
 |
 |
 |
 |
 |
Educação
|
|
| Simetria molecular e reações de dessimetrização em síntese orgânica# Molecular symmetry and desymmetrization reactions in organic synthesis |
|
Bruno Matos Paz*; Emilio C. de Lucca Júnior; Ronaldo A. Pilli
Instituto de Química, Universidade Estadual de Campinas, 13083-970 Campinas - SP, Brasil Recebido em 29/11/2020 *e-mail: brunopaz@unicamp.br Molecular Symmetry has found several applications in Organic Chemistry. The aim of this article is to showcase its relevance as a tool for synthetic planning. Natural Products and synthetic compounds with pharmaceutical applications were selected to highlight desymmetrization reactions that make use of organocatalysis, biocatalysis or by transition metals. The recognition of structural patterns in retrosynthetic analysis is discussed for compounds that exhibit explicit or hidden symmetry. Limitations of the aforementioned strategies are also described, as well as the untapped potential, challenges and frontiers yet to be explored regarding these concepts. Quim. Nova Simetria molecular e reações de dessimetrização em síntese orgânica# Molecular symmetry and desymmetrization reactions in organic synthesis
Bruno Matos Paz*; Emilio C. de Lucca Júnior; Ronaldo A. Pilli Instituto de Química, Universidade Estadual de Campinas, 13083-970 Campinas - SP, Brasil
*e-mail: brunopaz@unicamp.br Recebido em 29/11/2020
Molecular Symmetry has found several applications in Organic Chemistry. The aim of this article is to showcase its relevance as a tool for synthetic planning. Natural Products and synthetic compounds with pharmaceutical applications were selected to highlight desymmetrization reactions that make use of organocatalysis, biocatalysis or by transition metals. The recognition of structural patterns in retrosynthetic analysis is discussed for compounds that exhibit explicit or hidden symmetry. Limitations of the aforementioned strategies are also described, as well as the untapped potential, challenges and frontiers yet to be explored regarding these concepts. Keywords: molecular symmetry, desymmetrization, pattern recognition, pseudo C2-symmetry, hidden symmetry, total synthesis
INTRODUÇÃO Precursora do que viria a ser a definição moderna de Ciência, a Filosofia Natural preocupou-se com a estrutura da matéria desde os tempos dos filósofos pré-socráticos. A disposição espacial das unidades fundamentais da matéria foi associada as suas propriedades macroscópicas, mesmo no âmbito de correntes de pensamento que partiram de perspectivas distintas, como as de Leucipo e Empédocles.1 Na filosofia atomística de Leucipo e Demócrito, a matéria seria composta por entidades minúsculas e indivisíveis - átomos - que poderiam adotar geometrias diversas. Em Timeu (360 A.C.), Platão associou a existência dos elementos primordiais sugeridos por Empédocles aos poliedros regulares, hoje conhecidos como sólidos platônicos: tetraedro (fogo), octaedro (ar), cubo (terra) e icosaedro (água). Como parte da tentativa de desenvolver o que hoje seria chamada de uma "teoria de tudo",2 Platão associou a geometria às transformações que um elemento poderia sofrer. Como o tetraedro, octaedro e icosaedro apresentam faces triangulares, Platão deduziu que os elementos correspondentes eram interconversíveis. Já o cubo, que representava o elemento terra, não poderia ser transformado em fogo, água ou ar por não possuir faces triangulares. O dodecaedro, sólido platônico de faces pentagonais, foi o elo entre a teoria de partículas fundamentais e a cosmologia de Platão3 e seria o que, posteriormente, Aristóteles associou ao "éter". Embora a noção clássica de quatro elementos tenha entrado em descrédito com a contínua descoberta de novos elementos, teorias envolvendo o conceito de éter permaneceram populares até os experimentos de Michelson-Morley, em 1887.4 Nos primeiros anos do século XX, antes das revoluções no campo da Física que levariam ao modelo atômico atual, Lewis colocou em cena mais um sólido platônico ao propor um modelo de ligação baseado em um átomo cúbico.5 Com o advento da Mecânica Quântica, finalmente foram desenvolvidas as teorias sobre a ligação química capazes de descrever adequadamente o arranjo tridimensional de átomos em moléculas pequenas6 e macromoléculas,7 balizadas pelo descobrimento e refinamento da cristalografia de raios X8 e técnicas espectroscópicas.9 No âmbito da Síntese Orgânica, arranjos tridimensionais desafiadores10 sempre estiveram entre os alvos da comunidade sintética.11 Inspirado no dodecaedro - a quintessência, segundo Platão - o hidrocarboneto de estrutura esteticamente bela, representada pelo dodecaedrano (1), foi sintetizado em 1982, por Paquette e colaboradores.12 O biciclo[3.3.3]undecano (2) que lembra as pás de uma hélice e a estrutura 3, denominada de superfano, que remete às pás de uma roda d'água, foram sintetizados pelos grupos de Doyle, em 1970,13 e Boekelheide, em 1979,14 respectivamente. O alótropo de carbono C60 (4), conhecido como buckminsterfulereno apresenta um arranjo formado por 20 hexágonos e 12 pentágonos (60 átomos de carbono) que remete a uma bola de futebol, tendo sido sintetizado pela primeira vez em 1984, por Rohlfing, Cox e Kaldor15 através de vaporização a laser de uma amostra de carbono e sua estrutura foi determinada, em 1985, por Kroto, Heath, Curl e Smalley (Figura 1).16 Em particular, os fulerenos apresentam simetria icosaédrica e têm sido extensamente estudados nas últimas décadas.17
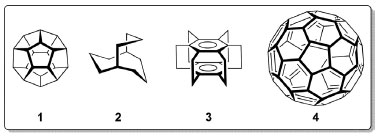 Figura 1. Compostos de simetria elevada. Dodecaedrano (1), biciclo[3.3.3] undecano (2), Superfano (3), Fulereno C60 (4)
Muitos dos praticantes da Síntese Química associam essa área do conhecimento a uma forma de arte que a remete à arquitetura e, de fato, alguns notáveis cientistas do passado lograram combinar aspectos das duas culturas em suas contribuições mais relevantes. Justus von Liebig, por exemplo, projetou os laboratórios de Química das universidades de Giessen e Munique e, posteriormente, atraiu para a área de Química o jovem talento August Kekulé que havia se mudado para Giessen para estudar arquitetura18 enquanto Robert Burns Woodward foi agraciado com o Prêmio Nobel de Química em 1965 por seus "feitos excepcionais na arte da síntese orgânica".19 Além da simples observação de padrões triviais na estrutura de objetos tridimensionais, o estudo da simetria tem um enfoque nas operações de simetria: operações realizadas no espaço tridimensional que, após aplicadas, não resultam em alteração observável na disposição espacial do objeto.20 Pode tratar-se de uma rotação em torno de um eixo ou reflexão em relação a um plano (Figura 2A), inversão em relação a um centro (Figura 2B) ou uma combinação delas (Figura 2C). Um plano de simetria pode estar coplanar (σv) ao eixo de rotação principal (Cn) ou perpendicular a ele (σh). Uma molécula apresenta um centro de inversão (i) quando os átomos em posições opostas a esse centro são idênticos. Ele pode, ou não, estar localizado sobre um átomo. Já um eixo de rotação impróprio (Sn) corresponde à combinação de uma rotação em torno de um eixo, seguida por uma reflexão em um plano perpendicular a este eixo. A teoria de grupos é a mais utilizada para descrever a simetria molecular. O conjunto de elementos de simetria obtido através da aplicação dessas operações de simetria determina o seu grupo pontual e possui uma descrição matemática rigorosa que está além do escopo desta revisão.21
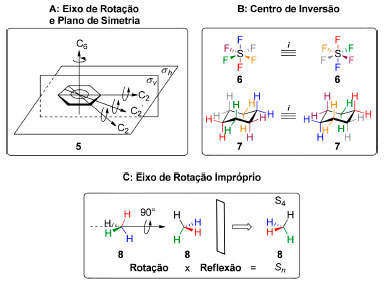 Figura 2. Operações e elementos de simetria em moléculas selecionadas: benzeno (5), hexafluoreto de enxofre (6), cicloexano (7) e metano (8). Nota: O uso de cores tem uma finalidade estritamente didática, uma vez que operações de simetria, quando aplicadas, não deixam alteração observável na disposição espacial do objeto
O estudo de simetria molecular possui aplicações e implicações em diversos ramos da Química: em espectroscopia eletrônica22 e vibracional (Infravermelho e Raman)23 a simetria é um fator determinante da quantidade de sinais esperados devido a sua influência nas regras de seleção. Em Ressonância Magnética Nuclear o número de sinais observados pode aumentar ou diminuir dependendo da presença de núcleos heterotópicos, diastereotópicos, enantiotópicos ou homotópicos.24 Ainda em uma perspectiva estrutural, a simetria de orbitais é fundamental na compreensão das ligações químicas, seja para determinar que orbitais podem se combinar ou para se avaliar a estabilidade relativa de diferentes arranjos geométricos.25 Quando aplicada aos orbitais de fronteira,26 as regras de simetria viabilizaram grandes avanços na compreensão de reações pericíclicas através do princípio da conservação da simetria de orbitais27 que entrou no léxico dos químicos orgânicos através das regras de Woodward-Hoffmann.28 Apesar de fundamentais para os praticantes de nossa disciplina, esses usos da simetria molecular estão além do escopo deste texto, uma vez que este trabalho tem como focos as aplicações de simetria às áreas de estereoisomeria, análise conformacional e planejamento sintético.
