, 2, 3133. 26. Morris, A. J.; Meyer, G. J.; Fujita, E.; Acc. Chem. Res. 2009, 42, 1983. 27. Prado, F.; Sousa, S.; Machado, A. E.; Patrocinio, A. O.; J. Braz. Chem. Soc. 2016. 28. Sousa, S. F.; Sampaio, R. N.; Barbosa Neto, N. M.; Machado, A. E. H.; Patrocinio, A. O. T.; Photochem. Photobiol. Sci. 2014, 13, 1213. 29. Müller, A. V.; Gonçalves, M. R.; Ramos, L. D.; Polo, A. S.; Frin, K. P. M.; Quim. Nova 2017, 40, 200. 30. Steinlechner, C.; Roesel, A. F.; Oberem, E.; Päpcke, A.; Rockstroh, N.; Gloaguen, F.; Lochbrunner, S.; Ludwig, R.; Spannenberg, A.; Junge, H.; Francke, R.; Beller, M.; ACS Catalysis 2019, 9, 2091. 31. Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O.; J. Am. Chem. Soc. 2008, 130, 2023. 32. Pellegrin, Y.; Odobel, F.; C. R. Chim. 2017, 20, 283. 33. Souza, S. F.; Patrocinio, A. O. T.; Quim. Nova 2014, 37, 886. 34. Manbeck, G. F.; Muckerman, J. T.; Szalda, D. J.; Himeda, Y.; Fujita, E.; J. Phys. Chem. B 2015, 119, 7457. 35. Smieja, J. M.; Kubiak, C. P.; Inorg. Chem. 2010, 49, 9283. 36. Grills, D. C.; Matsubara, Y.; Kuwahara, Y.; Golisz, S. R.; Kurtz, D. A.; Mello, B. A.; J. Phys. Chem. Lett. 2014, 5, 2033. 37. Li, X.; He, X.; Liu, X.; He, L.-N.; Sci. China Chem. 2017, 60, 841. 38. Tanaka, K.; Morimoto, M.; Tanaka, T.; Chem. Lett. 1983, 12, 901. 39. Ishida, H.; Tanaka, K.; Tanaka, T.; Chem. Lett. 1985, 14, 405. 40. Ishida, H.; Tanaka, K.; Tanaka, T.; Organometallics 1987, 6, 181. 41. Ishida, H.; Tanaka, H.; Tanaka, K.; Tanaka, T.; J. Chem. Soc., Chem. Commun. 1987, 131. 42. Nagao, H.; Mizukawa, T.; Tanaka, K.; Inorg. Chem. 1994, 33, 3415. 43. Nakajima, H.; Tanaka, K.; Chem. Lett. 1995, 24, 891. 44. Mizukawa, T.; Tsuge, K.; Nakajima, H.; Tanaka, K.; Angew. Chem., Inter. Ed. 1999, 38, 362. 45. Elgrishi, N.; Chambers, M. B.; Wang, X.; Fontecave, M.; Chem. Soc. Rev. 2017, 46, 761. 46. Chardon-Noblat, S.; Collomb-Dunand-Sauthier, M.-N.; Deronzier, A.; Ziessel, R.; Zsoldos, D.; Inorg. Chem. 1994, 33, 2961 47. Wang, W.-H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; & Fujita, E.; Chem. Rev. 2015, 115, 12936. doi:10.1021/acs.chemrev.5b00197 48. Gonell, S.; Massey, M. D.; Moseley, I. P.; Schauer, C. K.; Muckerman, J. T.; Miller, A. J. M.; J. Am. Chem. Soc. 2019, 141, 6658. 49. Pugh, J. R.; Bruce, M. R. M.; Sullivan, B. P.; Meyer, T. J.; Inorg. Chem. 1991, 30, 86. 50. Chen, Z. F.; Concepcion, J. J.; Brennaman, M. K.; Kang, P.; Norris, M. R.; Hoertz, P. G.; Meyer, T. J.; Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15606. 51. Tamaki, Y.; Tokuda, K.; Yamazaki, Y.; Saito, D.; Ueda, Y.; Ishitani, O.; Front. Chem. 2019, 7, 327. 52. Leite, M.; Duarte, L.; da Silva David, A. L. O.; Carlos, H.; Quim. Nova 2017, 40, 200. 53. Fujita, E.; Coord. Chem. Rev. 1999, 185-186, 373. 54. Church, T. L.; Andersson, P. G.; Coord. Chem. Rev. 2008, 252, 513. 55. Hanasaka, F.; Fujita, K.; Yamaguchi, R.; Organometallics 2004, 23, 1490. 56. Fujita, K.; Li, Z.; Ozeki, N.; Yamaguchi, R.; Tetrahedron Lett. 2003, 44, 2687. 57. Herskovitz, T.; J. Am. Chem. Soc. 1977, 99, 2391. 58. Inoue, Y.; Izumida, H.; Sasaki, Y.; Hashimoto, H.; Chem. Lett. 1976, 5, 863. 59. Himeda, Y.; Onozawa-Komatsuzaki, N.; Sugihara, H.; Kasuga, K.; Organometallics 2007, 26, 702. 60. Tanaka, R.; Yamashita, M.; Nozaki, K.; J. Am. Chem. Soc 2009, 131, 14168. 61. Liu, C.; Xie, J.