
 |
 |
 |
 |
 |
Revisão
|
|
| Enganando o citocromo P450BM3: catálise de várias transformações de substratos não nativos usando moléculas-traiçoeiras Tricking cytochrome P450BM3: catalysis of various non-native substrate transformations using decoy molecules |
|
Joshua K. StanfieldI; Kazuto SuzukiI; Kai YonemuraI; Talita MalewschikI,II; Osami ShojiI,*
I. Departamento de Química, Universidade de Nagóia, 464-8602 Nagóia - Aichi, Japão Recebido em 29/01/2021 *e-mail: shoji.osami@a.mbox.nagoya-u.ac.jp In order to accomplish a greener chemistry, enzymes, such as the fatty-acid hydroxylase cytochrome P450BM3, have garnered increasing attention as potential candidates for the development of potent biocatalysts in recent years. However, one of the biggest issues hampering the quick and efficient application of P450BM3 as a biocatalyst lies in its stringent substrate specificity. Consequently, diverse mutagenesis-based approaches have been successfully employed as a means to alter the substrate specificity of P450BM3, leading to the generation of a myriad of highly specialised mutant variants. Nevertheless, repeated exhaustive mutagenesis is a laborious process with no guarantee for success, thus, alternative methods to more easily alter the enzyme's substrate specificity have become increasingly desirable. In recent years, decoy molecules, which possess the ability to deceive wild-type P450BM3 into hydroxylating a range of non-native substrates, have emerged as such a "simpler" alternative. Within this review, focus will be placed upon the process underlying the development of these decoy molecules, which will be discussed in great detail. Furthermore, a summary of recent developments pertaining to the potential applications of decoy molecules from the development of a whole-cell biocatalyst to their use in crystallography will be discussed. INTRODUÇÃO As enzimas citocromo P450 (CIPs ou P450s) estão presentes em uma ampla gama de organismos e representam uma superfamília de proteínas hemetiolato,1 nas quais o sítio ativo possui o grupo prostético ferroprotoporfirina IX (heme), que está ligado axialmente ao átomo de enxofre de um resíduo de cisteína (Cys) proximal.2,3 Esse fato as diferencia de outras hemoproteínas, como mioglobina4 e peroxidase de rábano,5 que estão ligadas axialmente a um átomo de nitrogênio de um resíduo de histidina. P450s desempenham inumeráveis transformações oxidativas nos sistemas biológicos, como, por exemplo, a biossíntese de hormônios, o metabolismo de drogas e a desintoxicação de xenobióticos, através da oxifuncionalização desses substratos, tal como hidroxilações,6 epoxidações7 e sulfoxidações,8 e também por reações de decomposição, como a desmetilação de éteres9 e a descarboxilação de ácidos graxos10,11 (Esquema 1a).
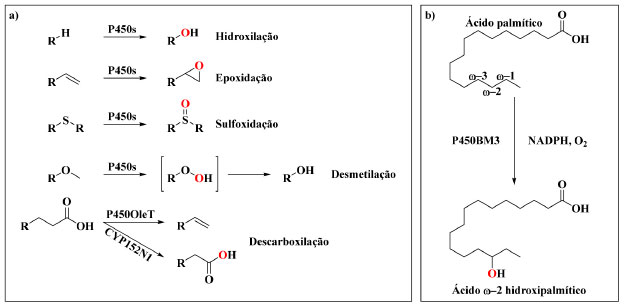 Esquema 1. a) Reações enzimáticas comumente catalisadas pelas enzimas P450. b) Hidroxilação do ácido palmítico pelo P450BM3 na posição subterminal ω−2
Na busca de reações químicas ambientalmente amigáveis, essas enzimas têm atraído atenção de pesquisadores na área de química verde pelo motivo de que elas possuem a capacidade de catalisar, sob condições amenas (temperatura ambiente, solução-tampão aquosa e pressão atmosférica),12 a inserção de um átomo de oxigênio em ligações inativas de carbono-hidrogênio (C-H), permitindo transformações desafiadoras que permanecem inacessíveis através de ferramentas químicas atualmente disponíveis.13-17 A transformação de substratos orgânicos pelas enzimas P450 são representadas pela equação abaixo, onde RH é um substrato genérico: O ciclo catalítico geral de oxidação de substratos, como detalhado no Esquema 2, pode ser dividido em nove etapas principais. Inicialmente, as enzimas do citocromo P450 são encontradas em seu estado de repouso, em que o ferro férrico (FeIII) do grupo prostético heme está no estado de spin baixo hexacoordenado com uma molécula de água como o sexto ligante (I). Após a ligação do substrato (RH), a molécula de água é eliminada do sítio ativo, levando à formação de uma espécie férrica pentacoordenada no estado de spin alto (II). Isso resulta em uma mudança positiva no potencial de redução do átomo de ferro, permitindo que um elétron da coenzima NADPH, a forma reduzida de fosfato de dinucleotídeo de nicotinamida e adenina (NADP+), seja transferido para o FeIII gerando uma espécie ferrosa (FeII) (III). Nessas condições, o oxigênio molecular (O2) pode ligar-se a essa espécie, formando um oxicomplexo (IV), que é reduzido por outro elétron proveniente do NADPH para formar um complexo férrico peróxido (V). Em seguida a protonação de um dos átomos de oxigênio, leva à formação de um complexo férrico hidroperóxido (VI), comumente referido como composto 0. Ele é protonado novamente seguido da ruptura heterolítica da ligação O-O, liberando uma molécula de água e gerando a espécie ativa, ferro-oxo de alta valência FeIV=O, composto I (VII), denominada de oxiferrilporfirina π-cátion radical. O composto I é altamente reativo e abstrai um radical de hidrogênio (H•) do substrato (RH) para criar um substrato radicalar (R•) e o complexo alcalino hidroxiferrilporfirina chamado composto II (VIII). Ao final, o radical hidroxila OH• é então rapidamente transferido do composto II ao R•, por um mecanismo conhecido como rebote de radical (do inglês radical rebound), para formar o produto hidroxilado (ROH), que é prontamente eliminado do sítio ativo. Existem duas possibilidades para a continuação do ciclo catalítico: I) uma molécula de água pode coordenar-se ao átomo de ferro, restaurando o estado de repouso (IX → I), ou, II) um novo substrato pode ligar-se diretamente ao sítio ativo, iniciando um novo ciclo de hidroxilação (IX → II). Entretanto, o ciclo catalítico das enzimas do citocromo P450 não é isento de falhas e nem sempre avança com 100% de eficiência. Existem múltiplas vias de desacoplamento (Esquema 2; linhas pontilhadas), que podem surgir devido às complicações durante o ciclo catalítico. O mais importante para esta revisão é o desacoplamento oxidase (VII → II), que ocorre quando o substrato é ligado de forma que nenhum átomo de hidrogênio esteja disponível para abstração pelo composto I, o que pode ocorrer na presença de um substrato não natural. Portanto, o composto I não tem outra escolha senão se reduzir ao complexo (II), liberando uma molécula de água, por meio da transferência de elétrons e prótons adicionais provenientes de uma molécula de NADPH. Embora na presença de um substrato nativo o acoplamento esteja próximo de 100%, quando as enzimas do citocromo P450 e suas variantes mutantes são utilizadas para a oxidação de substratos não nativos o desacoplamento oxidase representa um fenômeno problemático que ocorre com frequência.
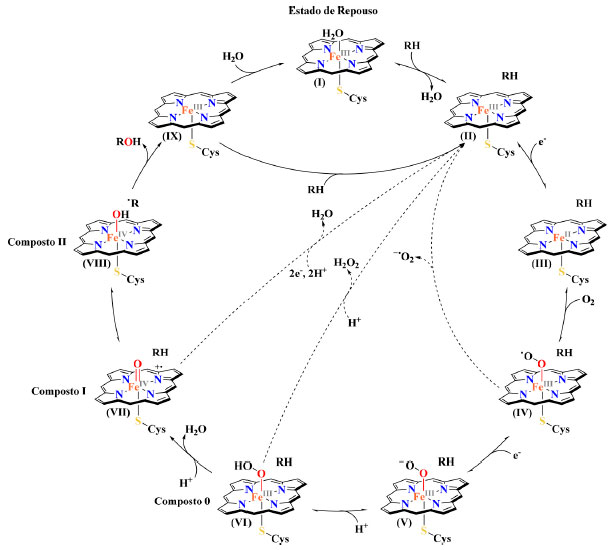 Esquema 2. Representação esquemática do ciclo catalítico geral das enzimas do citocromo P450 na presença de um substrato genérico (RH). As linhas pontilhadas representam vias de desacoplamento. Adaptada de Whitehouse e col. (2011)18
O Citocromo P450BM3 O foco desta revisão será o citocromo bacteriano CIP102A1 (P450BM3), uma enzima do tipo monooxigenase isolada do microrganismo Bacillus megaterium, devido ao fato de que tem chamado muita atenção por sua função como biocatalisador, uma vez que possui dois atributos especiais que o distinguem dos demais: I) P450BM3 possui uma das mais altas atividades catalíticas quando comparado com outras monooxigenases do citocromo P450 relatadas na literatura19 e, II) P450BM3 é uma proteína solúvel autossuficiente com seu domínio hémico expresso em conjunto com seu domínio redutase em uma única cadeia polipeptídica,20,21 tornando sua expressão e manuseio acessíveis. Isto contrasta com a maioria das enzimas do citocromo P450 que são expressas separadamente do seu domínio redutase, o qual pode ser constituído por múltiplos componentes complexos e, no pior caso, insolúveis. Os substratos nativos do P450BM3 são ácidos graxos, onde a enzima insere um átomo de oxigênio em uma das posições subterminais ω−1, ω−2 e ω−3 de ácidos graxos de cadeia longa (Esquema 1b).22 Substratos potenciais que não possuem o grupo ácido carboxílico, tais como o alcano de cadeia longa cetano, não são hidroxilados pelo P450BM3.23 Isso ocorre devido ao fato de que a elevada atividade catalítica do P450BM3 vem com o preço de uma especificidade rigorosa pelo substrato, que representa uma enorme desvantagem para sua utilidade como um biocatalisador versátil. Portanto, qualquer composto que seja estruturalmente diferente do substrato nativo provavelmente não será hidroxilado. Por esse motivo o P450BM3 tem sido alvo popular de inúmeros estudos de mutagênese para superar essa alta especificidade de substrato, para que essa enzima passe a reagir com uma grande variedade de substratos não nativos. Por exemplo, Meinhold e col. conseguiram transformar o P450BM3 de uma hidroxilase de ácido graxo subterminal em uma hidroxilase de alcano terminal através da engenharia de uma variante do P450BM3 contendo quinze mutações. Esse mutante pode realizar a desafiadora hidroxilação terminal do octano com seletividade de 52%.24 Recentemente, Li e col. conseguiram acessar uma rica variedade de diferentes produtos de oxidação de quinolinas e tetraidroquinolinas através do emprego de uma "enzimateca" do P450BM3 contendo um grande número de mutantes.25 Entretanto, o processo da evolução seguido da triagem de tais enzimas mutantes é imensamente trabalhoso e oneroso, exige múltiplos ciclos de mutagênese e necessita de uma gama de técnicas especializadas. Por mais poderosa que a técnica de mutagênese haja sido e continuará a ser para a engenharia de novos e potentes biocatalisadores, alternativas mais simples, que possam complementar a mutagênese, são desejadas para ajustar e aperfeiçoar as propriedades de reatividade do P450BM3. Uma alternativa com grande potencial será o assunto principal desta breve revisão. Há outras revisões que discutem este tópico sob outro ponto de vista e podem ser encontradas aqui.26-29
MOLÉCULAS-TRAIÇOEIRAS Descoberta O ciclo catalítico das enzimas P450, descrito no Esquema 2, mostra que, no estado de repouso da enzima, uma molécula de água está coordenada axialmente ao átomo central de ferro (FeIII) e o seu deslocamento, que ocorre pela introdução do substrato (RH) no meio da reação, atua como um "interruptor" dando início ao ciclo. Além disso, olhando a estrutura cristalina do P450BM3 complexado com o substrato ácido palmitoleico (Figura 1a), disponível no Banco de Dados de Proteínas (do inglês Protein Data Bank (PDB); www.rcsb.org/),30 entende-se que o substrato interage com a enzima em duas maneiras principais, I) interações iônicas do grupo carboxila do substrato com os resíduos Arg47 e Tyr51 do P450BM3 (Figura 1b), e II) interações hidrofóbicas da cadeia alquílica do substrato com aminoácidos que circundam o sítio de ligação do substrato.31
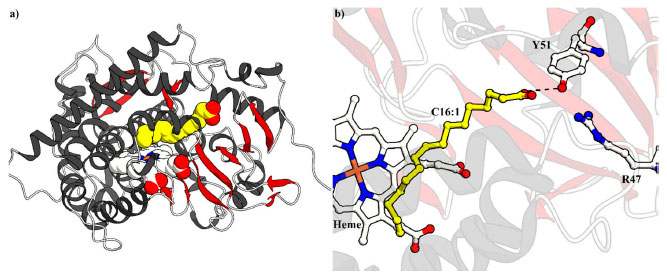 Figura 1. Estrutura cristalina a) geral e b) do sítio ativo do P450BM3 complexando o ácido palmitoleico (C16:1, amarelo) (código PDB: 1FAG). Interações iônicas são representadas como linhas pontilhadas
Baseado nessas interações, em 2011, dois trabalhos independentes foram publicados pelos grupos Reetz32 e Watanabe,33 os quais adotaram a mesma estratégia para forçar a ativação do ciclo catalítico da enzima citocromo P450BM3 (Esquema 2, etapa I → II). Como estratégia simples para evitar o uso da técnica de mutagênese, ambos os grupos propuseram o emprego de uma molécula inerte com estrutura similar aos substratos nativos da enzima (ácidos graxos), a qual teoricamente, levaria à formação da espécie catalítica ativa, que por sua vez iria hidroxilar substratos não nativos. O planejamento da estrutura dessa molécula mimética foi baseado em ácidos perfluorograxos (APFGs de ácidos perfluorograxos; uma sigla criada para esta revisão, do termo original em inglês PFCs de perfluoro carboxylic acids) (Esquema 3), que são estruturalmente similares ao substrato natural de ácido graxo e, devido à alta energia da ligação carbono-flúor (C-F), compostos perfluorados são quimicamente inertes e, subsequentemente, não seriam hidroxilados pelo composto I. Ambos os grupos de pesquisa previram que tais moléculas simples e inertes deveriam ser capazes de "enganar" o P450BM3, o qual pensaria que um substrato viável estivesse presente no sítio ativo, e assim, dando início ao ciclo catalítico.