ELEMENTOS DE SIMETRIA EM ESTEREOISOMERIA E ANÁLISE CONFORMACIONAL Simetria e quiralidade A molécula de metano (8), o composto orgânico mais simples, possui um átomo de carbono ligado a quatro substituintes idênticos e pertence ao grupo pontual tetraédrico (Td, Figura 3A). Outros compostos que apresentam simetria tetraédrica são o adamantano (9) e o tetraedrano (10). Enquanto o primeiro é um composto estável, o segundo é termodinamicamente instável (10a, R=H) e ainda não foi sintetizado. Entretanto, derivados contendo substituintes volumosos (10b, R=CMe329 ou 10c, R=SiMe3)30 são cineticamente estáveis e já foram sintetizados. O cubano (11, simetria octaédrica, Oh)31 e o dodecaedrano (1, simetria icosaédrica, Ih)12 são hidrocarbonetos inspirados em dois outros sólidos platônicos e também já tiveram suas sínteses totais concluídas. Derivados de cubano apresentam aplicação em Química Medicinal32 e na química de materiais com alta densidade energética.33
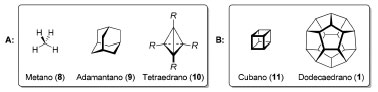 Figura 3. A: Compostos de simetria tetraédrica Td. B: Hidrocarbonetos inspirados em sólidos platônicos. Para o tetraedrano: 10a, R=H, 10b, R=CMe3 e 10c, R=SiMe3
Apesar de apresentarem carbonos tetraédricos devido a sua hibridização sp3, o agente alquilante iodometano (12), pertencente ao grupo pontual C3v, e o diclorometano (13), um solvente orgânico de simetria C2v, são mais rigorosamente descritos por outros grupos pontuais por não possuírem exatamente os mesmos elementos de simetria que um tetraedro perfeito, como o metano (8, Figura 4). O clorofluorometano (14, Cs), um gás refrigerante conhecido como Freon 31, possui como único elemento de simetria um plano que passa pelos dois halogênios e pela bissetriz do ângulo formado pelas ligações H-C-H. Já um carbono ligado a quatro substituintes diferentes, como no bromoclorofluorometano (15), é um centro estereogênico e possui dois possíveis arranjos espaciais distintos (grupo pontual C1) que são imagens especulares um do outro por não possuírem um plano de simetria, centro de inversão ou eixo de rotação impróprio, sendo descritos como R ou S, segundo as regras de Cahn-Ingold-Prelog.34
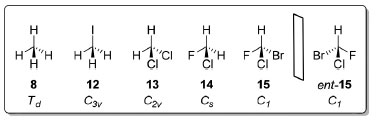 Figura 4. Compostos contendo carbono sp3 com diferentes padrões de substituição
Um objeto quiral não possui um eixo de rotação impróprio, centro de inversão ou plano de simetria em nenhuma de suas conformações. Vários arranjos satisfazem esta definição, seja em moléculas orgânicas ou complexos de metais de transição (Figura 5).35 Utilizando-se uma definição visualmente simples, o termo quiralidade foi cunhado por W. Thomson (Lord Kelvin), ainda no século XIX: um objeto é quiral caso não coincida exatamente com sua imagem especular.36
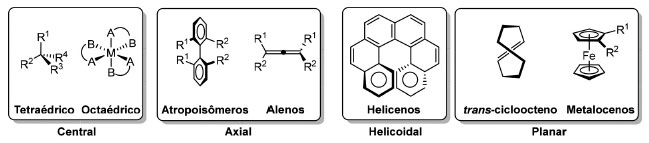 Figura 5. Compostos contendo diferentes elementos de quiralidade
Em uma perspectiva histórica, a constatação da presença de quiralidade em compostos de origem biológica pode ser atribuída às observações de Biot e Pasteur.37 Em 1817, Biot observou o desvio da luz plano polarizada quando estudava amostras de produtos naturais e, em 1832, que a forma natural do ácido tartárico, hoje denominado de ácido (R,R)-tartárico (16), era opticamente ativa em solução aquosa (dextrorotatória) mas o ácido paratartárico, hoje denominado de mistura racêmica do ácido tartárico (±16), não. Em 1848, Pasteur observou que o paratartarato de sódio e amônio tetraidratado era composto por dois tipos distintos de cristais que eram imagens especulares não-sobreponíveis um do outro. Após a separação manual dos cristais, Pasteur percebeu que ambas as soluções desses sais eram opticamente ativas, desviando a luz com a mesma magnitude, porém para sentidos opostos. Ao preparar os ácidos a partir dos sais de paratartarato, Pasteur descobriu que um deles era idêntico ao ácido tartárico natural (dextrorotatório) em todos os aspectos e o outro desviava o plano da luz no sentido anti-horário (levorotatório). Na nomenclatura atual, classificamos os cristais formados a partir das formas racêmicas dos tartaratos duplos de sódio e amônio (e de sódio e potássio) como conglomerados (i.e., uma mistura equimolar de dois tipos de cristais enantioméricos, cada um deles formado por um único enantiômero, correspondendo a uma resolução espontânea) enquanto as misturas racêmicas de outros sais do ácido tartárico cristalizavam-se na forma de compostos racêmicos nos quais ambos os enantiômeros estão presentes em quantidades idênticas nos cristais formados. O modelo em cortiça feito por Pasteur mostrado na Figura 6D permite uma melhor visualização de que, nos próprios cristais, as faces menores dos cristais dextro- e levorotatórios guardam relação de imagens especulares entre si.
 Figura 6. A: Diânions dos ácidos (-)- e (+)-tartárico (ent-16' e 16'); B: Hemiedros enantiomórficos esquematizados por Pasteur; C: Hemiedros imperfeitos cristalizados de tartarato de sódio e amônio;39 D: Modelo em cortiça confeccionado por Pasteur e cristais dos enantiômeros do tartarato de sódio e amônio38
Em realidade, além de sua capacidade aguçada de observação, Pasteur teve a sorte de encontrar dois racematos (tartaratos duplos de sódio e amônio e de sódio e potássio) que se cristalizam na forma de conglomerados o que permitiu que ele estabelecesse, de forma inequívoca, a existência da quiralidade molecular, isso em um momento histórico em que, apesar de existir, a teoria atômica da matéria ainda não era amplamente aceita pela comunidade científica.38 As consequências em nível molecular de tais observações macroscópicas não são triviais e tiveram um amplo alcance na Ciência. Quando colocadas ao lado dos avanços na química estrutural40 proporcionadas por Kekulé,41 Couper42 e Butlerov,43 como a tetravalência do carbono, por exemplo, deram suporte às proposições de van't Hoff44 e Le Bel,45 em 1874, sobre a disposição tetraédrica em torno do átomo de carbono em compostos orgânicos saturados. Exceto pelas interações com a luz plano polarizada, dois enantiômeros possuem propriedades físicas e químicas idênticas quando não estão em um ambiente quiral. A quase onipresença de compostos de origem biológica46 enantiomericamente puros é um fenômeno ainda não completamente elucidado47 mas central para a compreensão da química pré-biótica. Isso implica que todos os organismos - e suas respectivas biomacromoléculas - constituem ambientes quirais, de modo que dois enantiômeros podem ter propriedades biológicas completamente diferentes dentro de um mesmo organismo. Essa é uma consequência macroscópica das diferentes interações (diastereoméricas) que cada enantiômero tem com diversas biomacromoléculas do organismo em questão. Análise conformacional e simetria É possível que compostos com conectividade consideravelmente distintas pertençam ao mesmo grupo pontual. Na Figura 7A, temos vários exemplos de compostos que pertencem ao grupo pontual C2v. Entretanto, caso os dois hidrogênios das moléculas de água, formaldeído ou diclorometano sejam substituídos por grupos metila (Figura 7B), temos compostos com múltiplas conformações. O mesmo vale caso os grupos metila vierem a substituir os átomos de halogênio nos demais exemplos. A rigor, a simetria de cada rotâmero precisa ser analisada separadamente. Como a barreira rotacional em torno da ligação σC-C é de 12-40 kJ.mol-1 (Figura 7C),48 a interconversão entre os rotâmeros é extremamente rápida, o suficiente para que os sinais dos hidrogênios dos grupos metílicos de diferentes confôrmeros coalesçam quando observados por Ressonância Magnética Nuclear de Hidrogênio.49 Isso simplifica a análise, viabilizando, em uma primeira aproximação,50 considerar a média dos rotâmeros51 desses compostos como uma descrição satisfatória do sistema.