-H.; Tian, G.-L.; Li, W.; Zhou, Q.-L.; Chem. Sci.2015, 6, 2928. 62. Wang, W.-H.; Hull, J. F.; Muckerman, J. T.; Fujita, E.; Himeda, Y.; Energy Environ. Sci. 2012, 5, 7923. 63. Ogo, S.; Kabe, R.; Hayashi, H.; Harada, R.; Fukuzumi, S.; Dalton Trans. 2006, 4657. 64. Sato, S.; Morikawa, T.; Kajino, T.; Ishitani, O.; Angew. Chem., Int. Ed. 2013, 52, 988. 65. Sato, S.; Morikawa, T.; ChemPhotoChem 2018, 2, 207. 66. Manbeck, G. F.; Polyansky, D. E.; Fujita, E.; ACS Catal. 2020, 10, 6497. 67. Kang, P.; Cheng, C.; Chen, Z.; Schauer, C. K.; Meyer, T. J.; Brookhart, M.; J. Am. Chem. Soc. 2012, 134, 5500. 68. Bi, J.; Hou, P.; Kang, P.; ChemCatChem 2019, 11, 2069. 69. Ahn, S. T.; Bielinski, E. A.; Lane, E. M.; Chen, Y.; Bernskoetter, W. H.; Hazari, N.; Palmore, G. T. R.; Chem. Commun. 2015, 51, 5947. 70. Worl, L. A.; Duesing, R.; Chen, P.; Ciana, L. Della; Meyer, T. J.; J. Chem. Soc., Dalton Trans. 1991, 849. 71. Itokazu, M. K.; Polo, A. S.; de Faria, D. L. A.; Bignozzi, C. A.; Iha, N. Y. M.; Inorg. Chim. Acta 2001, 313, 149. 72. Polo, A. S.; Itokazu, M. K.; Frin, K. M.; de Toledo Patrocínio, A. O.; Murakami Iha, N. Y.; Coord. Chem. Rev. 2006, 250, 1669. 73. Polo, A. S.; Itokazu, M. K.; Murakami Iha, N. Y.; J. Photochem. Photobiol., A 2006, 181, 73. 74. Lin, R.; Fu, Y.; Brock, C. P.; Guarr, T. F.; Inorg. Chem. 1992, 31, 4346. 75. Morimoto, T.; Nakajima, T.; Sawa, S.; Nakanishi, R.; Imori, D.; Ishitani, O.; J. Am. Chem. Soc. 2013, 135, 16825. 76. Christ, C. S.; Yu, J.; Zhao, X.; Palmore, G. T. R.; Wrighton, M. S.; Inorg. Chem. 1992, 31, 4439. 77. Argazzi, R.; Bertolasi, E.; Chiorboli, C.; Bignozzi, C. A.; Itokazu, M. K.; Murakami Iha, N. Y.; Inorg. Chem. 2001, 40, 6885. 78. Kutal, Charles.; Weber, M. A.; Ferraudi, Guillermo.; Geiger, David.; Organometallics 1985, 4, 2161. 79. Nakada, A.; Ishitani, O.; ACS Catal. 2018, 8, 354. 80. Cabrera, C. R.; Abruña, H. D.; J. Electroanal. Chem. Interf. Electrochem. 1986, 209, 101. 81. Johnson, F. P. A.; George, M. W.; Hartl, F.; Turner, J. J.; Organometallics 1996, 15, 3374. 82. Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A.; Angew. Chem., Int. Ed. 2011, 50, 9903. 83. Hartl, F.; Rossenaar, B. D.; Stor, G. J.; Stufkens, D. J.; Recl. Trav. Chim. Pays-Bas 1995, 114, 565. 84. Takeda, H.; Koizumi, H.; Okamoto, K.; Ishitani, O.; Chem. Commun. 2014, 50, 1491. 85. Asay, M.; Jones, C.; Driess, M.; Chem. Rev. 2011, 111, 354. 86. Kuo, H.-Y.; Tignor, S. E.; Lee, T. S.; Ni, D.; Park, J. E.; Scholes, G. D.; Bocarsly, A. B.; Dalton Trans. 2020, 49, 891. 87. Machan, C. W.; Stanton, C. J.; Vandezande, J. E.; Majetich, G. F.; Schaefer, H. F.; Kubiak, C. P.; Agarwal, J.; Inorg. Chem. 2015, 54, 8849. 88. Agarwal, J.; Shaw, T. W.; Stanton, C. J.; Majetich, G. F.; Bocarsly, A. B.; Schaefer, H. F.; Angew. Chem., Int. Ed. 2014, 53, 5152. 89. Cheung, P. L.; Machan, C. W.; Malkhasian, A. Y. S.; Agarwal, J.; Kubiak, C. P.; Inorg. Chem. 2016, 55, 3192. 90. Zhang, J.-X.; Hu, C.-Y.; Wang, W.; Wang, H.; Bian, Z.-Y.; Appl. Catal., A 2016, 522, 145. 91. Wang, X.; Thiel, I.; Fedorov, A.; Copéret, C.; Mougel, V.; Fontecave, M.; Chem. Sci. 2017, 8, 8204. 92. Sampson, M. D.; Nguyen, A. D.; Grice, K. A.; Moore, C. E.; Rheingold, A. L.; Kubiak, C. P.; J. Am. Chem. Soc. 2014, 136, 5460. 