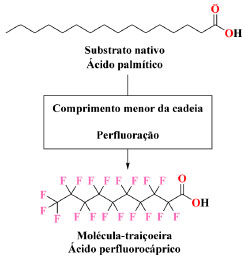 Esquema 3. Comparação da estrutura do substrato nativo com a primeira geração de moléculas-traiçoeiras. Essas moléculas possuem uma cadeia perfluoroalquílica mais curta do que substratos nativos
Estudos preliminares confirmaram o consumo de NADPH após a adição de APFGs a uma solução contendo a enzima, o que indica que a indução da primeira etapa do ciclo catalítico (I → II), necessária para a formação do composto I, pode ser estimulada por esses substratos miméticos inertes, doravante referidos como "moléculas-traiçoeiras" (MT), um termo que criamos para o termo em inglês "decoy molecules" e em alemão "Täuschmoleküle". Com essa descoberta, uma série de moléculas-traiçoeiras foi planejada e estudada, variando o comprimento da cadeia perfluorocarbônica, onde a molécula mais curta estudada foi o ácido perfluorocaprílico (APFG8; onde o número indica o tamanho da cadeia perfluorocarbônica) e a mais longa foi o ácido perfluoromirístico (APFG14). O substrato não nativo escolhido para investigação do potencial catalítico desse sistema foi o alcano gasoso propano, um dos componentes do gás natural, sendo útil como combustível, solvente e matéria-prima para derivados.34 Essa reação é interessante, pois a alta energia das ligações carbono-hidrogênio torna a hidroxilação direta de alcanos gasosos curtos desafiadora, devido ao fato de que o produto hidroxilado possui maior propensão a uma oxidação subsequente, levando à formação de produtos indesejados, como, por exemplo, COx. Como previsto, o gás propano só foi hidroxilado pelo P450BM3 quando uma molécula-traiçoeira foi adicionada à mistura de reação, sendo detectada a formação de álcool isopropílico (i-propanol) como produto principal (Figura 2 esquerda, barras vermelhas). Uma correlação interessante entre o tamanho da cadeia dos APFGs e a atividade catalítica foi observada, em que houve um aumento da atividade catalítica do P450BM3 conforme a cadeia do APFG foi alongada. Contudo, também foi observado que se a cadeia perfluorocarbônica for muito longa, esta iria obstruir a entrada de um composto exógeno (substrato não nativo) no sítio ativo. Seguindo a mesma lógica, APFGs curtos, embora permitissem mais espaço para entrada de outros compostos no sítio ativo, mostraram não ser capazes de ativar a enzima de forma eficiente. A frequência de rotatividade (FR; em inglês turnover frequency (TOF)), que significa o número das moléculas de produto produzidas por uma única enzima a cada minuto) máxima para a hidroxilação do propano ao i-propanol em uma reação de dez minutos chegou a 67 min−1 P450−1 na presença de ácido perfluorocáprico (APFG10). A eficiência de acoplamento (EA; em inglês coupling efficiency (CE)) com NADPH para essa reação, que pode ser calculada usando equação (2), estagnou em 18% (Figura 2, esquerda, círculos ocos roxos), embora a EA da reação nativa (hidroxilacão de ácidos graxos) pudesse atingir quase 100%. Esse resultado indica que o desacoplamento oxidase do composto I (Esquema 2 VII → II) provavelmente esteja competindo com a hidroxilação do propano.35
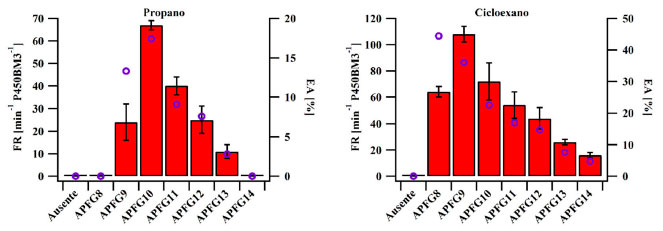 Figura 2. Frequência de rotatividade de hidroxilação dos substratos não nativos propano (esquerda) e cicloexano (direita) pelo P450BM3 na ausência e na presença de moléculas-traiçoeiras de comprimentos variados (barras vermelhas indicam a FR e círculos ocos roxos indicam a EA). Condições de reação: P450BM3 do tipo selvagem (0,5 µmol L-1), molécula-traiçoeira (100 µmol L-1), tampão saturado com gás propano e oxigênio (4:1) ou cicloexano (10 mmol L-1), NADPH (5 mmol L-1), durante dez minutos em tampão Tris-HCl 20 mmol L-1, pH 7,4 contendo KCl (100 mmol L-1)
Na presença de um substrato não nativo mais volumoso, como, por exemplo, o cicloalcano cicloexano, o produto cicloexanol foi obtido (Figura 2, direita). Essa reação prosseguiu com uma EA superior de até 44% na presença de APFG8, mostrando que o desacoplamento oxidase foi significativamente reduzido, o que pode indicar que o cicloexano provavelmente esteja ligado de forma mais estável ao sítio ativo quando comparado ao gás propano. Curiosamente, também foi observada uma correlação entre o comprimento da cadeia da molécula-traiçoeira e o tamanho do substrato. Como exemplo, APFG10 foi identificado como a molécula-traiçoeira mais adequada para a hidroxilação do propano, enquanto o ácido perfluoropelargônico (APFG9), o qual possui uma unidade perfluorocarbônica a menos, resultou em uma melhor atividade para o cicloexano. Da mesma forma, a mais curta molécula-traiçoeira testada, APFG8, não exibiu nenhuma atividade com propano, mas sua atividade com cicloexano foi significativa (Figura 2 esquerda e direita). Além disso, os ácidos perfluorograxos de cadeia média APFG9 e APFG10 mostraram ser as moléculas-traiçoeiras mais adequadas para a hidroxilação dos substratos cicloexano e propano, respectivamente, onde o tamanho do substrato determinou o comprimento ideal da cadeia da molécula-traiçoeira. Esses resultados demonstraram que o P450BM3 pode ser facilmente enganado para realizar a hidroxilação de substratos não nativos, através da fácil adição de moléculas-traiçoeiras à mistura de reação. Esses simples ácidos perfluorograxos são comumente chamados de moléculas-traiçoeiras de "primeira geração". Baseado nesses resultados, o ciclo catalítico introduzido anteriormente no Esquema 2 foi simplificado, onde somente as três etapas mais relevantes são ressaltadas (etapas I → II, II → VII e VII → I). O novo ciclo catalítico (Esquema 4), na presença de uma molécula-traiçoeira e cicloexano como substrato não nativo, é comparado ao ciclo catalítico na ausência de moléculas-traiçoeiras e na presença de um substrato nativo, como, por exemplo, ácido palmítico.
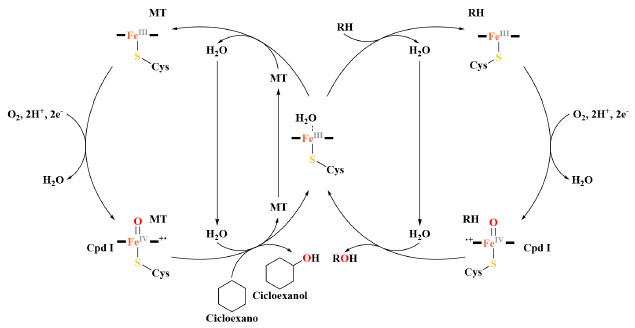 Esquema 4. Comparação entre o ciclo catalítico na presença de um substrato nativo (RH) (direita) e o ciclo catalítico na presença de uma molécula-traiçoeira (MT) e o substrato não nativo cicloexano (esquerda)
A comparação entre esses ciclos mostra de maneira clara que tanto a molécula-traiçoeira (MT) como o ácido graxo (RH) ativam a enzima de maneira semelhante, removendo a água do sítio ativo. No entanto, no caso da molécula-traiçoeira, o cicloexano pode entrar no sítio ativo e é hidroxilado em seu lugar. Em busca de uma reação de hidroxilação mais eficaz Embora a hidroxilação de substratos não nativos na presença de moléculas-traiçoeiras apresente uma nova abordagem muito promissora, sua eficácia ainda é significativamente inferior, quando comparada com os elevados valores de FR obtidos com os substratos nativos de ácidos graxos. Isso significa que as moléculas-traiçoeiras de primeira geração precisariam ser aprimoradas, para que o sistema P450BM3 na presença dessas moléculas alcançasse ou até mesmo superasse o desempenho do sistema nativo. No entanto, ao invés de focar no aprimoramento de moléculas-traiçoeiras, o grupo de pesquisa Watanabe, inicialmente, resolveu focar em estratégias para aumentar a eficiência da reação de hidroxilação do gás propano, concentrando-se em maneiras de melhorar a baixa EA observada para esse sistema com moléculas-traiçoeiras de primeira geração. Kawakami e col. sugeriram que o fator responsável seria a baixa afinidade de alcanos gasosos com o sítio ativo da enzima. Uma alternativa para superar esse problema seria o acréscimo da concentração dos alcanos gasosos na solução tampão utilizada na reação, o que poderia levar a uma alta taxa de formação de produto e, ao mesmo tempo, reduziria a quantidade de NADPH desperdiçada devido ao desacoplamento. Duas abordagens foram avaliadas para aumentar a concentração de alcanos gasosos em solução: I) reações à baixa temperatura36 e, II) reações sob alta pressão.35 Em geral, o aumento da solubilidade de alcanos gasosos é inversamente proporcional à diminuição da temperatura, ou seja, quanto mais baixa a temperatura da solução tampão, uma maior quantidade de gás pode ser dissolvida. Por exemplo, a 20 °C a solubilidade do propano na água é 1,75 mmol L-1 e a 4 °C a concentração dobra para 3,46 mmol L-1.37 Como esperado, ao reduzir a temperatura de reação de 20 °C para 4 °C, a EA para hidroxilação do propano aumentou de 38% para 44% na presença de APFG10 e, ainda mais notavelmente, de 31% para 51% com APFG9 após uma hora de reação (Tabela 1). No entanto, a FR com APFG10, por exemplo, diminuiu de 54 ± 3 min−1 P450−1 para 16 ± 1 min−1 P450−1, devido à dificuldade da enzima em superar a barreira de energia para a catálise sob condições de baixa temperatura. Com o intuito de melhorar os resultados obtidos, reações de 24 horas foram realizadas com a temperatura a 0 e 20 °C e, notavelmente, efeitos mais drásticos foram observados (Tabela 2). Quando APFG9 foi usado como molécula-traiçoeira e baixando a temperatura de reação de 20 °C para 0 °C, o número total de rotatividade (NTR; em inglês total turnover number (TTN)) em 24 horas foi dobrado de 4760 P450−1 para 9840 P450−1 e a EA também aumentou de 17% para 40%.