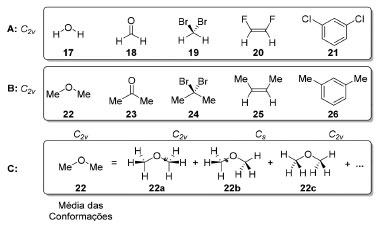 Figura 7. A: Compostos de simetria C2v. B: Compostos em que a média de seus rotâmeros pertencem ao grupo pontual C2v. C: conformações discretas para os rotâmeros do éter dimetílico 22
Quando diferentes arranjos espaciais de um mesmo composto precisam ser levados em consideração é necessário recorrer à análise conformacional. A importância da simetria para um estudo de análise conformacional reside no fato de as principais evidências espectroscópicas para estudar as espécies envolvidas serem as espectroscopias de Infravermelho e Raman. As absorções de diferentes rotâmeros - e confôrmeros - de uma mesma molécula são distintas nesse tipo de espectroscopia, justamente por não apresentarem o mesmo tipo de simetria, ou seja, pertencerem a grupos pontuais diferentes.52 Comecemos com o etano, o hidrocarboneto de estrutura flexível mais simples. Para o etano (27) existem vários rotâmeros que se encontram em equilíbrio dinâmico. O rotâmero de mais baixa energia é o de conformação alternada 27a (D3d), 12,1 kJ mol-1 mais estável que o rotâmero de mais alta energia de conformação eclipsada 27b (D3h, Esquema 1).
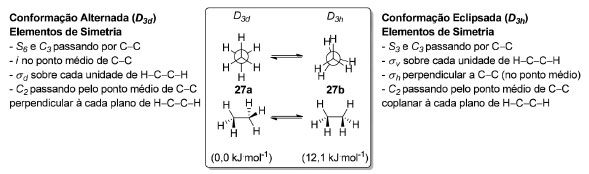 Esquema 1. Simetria e energias das conformações do etano (27)
Essa barreira energética foi determinada por cálculos ab initio, a partir da aplicação da Termodinâmica Estatística à análise espectroscópica vibracional do etano.53 Uma intensa discussão na comunidade de Química Teórica foi dedicada à origem dessa barreira rotacional atribuída às contribuições de efeitos estéricos e estereoeletrônicos.54 O componente estérico deriva da repulsão entre as nuvens eletrônicas das ligações σC-H vicinais (repulsão de Pauli) que é maximizada na conformação eclipsada 27b (D3h). Já o componente estereoeletrônico estabilizante deriva da interação entre um orbital σC-H e o orbital σ*C-H vicinal (hiperconjugação).55 Uma vez que essa interação é máxima quando ambas as ligações estão em uma conformação anti-periplanar (180° em relação uma a outra), esse efeito estabilizante é maximizado na conformação alternada do etano (27a, D3d, Esquema 1 e Figura 8).
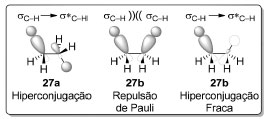 Figura 8. Efeitos estérico e estereoeletrônico nas conformações do etano (27).56 Representação localizada dos orbitais moleculares σC-H e σ*C-H. Os hidrogênios dos orbitais σ e σ* da ligação σC-H foram omitidos por clareza
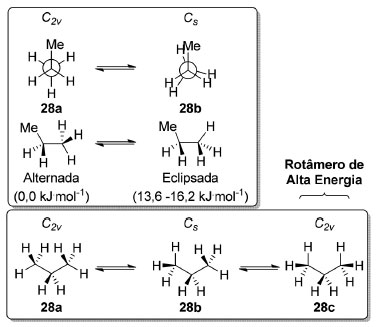
Rigorosamente, os efeitos estéricos e estereoeletrônicos não são "observáveis físicas" per se.57 A dificuldade de se avaliar experimentalmente a influência relativa dos dois efeitos em um mesmo sistema é consequência de os resultados experimentais tipicamente só expressarem a resultante. Isso vale mesmo para experimentos realizados na escala de tempo das rotações em torno das ligações σ (fentossegundos).58 Entretanto, o balanço desses efeitos é de extrema relevância para a química estrutural por ser fundamental para a compreensão das conformações de compostos mais complexos.59 Para compostos polares, efeitos eletrostáticos também são relevantes.60 A análise conformacional para o propano (28) é similar à do etano (27), sendo a principal diferença os grupos pontuais dos rotâmeros envolvidos (Esquema 2). O rotâmero de conformação alternada 28a (C2v) é 13,6-16,2 kJ.mol-1 mais estável que o rotâmero de conformação eclipsada 28b (Cs), dependendo da técnica experimental utilizada para obter o valor da barreira rotacional (Esquema 2).61
 Esquema 2. Simetria e energias das conformações do propano (28)
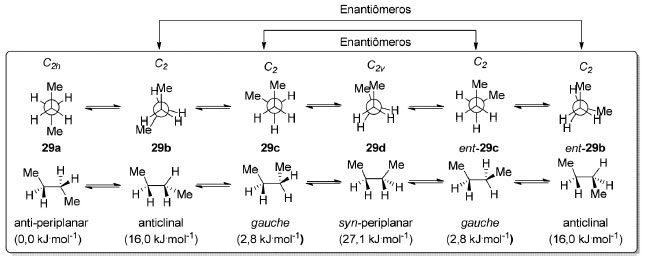
A barreira rotacional para o propano (28) é um pouco mais elevada que para o etano (27, 12-35%), já que o rotâmero eclipsado apresenta repulsão entre um grupo metila e uma ligação σC-H. Em princípio, há ainda a possibilidade de um rotâmero de alta energia adotar uma conformação duplamente eclipsada 28c (C2v) mas, até onde sabemos, não há evidências espectroscópicas para o mesmo.62 As conformações eclipsadas correspondem a máximos na superfície de energia potencial. O n-butano (29) é o primeiro alcano linear a ter o que podemos chamar de confôrmeros, rotâmeros de conformação alternadas correspondentes a mínimos na superfície de energia potencial, mas que apresentam energias diferentes (Esquema 3). A população relativa de um dado confôrmero é determinada por uma distribuição de Boltzmann. Assim, o confôrmero presente em maior proporção no equilíbrio corresponde à estrutura de menor energia livre de Gibbs.
 Esquema 3. Simetria e energias das conformações do n-butano (29). Nível de teoria utilizado para os cálculos de ΔG°: QCISD-(T)/6-311+G(3df,2pd)63
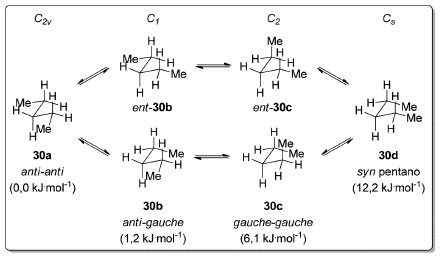
Para o n-butano (29), a conformação anticlinal (C2) é observada na forma de um par de enantiômeros 29b e ent-29b e corresponde a um máximo de energia de 16,0 kJ.mol-1 em relação ao confôrmero anti-periplanar 29a (C2h), o mais estável.63 Esse valor é comparável à barreira rotacional do propano (28) uma vez que corresponde ao mesmo tipo de repulsão: entre um grupo metila e uma ligação σC-H vicinal. O confôrmero anti-periplanar do n-butano (29) é 2,8 kJ.mol-1 mais estável que os confôrmeros enantioméricos gauche 29c e ent-29c (Esquema 3). Já a conformação syn-periplanar 29d corresponde a um máximo de energia de 27,1 kJ.mol-1 em relação ao confôrmero mais estável 29a (C2h). Esse valor relativamente elevado se deve à repulsão entre dois grupos metila vicinais.64 Para o n-pentano (30) temos uma análise conformacional focada em conformações alternadas (Esquema 4). O confôrmero mais estável é o anti-anti 30a (C2v) no qual todas as ligações C-C são anti-periplanares entre si, correspondendo a uma conformação em "zig-zag". Essa é a conformação de mais baixa energia para alcanos lineares de até 17 carbonos.65
 Esquema 4. Simetria e energias das conformações do n-pentano (30). Nível de teoria utilizado para os cálculos de ΔG°: CCSD(T)(full)/cc-pVTZ)64
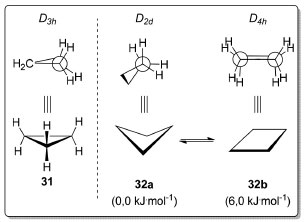
Para analisar os demais confôrmeros do n-pentano (30) é preciso tomar o metileno central como ponto de referência e observar a variação de energia ao se mudar a disposição espacial relativa das ligações σC-C(CH2-CH3) após uma rotação de 60° em torno de outra ligação σC-C(CH2-CH2). Quando uma ligação σC-C(CH2-CH3) apresenta relação anti e uma segunda encontra-se em relação gauche, temos os confôrmeros enantioméricos anti-gauche 30b e ent-30b (C1) que são 1,2 kJ.mol-1 menos estáveis em relação ao confôrmero mais estável 30a (C2v).66 A transição correspondente ao equilíbrio entre estas duas conformações pôde ser observada experimentalmente.67 Quando ambas as ligações σC-C(CH2-CH3) estão em relação gauche em um arranjo de simetria C2, temos o par de enantiômeros gauche-gauche 30c e ent-30c, 6,1 kJ.mol-1 menos estável que o confôrmero anti-anti. Na conformação em que ambas as ligações σC-C(CH2-CH3) estão em relação gauche em um arranjo de simetria Cs, temos as chamadas interações syn-pentano presentes no confôrmero de mais alta energia 30d, 12,2 kJ.mol-1 acima em relação à energia do confôrmero mais estável 30a (C2v). Nos alcanos lineares os ângulos entre as ligações σC-C e σC-H são próximos do ângulo observado em um tetraedro (~109,5 °), esperado para carbonos de hibridização sp3. Entretanto, em cicloalcanos o ângulo entre essas ligações é extremamente dependente do tamanho do anel. Caso os cicloalcanos adotassem uma estrutura planar, ou seja, se todas as ligações σC-C estivessem coplanares, o ângulo entre elas seria igual ao observado no respectivo polígono equilátero. O desvio em relação ao ângulo tetraédrico decorrente da tensão angular é de 49,5° para o ciclopropano (31), 19,5° para o ciclobutano (32) e apenas de 1,5° para o ciclopentano (33). Nessa disposição espacial, todas as ligações σC-H estariam eclipsadas (tensão torsional), o que aumentaria ainda mais sua energia relativa. Essa conformação só corresponde a um mínimo de energia para o ciclopropano (31, D3h, Esquema 5), respondendo pela sua alta reatividade quando comparada a outros cicloalcanos.