93. Chen, Z.; Chen, C.; Weinberg, D. R.; Kang, P.; Concepcion, J. J.; Harrison, D. P.; Brookhart, M. S.; Meyer, T. J.; Chem. Commun. 2011, 47, 12607. 94. Johnson, B. A.; Maji, S.; Agarwala, H.; White, T. A.; Mijangos, E.; Ott, S.; Angew. Chem., Int. Ed. 2016, 55, 1825. 95. Grills, D. C.; Matsubara, Y.; Kuwahara, Y.; Golisz, S. R.; Kurtz, D. A.; Mello, B. A.; The J. Phys. Chem. Lett. 2014, 5, 2033. 96. Sampson, M. D.; Froehlich, J. D.; Inorg. Chem. 2010, 49, 9283. 97. Souza, B. L.; Faustino, L. A.; Prado, F. S.; Sampaio, R. N.; Maia, P. I. S.; Machado, A. E. H.; Patrocinio, A. O. T.; Dalton Trans. 2020, 49, 16368. 98. Talukdar, K.; Sinha Roy, S.; Amatya, E.; Sleeper, E. A.; Le Magueres, P.; Jurss, J. W.; Inorg. Chem. 2020, 59, 6087. 99. Liang, Y.; Nguyen, M. T.; Holliday, B. J.; Jones, R. A.; Inorg. Chem. Commun. 2017, 84, 113. 100. Sampson, M. D.; Kubiak, C. P.; J. Am. Chem. Soc. 2016, 138, 1386. 101. Sun, C.; Prosperini, S.; Quagliotto, P.; Viscardi, G.; Yoon, S. S.; Gobetto, R.; Nervi, C.; Dalton Trans. 2016, 45, 14678. 102. Lehn, J.-M.; Ziessel, R.; J. Organomet. Chem. 1990, 382, 157. 103. Tamaki, Y.; Koike, K.; Ishitani, O.; Chem. Sci. 2015, 6, 7213. 104. Kamada, K.; Jung, J.; Taku, W.; Sekizawa, K.; Sato, S.; Morikawa, T.; Fukuzumi, S.; Saito, S.; J. Am. Chem. Soc. 2020, 142, 23 105. Kuramochi, Y.; Itabashi, J.; Fukaya, K.; Enomoto, A.; Yoshida, M.; Ishida, H.; Chem. Sci. 2015, 6, 3063. 106. Genoni, A.; Chirdon, D. N.; Boniolo, M.; Sartorel, A.; Bernhard, S.; Bonchio, M.; ACS Catal. 2017, 7, 154. 107. Faustino, L. A.; Souza, B. L.; Nunes, B. N.; Duong, A.-T.; Sieland, F.; Bahnemann, D. W.; Patrocinio, A. O. T.; ACS Sustainable Chem. Eng. 2018, 6, 6073. 108. Shakeri, J.; Farrokhpour, H.; Hadadzadeh, H.; Joshaghani, M.; RSC Adv. 2015, 5, 41125. 109. Ching, H. Y. V.; Wang, X.; He, M.; Perujo Holland, N.; Guillot, R.; Slim, C.; Griveau, S.; Bertrand, H. C.; Policar, C.; Bedioui, F.; Inorg. Chem. 2017, 56, 2966. 110. Gholamkhass, B.; Mametsuka, H.; Koike, K.; Tanabe, T.; Furue, M.; Ishitani, O.; Inorg. Chem. 2005, 44, 2326. 111. Sampson, M. D.; Kubiak, C. P.; Inorg. Chem. 2015, 54, 6674. 112. Singh, H.; Tomer, V. K.; Jena, N.; Bala, I.; Sharma, N.; Nepak, D.; De Sarkar, A.; Kailasam, K.; Pal, S. K.; J. Mater. Chem. A 2017, 5, 21820. 113. Cheung, P. L.; Machan, C. W.; Malkhasian, A. Y. S.; Agarwal, J.; Kubiak, C. P.; Inorg. Chem. 2016, 55, 3192. 114. White, J. L.; Baruch, M. F.; Pander, J. E.; Hu, Y.; Fortmeyer, I. C.; Park, J. E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; Shaw, T. W.; Abelev, E.; Bocarsly, A. B.; Chem. Rev. 2015, 115, 12888. 115. Fujishima, A.; Honda, K.; Nature 1972, 238, 37. 116. Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K.; Nature 1979, 277, 637. 117. Sun, Z.; Talreja, N.; Tao, H.; Texter, J.; Muhler, M.; Strunk, J.; Chen, J.; Angew. Chem., Int. Ed. 2018, 57, 7610. 118. Nunes, B. N.; Lopes, O. F.; Patrocinio, A. O. T.; Bahnemann, D. W.; Catalysts 2020, 10, 126. 119. Bueno, R.; Lopes, O.; Carvalho, K.; Ribeiro, C.; Mourão, H.; Quim. Nova 2019, 42, 661. 120. Zeng, S.; Kar, P.; Thakur, U. K.; Shankar, K.; Nanotechnology 2018, 29, 052001. 121. Takata, T.; Pan, C.; Domen, K.; Sci. Technol. Adv. Mater. 2015, 16, 033506. 122. Karamian, E.; Sharifnia, S.; J. CO2 Util. 2016, 16, 194. 123. Li, P.; Hu, H.; Luo, G.; Zhu, S.; Guo, L.; Qu, P.; Shen, Q.; He, T.; ACS Appl. Mater. Interfaces 2020, 12, 56039. 