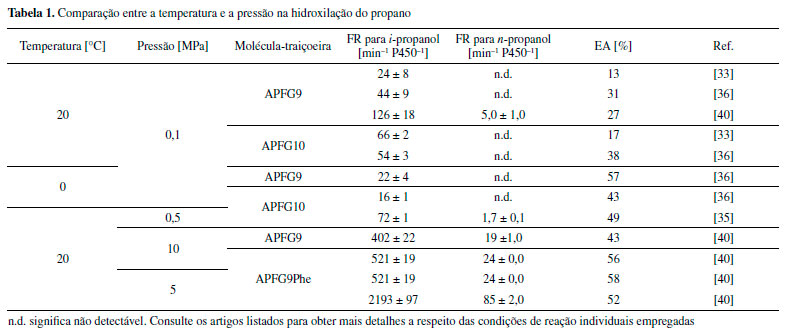

Como segunda abordagem, a solubilidade do propano foi aumentada pela pressurização. Kawakami e col. fizeram uso de um reator de alta pressão, onde a solução tampão de reação foi pressurizada a 0,5 MPa (cerca 5 atm) com gás propano na presença de APFG10 (Tabela 1). Os resultados obtidos revelaram que a EA alcançou 49 % com uma FR de 72 min−1 P450−1 para a formação de i-propanol. Além disso, pela primeira vez, o álcool n-propílico (n-propanol) também pôde ser detectado como produto nas condições contendo moléculas-traiçoeiras (FR: 1,7 min−1 P450−1), indicando que a pressurização permite a hidroxilação desafiadora de um carbono primário. Portanto, os autores também resolveram examinar a hidroxilação do etano sob alta pressão de 0,5 MPa, e após duas horas de reação, o produto etanol pôde ser detectado por cromatografia gasosa (CG), onde o rendimento do produto foi estimado em 80 µmol L-1 (uma FR de 40 h−1 P450−1). Entretanto, a hidroxilação do metano, a qual é extremamente desafiadora e conhecida como uma das reações de "Santo Graal" na química orgânica, não foi observada na presença de moléculas-traiçoeiras, mesmo sob condições de alta pressão, indicando que modificações adicionais serão necessárias para atingir esse objetivo no futuro.32,35
EVOLUÇÃO DE MOLÉCULAS-TRAIÇOEIRAS Embora houvesse sido observado um aumento nos valores na FR da hidroxilação dos alcanos gasosos ao ajustar as condições de reação na presença de moléculas-traiçoeiras, a eficiência da reação foi estagnada ao usar ácidos perfluorograxos simples (moléculas-traiçoeiras de primeira geração) indicando que um incremento adicional na atividade usando essas moléculas não seria possível. Assim, tornou-se necessário descobrir novos tipos de moléculas-traiçoeiras com maior diversidade estrutural para que o P450BM3 atingisse o seu pleno potencial catalítico em reações na presença dessas novas moléculas. Nesta seção a evolução de moléculas-traiçoeiras será detalhada e a representação dos diferentes estágios desse desenvolvimento pode ser observado no Esquema 5.
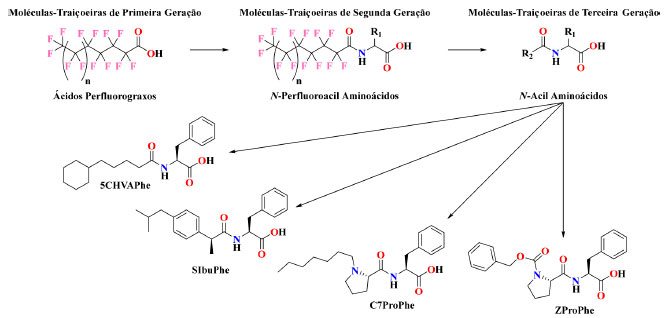 Esquema 5. Esquema mostrando a evolução de moléculas-traiçoeiras de primeira geração a terceira geração. n representa o comprimento da cadeia perfluoroalquílica, sendo n = 2 correspondente ao APFG8. R1 significa um dos vinte aminoácidos canônicos. R2 significa um ácido carboxílico. Quatro exemplos de moléculas-traiçoeiras de terceira geração que possuem boas propriedades, 5CHVAPhe, SIbuPhe, C7ProPhe e ZProPhe, também são apresentadas
Moléculas-traiçoeiras de segunda geração Design e aprimoramento Uma pista sobre como melhorar a eficácia de moléculas-traiçoeiras pôde ser obtida a partir de um trabalho publicado por Haines e col., em que o substrato nativo, ácido palmítico, foi modificado com o aminoácido glicina (N-palmitoilglicina; C16Gly). Essa modificação levou a uma melhora na afinidade de ligação do substrato com o P450BM3 com uma ordem de grandeza de 2 µmol L-1 para 262 nmol L-1 (dado como o valor da constante de dissociação (Kd)), o que provocou um aumento considerável na FR. Uma explicação para este efeito pôde ser extraída da estrutura cristalina deste complexo (Figura 3a e b), a qual revelou que a modificação com glicina aumenta o número de ligações de hidrogênio formadas (compare as Figuras 1b e 3b) o que por sua vez, leva a uma fixação do substrato mais forte.38 Em comparação, Kawakami e col. estimaram, por meio da técnica de titulação espectrofotométrica, que APFG9 se liga fracamente ao P450BM3 com um valor Kd em torno de 980 µmol L-1.33
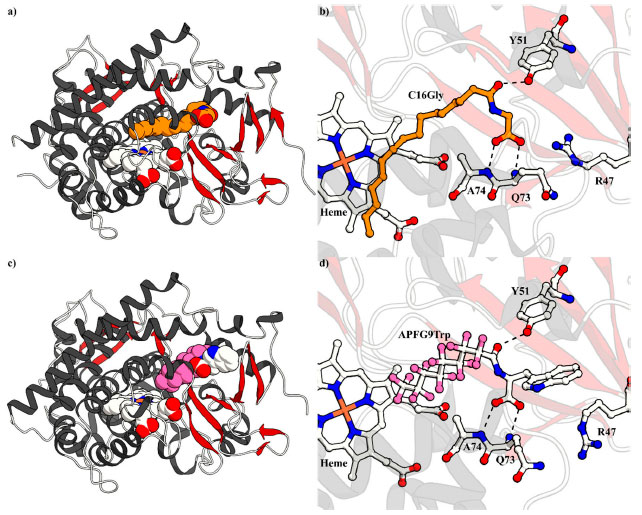 Figura 3. a) e b) Estruturas de raio X do P450BM3 em complexo com N-palmitoilglicina (C16Gly, laranja) (código PDB: 1JPZ); e c) e d) com N-perfluoropelargonoil-l-triptofano (APFG9Trp, rosa e branco) (código PDB: 3WSP)
Cong e col. conjecturaram que o fenômeno relatado por Haines e col. poderia ser potencialmente utilizado para aumentar a eficácia de moléculas-traiçoeiras ao melhorar sua afinidade de ligação ao P450BM3. A modificação de APFG9 com l-triptofano (N-perfluoropelargonoil-l-triptofano; APFG9Trp) melhorou a afinidade de ligação por mais de duas ordens de grandeza apresentando um valor de 1,6 µmol L-1 para Kd. Como previsto, este incremento na afinidade de moléculas-traiçoeiras por P450BM3 traduziu-se em um aumento significativo na FR da hidroxilacão de substratos não nativos. Um exemplo ilustrativo é a hidroxilação do propano, onde a taxa pôde ser quadruplicada de 67 min−1 P450−1 (molécula-traiçoeira de primeira geração APFG10) para 256 min−1 P450−1 (molécula-traiçoeira de segunda geração N-perfluoropelargonoil-l-leucina (APFG9Leu)). Curiosamente, a atividade catalítica também foi influenciada pela combinação do aminoácido e do substrato empregada, com atividade catalítica para hidroxilação do propano sendo mais alta para l-leucina e mais baixa para glicina entre os oitos aminoácidos testados. Já nas reações de hidroxilação do cicloexano e do etano, as modificações com l-leucina exibiram os melhores resultados, enquanto as moléculas-traiçoeiras com l-alanina e l-isoleucina apresentaram os piores, respectivamente (dados não mostrados).39 Caracterização estrutural por meio de cristalografia Embora a cristalização do P450BM3 houvesse se mostrado infrutífera na presença de moléculas-traiçoeiras de primeira geração, as moléculas-traiçoeiras de segunda geração modificadas com l-triptofano produziram cristais de qualidade para o estudo estrutural por difração, o qual revelou a maneira como elas se ligam ao sítio de ligação do substrato. Como pode ser observado na estrutura cristalina do P450BM3 em complexo com APFG9Trp (Figuras 3c e 3d), a molécula-traiçoeira de segunda geração está ligada da mesma forma que a C16Gly (Figura 3b), com os resíduos Tyr51, Gln73 e Ala74 interagindo com a molécula-traiçoeira, o que a mantém ligada fortemente no sito de ligação do substrato através de ligações de hidrogênio. Por causa dessas interações fortes, o átomo de carbono terminal da cadeia perfluoroalquílica da molécula-traiçoeira é mantido a 8,5 Å de distância do átomo central de ferro do grupo heme, mostrando que há muito espaço para ligação de um substrato exógeno pequeno (compare Figura 4a e b).39 Avanços na atividade catalítica
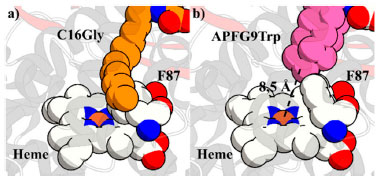 Figura 4. Comparação entre o espaço no sítio ativo do P450BM3 complexando com a) o substrato C16Gly (código PDB: 1JPZ) e b) a molécula-traiçoeira de segunda geração APFG9Trp (código PDB: 3WSP). No caso de C16Gly, observa-se que F87 se encontra rotacionado de tal forma que o sítio ativo é completamente obstruído. Entretanto, no caso de APFG9Trp, há espaço suficiente para acessar o sítio ativo
Considerando os resultados promissores obtidos com as moléculas-traiçoeiras de primeira geração por pressurização no segmento anterior, o mesmo foi tentado para as moléculas-traiçoeiras de segunda geração. Recentemente, Ariyasu e col. desenvolveram um novo tipo de reator de alta pressão que pode alcançar pressões muito altas de até 10 MPa com segurança (cerca 100 atm, o que representa o vigíntuplo da pressão aplicada na seção anterior), sem a necessidade de cilindros de gás de alta pressão.40 O reator foi construído utilizando uma bomba de cromatografia líquida de alta eficiência (CLAE) para que comprima o substrato gasoso a altas pressões com base em um sistema de pressurização de fluxo líquido (Esquema 6). Ao elevar a pressão em cem vezes, de 0,1 MPa para 10 MPa, na presença da molécula-traiçoeira de primeira geração APFG9, observou-se que a FR da formação de n-propanol foi triplicada, a formação de i-propanol foi sextuplicada e a EA melhorou em 59% (Tabela 1). Além disso, a combinação de pressões extremamente altas com moléculas-traiçoeiras de segunda geração levou a uma maior atividade de hidroxilação do propano quando comparada com a da primeira geração. Pressurizando o tampão a 10 MPa na presença de N-perfluoropelargonoil-l-fenilalanina (APFG9Phe), as FRs alcançaram 521 ± 19 min−1 P450−1 para i-propanol e 24 ± 0,0 min−1 P450−1 para n-propanol com uma EA de 56% (Tabela 1). Surpreendentemente, mesmo com 5 MPa, quase os mesmos resultados foram obtidos, o que indica que a solubilidade do propano provavelmente já esteja maximizada em 5 MPa. Ariyasu e col. obtiveram grande sucesso na melhoria da atividade de hidroxilação do propano, porém, sob as condições de reação empregadas (0,5 µmol L-1 P450BM3, 5 mmol L-1 NADPH, 10 min de reação), foi notado que quase todo o NADPH foi consumido dentro de três minutos de reação. Por isso, eles propuseram a hipótese de que o esgotamento do NADPH pudesse ter limitado a FR. Assim, a fim de evitar que o NADPH se esgotasse durante a reação permitindo uma estimativa mais precisa da FR, eles realizaram uma reação de um minuto na presença da APFG9Phe, a qual apresentou uma taxa inicial de cerca de 2200 min−1 P450−1,40 o que é muito próxima da taxa de hidroxilação determinada para os ácidos graxos nativos.41 Ademais, essa FR é o valor mais alto relatado entre todas as monooxigenases, incluindo a monooxigenase do propano P450PMOR1, um mutante do P450BM3 que carrega um total de 24 mutações cuja evolução foi especificamente desenvolvida para a hidroxilação do propano.42 A combinação de APFG9Phe com o reator de alta pressão também demonstrou ser eficaz para a hidroxilação mais desafiadora do etano ao etanol, que rendeu uma FR de 28,2 ± 1,8 min−1 P450−1.43