 Esquema 5. Simetria e energias das conformações do ciclopropano (31) e do ciclobutano (32). Nível de teoria utilizado para os cálculos da barreira rotacional do ciclobutano (32) CCSD(T)/cc-pVQZ.68
Para o ciclobutano (32), a conformação planar 32b (D4h) corresponde a um máximo de energia em relação à conformação mais estável 32a de simetria D2d,69 onde um dos carbonos do anel está acima do plano formado pelos três outros carbonos em uma inclinação de aproximadamente 30°, adotando uma conformação 6,0 kJ.mol-1 mais estável que a conformação planar 32b (Esquema 5).70 Apesar do ciclopentano (33) possuir uma tensão angular muito pequena, a tensão torsional é suficiente para fazer com que a diferença de energia entre a conformação planar 33c e suas conformações mais estáveis seja de 25,1 kJ.mol-1(Esquema 6).71 O ciclopentano (33) possui duas conformações estáveis, a primeira denominada de envelope 33b (Cs), em que quatro átomos de carbono estão coplanares e um deles está ligeiramente acima (ou abaixo) do plano (D5h). A outra conformação é chamada de meia-cadeira (C2) na qual um carbono está ligeiramente acima e outro ligeiramente abaixo do plano formado pelos outros três carbonos do anel. Essa conformação (C2) existe na forma de dois enantiômeros 33a e ent-33a. As energias dos confôrmeros meia-cadeira e envelope são praticamente idênticas e a barreira de interconversão (pseudorotação) entre os dois é quase nula.72
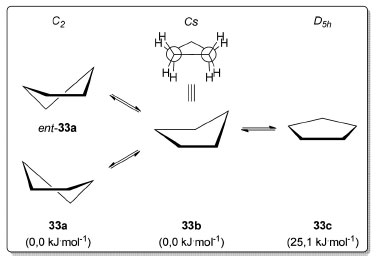 Esquema 6. Simetria e energias das conformações do ciclopentano (33). Nível de teoria utilizado para os cálculos da barreira rotacional do ciclopentano CCSD(T)/cc-pVTZ71
Diferente dos cicloalcanos menores, o cicloexano (34) não apresenta tensão angular na sua conformação cadeira 34a (D3d) que é a mais estável (Esquema 7). Neste confôrmero, todos os carbonos apresentam uma conformação alternada o que minimiza a tensão torsional. À temperatura ambiente, praticamente todas as moléculas de cicloexano se encontram na conformação cadeira (> 99,9%).73 O outro confôrmero que corresponde a um mínimo de energia é o barco torcido (D2), observado na forma de dois enantiômeros, 34c e ent-34c, sendo 23,9 kJ.mol-1 mais elevado em energia que a conformação cadeira 34a (D3d).74 A interconversão entre os confôrmeros cadeira 34a e barco torcido (34c e ent-34c) passa por um máximo de energia (45,3 kJ.mol-1) referente à conformação meia cadeira, 34b e ent-34b, de simetria C2.75 Nessa conformação, quatro carbonos estão coplanares enquanto os dois carbonos restantes se encontram acima e abaixo deste plano.76 A conformação barco 34d, de simetria C2v corresponde a um máximo de energia (30,4 kJ.mol-1) referente à barreira de interconversão entre os confôrmeros barco torcido 34c e ent-34c (D2).
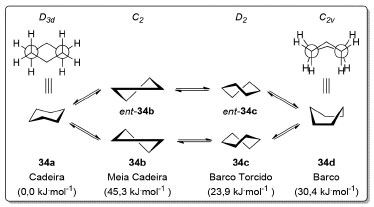 Esquema 7. Simetria e energias das conformações do cicloexano (34). Nível de teoria utilizado para os cálculos de ΔG°: CCSD(T)/CBS-aTQ limit (xB97X-D/cc-pVTZ)74
Os hidrogênios no cicloexano (34) não são todos equivalentes. A -90 °C, dois sinais são observados no espectro de RMN 1H do cicloexano-d11 (34-d11) referentes aos hidrogênios em posição axial e equatorial (Esquema 8).77 Entretanto, à medida que a temperatura aumenta, os sinais coalescem, uma vez que a escala de tempo do experimento é muito longa em relação à taxa de interconversão entre os confôrmeros (cadeira 34a e cadeira invertida 34a'). Em cicloexanos monossubstituídos (34e), a repulsão 1,3-diaxial faz com que o confôrmero em que o substituinte está em posição equatorial seja mais estável. Essa preferência conformacional devida a efeitos estéricos pode ser parcial ou completamente revertida na presença de efeitos estereoeletrônicos.78
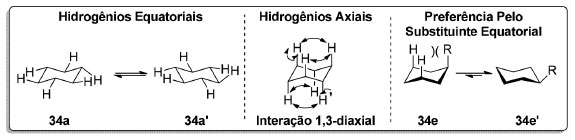 Esquema 8. Posições axial e equatorial no cicloexano (34)
DESSIMETRIZAÇÃO EM SÍNTESE ORGÂNICA Fundamentos de reações de dessimetrização A IUPAC define dessimetrização como sendo a "modificação de um objeto que resulta na perda de um ou mais elementos de simetria, como aqueles que impedem a quiralidade (plano de simetria, centro de inversão e eixo de rotação-reflexão), como ocorre na conversão de uma entidade molecular proquiral em uma quiral."79 A partir dessa definição, entende-se que a dessimetrização não se limita à remoção de elementos de simetria que impedem a quiralidade. Dessa forma, podemos dividir a dessimetrização em dois diferentes tipos: a dessimetrização não-estereosseletiva e a dessimetrização estereosseletiva, essa última podendo ainda ser subdividida em dessimetrização diastereo- e enantiosseletiva (Esquema 9).
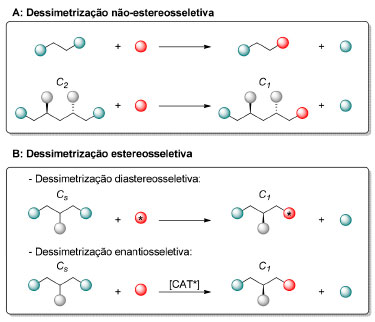 Esquema 9. Diferentes tipos de dessimetrização. "*" se refere a um reagente ou catalisador quiral
Enquanto as reações de dessimetrização apresentadas no esquema 9B são facilmente classificadas como estereosseletivas,79 ou seja, são reações em que há a formação preferencial de um estereoisômero (diastereoisômero ou enantiômero) em detrimento do outro, as reações apresentadas no esquema 9A não. Essas reações não podem ser classificadas como regiosseletivas,79 uma vez que não há direção de formação ou quebra preferencial de ligação. Além disso, elas também não podem ser classificadas como quimiosseletivas,79 pois essas transformações não implicam em preferência por um grupo funcional em detrimento de outro. Um ponto que merece atenção é o uso inadvertido da expressão dessimetrização. Na síntese total da (-)-maximiscina (35), Baran e colaboradores tratam a conversão de (+)-38 em (+)-39 como uma metoxilação dessimetrizante via ativação C-H. É inquestionável que a etapa de diferenciação das duas metilas no composto (+)-38 é desafiadora, no entanto, uma vez que tanto a molécula (+)-38 quanto a molécula (+)-39 pertencem ao grupo pontual C1, essa transformação não corresponde a uma etapa de dessimetrização (Esquema 10).80 Vale a pena destacar que a conversão do ácido carboxílico meso 36 (Cs) na amida (+)-38 (C1) é, de fato, uma etapa de dessimetrização diastereosseletiva que não foi apreciada pelos autores.