124. Ong, W.-J.; Tan, L.-L.; Chai, S.-P.; Yong, S.-T.; Mohamed, A. R.; Nano Energy 2015, 13, 757. 125. Jung, H.; Cho, K. M.; Kim, K. H.; Yoo, H.-W.; Al-Saggaf, A.; Gereige, I.; Jung, H.-T.; ACS Sustainable Chem. Eng. 2018, 6, 5718. 126. Liu, Y.; Shen, D.; Zhang, Q.; Lin, Y.; Peng, F.; Appl. Catal., B 2021, 283, 119630. 127. Han, Q.; Bai, X.; Man, Z.; He, H.; Li, L.; Hu, J.; Alsaedi, A.; Hayat, T.; Yu, Z.; Zhang, W.; Wang, J.; Zhou, Y.; Zou, Z.; J. Am. Chem. Soc. 2019, 141, 4209. 128. Liang, L.; Li, X.; Sun, Y.; Tan, Y.; Jiao, X.; Ju, H.; Qi, Z.; Zhu, J.; Xie, Y.; Joule 2018, 2, 1004. 129. Nogueira, A. E.; da Silva, G. T. S. T.; Oliveira, J. A.; Torres, J. A.; da Silva, M. G. S.; Carmo, M.; Ribeiro, C.; Catal. Commun. 2020, 137, 105929. 130. Kar, P.; Zeng, S.; Zhang, Y.; Vahidzadeh, E.; Manuel, A.; Kisslinger, R.; Alam, K. M.; Thakur, U. K.; Mahdi, N.; Kumar, P.; Shankar, K.; Appl. Catal., B 2019, 243, 522. 131. Kwak, B. S.; Do, J. Y.; Park, N.-K.; Kang, M.; Sci Rep 2017, 7, 16370. 132. Zhang, T.; Low, J.; Koh, K.; Yu, J.; Asefa, T.; ACS Sustainable Chem. Eng. 2018, 6, 531. 133. Mateo, D.; Albero, J.; García, H.; Energy Environ. Sci. 2017, 10, 2392. 134. Ribeiro, L. S.; Pinatti, I. M.; Torres, J. A.; Giroto, A. S.; Lesse, F.; Longo, E.; Ribeiro, C.; Nogueira, A. E.; Mater. Lett. 2020, 275, 128165. 135. Nogueira, A. E.; Silva, G. T. S. T.; Oliveira, J. A.; Lopes, O. F.; Torres, J. A.; Carmo, M.; Ribeiro, C.; ACS Appl. Energy Mater. 2020, 3, 7629. 136. Bafaqeer, A.; Tahir, M.; Amin, N. A. S.; Appl. Surf. Sci. 2018, 435, 953. 137. Zhao, Z.; Fan, J.; Wang, J.; Li, R.; Catal. Commun. 2012, 21, 32. 138. Zhang, Q.; Lin, C.-F.; Jing, Y. H.; Chang, C.-T.; J. Air Waste Manage. Assoc. 2014, 64, 578. 139. Madhusudan, P.; Wageh, S.; Al-Ghamdi, A. A.; Zhang, J.; Cheng, B.; Yu, Y.; Appl. Surf. Sci. 2020, 506, 144683. 140. Olowoyo, J. O.; Kumar, M.; Singh, B.; Oninla, V. O.; Babalola, J. O.; Valdés, H.; Vorontsov, A. V.; Kumar, U.; Carbon 2019, 147, 385. 141. Liu, S.-H.; Lu, J.-S.; Chen, Y.-C.; Sustainability 2018, 10, 4145. 142. Tu, W.; Li, Y.; Kuai, L.; Zhou, Y.; Xu, Q.; Li, H.; Wang, X.; Xiao, M.; Zou, Z.; Nanoscale 2017, 9, 9065. 143. Li, A.; Wang, T.; Li, C.; Huang, Z.; Luo, Z.; Gong, J.; Angew. Chem., Int. Ed. 2019, 58, 3804. 144. Gao, S.; Gu, B.; Jiao, X.; Sun, Y.; Zu, X.; Yang, F.; Zhu, W.; Wang, C.; Feng, Z.; Ye, B.; Xie, Y.; J. Am. Chem. Soc. 2017, 139, 3438. 145. Hori, Y. In Modern Aspects of Electrochemistry; Vayenas, C. G., White, R. E., Gamboa-Aldeco, M. E., eds.; Springer: New York, 2008; Vol. 42, pp. 89-189. 146. Neatu, T.; Maciá-Agullò, J. A.; Concepciòn, P.; Garcia, H.; J. Am. Chem. Soc. 2014, 136, 15969. 147. Mishra, A. K.; Ramaprabhu, S.; AIP Adv. 2011, 1, 032152 148. Ong, W. J.; Tan, L. L.; Chai, S. P.; Yong, S. T.; Mohamed, A. R.; Nano Energy 2015, 13, 757. 149. Tan, L. L.; Ong, W. J.; Chai, S. P.; Mohamed, A. R.; Chem. Eng. J. 2017, 308, 248. 150. Crake, A.; Christoforidis, K. C.; Godin, R.; Moss, B.; Kafizas, A.; Zafeiratos, S.; Durrant, J. R.; Petit, C.; Appl. Catal., B 2019, 242, 369. 151. da Silva, G. T. S. T.; Carvalho, K. T. G.; Lopes, O. F.; Ribeiro, C.; Appl. Catal., B 2017, 216, 70. 152. Wang, C.; Thompson, R. L.; Baltrus, J.; Matranga, C.; J. Phys. Chem. Lett. 2010, 1, 48. 