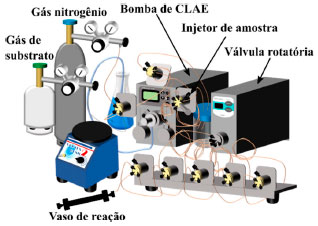 Esquema 6. Reator de alta pressão baseado em pressurização de fluxo líquido (patente número 2019-69404).44 Empregando esse reator, pressões de até 10 MPa podem ser alcançadas
Moléculas-traiçoeiras de terceira geração Shoji e col. observaram, a partir da estrutura cristalina do APFG9Trp (Figura 4b), que a modificação das moléculas-traiçoeiras com aminoácidos ajuda a manter a cadeia perfluoroalquílica dessa molécula suficientemente distante do grupo prostético heme (8,5 Å), o que evita que essa molécula seja oxidada pelo composto I (Esquema 2, VII), mesmo na ausência de perfluoração. Assim, eles supuseram que as moléculas-traiçoeiras de primeira geração exigiam a perfluoração da cadeia carbônica, já que possuíam um menor número de interações com o sítio de ligação do substrato na enzima e, consequentemente, elas deslizariam inadvertidamente para dentro do sítio ativo sendo hidroxiladas. Já as moléculas-traiçoeiras de segunda geração permanecem mais firmemente ligadas ao P450BM3 devido às interações adicionais introduzidas pela modificação com aminoácidos. Isso significa que mesmo os ácidos graxos não perfluorados devam ser capazes de servir como moléculas-traiçoeiras e, do ponto de vista econômico, ambiental e facilidade de síntese, os compostos perfluorados são menos do que ideais e sua substituição seria desejável. Portanto, uma série de ácidos graxos modificados com aminoácidos foi preparada, e, como previsto, N-acil aminoácidos simples atuaram como moléculas-traiçoeiras, sendo assim denominados como moléculas-traiçoeiras de terceira geração. Empregando N-pelargonoil-l-fenilalanina (AG9Phe), a atividade catalítica na hidroxilação direta do benzeno a fenol melhorou 22% para 192 ± 8 min−1 P450−1, de 157 ± 4 min−1 P450−1 obtida anteriormente com APFG9Phe. Assim como foi observado com moléculas-traiçoeiras de segunda geração, a atividade catalítica do P450BM3 na presença das moléculas-traiçoeiras de terceira geração é altamente dependente do comprimento da cadeia e do tipo de aminoácido. Ademais, a exigência de perfluoração imposta às moléculas-traiçoeiras de primeira e segunda geração, a qual limitava a diversidade estrutural dessas moléculas, foi removida, portanto, qualquer ácido carboxílico que preencha certos requisitos estruturais, como, por exemplo, comprimento e volume, pode servir como bloco de construção para novas moléculas-traiçoeiras. Uma série dessas moléculas foram sintetizadas, das quais as quatro mais promissoras encontradas nesta fase são destacados no Esquema 5. Essas novas moléculas-traiçoeiras de terceira geração levaram a uma melhoria significativa na atividade catalítica. Por exemplo, na presença da molécula-traiçoeira dipeptídica N-enantil-l-prolil-l-fenilalanina (C7ProPhe), a hidroxilação direta do benzeno a fenol aumentou para 259 ± 12 min−1 P450−1 e apresentou uma EA de 43%, mais que o dobro obtido com APFG9.45,46 Agora, ao imaginar todas as combinações possíveis de ácidos carboxílicos disponíveis comercialmente em combinação com pelo menos vinte aminoácidos canônicos, o número de moléculas-traiçoeiras concebíveis aumentou exponencialmente. Evolução adicional de moléculas-traiçoeiras por abordagem de triagem Moléculas-traiçoeiras mais eficazes ainda podem ser descobertas entre as enormes combinações possíveis de blocos de construção, todavia, mesmo que o foco seja colocado apenas em moléculas-traiçoeiras dipeptídicas facilmente sintetizadas através da técnica de síntese de peptídeos em fase sólida (SPFS), tais como C7ProPhe e N-benziloxicarbonil-l-prolil-l-fenilalanina (ZProPhe), o processo de triagem até mesmo de uma fração das combinações possíveis ainda representaria um esforço sintético absurdo. Yonemura e col. criaram assim um método de triagem em etapas, com foco em moléculas-traiçoeiras com esqueleto dipeptídico N-substituído. A abordagem adotada pelos autores foi baseada em uma triagem lógica em seis etapas (Esquema 7) focada apenas nas combinações mais promissoras de subestruturas eficazes, as quais foram identificadas usando um método rápido de triagem colorimétrica baseado na detecção do fenol a partir de benzeno.47 Usando esse método, o número de moléculas-traiçoeiras sintetizadas foi reduzido para 613, apenas 0,6% do total de 98.072 possibilidades provenientes da combinação teórica de todos os possíveis blocos de construção empregados.43 Nessa quimioteca composta de moléculas-traiçoeiras dipeptídicas N-substituídas, a molécula denominada N-(3-cilopentil)propanoil-l-pipecolil-l-fenilalanina (3CPPAPipPhe), apresentou resultados muito promissores na hidroxilação direta do benzeno ao fenol, com a FR atingindo 405 ± 8 min−1 P450−1, um aumento de 57% comparado com a molécula-traiçoeira ZProPhe. Este resultado também foi superior a qualquer valor relatado anteriormente, incluindo mutantes do P450BM3 desenvolvidos pela técnica de evolução dirigida.48 Além disso, a hidroxilação desafiadora do etano foi acelerada com a adição destas moléculas-traiçoeiras sob pressurização, com a FR atingindo 82,7 min−1 P450−1 na presença de N-enantoil-l-pipecolil-l-fenilalanina (C7AMPipPhe), que representou quase o triplo do valor obtido para APFG9Phe,40 e o maior entre os valores relatados catalisados pelos mutantes do P450s (Tabela 3).49-52
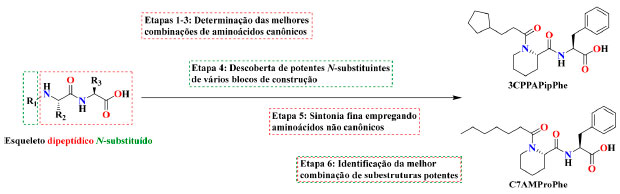 Esquema 7. Visão geral da evolução sistemática de moléculas-traiçoeiras dipeptídicas N-substituídas provenientes da triagem executada em seis etapas. As duas moléculas mais eficazes descobertas por esta abordagem são mostradas à direita
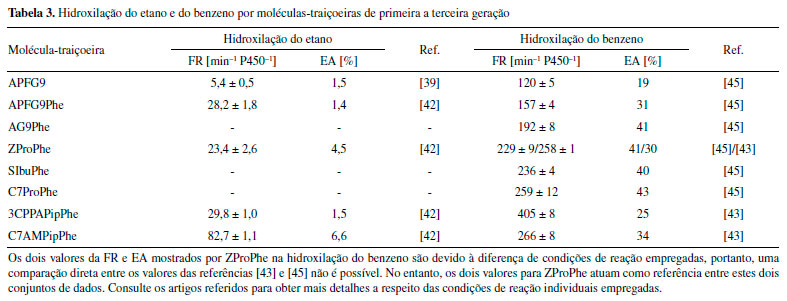
APLICAÇÕES DE MOLÉCULAS-TRAIÇOEIRAS Na seção anterior, a descoberta de moléculas-traiçoeiras e seu subsequente aprimoramento foram detalhados com foco na hidroxilação de alcanos gasosos. Nesta seção será abordado o uso de moléculas-traiçoeiras em diversas aplicações práticas, desde sua utilização in vivo até a cristalização. Ao final da seção, outros sistemas de moléculas-traiçoeiras também serão discutidos. Rumo à aplicação prática: Aplicação de moléculas-traiçoeiras em biocatálise com células inteiras A produção industrial de fenol e acetona a partir de benzeno e propileno pelo processo Hock é um processo que requer muita energia e, portanto, uma alternativa mais verde e energeticamente eficiente é desejável. A potente enzima P450BM3, juntamente com o sistema de moléculas-traiçoeiras, poderia representar uma alternativa verde para esse problema. Porém, para que esse sistema possa ser aplicado de maneira prática, há uma séria limitação a ser resolvida, nomeadamente a exigência de uma quantidade estequiométrica do cofator NADPH como doador de elétrons. O custo exorbitante deste reagente apresenta-se como um obstáculo que inviabiliza a aplicação em larga escala de moléculas-traiçoeiras in vitro, a menos que outros meios para gerar e reabastecer economicamente o NADPH necessário sejam encontrados. Karasawa e col. propuseram uma solução elegante para esse problema, na qual se fez o uso de um sistema biocatalítico com células inteiras de Escherichia coli com moléculas traiçoeiras, que exige apenas glicose, um reagente acessível, no meio de cultura como a única fonte energética, sem a necessidade de fornecer o dispendioso NADPH, como mostra o Esquema 8. Glicose adicionada ao meio de reação é metabolizada para estimular a produção de NADPH dentro de células inteiras de E. coli. Um benefício adicional de um biocatalisador de células inteiras comparado com os sistemas in vitro é que não há necessidade de isolar ou purificar nenhuma proteína. Como a expressão do P450BM3 em E. coli é simples, o único problema a ser resolvido para obter um biocatalisador de células inteiras é a permeabilidade da membrana bacteriana pelas moléculas-traiçoeiras (Esquema 8, passo 2). A bactéria E. coli, abrigando o gene responsável pela produção da enzima P450BM3, foi suspendida em tampão fosfato contendo 10 mmol L-1 de benzeno e 100 µmol L-1 de molécula-traiçoeira e o sistema foi incubado por cinco horas em temperatura ambiente. Como esperado, na ausência de moléculas-traiçoeiras, a produção de fenol foi mínima. A adição de moléculas-traiçoeiras de primeira e segunda geração rendeu uma pequena quantidade de fenol, quando comparada com os rendimentos obtidos in vitro (Tabela 4), destacando que ácidos perfluorograxos provavelmente não atravessaram a membrana bacteriana de maneira eficiente. Todavia, o uso de moléculas-traiçoeiras de terceira geração, como por exemplo, N-(S)-ibuprofenoil-l-fenilalanina (SIbuPhe) e C7ProPhe, mostrou melhores rendimentos atingindo 38%. Curiosamente, N-(R)-ibuprofenoil-l-fenilalanina (RIbuPhe) exibiu rendimentos significativamente mais baixos in vivo do que SIbuPhe, embora ambas as moléculas houvessem exibido resultados in vitro semelhantes, destacando que pequenas mudanças na estrutura das moléculas-traiçoeiras, tal como estereoquímica, podem causar uma diferença na permeabilidade dessas moléculas na membrana bacteriana. Sob condições otimizadas (DO600 = 12,6 de biocatalisador de células inteiras), o rendimento de fenol atingiu 59%, com 75% de benzeno sendo convertido em fenol ou hidroquinona (seletividade para fenol de 78%).53 Como exemplificado por esses resultados, um avanço significativo no sistema de moléculas-traiçoeiras foi alcançado, o que aproxima este sistema de sua aplicação prática em um futuro não tão distante.