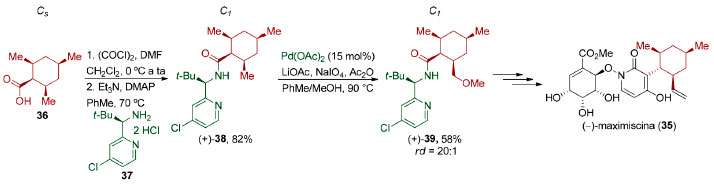 Esquema 10. Funcionalização estereosseletiva na síntese da (-)-maximiscina (35) por Baran e colaboradores80
Outro ponto que merece ser destacado é o uso da expressão dessimetrização local. Sharpe e Johnson, na síntese total da (-)-paspalina (40),81 fazem uso da expressão "dessimetrização global" para se referir a dessimetrização enantiosseletiva e "dessimetrização local" para uma etapa de diferenciação de duas metilas diastereotópicas (Esquema 11). Como na segunda etapa substrato e produto apresentam simetria C1, o emprego de tal terminologia não está de acordo com a definição da IUPAC e, portanto, o seu uso não é recomendado.
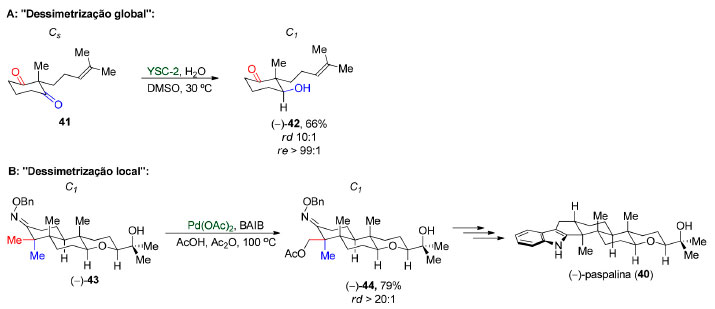 Esquema 11. Terminologia adotada por Sharpe e Johnson na sintese da (-)-paspalina (40)81
Na dessimetrização não-estereosseletiva, encontramos situações onde a quebra de simetria de uma molécula aquiral não leva à formação de uma molécula quiral como, por exemplo, na monoproteção do 1,3-propanodiol (45), (grupo pontual C2v), com TBSCl levando à formação do álcool 46 (grupo pontual: Cs, Esquema 12A).82
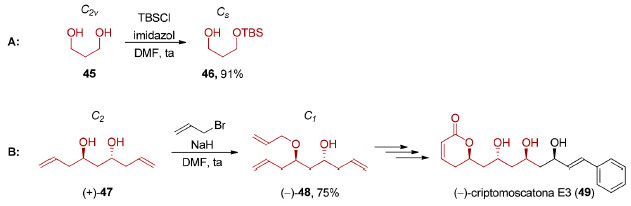 Esquema 12. Exemplos de dessimetrizações não-estereosseletivas. A: Carreira,82 B: Pilli85
Outra possibilidade é a diferenciação83 de sítios homotópicos em moléculas quirais. A monofuncionalização do diol 4784 que possui eixo de simetria C2, com brometo de alila, forneceu o composto 48 que pertence ao grupo pontual C1, um intermediário na síntese total da (−)-criptomoscatona E3 (49), em 75% de rendimento (Esquema 12B).85 As reações de dessimetrização não-estereosseletivas são processos de monofuncionalização e, portanto, não serão abordadas neste artigo. Já nas dessimetrizações estereosseletivas, a quebra de simetria leva à transformação de uma molécula aquiral em uma molécula quiral. Este tipo de processo pode envolver o emprego de quantidades estequiométricas de um agente de dessimetrização quiral (dessimetrização diastereosseletiva) ou de quantidade estequiométrica de um reagente aquiral na presença de um catalisador quiral (dessimetrização enantiosseletiva, Esquema 9). A dessimetrização estereosseletiva de compostos meso, aquirais devido à presença de um elemento de simetria (σ, i ou Sn), por catálise metálica, organocatálise ou mediante o uso de enzimas aumenta significativamente a brevidade e a eficiência de uma rota sintética. Em uma típica síntese estereosseletiva, um processo enantiosseletivo precede às etapas diastereosseletivas que instalam os centros estereogênicos remanescentes na estrutura desejada. A dessimetrização de compostos meso (Cs ou Ci) inverte a sequência típica dessas etapas (Esquema 13). Por outro lado, a dessimetrização de compostos proquirais ocorre, geralmente, em etapas iniciais da síntese.
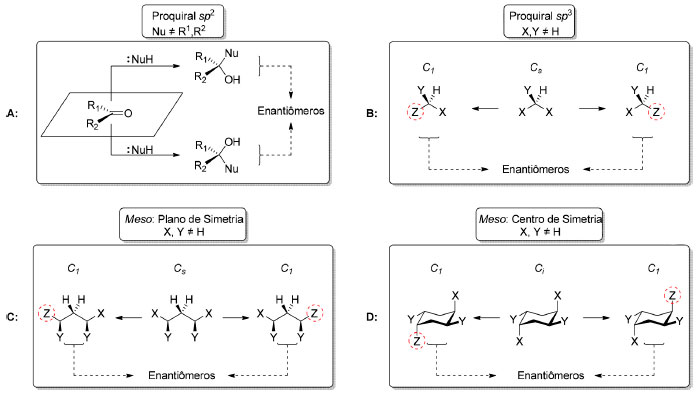 Esquema 13. Dessimetrizações. A: Compostos proquirais sp2 (não tratados neste trabalho); B: Compostos proquirais sp3; C: Compostos meso contendo plano de simetria (σ); D: Compostos meso contendo centro de inversão (i)
Os compostos proquirais incluem aqueles que apresentam um carbono sp2 ou carbono sp3 proquiral. Por exemplo, cetonas contendo dois substituintes diferentes ligados à carbonila ou alcenos com dois substituintes distintos ligados ao carbono sp2 são proquirais. Já compostos em que o centro proquiral situa-se em um carbono sp3 ligado a dois grupos enantiotópicos são aquirais devido à presença de um plano de simetria. Enquanto a quebra de simetria em compostos com carbono sp3 proquiral leva à formação de um centro estereogênico onde antes residia o carbono sp3 (Esquema 13B), a dessimetrização de um composto meso remove os elementos de simetria que impediam a quiralidade, mesmo o composto já possuindo centros estereogênicos (Esquema 13C-D). Esses dois casos diferenciam-se da situação de obtenção de compostos quirais a partir de reações entre compostos com faces proquirais em carbono sp2 que não serão tratados neste trabalho (Esquema 13A).86 Visando familiarizar o leitor com as recomendações mais recentes, será adotada a denominação de re para descrever a razão enantiomérica e rd para a razão diastereomérica em processos estereosseletivos.87 A título de exemplo de um processo de dessimetrização diastereosseletiva (Esquema 14) podemos citar a dessimetrização do anidrido cíclico 50 que apresenta carbono sp3 proquiral (Cs) pelo álcool (R)-feniletílico (51), empregado como agente de esterificação do anidrido glutárico nos estágios iniciais da síntese da (+)-compactina (53), por Heathcock e colaboradores, em 1985.88 Esse processo forneceu preferencialmente o composto 52 de configuração R como produto principal (75%, rd 15:1).