153. Sun, Z.; Wang, H.; Wu, Z.; Wang, L.; Catal. Today 2018, 300, 160. 154. Crake, A.; Christoforidis, K. C.; Godin, R.; Moss, B.; Kafizas, A.; Zafeiratos, S.; Durrant, J. R.; Petit, C.; Appl. Catal., B 2019, 242, 369. 155. Xin, C.; Hu, M.; Wang, K.; Wang, X.; Langmuir 2017, 33, 6667. 156. Nogueira, A. E.; Oliveira, J. A.; da Silva, G. T. S. T.; Ribeiro, C.; Sci. Rep. 2019, 9, 1316. 157. Olivo, A.; Ghedini, E.; Signoretto, M.; Compagnoni, M.; Rossetti, I.; Energies 2017, 10, 1. 158. Grigioni, I.; Dozzi, M. V.; Bernareggi, M.; Chiarello, G. L.; Selli, E.; Catal. Today 2017, 281, 214. 159. Yuan, L.; Xu, Y. J.; Appl. Surf. Sci. 2015, 342, 154. 160. Vu, N. N.; Kaliaguine, S.; Do, T. O.; Adv. Funct. Mater. 2019, 29, 1. 161. Shi, W.; Li, M.; Ren, H.; Guo, F.; Huang, X.; Shi, Y.; Tang, Y.; Beilstein J. Nanotechnol. 2019, 10, 1360. 162. Guo, S.; Yang, P.; Zhao, Y.; Yu, X.; Wu, Y.; Zhang, H.; Yu, B.; Han, B.; George, M. W.; Liu, Z.; ChemSusChem 2020, 13, 6278. 163. Nematollahi, R.; Ghotbi, C.; Khorasheh, F.; Larimi, A.; J. CO2 Util. 2020, 41, 101289. 164. Lin, L. Y.; Nie, Y.; Kavadiya, S.; Soundappan, T.; Biswas, P.; Chem. Eng. J. 2017, 316, 449. 165. Khan, M. M.; Multifunctional Photocatalytic Materials for Energy; Elsevier: Amsterdam, 2018; pp. 5-18. 166. Egorova, K. S.; Ananikov, V. P.; Organometallics 2017, 36, 4071. 167. Tahir, M.; Amin, N. A. S.; Chem. Eng. J. 2016, 285, 635. 168. Velasco-Hernández, A.; Esparza-Muñoz, R. A.; de Moure-Flores, F. J.; Santos-Cruz, J.; Mayén-Hernández, S. A.; Mater. Sci. Semicond. Process. 2020, 114. 169. Chen, H.; Chu, F.; Yang, L.; Ola, O.; Du, X.; Yang, Y.; Appl. Energy 2018, 230, 1403. 170. Mohd Adnan, M. A.; Muhd Julkapli, N.; Amir, M. N. I.; Maamor, A.; Int. J. Environ. Sci. Technol. 2019, 16, 547. 171. Porley, V.; Robertson, N. In Nanostructured Photocatalysts; Boukherroub, R., Ogale, S. B., Robertson, N., eds.; Elsevier: Amsterdam, 2020. 172. Long, S.; Wang, H.; He, K.; Zhou, C.; Zeng, G.; Lu, Y.; Cheng, M.; Song, B.; Yang, Y.; Wang, Z.; Luo, X.; Xie, Q.; Colloids Surf., A 2020, 594, 124666. 173. Yang, L.; Liu, Y.; Zhang, R.; Li, W.; Li, P.; Wang, X.; Zhou, Y.; Chin. J. Catal. 2018, 39, 646. 174. Chen, C. Y.; Yu, J. C. C.; Nguyen, V. H.; Wu, J. C. S.; Wang, W. H.; Kocí, K.; Catalysts 2017, 7, 63. 175. Khalil, M.; Gunlazuardi, J.; Ivandini, T. A.; Umar, A.; Renewable Sustainable Energy Rev. 2019, 113, 109246. 176. Khalilzadeh, A.; Shariati, A.; Solar Energy 2018, 164, 251. 177. Zhao, H.; Pan, F.; Li, Y.; Journal of Materiomics 2017, 3, 17. 178. Das, B.; Jia, C.; Ching, K.; Bhadbhade, M.; Chen, X.; Ball, G. E.; Colbran, S. B.; Zhao, C.; ChemCatChem 2020, 12, 1292. 179. Hartley, F. R.; Vezey, P. N. Stone, F. G. A.; West, R. B. T.-A. In Advances in Organometallic Chemistry; Stone, F. G. A., West, R. B. T.-A., eds.; Academic Press, 1977; Vol. 15, pp. 189-234. 180. Kuriki, R.; Yamamoto, M.; Higuchi, K.; Yamamoto, Y.; Akatsuka, M.; Lu, D.; Yagi, S.; Yoshida, T.; Ishitani, O.; Maeda, K.; Angew. Chem., Int. Ed. 2017, 56, 4867. 181. Kuriki, R.; Sekizawa, K.; Ishitani, O.; Maeda, K.; Angew. Chem., Int. Ed. 2015, 54, 2406. 182. Kuriki, R.; Matsunaga, H.; Nakashima, T.; Wada, K.; Yamakata, A.; Ishitani, O.; Maeda, K.; J. Am. Chem. Soc. 2016, 138, 5159. 183. Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J. M.; Domen, K.; Antonietti, M.; Nat. Mater. 