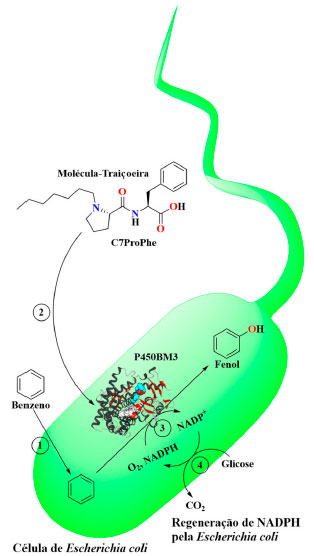 Esquema 8. Ilustração simplificada que apresenta a aplicação de moléculas-traiçoeiras em biocatálise com células inteiras. A bactéria E. coli geneticamente modificada, a qual expressa a enzima citocromo P450BM3 do tipo selvagem, foi cultivada em meio de cultura contendo glicose. (1) A molécula de Benzeno permeia a membrana da E. coli. (2) A molécula-traiçoeira penetra a membrana bacteriana. (3) O benzeno presente no interior da célula é hidroxilado pelo P450BM3 produzido dentro da E. coli usando NADPH como doador de elétrons. (4) O cofator NADPH é regenerado pela E. coli através do metabolismo da glicose
Controlando a enantiosseletividade da hidroxilação benzílica com moléculas-traiçoeiras Até esse momento, as moléculas-traiçoeiras vinham sendo empregadas exclusivamente no esforço de ativar o P450BM3 para hidroxilações de substratos não nativos com taxas de rotatividade catalítica cada vez mais altas. Entretanto, a alta rotatividade catalítica não é o único aspecto importante de uma reação catalítica, onde a alta regio- e estereosseletividade também são desejáveis. A análise da estrutura cristalina de raios X revelou que a ligação de moléculas-traiçoeiras poderia induzir mudanças conformacionais do P450BM3 e remodelar seu sítio ativo.54 Portanto, a estrutura da molécula-traiçoeira é considerada ser capaz de modular a forma como os substratos não nativos aproximam-se da espécie catalítica ativa (Esquema 1, etapa VII) durante a catálise e, assim, controlar a enantiosseletividade. Um relatório de Suzuki e col. de 2017 demonstrou que a estereosseletividade da hidroxilação benzílica dos compostos aromáticos pode ser influenciada pela adição de moléculas-traiçoeiras. Na ausência de qualquer molécula-traiçoeira, o excesso enantiomérico (ee) para a hidroxilação do etilbenzeno foi 45% (R) ee, o qual aumentou para 85% (R) ee na presença de N-(5-cicloexil)valeroil-l-fenilalanina (5CHVAPhe) (Tabela 5). O uso de moléculas-traiçoeiras na reação de hidroxilação do indano levou a uma inversão da estereosseletividade na formação dos produtos (R) e (S) (Esquema 9), sendo que na presença de 5CHVAPhe observou-se 53% (R) ee, comparado com ZProPhe que apresentou 56% (S) ee (Tabela 5). Na ausência de uma molécula-traiçoeira na mistura de reação, a enantiosseletividade foi ligeiramente a favor do enantiômero (S) com 16% ee. Notavelmente, a adição de moléculas-traiçoeiras aumentou a taxa de rotatividade (dados não mostrados), bem como a enantiosseletividade, o que mostra que é possível atingir um sistema catalítico quase perfeito, em que se pode alcançar tanto altos rendimentos quanto seletividade, o que é um desafio frequentemente encontrado na área de catálise.

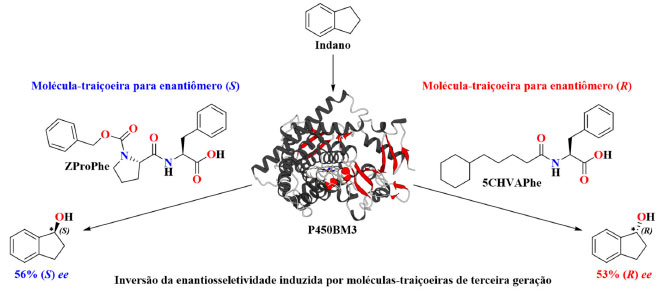 Esquema 9. Hidroxilação enantiodivergente na posição benzílica do indano induzida por moléculas-traiçoeiras
Aplicação de moléculas-traiçoeiras com mutagênese Os trabalhos supracitados comprovaram que as moléculas-traiçoeiras imitam ácidos graxos, e são capazes de alterar a seletividade da enzima P450BM3 do tipo selvagem para o substrato, de ácidos graxos para substratos não nativos. Embora a maior vantagem do sistema de moléculas-traiçoeiras seja o uso da enzima do tipo selvagem, a utilização da técnica de mutagênese também apresenta benefícios para a melhoria da atividade catalítica de enzimas. Por essa razão, e com o objetivo de obter um catalisador mais potente, o sistema de moléculas-traiçoeiras foi combinado com a técnica de mutagênese. Em 2016, o grupo de pesquisa Bell demonstrou que as hidroxilações de substratos não nativos catalisadas por enzimas P450BM3 mutantes, que aumentam a taxa de reação, poderiam ser significativamente aceleradas com a ajuda de moléculas-traiçoeiras.55 Usando a variante K2 do P450BM3, a qual continha uma mutação quíntupla (A191T/N239H/I259V/A276T/L353I), houve uma melhoria na hidroxilação aromática na presença de moléculas-traiçoeiras. Como exemplo, esse mutante apresentou uma FR de apenas 7 min−1 para a hidroxilação do tolueno, enquanto na presença de APFG10, a FR observada aumentou em quase o quadragíntuplo para 271 min−1 (Figura 5 amarelo). Em um estudo sucessivo, eles mostraram que outras mutantes do P450BM3, R19 (R47L/Y51F/H171L/Q307H/N319Y), GVQ (A74G/F87V/L188Q) e RLYFIP (R47L/Y51F/I401P), também poderiam ser ativadas pela adição de moléculas-traiçoeiras.56-58 Na hidroxilação do alcano cíclico ciclopentano, o uso das variantes mencionadas acima na presença de moléculas traiçoeiras, resultou em um aumento na FR de 8 a 48 vezes, comparado com a taxa observada na ausência das moléculas-traiçoeiras. Além disso, os mesmos mutantes do P450BM3 apresentaram uma melhoria de aproximadamente 3, 4, 10 e 11 vezes na FR quando comparada com os resultados obtidos com a enzima P450BM3 tipo selvagem com APFG10.
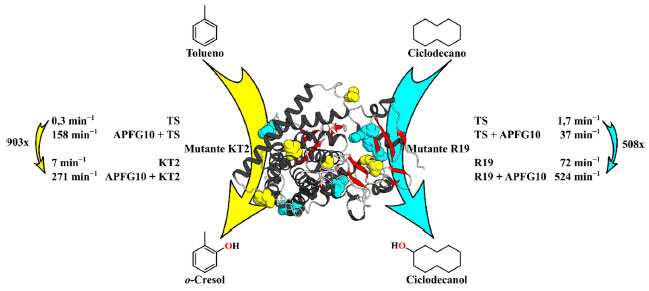 Figura 5. Hidroxilação do tolueno e ciclodecano pelas variantes do citocromo P450BM3, onde TS é o tipo selvagem, e KT2 (amarelo) e R19 (ciano) são mutantes do citocromo P450BM3, respectivamente. Os aminoácidos que foram submetidos a mutagênese são coloridos em ciano para KT2 e em amarelo para R19
Um benefício do emprego de moléculas-traiçoeiras com variantes P450BM3 é exemplificado pela hidroxilação de substratos grandes e volumosos que são difíceis de catalisar com a enzima do tipo selvagem. Por exemplo, enquanto a enzima P450BM3 tipo selvagem com APFG10 produziu ciclodecanol a partir de ciclodecano com uma FR de apenas 37 min−1, a combinação da variante R19 com moléculas-traiçoeiras levou a uma maior taxa de formação de produtos, atingindo 524 min−1. Na ausência de uma molécula-traiçoeira, a FR era de apenas 72 min−1, o que demonstra que essa variante foi ativada eficientemente pela molécula-traiçoeira e elas trabalham em conjunto sinergicamente. Esses estudos confirmam e ampliam a aplicabilidade do sistema de moléculas-traiçoeiras em conjunto com mutantes da enzima P450BM3, potencialmente levando a uma catálise mais eficiente, de difícil acesso pela enzima do tipo selvagem. Aplicação de moléculas-traiçoeiras em "reações não naturais" Em 2013, a laureada do prêmio Nobel de Química 2018 Prof.I Frances H. Arnold foi pioneira na utilização das enzimas do citocromo P450 na catálise de reações não naturais. O grupo de pesquisa Arnold demonstrou que as enzimas P450BM3 planejadas puderam catalisar a ciclopropanação de olefinas através da transferência de carbenos do diazoacetato de etila (DAE).59 Inspirada em suas pesquisas, Suzuki e col. exploraram a possibilidade de se empregar moléculas-traiçoeiras para ajudar outros tipos de reações, além das oxidações, tais como a ciclopropanação do estireno. Os autores foram capazes de mostrar com sucesso que moléculas-traiçoeiras modificam e remodelam o sítio ativo do P450BM3, e trabalham como auxiliares para guiar a estereosseletividade mesmo para reações que não sejam oxidações.60 O alvo deste sistema foi o mutante duplo F87A/T268A do P450BM3, o qual apresentou estereosseletividades moderadas (seletividade cis de 81% e enantiosseletividade pelo isômero (1S,2R) de 75%) na ciclopropanação do estireno usando DAE como um precursor de carbeno na ausência de uma molécula-traiçoeira (Esquema 10). A simples adição do composto RIbuPhe como uma molécula-traiçoeira neste sistema aperfeiçoou ambas as diastereo- e enantiosseletividade do produto da ciclopropanação do estireno (seletividade cis de 91% e enantiosseletividade pelo isômero (1S,2R) de 94%) (Esquema 10).