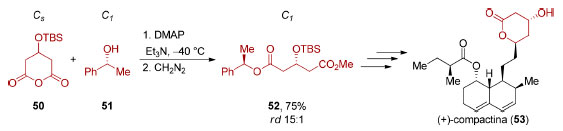 Esquema 14. Dessimetrização aplicada à síntese da (+)-compactina (53), por Heathcock e Rosen88
Desde a descoberta que metabólitos de fungos como, por exemplo, compactina89 e lovastatina (Mevinolin® ou Mevacor®),90 juntamente com a estatina semissintética sinvastatina (Zocor®),91 eram capazes de inibir a enzima 3-Hidroxi-3-MetilGlutarato redutase (HMG-CoA), responsável pela redução da (S)-HMG-CoA a (R)-mevalonato e, dessa maneira, inibir a biossíntese de colesterol, essa família de fármacos, conhecida como estatinas, passou a ser amplamente empregada no controle da hipercolesteremia e na dislipidemia. Posteriormente, estatinas sintéticas mais potentes foram desenvolvidas, culminando com a introdução no mercado da atorvastatina (54, Lipitor®), em 1997. Do ponto de vista estrutural, todos esses compostos apresentam uma unidade relacionada ao ácido (R)-3-hidroxiglutárico que é o grupo farmacofórico responsável pela inibição enzimática. A síntese industrial da atorvastina (54) emprega a reação de Paal-Knorr entre uma 1,4-dicetona 55 e uma amina primária 56 contendo os dois centros estereogênicos presentes na estrutura do fármaco. Uma das rotas de preparação da amina 56 envolve a dessimetrização enzimática da 3-hidroxiglutaronitrila 58 para fornecer o ácido (R)-4-ciano-3-hidroxibutírico que foi transformado no éster etílico correspondente 57, permitindo obter a amina primária 56 necessária para a condensação de Paal-Knorr (Esquema 15).92
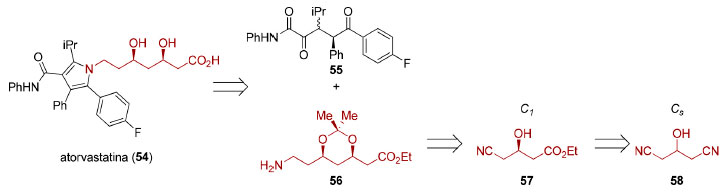 Esquema 15. Dessimetrização aplicada à síntese da atorvastatina (54)
A atorvastatina (54), patenteada como Lipitor® pela Pfizer em 1997, acumulou um total de US$130 bilhões em vendas ao longo de 14 anos, antes de a patente expirar, chegando a ocupar o posto de fármaco mais vendido no mundo.93 Dessimetrização de compostos contendo carbonos sp3 proquirais O glicerol (59) é uma molécula que apresenta um carbono sp3 proquiral com potencial de uso industrial como matéria-prima para a obtenção de compostos quirais uma vez que é um subproduto da reação de transesterificação utilizada para a produção de biodiesel (Figura 9A). Apesar de possuir diversas aplicações industriais, no Brasil a demanda é consideravelmente inferior à quantidade produzida, uma vez que a produção nacional já alcança centenas de milhares de toneladas ao ano.94
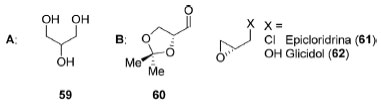 Figura 9. A: Glicerol (59). B: Derivados (60-62) do glicerol (59) mais utilizados em síntese orgânica
Diversos intermediários sintéticos (60-62) utilizados em sínteses assimétricas podem ser facilmente obtidos, de forma racêmica, a partir do glicerol o que tem incentivado o desenvolvimento de metodologias de dessimetrização desse composto (Figura 9B).95 Várias estratégias foram desenvolvidas para dessimetrização de 1,3-dióis proquirais através de reação de acilação96 ou de proteção via grupos de silício.97 A dessimetrização do próprio glicerol (59) se mostrou desafiadora, tipicamente necessitando funcionalização prévia.98 A dessimetrização direta do glicerol (59) foi obtida de forma enantiosseletiva por Asano, Ubukata e colaboradores99 através de uma reação de acilação enzimática (Esquema 16), utilizando anidridos de ácidos carboxílicos 63 contendo cadeias laterais aromáticas ou estirênicas. Quando anidridos derivados de ácidos carboxílicos alifáticos foram utilizados, o enantiômero oposto foi obtido, porém em baixa enantiosseletividade.
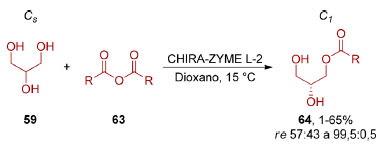 Esquema 16. Dessimetrização enzimática enantiosseletiva do glicerol (59) via acilação
Tan e colaboradores100 desenvolveram uma reação de dessimetrização enantiosseletiva do glicerol (59, Esquema 17A) utilizando um organocatalisador capaz de se ligar reversivelmente a umas das hidroxilas do glicerol (59, Esquema 17B) e dirigir a etapa de sililação de forma intramolecular. A etapa de dessimetrização fornece o enantiômero 67 com uma seletividade de aproximadamente 9:1 (já que k1 é maior do que k2), mas que pode ser enriquecido por uma resolução cinética na qual o enantiômero minoritário (ent-67) é parcialmente consumido (já que k4 é maior do que k3, Esquema 17C), formando o subproduto bis-sililado 68.
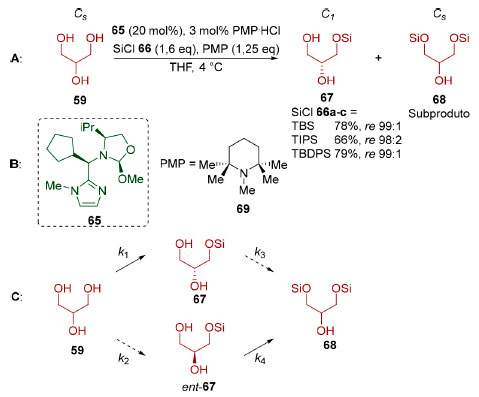 Esquema 17. A: Dessimetrização enantiosseletiva de glicerol (59). B: Estrutura do catalisador 65 e da 1,2,2,6,6-pentametilpiperidina (69). C: Dessimetrização seguida por resolução cinética
Os ácidos (R)- e (S)-β-hidroxi isobutíricos (70 e ent-70) são compostos muito utilizados como materiais de partida na síntese de produtos naturais e fármacos, sendo que suas preparações a partir da dessimetrização enzimática do ácido isobutírico (71) foram adaptadas à escala industrial (Esquema 18).
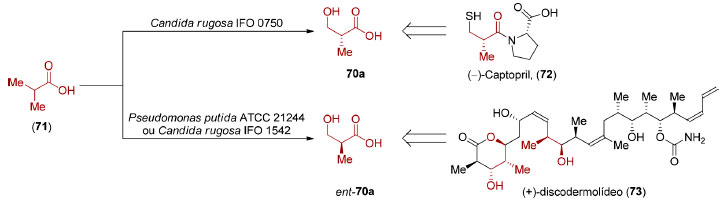 Esquema 18. Dessimetrização enzimática do ácido isobutírico (71)105,106
A reação mediada por Candida rugosa IFO 0750 fornece o enantiômero (R)-70,101 utilizado na síntese do Captopril® (72),102 um fármaco mundialmente conhecido no tratamento de hipertensão arterial. Seu mecanismo de ação, descoberto por Sir John R. Vane, ganhador do Prêmio Nobel de Medicina ou Fisiologia em 1982, envolve a inibição da enzima conversora de angiotensina I (ECA I).103 Deve-se ressaltar que os estudos de Vane só foram possíveis graças a descoberta do fator de potenciação da bradicinina, um peptídeo extraído do veneno da Bothrops jararaca, por Sérgio Henrique Ferreira.104 Já a dessimetrização das metilas do ácido isobutírico (71) por Pseudomonas putida ATCC 21244 ou Candida rugosa IFO 1542 leva à formação do ácido (S)-β-hidroxi isobutírico (ent-70a),105 precursor do éster de Roche de configuração S (ent-70b), utilizado com sucesso, entre outros, no memorável escalonamento realizado pela Novartis, em 2004, que culminou com a preparação de 60 gramas do produto natural (+)-discodermolídeo (73).106 Graças à quantidade obtida, o (+)-discodermolídeo (73) chegou à fase de ensaios clínicos I, sendo descartado devido a sua toxicidade. A hidroxilação enzimática do ácido isobutírico (71), formalmente uma reação de oxidação de ligação σC-H, tem inspirado grupos de pesquisa dedicados ao desenvolvimento de métodos catalíticos baseados em metais de transição. Em 2017, Yu e colaboradores reportaram reações de arilação, alquenilação e alquinilação, catalisadas por paládio, de um derivado do ácido isobutírico (71) levando à formação de compostos com razão enantiomérica de até 99:1 (Esquema 19).107
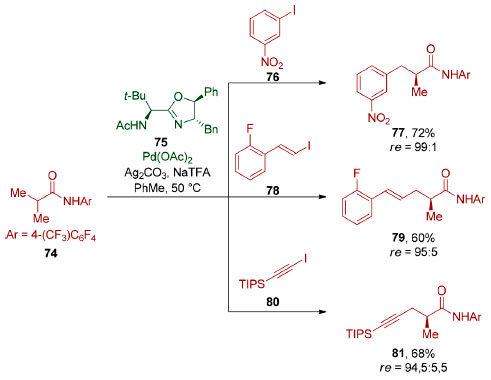 Esquema 19. Dessimetrização organometálica de um derivado do ácido isobutírico (71), por Yu e colaboradores107
Em 2018, Correia e colaboradores relataram uma dessimetrização enantiosseletiva do álcool proquiral 82 através de uma reação de Heck−Matsuda (Esquema 20A).108 O produto 86 foi obtido como o diastereoisômero cis (rd=7:1-20:1), em excelente enantiosseletividade (re=89,5:10,5-98:2). Cálculos DFT (M06/6-31+G(d,p),SDD(Pd)) sugerem que a diastereosseletividade cis observada resulta de uma interação não-covalente entre a hidroxila do substrato e o centro metálico do catalisador de paládio catiônico (Esquema 20B).
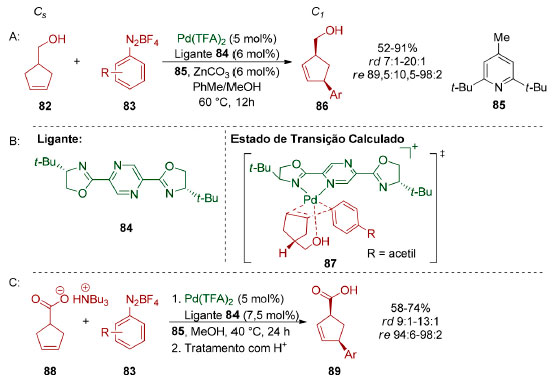 Esquema 20. Dessimetrização através de catálise via paládio, por Correia e colaboradores108
O grupo de Correia também demonstrou que a interação eletrostática de carboxilatos com complexos catiônicos de paládio torna esses grupos diretores viáveis para dessimetrização via reação de Heck-Matsuda (Esquema 20C). O diastereoisômero cis (re = 9:1-13:1) também foi majoritário e alta enantiosseletividade (re=94:6-98:2) foi observada. A aplicabilidade e complementaridade das duas transformações foi demonstrada na síntese do inibidor de fosfodiesterase 4 89a (Esquema 21). O inibidor foi obtido através de uma hidrogenação da dupla ligação de 86a, seguida por oxidação do álcool primário ao ácido carboxílico, sendo, no entanto, observada uma erosão da estereosseletividade a cada etapa. Essa limitação pôde ser contornada empregando-se o carboxilato 88 como substrato na dessimetrização via reação de Heck-Matsuda.