2009, 8, 76. 184. Windle, C. D.; Pastor, E.; Reynal, A.; Whitwood, A. C.; Vaynzof, Y.; Durrant, J. R.; Perutz, R. N.; Reisner, E.; Chem. Eur. J. 2015, 21, 3746. 185. Rosser, T. E.; Windle, C. D.; Reisner, E.; Angew. Chem., Int. Ed. 2016, 55, 7388. 186. Kumar, P.; Sain, B.; Jain, S. L.; J. Mater. Chem. A 2014, 2, 11246. 187. Gunasekar, G. H.; Yoon, S.; J. Mater. Chem. A 2019, 7, 14019. 188. Shao, X.; Yang, X.; Xu, J.; Liu, S.; Miao, S.; Liu, X.; Su, X.; Duan, H.; Huang, Y.; Zhang, T.; Chem 2019, 5, 693. 189. Gunasekar, G. H.; Park, K.; Ganesan, V.; Lee, K.; Kim, N.-K.; Jung, K.-D.; Yoon, S.; Chem. Mater. 2017, 29, 6740. 190. Li, L.; Zhang, S.; Xu, L.; Wang, J.; Shi, L.-X.; Chen, Z.-N.; Hong, M.; Luo, J.; Chem. Sci. 2014, 5, 3808. 191. Wang, Y.; Marquard, S. L.; Wang, D.; Dares, C.; Meyer, T. J.; ACS Energy Lett. 2017, 2, 1395. 192. Kang, P.; Zhang, S.; Meyer, T. J.; Brookhart, M.; Angew. Chem., Int. Ed. 2014, 53, 8709. 193. Wang, Y.; Wang, Z.; Dinh, C. T.; Li, J.; Ozden, A.; Golam Kibria, M.; Seifitokaldani, A.; Tan, C. S.; Gabardo, C. M.; Luo, M.; Zhou, H.; Li, F.; Lum, Y.; McCallum, C.; Xu, Y.; Liu, M.; Proppe, A.; Johnston, A.; Todorovic, P.; Zhuang, T. T.; Sinton, D.; Kelley, S. O.; Sargent, E. H.; Nat. Catal. 2020, 3, 98. 194. Garg, S.; Li, M.; Weber, A. Z.; Ge, L.; Li, L.; Rudolph, V.; Wang, G.; Rufford, T. E.; J. Mater. Chem. A 2020, 8, 1511. 195. Chen, Y.; Vise, A.; Klein, W. E.; Cetinbas, F. C.; Myers, D. J.; Smith, W. A.; Smith, W. A.; Smith, W. A.; Deutsch, T. G.; Neyerlin, K. C.; ACS Energy Lett. 2020, 5, 1825. 196. Liang, S.; Altaf, N.; Huang, L.; Gao, Y.; Wang, Q.; J. CO2 Util. 2020, 35, 90. 197. Gurudayal, G.; Bullock, J.; Srankó, D. F.; Towle, C. M.; Lum, Y.; Hettick, M.; Scott, M. C.; Javey, A.; Ager, J.; Energy Environ. Sci. 2017, 10, 2222. 198. Zhu, S.; Li, T.; Cai, W. Bin; Shao, M.; ACS Energy Lett. 2019, 4, 682. 199. Nguyen, T. N.; Dinh, C.-T.; Chem. Soc. Rev. 2020, 49, 7488. 200. Merino-Garcia, I.; Alvarez-Guerra, E.; Albo, J.; Irabien, A.; Chem. Eng. J. 2016, 305, 104. 201. Kibria, M. G.; Edwards, J. P.; Gabardo, C. M.; Dinh, C. T.; Seifitokaldani, A.; Sinton, D.; Sargent, E. H.; Adv. Mater. 2019, 31, 1. 202. Van Rysselberghe, P.; J. Am. Chem. Soc. 1946, 68, 2047. 203. Hori, Y.; Kikuchi, K.; Murata, A.; Suzuki, S.; Chem. Lett. 1986, 897. 204. Hori, Y.; Kikuchi, K.; Suzuki, S.; Chem. Lett. 1985, 1695. 205. Hori, Y.; Handbook Fuel Cells 2010, 1. 206. Kondratenko, E. V.; Mul, G.; Baltrusaitis, J.; Larrazabal, G. O.; Perez-Ramirez, J.; Larrazábal, G. O.; Pérez-Ramírez, J.; Energy Environ. Sci. 2013, 6, 3112. 207. Burdyny, T.; Smith, W. A.; Energy Environ. Sci. 2019, 12, 1442. 208. Verma, S.; Lu, X.; Ma, S.; Masel, R. I.; Kenis, P. J. A.; Phys. Chem. Chem. Phys. 2015, 18, 7075. 209. Ross, M. B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E. H.; Nat. Catal. 2019, 2, 648. 210. Vennekoetter, J. B.; Sengpiel, R.; Wessling, M.; Chem. Eng. J. 2019, 364, 89. 211. Gao, D.; Cai, F.; Wang, G.; Bao, X.; Curr. Opin. Green Sustain. Chem. 2017, 3, 39 212. Kortlever, R.; Shen, J.; Schouten, K. J. P.; Calle-Vallejo, F.; Koper, M. T. M.; J. Phys. Chem. Lett. 2015, 6, 4073. 213. Baruch, M. F.; Pander, J. E.; White, J. L.; Bocarsly, A. B.; ACS Catal. 2015, 5, 3148. 214. Jones, J.