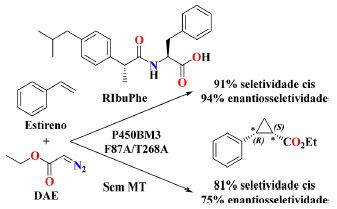 Esquema 10. Comparação entre a ciclopropanação do estireno mediada pelo mutante duplo F87A/T268A do P450BM3 na ausência e na presença da molécula-traiçoeira RIbuPhe
Essas seletividades estão ao mesmo nível da variante BM3-CIS, um mutante obtido pela evolução dirigida que contém treze mutações,59 enfatizando a simplicidade da abordagem usando moléculas traiçoeiras. É notável que as moléculas-traiçoeiras não serviram como um inibidor do mutante para a reação, embora, provavelmente, houvessem se mantido ligadas no sítio de ligação do substrato do P450BM3. Considerando que vários tipos de reações de transferência de carbeno e nitreno de utilidade sintética foram desenvolvidos através da evolução dirigida do P450BM3,61,62 a combinação de um mutante que exibe uma seletividade moderada com moléculas-traiçoeiras, como um toque final, pode ser uma opção atrativa para render maiores estereosseletividades nas reações de interesse. Usando moléculas-traiçoeiras em combinação com P450BM3 incorporando metaloporfirinas sintéticas Além da mutagênese, outra alternativa promissora para ajustar a reatividade da enzima é a troca do cofator heme por uma metaloporfirina sintética. Todavia, devido à complexidade estrutural do P450BM3, a reconstituição bem sucedida da enzima de comprimento total (domínios hémico e redutase juntos) ainda não foi relatada. Apenas seu domínio hémico foi reconstituído com sucesso, incorporando uma pequena seleção de metaloporfirinas sintéticas, como, por exemplo, manganêsprotoporfirina IX63 e metilirídiodeuteroporfirina IX,64 entre outros.65,66 Omura e col. elaboraram um protocolo, no qual o domínio hémico reconstituído pôde ser posteriormente ligado ao domínio redutase para produzir a enzima de comprimento total, utilizando a atividade da transpeptidase Sortase A (SrtA) da bactéria Staphylococcus aureus (Esquema 11).67 Nesse trabalho, o domínio hémico do P450BM3 sem o grupo heme (apo-P450BM3) foi primeiro reconstituído com a manganêsprotoporfirina IX (MnPPIX) empregando um método suave (Esquema 11a)68 e, posteriormente, foi combinado ao seu domínio redutase para gerar MnPPIX-P450BM3 (Esquema 11b). Embora a atividade catalítica geral do MnPPIX-P450BM3 fosse inferior à da enzima tipo selvagem (FePPIX-P450BM3), um efeito interessante pôde ser observado. Na presença da molécula-traiçoeira de segunda geração APFG9Trp, a proporção dos produtos de hidroxilação do propano, i-propanol e n-propanol, obtidas na presença do FePPIX-P450BM3 foi 19,8 : 1. Porém, quando o MnPPIX-P450BM3 foi empregado, a proporção mudou para 4,7 : 1, com aproximadamente 3,6 vezes mais n-propanol sendo produzido.67 Esses resultados não só mostram que moléculas-traiçoeiras e metaloporfirinas sintéticas podem ser efetivamente combinadas, mas também que a metaloporfirina pode exercer um efeito significativo sobre a reatividade das enzimas e, neste caso, a regiosseletividade.
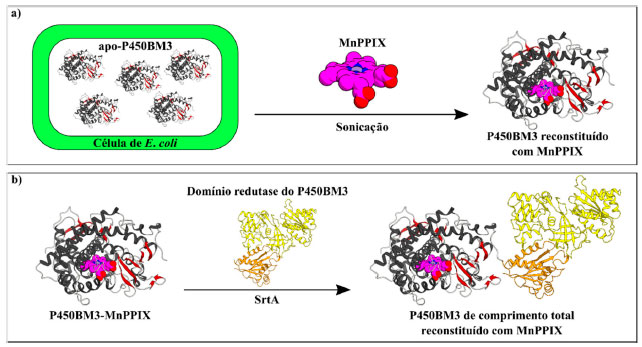 Esquema 11. a) Através de um método suave, em que E. coli são cultivadas em condições limitação de ferro, para obter o domínio hémico do P450BM3 sem heme (apo-P450BM3). A metaloporfirina sintética (aqui MnPPIX) é suplementada na etapa de disrupção celular por sonicação para obter P450BM3 reconstituído. b) Utilizando a atividade de Sortase A, o domínio hémico do P450BM3 reconstituído com MnPPIX é ligado ao domínio redutase
A expectativa é que metaloporfirinas sintéticas, que abrigam metais diferentes do ferro ou manganês, deveriam possibilitar um maior ajuste das propriedades reativas da enzima e, também, permitir o acesso a produtos e a estereosseletividades até então inalcançáveis. Acelerando a cristalização do domínio hémico do P450BM3 com uma nova molécula-traiçoeira Como já mencionado nas seções anteriores, moléculas-traiçoeiras têm sido vistas apenas como ativadoras e aceleradoras do ciclo catalítico do P450BM3 para hidroxilação de substratos não nativos. Stanfield e col. desvendaram um novo tipo de molécula-traiçoeira de terceira geração que cumpre uma função inovadora: a aceleração da cristalização do domínio hémico do P450BM3.69 Na presença da molécula N-abietoil-l-triptopfano (AbiATrp), na qual a estrutura do composto natural ácido abiético foi modificado com l-triptofano, macrocristais visíveis do domínio hémico do P450BM3 complexado com AbiATrp puderam crescer dentro de duas horas, e a técnica de difração de raios X permitiu a determinação da estrutura tridimensional com uma resolução quase atômica de 1,22 Å (Figura 6a). Ademais, os autores demonstraram que estes macrocristais também podem ser utilizados como cristais sementes para estimular o crescimento de novos cristais. Os cristais resultantes possuíam ótima difração de raios X com alta resolução. O procedimento desenvolvido por Stanfield e col. é detalhado no Esquema 12, onde macrocristais do domínio hémico do P450BM3 são macerados gerando cristais sementes, os quais são adicionados a uma solução contendo a enzima citocromo P450BM3 desejada (tipo selvagem, mutantes, na presença de moléculas-traiçoeiras ou reconstituída com diferentes metaloporfirinas) e o agente precipitante. Usando esta abordagem, em alguns casos, microcristais foram observados em questão de minutos.69
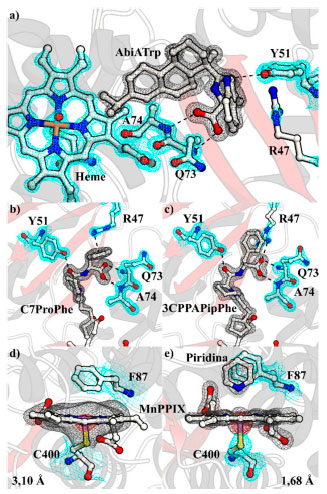 Figura 6. a) Estrutura cristalina do domínio hémico do P450BM3 em complexo com AbiATrp com resolução de 1,22 Å (código PDB: 6JLV). O mapa de densidade eletrônica 2Fo−Fc contornado a 2,5σ (malha ciana) e o mapa Fo−Fc omitindo AbiATrp contornado a 5,0σ (malha cinza). b) Estrutura cristalina do domínio hémico do P450BM3 em complexo com C7ProPhe com resolução de 1,41 Å (código PDB: 6K58). O mapa de densidade eletrônica 2Fo−Fc contornado a 2,0σ (malha ciana) e o mapa Fo−Fc omitindo C7ProPhe contornado a 4,5σ (malha cinza). c) Estrutura cristalina do domínio hémico do P450BM3 em complexo com 3CPPAPipPhe com resolução de 1,74 Å (código PDB: 6L1B). O mapa de densidade eletrônica 2Fo−Fc contornado a 2,0σ (malha ciana) e o mapa Fo−Fc omitindo 3CPPAPipPhe contornado a 3,5σ (malha cinza). d) Estrutura cristalina do domínio hémico do P450BM3 reconstruído com MnPPIX com resolução de 3,10 Å obtida por um protocolo padrão (código PDB: 5ZIS). O mapa de densidade eletrônica 2Fo−Fc contornado a 1,0σ (malha ciana) e o mapa Fo−Fc omitindo MnPPIX contornado a 3,0σ (malha cinza). O mapa anómalo de Fourier-diferença coletado a 1,75 Å contornado a 5,0σ (malha vermelha). e) Estrutura cristalina do domínio hémico do P450BM3 reconstruído com MnPPIX ligada piridina com resolução de 1,68 Å obtida pelo novo método (código PDB: 6JZS). O mapa de densidade eletrônica 2Fo−Fc contornado a 2,5σ (malha ciana) e o mapa Fo−Fc omitindo MnPPIX contornado a 5,0σ (malha cinza). O mapa anómalo de Fourier-diferença coletado a 1,75 Å contornado a 10,0σ (malha vermelha). Interações iônicas são representadas como linhas pontilhadas
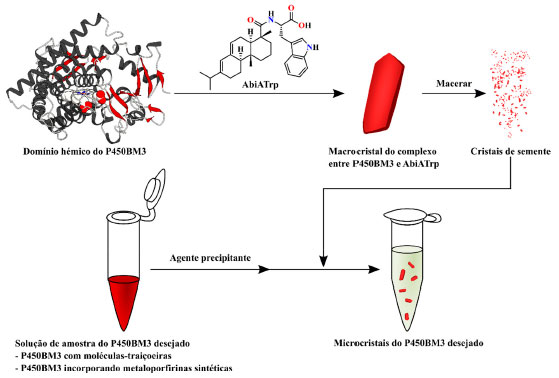 Esquema 12. Visão geral do processo de microcristalização. O domínio hémico do P450BM3 é misturado com a molécula-traiçoeira AbiATrp e é cristalizado para gerar macrocristais. Os macrocristais formados são rigorosamente macerados resultando em cristais microscópicos, os quais podem ser utilizados como cristais de semente para estimular o crescimento de outros cristais desejados do domínio hémico do P450BM3, incorporando outras moléculas-traiçoeiras, metaloporfirinas sintéticas ou até mesmo, em alguns casos, ambas
Esse novo protocolo de microcristalização para o domínio hémico do P450BM3 utilizando uma molécula-traiçoeira de terceira geração pôde ser aplicado com sucesso na cristalização dessa enzima na presença de moléculas-traiçoeiras potentes, como C7ProPhe69 e 3CPPAPipPhe43 (Figura 6b e d), as quais não puderam ser cristalizadas empregando protocolos de cristalização padrão. Adicionalmente, o P450BM3 reconstituído com diferentes metalocomplexos também pôde ser cristalizado com uma qualidade de difração excelente, mesmo que a troca do cofator heme pudesse tornar a cristalização do domínio hémico do P450BM3 quase impossível. Por exemplo, em uma tentativa de obter cristais da enzima reconstituída com a MnPPIX, Omura e col. tiveram que conduzir um processo extensivo de triagem de condições de cristalização, e embora os autores conseguissem obter cristais, a resolução da análise de difração foi somente 3,10 Å (Figura 6d).67 Ao utilizar o método delineado no Esquema 12, cristais de alta qualidade foram obtidos, o que permitiu a obtenção de uma estrutura cristalina com resolução de 1,68 Å. A observação de pequenas perturbações na estrutura proteica, por exemplo, o átomo central de manganês distorcido fora do plano do anel porfirínico em direção a Cys400, só foi possível devido a essa alta resolução, as quais seriam invisíveis em resoluções menores (Figura 6e). Através da observação cuidadosa das estruturas cristalinas de alta resolução do domínio hémico do P450BM3 incorporando vários metalocomplexos sintéticos, moléculas-traiçoeiras e substratos analógicos, viabilizada por esta técnica de microcristalização, Stanfield e col. também foram capazes de identificar perturbações estruturais transitórias específicas que podem ocorrer durante o ciclo catalítico do P450BM3.69