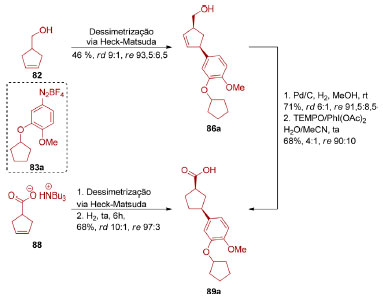 Esquema 21. Dessimetrização através de catálise via paládio aplicada à síntese de um inibidor de fosfodiesterase 4 (89a), pelo grupo de Correia108
Em 2008, Hoveyda, Schrock e colaboradores utilizaram uma reação de dessimetrização baseada em metátese intramolecular de olefinas na síntese total do alcaloide da família Aspidosperma, (+)-quebrachamina (90, Esquema 22).109 O catalisador de molibdênio utilizado (92) possui quiralidade no metal e no ligante. A metátese de fechamento de anel enantiosseletiva do trieno 91 forneceu o dieno tetracíclico 93 (84%), em excelente enantiosseletividade (re 98:2). Reação de hidrogenação das duplas ligações (97%), empregando-se o catalisador de Adams (PtO2), forneceu o produto natural (+)-quebrachamina (90).
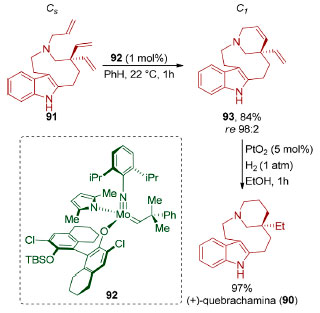 Esquema 22. Síntese total da (+)-quebrachamina (90) através de uma reação de dessimetrização via metátese de olefinas pelo grupo de Schrock e Hoveyda109
Mesmo reações fundamentais, como a enolização, podem ser utilizadas para viabilizar reações de dessimetrização. Em 2005, Aggarwal e Olofsson demonstraram que a cicloexanona proquiral 94 poderia sofrer desprotonação enantiosseletiva (Esquema 23),110 na presença de uma amida de lítio quiral 95, e o enolato formado 96 interceptado in situ, utilizando-se o sal de diaril-iodônio 97, resultando no produto de α-arilação 98 (41%), em alta estereosseletividade (rd > 20:1, re 93:7), que foi submetido a uma redução diastereosseletiva da carbonila (rd 7:1, 83%). A conversão do álcool 99 em um mesilato, seguida por uma clivagem ácida do grupo de proteção, viabilizou a formação de uma amina livre. Uma reação de substituição nucleofílica transanular (90%) completou a síntese da (−)-epibatidina (100).
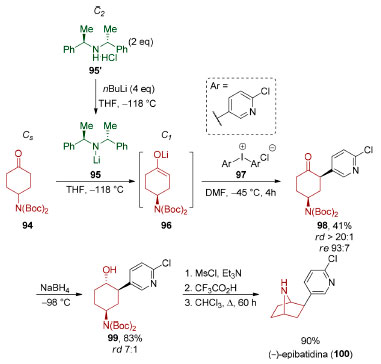 Esquema 23. Síntese da (−)-epibatidina (100) por Aggarwal e Olofsson110
Em 2015, o grupo de Dixon desenvolveu uma reação de dessimetrização enantiosseletiva, via catálise por enamina, na aminocetona cíclica proquiral 101 (Esquema 24).111 A etapa de dessimetrização consistiu em uma adição de Michael de uma enamina, formada in situ, a um éster α,β-insaturado levando à construção de três estereocentros (rd > 98:2 e re > 91,5:8,5). O heterociclo 103 formado apresenta o esqueleto 2-azabiciclo[3.3.1]nonano, fragmento estrutural presente em vários produtos naturais como os da família dos morfanos. É interessante notar que quando um éster α,β-insaturado de geometria Z foi utilizado, o mesmo diastereoisômero foi obtido, porém na forma racêmica. Apesar desse controle do substrato, a enantiosseletividade é resultado das múltiplas interações covalentes e não-covalentes com o catalisador 102.
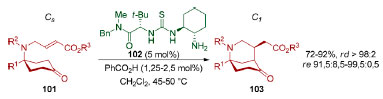 Esquema 24. Dessimetrização organocatalisada por Dixon e colaboradores111
A reação de ciclização entre uma 1,3-dicetona cíclica 105 e uma cetona α,β-insaturada 104, um caso particular de uma anelação de Robinson que envolve uma adição de Michael, seguida por uma condensação aldólica intramolecular para formar o sistema bicíclico 107 contendo um estereocentro quaternário que oferece a oportunidade de dessimetrização.112 Em 2000, Barbas e colaboradores demonstraram a generalidade de uma anelação de Robinson assimétrica catalisada por prolina (106, Esquema 25).113
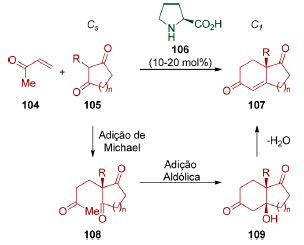 Esquema 25. Anelação de Robinson assimétrica organocatalisada na síntese da cetona de Hajos-Parrish-Eder-Sauer-Wiechert (n=1) e de Wieland−Miescher (n=2)113
Para n=1 (Esquema 26), a cetona obtida é conhecida como cetona de Hajos-Parrish-Eder-Sauer-Wiechert (107a)114 e foi empregada na síntese total de diversos produtos naturais, especialmente terpenóides como, por exemplo, (+)-cortistatina A (110),115 aplicurodinona-1 (111),116 (−)-nitidasina (112),117 (+)-ciantiwigina AC (113),118 entre vários outros.119 Uma razão para tamanha aplicabilidade é a presença do sistema de anéis de cinco e seis membros fundidos em um vasto número de produtos naturais.
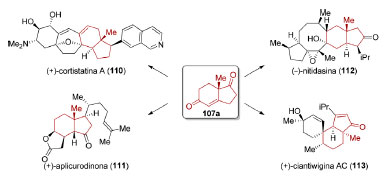 Esquema 26. Aplicações sintéticas da cetona de Hajos-Parrish-Eder-Sauer-Wiechert (107a)
As várias aplicações sintéticas da cetona de Hajos-Parrish-Eder-Sauer-Wiechert (107a) motivaram estudos acerca do mecanismo da sua formação via organocatálise. Um estudo realizado por List e colaboradores utilizando marcação isotópica in situ (H2O18), ressonância magnética nuclear e espectrometria de massas deu suporte à presença de catálise via enaminas,120 em contraposição às especulações iniciais acerca desta reação.121 Cálculos DFT (B3LYP/6-31+G(d,p)) realizados pelo grupo de Houk identificaram que a formação da ligação σC-C é a etapa estereodeterminante,122 ou seja, é a etapa responsável pela dessimetrização observada (Esquema 27). Foi identificado um estado de transição 14 kJ mol-1 menos energético em relação ao que leva à formação do enantiômero minoritário, coerente com a alta enantiosseletivade observada experimentalmente. Estudos de efeitos isotópicos em abundância natural também dão suporte a este mecanismo.123
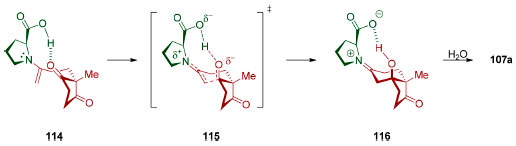 Esquema 27. Etapa estereodeterminante para formação organocatalisada da cetona de Hajos-Parrish-Eder-Sauer-Wiechert (107a). Nível de teoria utilizado para os cálculos das energias do estado de transição (B3LYP/6-31+G(d,p))122
Para n=2 (Esquema 27), obtém-se a chamada cetona de Wieland−Miescher (107b) que foi utilizada na síntese total de vários produtos naturais como o (−)-penitrem D (117),124 (+)-halenaquinol (118),125 (−)-glaucarubolona (119),126 a (−)-escabronina G (120)127 e o Taxol® (121).128,129 A presença de sub-estruturas decalínicas em vários desses produtos naturais e a disposição das carbonilas na cetona de Wieland-Miescher 107b viabilizou a conversão desta em sistemas tri-, tetra- ou pentacíclicos (Esquema 28).