-P.; Prakash, G. K. S.; Olah, G. A.; Isr. J. Chem. 2014, 54, 1451. 215. Feaster, J. T.; Shi, C.; Cave, E. R.; Hatsukade, T.; Abram, D. N.; Kuhl, K. P.; Hahn, C.; Nørskov, J. K.; Jaramillo, T. F.; ACS Catal. 2017, 7, 4822. 216. Li, C. W.; Ciston, J.; Kanan, M. W.; Nature 2014, 508, 504. 217. Li, C. W.; Kanan, M. W.; J. Am. Chem. Soc. 2012, 134, 7231. 218. Kuhl, K. P.; Hatsukade, T.; Cave, E. R.; Abram, D. N.; Kibsgaard, J.; Jaramillo, T. F.; J. Am. Chem. Soc. 2014, 136, 14107. 219. Gattrell, M.; Gupta, N.; Co, A.; J. Electroanal. Chem. 2006, 594, 1. 220. Ma, M.; Trześniewski, B. J.; Xie, J.; Smith, W. A.; Angew. Chem., Int. Ed. 2016, 55, 9748 221. Hsieh, Y.-C.; Senanayake, S. D.; Zhang, Y.; Xu, W.; Polyansky, D. E.; ACS Catal. 2015, 5349. 222. Kim, C.; Jeon, H. S.; Eom, T.; Jee, M. S.; Kim, H.; Friend, C. M.; Min, B. K.; Hwang, Y. J.; J. Am. Chem. Soc. 2015, 137, 13844. 223. Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G. S.; Kimmel, Y. C.; Chen, J. G.; Jiao, F.; Nat. Commun. 2014, 5, 1. 224. Qiao, J.; Liu, Y.; Hong, F.; Zhang, J.; Chem. Soc. Rev. 2014, 43, 631. 225. Ganesh, I.; Renewable Sustainable Energy Rev. 2016, 59, 1269. 226. Mistry, H.; Varela, A. S.; Kühl, S.; Strasser, P.; Cuenya, B. R.; Nat. Rev. Mater. 2016, 16009. 227. Tang, W.; Peterson, A. a.; Varela, A. S.; Jovanov, Z. P.; Bech, L.; Durand, W. J.; Dahl, S.; Nørskov, J. K.; Chorkendorff, I.; Phys. Chem. Chem. Phys. 2012, 14, 76. 228. Ren, D.; Wong, N. T.; Handoko, A. D.; Huang, Y.; Yeo, B. S.; J. Phys. Chem. Lett. 2015, 20. 229. Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B. R.; Strasser, P.; J. Am. Chem. Soc. 2014, 136, 6978. 230. Lopes, O. F.; Varela, H.; ChemistrySelect 2018, 3, 9046. 231. Ren, D.; Deng, Y.; Handoko, A. D.; Chen, C. S.; Malkhandi, S.; Yeo, B. S.; ACS Catal. 2015, 5, 2814. 232. Mistry, H.; Varela, A. S.; Bonifacio, C. S.; Zegkinoglou, I.; Sinev, I.; Choi, Y. W.; Kisslinger, K.; Stach, E. A.; Yang, J. C.; Strasser, P.; Cuenya, B. R.; Nat. Commun. 2016, 7, 12123. 233. Kim, D.; Lee, S.; Ocon, J. D.; Jeong, B.; Lee, J. K.; Lee, J.; Phys. Chem. Chem. Phys. 2015, 17, 824. 234. Pavesi, D.; Ali, F. S. M.; Anastasiadou, D.; Kallio, T.; Figueiredo, M.; Gruter, G. J. M.; Koper, M. T. M.; Schouten, K. J. P.; Catal. Sci. Technol. 2020, 10, 4264. 235. Huang, J.; Mensi, M.; Oveisi, E.; Mantella, V.; Buonsanti, R.; J. Am. Chem. Soc. 2019, 141, 2490. 236. Kim, D.; Resasco, J.; Yu, Y.; Asiri, A. M.; Yang, P.; Nat. Commun. 2014, 5, 4948. 237. Hansen, H. A.; Shi, C.; Lausche, A. C.; Peterson, A. A.; Nørskov, J. K.; Phys. Chem. Chem. Phys. 2016, 18, 9194. 238. Kortlever, R.; Peters, I.; Balemans, C.; Kas, R.; Kwon, Y.; Mul, G.; Koper, M. T. M.; Chem. Commun. 2016, 52, 10229. 239. Gurudayal, G.; Perone, D.; Malani, S.; Lum, Y.; Haussener, S.; Ager, J. W.; ACS Appl. Energy Mater. 2019, 2, 4551. 240. Lum, Y.; Ager, J. W.; Energy Environ. Sci. 2018, 11, 2935. 241. Del Castillo, A.; Alvarez-Guerra, M.; Solla-Gullón, J.; Sáez, A.; Montiel, V.; Irabien, A.; J. CO2 Util. 2017, 18, 222. 242. Kas, R.; Kortlever, R.; Yilmaz, H.; Koper, M. T. M.; Mul, G.; ChemElectroChem 2015, 2, 354. 243. Rosen, B. A.; Salehi-Khojin, A.; Thorson, M. R.; Zhu, W.; Whipple, D. T.; Kenis, P. J. A.; Masel, R. I.; Science 2011, 334, 643. 244. Shi, J.; Li, Q.-Y.; Shi, F.; Song, N.; Jia, Y.-J.; Hu, Y.