OUTROS SISTEMAS DE MOLÉCULAS-TRAIÇOEIRAS Além do P450BM3, que representa o exemplo mais amplamente estudado, outros sistemas de moléculas-traiçoeiras, usando diferentes enzimas também foram estabelecidos e serão abordados brevemente nesta última seção. Um dos primeiros sistemas de moléculas-traiçoeiras foi desenvolvido pelo grupo de pesquisa Watanabe em 2007, em que Shoji e col. demonstraram que a peroxigenase de ácidos graxos CIP152A1 (P450BSβ) isolada da bactéria Bacillus subtilis pode ser enganada para hidroxilar substratos não nativos. O sistema de moléculas-traiçoeiras elaborado para o P450BSβ, no entanto, difere significativamente do sistema para o P450BM3, decorrente das pronunciadas diferenças entre estas famílias de enzimas na ligação do substrato e na progressão do ciclo catalítico. 1) Diferente do P450BM3, P450BSβ hidroxila os carbonos α e β de ácidos graxos de cadeia longa. 2) Ao passo que em P450BM3 o grupo ácido carboxílico se liga perto do exterior da enzima com a cadeia alquílica penetrando no sítio ativo (Figura 1), no caso das peroxigenases P450 da família CIP152, o grupo ácido carboxílico liga-se a um resíduo de arginina altamente conservado localizado acima do grupo prostético heme (Figura 7). Isso coloca o grupo ácido carboxílico em uma posição favorável para atuar como catalisador ácido-base geral na geração do composto I usando peróxido de hidrogênio como agente oxidante, o qual é um reagente econômico e atua como única fonte de oxigênio, invertendo a via de desacoplamento peróxido detalhado no Esquema 2 (II → IV, conhecido no inglês como "peroxide shunt").70 Devido ao fato que P450BSβ é uma peroxigenase de ácidos graxos de cadeia longa, ácidos graxos com menos de 10 átomos de carbono não são considerados como substratos adequados. Consequentemente, Shoji e col. propuseram que ácidos graxos com esse comprimento aproximado deveriam ser capazes de funcionar como moléculas-traiçoeiras, enganando a enzima para se tornar ativada, e oxidar compostos exógenos. Por exemplo, na presença do ácido enântico (sete átomos de carbono), a oxidação de um elétron do guaiacol por P450BSβ prosseguiu com uma FR de 3750 min−1 P450−1, onde o uso de ácidos graxos mais longos e mais curtos levou a uma queda na atividade catalítica.71 Ademais, Shoji e col. e Fujishiro e col. estabeleceram que a escolha do ácido carboxílico exerce um efeito na estereosseletividade da oxidação de substratos não nativos pelo P450BSβ.71,72 Um exemplo representativo é a sulfoxidação de tioanisol, na qual 29 % ee (R) pôde ser atingida com ácido enântico, enquanto com ácido p-tolilacético a seletividade pôde ser invertida a 11% ee (S).72 Além disso, Fujishiro e col. e Onoda e col. mostraram que uma outra enzima que é um parente próximo do P450BSβ, CIP152B1 (P450SPα) extraída da bactéria Sphingomonas paucimobilis, também pôde ser enganada pelo mesmo tipo de moléculas-traiçoeiras para alcançar a hidroxilação de substratos não nativos.73,74
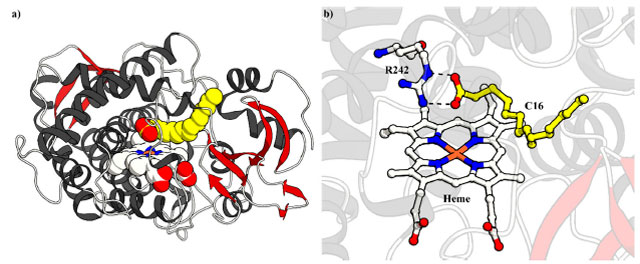 Figura 7. Estrutura cristalina a) geral e b) do sítio ativo do P450BSβ em complexo com ácido palmítico (C16, amarelo) (código PDB: 1IZO). Interações iônicas são representadas como linhas pontilhadas
Nos estudos supracitados, devido à baixa solubilidade da maioria dos ácidos graxos empregados, apenas concentrações de 20 mmol L-1 e abaixo foram testadas. No entanto, sob essas condições de baixa concentração, o ácido acético não funcionou como uma molécula-traiçoeira.71 Em um trabalho subsequente de Onoda e col., o ácido acético foi descoberto para servir como uma molécula-traiçoeira para as enzimas P450BSβ e P450SPα quando a concentração foi elevada, isto só foi possível devido a sua miscibilidade "ilimitada" em água. O emprego de uma solução tampão com uma concentração de ânions acetato mais elevada (1 mol L-1) levou a um aumento significativo da FR de epoxidação do estireno por P450BSβ de 590 min−1 P450−1 (compare com a FR de 330 min−1 P450−1 obtida com 20 mmol L-1 de ácido capróico - seis átomos de carbono).74 Recentemente foram desenvolvidos dois sistemas inteiramente novos de moléculas-traiçoeiras. Por exemplo, Demming e col. demonstram que o oleato hidratase oriundo da bactéria Elizabethkingia meningoseptica pode ser manipulado para a hidratação assimétrica de alcenos alifáticos, como α-deceno, na presença de um ácido graxo de cadeia curta como moléculas-traiçoeiras.75 Por outro lado, Zhang e col. empregaram uma fotodecarboxilase isolada da alga verde unicelular Chlorella variabilis CN64A com alcanos de cadeia longa como moléculas-traiçoeiras para melhorar a descarboxilação fotobiocatalítica de ácidos graxos de cadeia curta.76
CONCLUSÕES A beleza do sistema de moléculas-traiçoeiras, que foi introduzido em detalhes nesta revisão, reside em sua simplicidade. A fácil adição de um simples ácido carboxílico modificado com um aminoácido à mistura de reação é tudo o que é necessário para enganar o P450BM3 do tipo selvagem para hidroxilar substratos não nativos, transformações que não haveriam sido possíveis na ausência dessas moléculas. Até agora, isso exigia o uso da laboriosa técnica de mutagênese do P450BM3. O fato dessas transformações funcionarem sem a necessidade de qualquer mutagênese, ou seja, enzimas selvagens não adulteradas poderem ser empregadas, é um dos pontos mais fortes a favor desse sistema. Moléculas-traiçoeiras ativam o P450BM3 para a hidroxilação de uma gama de substratos não nativos, desde que eles possam se acomodar no espaço do sítio ativo deixado pela molécula-traiçoeira. Consequentemente, ao contrário da maioria das variantes do P450BM3 que são altamente especializadas para hidroxilar um determinado tipo de substrato, as moléculas-traiçoeiras exibiram aplicabilidade com um amplo leque de substratos, desde alcanos gasosos até diversos compostos aromáticos. Além disso, uma outra vantagem desse sistema é que também pode ser empregado em combinação com a mutagênese, a qual prevê superar diversas limitações impostas pela enzima do tipo selvagem que moléculas-traiçoeiras por si só não conseguiriam superar. A sofisticada combinação de moléculas-traiçoeiras com mutagênese e a troca do grupo prostético heme por metaloporfirinas sintéticas são técnicas que nos aproximam do desenvolvimento de um biocatalisador perfeito. O fato do sistema de moléculas-traiçoeiras ser compatível com células inteiras aproxima consideravelmente esse sistema das aplicações do mundo real, e se moléculas-traiçoeiras mais baratas e fáceis de sintetizar puderem ser produzidas, então até mesmo a aplicação industrial pode tornar-se realidade um dia.
LISTA DE SIGLAS 3CPPAPipPhe N-(3-Ciclopentil)propanoil-l-pipecolil-l-fenilalanina 5CHVAPhe N-(5-Cicloexil)valeroil-l-fenilalanina AbiATrp N-Abietoil-l-triptofano AG9Trp N-Pelargonoil-l-triptofano AG9Phe N-Pelargonoil-l-fenilalanina AG10Phe N-aproil-l-fenilalanina AG11Phe N-Undeciloil-l-fenilalanina APFG Ácido perfluorograxo APFG8 Ácido perfluorocaprílico APFG9 Ácido pelargônico APFG10 Ácido cáprico APFG11 Ácido undecílico APFG12 Ácido láurico APFG13 Ácido cáprico APFG14 Ácido perfluoromirístico APFG9Phe N-Perfluoropelargonoil-l-fenilalanina APFG9Leu N-perfluoropelargonoil-l-leucina APFG9Trp N-Perfluoropelargonoil-l-triptofano C7AMPipPhe N-Enantoil-l-pipecolil-l-fenilalanina C7ProPhe N-Enantil-l-prolil-l-fenilalanina C16 Ácido palmítico C16:1 Ácido palmitoleico C16Gly N-palmitoilglicina DAE Diazoacetato de etila EA Eficiência de acoplamento (sigla inglesa: CE) ee Excesso enantiomérico FePPIX Ferroprotoporfirina IX (heme) FR Frequência de rotatividade (sigla inglesa: TOF) MnPPIX Manganêsprotoporfirina IX MT Molécula-traiçoeira NTR Número total de rotatividade (sigla inglesa: TTN) R/SIbuPhe N-(R/S)-Ibuprofenoil-l-fenilalanina ZProPhe N-Benziloxicarbonil-l-prolil-l-fenilalanina
AGRADECIMENTOS O presente trabalho foi realizado com os apoios de JST CREST Nº de subsídio JPMJCR15P3 e JSPS KAKENHI Nº de subsídio JP15H05806 para Prof. Dr. O. Shoji, JP18J23340 para K. Suzuki e JP20J23653 para K. Yonemura. Dr. J. K. Stanfield agradece à Fundação TOBE MAKI pela bolsa concedida e à Sr.I Y. Watanabe por ensinar-lhe português. Os autores gostariam de agradecer a Dr.I K. C. M. Westrup pela cuidadosa revisão do português deste manuscrito e Sr. R. N. dos S. Ikeda e Sr. G. L. D. de Sousa por sua ajuda em questões linguísticas. Finalmente, Dr. J. K. Stanfield também gostaria de agradecer ao Prof. Dr. R. A. Ghiladi da Universidade Estadual da Carolina do Norte por enviar a doutoranda T. Malewschik ao Japão. Sem o apoio enérgico dela, esta revisão nunca haveria sido escrita.
REFERÊNCIAS 1. Nelson, D. R.; Phil. Trans. R. Soc. B 2013, 368, 20120430. 2. Poulos, T. L.; Finzel, B. C.; Gunsalus, I. C.; Wagner, G. C.; Kraut, J.; J. Biol. Chem. 1985, 260, 16122. 3. Stern, J. O.; Peisach, J.; J. Biol. Chem. 1974, 249, 7495. 4. Kendrew, J. C.; Dickerson, R. E.; Strandberg, B. E.; Hart, R. G.; Davies, D. R.; Phillips, D. C.; Shore, V. C.; Nature 1960, 185, 422. 5. Gajhede, M.; Schuller, D. J.; Henriksen, A.; Smith, A. T.; Poulos, T. L.; Nat. Struct. Biol. 1997, 4, 1032. 6. Cooper, D. Y.; Estabrook, R. W.; Rosenthal, O.; J. Biol. Chem. 1963, 238, 1320. 7. Ray, J. W.; Biochem. Pharmacol. 1967, 16, 99. 8. Fruetel, J.; Chang, Y. T.; Collins, J.; Loew, G.; Ortiz de Montellano, P. R.; J. Am. Chem. Soc. 1994, 116, 11643. 9. Dardas, A.; Gal, D.; Barrelle, M.; Sauret-Ignazi, G.; Sterjiades, R.; Pelmont, J.; Arch. Biochem. Biophys. 1985, 236, 585. 10. Onoda, H.; Shoji, O.; Suzuki, K.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; Catal. Sci. Technol. 2018, 8, 434. 11. Rude, M. A.; Baron, T. S.; Brubaker, S.; Alibaba, M.; Del Cardayre, S. B.; Schirmer, A.; Appl. Environ. Microbiol. 2011, 77, 1718. 12. da Silva, F. M.; de Lacerda, P. S. B.; Jones Junior, J.; Quim. Nova 2005, 28, 103. 13. Ciaramella, A.; Minerdi, D.; Gilardi, G.; Rend. Fis. Acc. Lincei 2017, 28, 169. 14. Bernhardt, R.; Urlacher, V. B.; Appl. Microbiol. Biotechnol. 2014, 98, 6185. 15. Fasan, R.; ACS Catal. 2012, 2, 647. 16. Teixeira, I. S.; Milagre, C. D. F.; Quim. Nova 2020, 43, 773. 17. Ortiz de Montellano, P. R.; Chem. Rev. 2010, 110, 932. 18. Whitehouse, C. J. C.; Bell, S. G. Wong, L. L.; Chem. Soc. Rev. 2012, 41, 1218. 19. Munro, A. W.; Leys, D. G.; McLean, K. J.; Marshall, K. R.; Ost, T. W. B.; Daff, S.; Miles, C. S.; Chapman, S. K.; Lysek, D. A.; Moser, C. C.; Page, C. C.; Dutton, P. L.; Trends Biochem. Sci. 2002, 27, 250. 20. Narhi, L. O.; Fulco, A. J.; J. Biol. Chem. 1987, 262, 6683. 21. Narhi, L. O.; Fulco, A. J.; J. Biol. Chem. 1986, 261, 7160. 22. Miura, Y.; Fulco, A. J.; J. Biol. Chem. 1974, 249, 1880. 23. Miura, Y.; Fulco, A. J.; Biochim. Biophys. Acta, Lipids Lipid Metab. 1974, 388, 305. 24. Meinhold, P.; Peters, M. W.; Hartwick, A.; Hernandez, A. R.; Arnold, F. H.; Adv. Synth. Catal. 2006, 348, 763. 25. Li, Y.; Wong, L. L.; Angew. Chem., Int. Ed. 2019, 58, 9551; Angew. Chem. 2019, 131, 9651. 26. Ariyasu, S.; Stanfield, J. K.; Aiba, Y.; Shoji, O.; Curr. Opin. Chem. Biol. 2020, 59, 155. 27. Shoji, O.; Aiba, Y.; Watanabe, Y.; Acc. Chem. Res. 2019, 52, 925. 28. Shoji, O.; Watanabe, Y.; Chem. Lett. 2017, 46, 278. 29. Shoji, O.; Watanabe, Y.; Isr. J. Chem. 2015, 55, 32. 30. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. 31. Li, H.; Poulos, T. L.; Nat. Struct. Biol. 1997, 4, 140. 32. Zilly, F. E.; Acevedo, J.P.; Augustyniak, W.; Deege, A.; Häusig, U. W.; Reetz, M.T.; Angew. Chem. Int. Ed. 2011, 50, 2720; Angew. Chem. 2011, 123, 2776. 33. Kawakami, N.; Shoji, O.; Watanabe, Y.; Angew. Chem., Int. Ed. 2011, 50, 5315; Angew. Chem. 2011, 123, 5427. 34. Klabunde, J.; Bischoff, C.; Papa, A. J.; Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH: Weinheim, 2018. 35. Kawakami, N.; Shoji, O.; Watanabe, Y.; Chem. Sci. 2013, 4, 2344. 36. Kawakami, N.; Cong, Z.; Shoji, O.; Watanabe. Y.; J. Porphyr. Phthalocyanines 2015, 19, 329. 37. Yalkowsky, S. H.; He, Y.; Jain, P.; Handbook of Aqueous Solubility Data, 2nd ed., CRC Press: Boca Raton, 2010. 38. Haines, D. C.; Tomchick, D. R.; Machius, M.; Peterson, J. A.; Biochemistry 2001, 40, 13456. 39. Cong, Z.; Shoji, O.; Kasai, C.; Kawakami, N.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; ACS Catal. 2015, 5, 150. 40. Ariyasu, S.; Kodama, Y.; Kasai, C.; Cong, Z.; Stanfield, J. K.; Aiba, Y.; Watanabe, Y.; Shoji, O.; ChemCatChem 2019, 11, 4709. 41. Rowlatt, B.; Yorke, J. A.; Strong, A. J.; Whitehouse, C. J. C.; Bell, S. G.; Wong, L. L.; Protein Cell 2011, 2, 656. 42. Fasan, R.; Chen, M. M.; Crook, N. C.; Arnold, F. H.; Angew. Chem., Int. Ed. 2007, 46, 8414; Angew. Chem. 2007, 119, 8566. 43. Yonemura, K.; Ariyasu, S.; Stanfield, J. K.; Suzuki, K.; Onoda, H.; Kasai, C.; Sugimoto, H.; Aiba, Y.; Watanabe, Y.; Shoji, O.; ACS Catal. 2020, 10, 9136. 44. Watanabe, Y.; Shoji, O.; Jpn. Kokai Tokkyo Koho P2019-69404A 2019. 45. Shoji, O.; Yanagisawa, S.; Stanfield, J. K.; Suzuki, K.; Cong, Z.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; Angew. Chem. Int. Ed. 2017, 56, 10324; Angew. Chem. 2017, 129, 10460. 46. Shoji, O.; Kunimatsu, T.; Kawakami, N.; Watanabe, Y.; Angew. Chem., Int. Ed. 2013, 52, 6606; Angew. Chem. 2013, 125, 6738. 47. Emerson, E.; J. Org. Chem. 1943, 8, 417. 48. Farinas, E. T.; Alcalde, M.; Arnold, F.; Tetrahedron 2004, 60, 525 49. Fasan, R.; Meharenna, Y. T.; Snow, C. D.; Poulos, T. L.; Arnold, F. H.; J. Mol. Biol. 2008, 383, 1069. 50. Meinhold, R.; Peters, M. W.; Chen, M. M. Y.; Takahashi, K.; Arnold, F. H.; ChemBioChem 2005, 6, 1765. 51. Xu, F.; Bell, S. G.; Lednik, J.; Insley, A.; Rao, Z.; Wong, L. L.; Angew. Chem., Int. Ed. 2005, 44, 4029; Angew. Chem. 2005, 117, 4097. 52. Chen, M. M. Y.; Snow, C. D.; Vizcarra, C. L.; Mayo, S. L.; Arnold, F. H.; Protein Eng. Des. Sel. 2012, 25, 171. 53. Karasawa, M.; Stanfield, J. K.; Yanagisawa, S.; Shoji, O.; Watanabe, Y.; Angew. Chem., Int. Ed. 2018, 57, 12264; Angew. Chem. 2018, 130, 12444. 54. Suzuki, K.; Stanfield, J. K.; Shoji, O.; Yanagisawa, S.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; Catal. Sci. Technol. 2017, 7, 3332. 55. Munday, S. D.; Shoji, O.; Watanabe, Y.; Wong, L. L.; Bell, S. G.; Chem. Commun. 2016, 52, 1036. 56. Munday. S. D.; Dezvarei, S.; Bell, S. G.; ChemCatChem 2016, 8, 2789. 57. Dezvarei, S.; Onoda, H.; Shoji, O.; Watanabe, Y.; Bell, S. G.; J. Inorg. Biochem. 2018, 183, 137. 58. Dezvarei, S.; Lee, J. H. Z.; Bell, S. G.; Enzyme Microb. Technol. 2018, 111, 29. 59. Coelho, P. S.; Brustad, E. M.; Kannan, A.; Arnold, F. H.; Science 2013, 339, 307. 60. Suzuki, K.; Shisaka, Y.; Stanfield, J. K.; Watanabe, Y.; Shoji, O.; Chem. Commun. 2020, 56, 11026. 61. Prier, C. K.; Zhang, R. K.; Buller, A. R.; Brinkmann-Chen, S.; Arnold, F. H.; Nat. Chem. 2017, 9, 629. 62. Zhang, R. K.; Chen, K.; Huang, X.; Wohlschlager, L.; Renata, H.; Arnold, F. H.; Nature 2019, 565, 67. 63. Lelyveld, V. S.; Brustad, E.; Arnold, F. H.; Jasanoff, A.; J. Am. Chem. Soc. 2011, 133, 649. 64. Reynolds, E. W.; Schwochert, T. D.; McHenry, M. W.; Watters, J. W.; Brustad, E. M.; ChemBioChem 2017, 18, 2380. 65. Davydov, D. R.; Ponomarev, G. V.; Bobrovnikova-Marjon, E.; Haines, D. C.; Peterson, J. A.; Biotechnol. Appl. Biochem. 2013, 60, 41. 66. Reynolds, E. W.; McHenry, M. W.; Cannac, F.; Gober, J. G.; Snow, C. D.; Brustad, E. M.; J. Am. Chem. Soc. 2016, 138, 12451. 67. Omura, K.; Aiba, Y.; Onoda, H.; Stanfield, J. K.; Ariyasu, S.; Sugimoto, H.; Shiro, Y.; Shoji, O.; Watanabe, Y.; Chem. Commun. 2018, 54, 7892. 68. Kawakami, N.; Shoji, O.; Watanabe, Y.; ChemBioChem 2012, 13, 2045. 69. Stanfield, J. K.; Omura, K.; Matsumoto, A.; Kasai, C.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; Shoji, O.; Angew. Chem. Int. Ed. 2020, 59, 7611; Angew. Chem. 2020, 132, 7681. 70. Lee, D. S.; Yamada, A.; Sugimoto, H.; Matsunaga, I.; Ogura, H.; Ichihara, K.; Adachi, S.; Park, S. Y.; Shiro, Y.; J. Biol. Chem. 2003, 278, 9761. 71. Shoji, O.; Fujishiro, T.; Nakajima, H.; Kim, M.; Nagano, S.; Shiro, Y.; Watanabe, Y.; Angew. Chem. Int. Ed. 2007, 46, 3656; Angew. Chem. 2007, 119, 3730. 72. Fujishiro, T.; Shoji, O.; Watanabe, Y.; Tetrahedron Lett. 2011, 52, 395. 73. Fujishiro, T.; Shoji, O.; Kawakami, N.; Watanabe, Y.; Sugimoto, H.; Shiro, Y.; Watanabe, Y.; Chem. Asian J. 2012, 7, 2286. 74. Onoda, H.; Shoji, O.; Watanabe, Y.; Dalton Trans. 2015, 44, 15316. 75. Demming, R. M.; Hammer, S. C.; Nestl, B. M.; Gergel, S.; Fademrecht, S.; Pleiss, J.; Hauer, B.; Angew. Chem. Int. Ed. 2019, 58, 173; Demming, R. M.; Hammer, S. C.; Nestl, B. M.; Gergel, S.; Fademrecht, S.; Pleiss, J.; Hauer, B.; Angew. Chem. 2019, 131, 179. 76. Zhang, W.; Ma, M.; Huijbers, M. M. E.; Filonenko, G. A.; Pidko, E. A.; van Shie, M; de Boer, S.; Burek, B. O.; Bloh, J. Z.; van Berkel, W. J. H.; Smith, W. A.; Hollmann, F.; J. Am. Chem. Soc. 2019, 141, 3116. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access