-Q.; Shen, F.; Yang, D.; Dai, Y.-N.; J. Electrochem. Soc. 2016, 163, G82. 245. Sun, L.; Ramesha, G. K.; Kamat, P. V.; Brennecke, J. F.; Langmuir 2014, 30, 6302. 246. Gabardo, C. M.; Seifitokaldani, A.; Edwards, J. P.; Dinh, C. T.; Burdyny, T.; Kibria, M. G.; O'Brien, C. P.; Sargent, E. H.; Sinton, D.; Energy Environ. Sci. 2018, 11, 2531. 247. Cook, R. L.; MacDuff, R. C.; Sammells, A. F.; J. Electrochem. Soc. 1990, 137, 607. 248. Ma, S.; Sadakiyo, M.; Luo, R.; Heima, M.; Yamauchi, M.; Kenis, P. J. A.; J. Power Sources 2016, 301, 219. 249. Ma, S.; Lan, Y.; Perez, G. M. J.; Moniri, S.; Kenis, P. J. A.; ChemSusChem 2014, 7, 866. 250. Luo, M.; Wang, Z.; Li, Y. C.; Li, J.; Li, F.; Lum, Y.; Nam, D. H.; Chen, B.; Wicks, J.; Xu, A.; Zhuang, T.; Leow, W. R.; Wang, X.; Dinh, C. T.; Wang, Y.; Wang, Y.; Sinton, D.; Sargent, E. H.; Nat. Commun. 2019, 10, 1. 251. Dinh, C. T.; Burdyny, T.; Kibria, G.; Seifitokaldani, A.; Gabardo, C. M.; Pelayo García De Arquer, F.; Kiani, A.; Edwards, J. P.; De Luna, P.; Bushuyev, O. S.; Zou, C.; Quintero-Bermudez, R.; Pang, Y.; Sinton, D.; Sargent, E. H.; Science 2018, 360, 783. 252. Dinh, C. T.; García De Arquer, F. P.; Sinton, D.; Sargent, E. H.; ACS Energy Lett. 2018, 3, 2835. 253. García de Arquer, F. P.; Dinh, C. T.; Ozden, A.; Wicks, J.; McCallum, C.; Kirmani, A. R.; Nam, D. H.; Gabardo, C.; Seifitokaldani, A.; Wang, X.; Li, Y. C.; Li, F.; Edwards, J.; Richter, L. J.; Thorpe, S. J.; Sinton, D.; Sargent, E. H.; Science 2020, 367, 661. 254. Baccaro, A.; Gutz, I.; Quim. Nova 2018, 41, 326. 255. Xie, S.; Zhang, Q.; Liu, G.; Wang, Y.; Chem. Commun. 2016, 52, 35. 256. Castro, S.; Albo, J.; Irabien, A.; ACS Sustainable Chem. Eng. 2018, 6, 15877. 257. Gurudayal, G.; Beeman, J. W.; Bullock, J.; Wang, H.; Eichhorn, J.; Towle, C.; Javey, A.; Toma, F. M.; Mathews, N.; Ager, J. W.; Energy Environ. Sci. 2019, 12, 1068. 258. Schreier, M.; Héroguel, F.; Steier, L.; Ahmad, S.; Luterbacher, J. S.; Mayer, M. T.; Luo, J.; Grätzel, M.; Nat. Energy 2017, 2, 17087. 259. Zhang, N.; Long, R.; Gao, C.; Xiong, Y.; Sci. China Mater. 2018, 61, 771. 260. Morikawa, T. In Lecture Notes in Energy; Sugiyama, M., Fujii, K., Nakamura, S., eds.; Springer International Publishing: Cham, 2016; Vol. 32, pp. 281-296. 261. Zhang, Z.; Yates, J. T.; Chem. Rev. 2012, 112, 5520. 262. Irtem, E.; Hernández-Alonso, M. D.; Parra, A.; Fàbrega, C.; Penelas-Pérez, G.; Morante, J. R.; Andreu, T.; Electrochim. Acta 2017, 240, 225. 263. Xu, S.; Carter, E. A.; Chem. Rev. 2019, 119, 6631. 264. Ding, P.; Jiang, T.; Han, N.; Li, Y.; Mater. Today Nano 2020, 10, 100077. 265. Sahara, G.; Kumagai, H.; Maeda, K.; Kaeffer, N.; Artero, V.; Higashi, M.; Abe, R.; Ishitani, O.; J. Am. Chem. Soc. 2016, 138, 14152. 266. Schreier, M.; Curvat, L.; Giordano, F.; Steier, L.; Abate, A.; Zakeeruddin, S. M.; Luo, J.; Mayer, M. T.; Grätzel, M.; Nat. Commun. 2015, 6, 7326. 267. Arai, T.; Tajima, S.; Sato, S.; Uemura, K.; Morikawa, T.; Kajino, T.; Chem. Commun. 2011, 47, 12664. 268. Arai, T.; Sato, S.; Uemura, K.; Morikawa, T.; Kajino, T.; Motohiro, T.; Chem. Commun. 2010, 46, 6944. 269. Pan, P.-W.; Chen, Y.-W.; Catal. Commun. 2007, 8, 1546. 270. Morikawa, T.; Saeki, S.; Suzuki, T.; Kajino, T.; Motohiro, T.; Appl. Phys. Lett. 2010, 96, 142111. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access