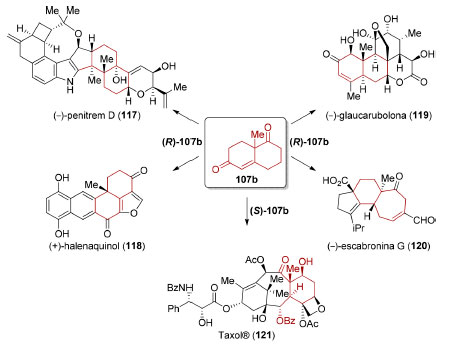 Esquema 28. Aplicações sintéticas da cetona de Wieland−Miescher (107b)
Dessimetrização de compostos meso A dessimetrização de anidridos cíclicos é uma estratégia importante para a formação enantiosseletiva de monoésteres de ácidos dicarboxílicos que pode ser levada a cabo, de forma diastereosseletiva, mediante o uso de nucleófilos quirais como álcoois e aminas (ver Esquema 14). Em 2000, Bolm e colaboradores utilizaram os alcaloides quinina (122) e quinidina (123) em uma reação de alcoólise enantiodivergente (re 99,5:0,5) do aduto endo 124 obtido da reação de Diels-Alder entre ciclopentadieno e anidrido maleico (Esquema 29).130 O reagente quiral pôde ser recuperado ao final deste processo.
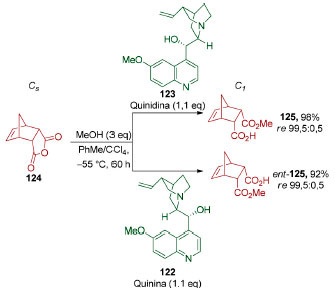 Esquema 29. Dessimetrização de anidridos cíclicos pelo grupo de Bolm130
Processos catalíticos também foram desenvolvidos para a abertura de anidridos cíclicos meso mediante o uso de nucleófilos aquirais. Em 1998, Seebach e colaboradores desenvolveram o uso de complexos de Ti(IV)-TADDOL (126), tanto em quantidades estequiométricas quanto catalíticas, para a dessimetrização de anidridos cíclicos (Esquema 30).131 Apesar de ser altamente enantiosseletiva (re 98:2), a versão catalítica ainda sofre com longos tempos reacionais (24 dias de reação).
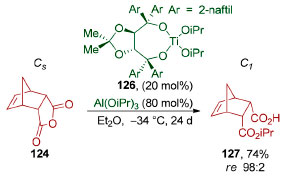 Esquema 30. Dessimetrização de anidridos cíclicos pelo grupo de Seebach131
Em 2018, Baran e colaboradores132 empregaram a dessimetrização de Bolm para a preparação enantiosseletiva de um intermediário da síntese da (+)-asenapina (128) descrita por Chandrasekhar.133 A (+)-asenapina (128) é um agente antipsicótico aprovado para uso clínico em sua forma racêmica, embora o isômero dextrorotatório apresente propriedades farmacocinéticas superiores (Esquema 31).
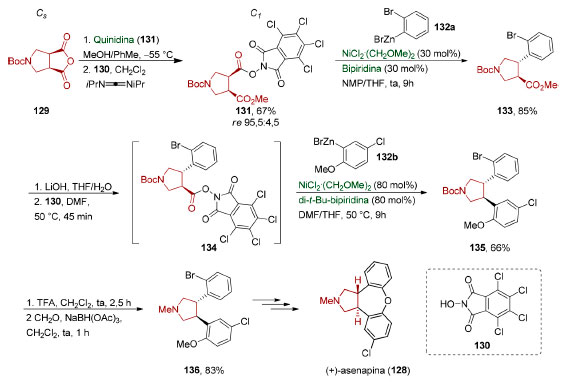 Esquema 31. Dessimetrização utilizada na síntese formal da (+)-asenapina (128), por Baran e colaboradores132
O grupo carboxílico do monoéster metílico enantiopuro obtido por dessimetrização do anidrido 129 foi convertido no éster redox-ativo 131 (67%). Dois acoplamentos descarboxilativos mediados por níquel (85% e 66% de rendimento) viabilizaram a incorporação estereosseletiva dos dois substituintes arila. Desproteção, seguida por uma aminação redutiva (83%), permitiu obter o intermediário 136 em oito etapas e 31% de rendimento global, completando uma síntese formal enantiosseletiva da (+)-asenapina (128).133 Em 2006, Trost e colaboradores134 utilizaram uma reação de substituição alílica enantiosseletiva, via catálise de paládio, na dessimetrização do carbonato duplo do meso-1,3-ciclopentenodiol (137, Esquema 32). O éster pirrólico 138 foi utilizado como nucleófilo e o produto 140 foi obtido em excelente enantiosseletividade (83%, re 96:4). O éster 140 foi convertido em um derivado de ácido hidroxâmico 141 (73%) de forma a viabilizar uma etapa de substituição alílica intramolecular (91%). O produto tricíclico 142 obtido foi então convertido em quatro etapas na (+)-agelastatina A (143).
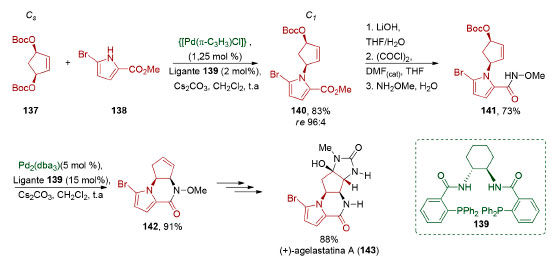 Esquema 32. Dessimetrização através de catálise via paládio aplicada à síntese da (+)-agelastatina A (143), por Trost e colaboradores134
O grupo de Trost também demonstrou uma variação desse processo de dessimetrização ao utilizar o éster do ácido hidroxâmico 144 (Esquema 33).135 Nesse processo, mediado pelo intermediário 146, ocorrem duas substituições alílicas one-pot (82%, re 99:1) formando-se 145, um isômero de posição do produto obtido no procedimento em duas etapas (142). O produto tricíclico 145 foi convertido na (-)-agelastatina A (ent-143) em cinco etapas.
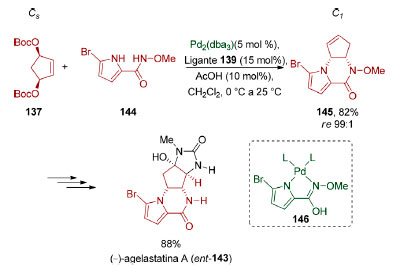 Esquema 33. Dessimetrização através de catálise via paládio aplicada à síntese da (-)-agelastatina A (ent-143), pelo grupo de Trost135
Em 2015, Hoveyda, Schrock e colaboradores utilizaram a dessimetrização enantiosseletiva do composto 148 na síntese de um tetraidropirano trissubstituído 150 (Esquema 34).136 Essa estratégia de dessimetrização utiliza o catalisador de molibdênio 149 em uma reação de metátese de olefinas que a um só tempo apresenta as características de um metátese cruzada, de abertura de anel e Z-seletiva (88%, >Z/E 99:1, re 99:1). O éter vinílico do tetraidropirano 150 formado foi hidrolisado e o aldeído correspondente foi oxidado nas condições de Pinnick (86%). O ácido carboxílico 151 foi então aplicado na síntese total do (+)-neopeltolídeo (152).
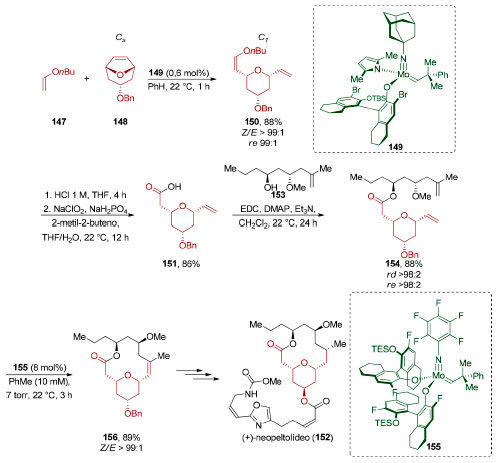 Esquema 34. Síntese total do (+)-neopeltolídeo (152) através de uma dessimetrização via metátese de olefinas, por Hoveyda, Schrock e colaboradores136
Esterificação de 151 com o álcool secundário 153 (88%) viabilizou a formação do éster 154 contendo todos os carbonos do macrociclo presente no produto final 152 (Esquema 34). Visando otimizar uma reação de macrociclização via metátese de fechamento de anel, os grupos de pesquisa de Hoveyda e Schrock desenvolveram um novo catalisador de molibdênio 155 que permitiu reduzir a carga de catalisador mesmo frente à baixa reatividade da olefina gem-dissubstituída de 154. O produto de macrociclização 156 (Z/E >99:1, 89%) foi então convertido no (+)-neopeltolídeo (152). A reação de di-hidroxilação enzimática de anéis aromáticos permite a obtenção de blocos de construção quirais de grande utilidade sintética. Em 1991, Johnson e colaboradores utilizaram o diol obtido por di-hidroxilação do benzeno (5) através de um processo quimioenzimático, promovido por Pseudomonas putida (Esquema 35).137 Após a conversão do diol 157 no acetonídeo meso 159 (53%), este foi submetido a uma quebra de simetria por acetilação promovida por P. cepacia (Amano P-30) que forneceu 161 (90%, re > 97,5:2,5). Esterificação de Mitsunobu (100%), seguida por hidrólise do acetonídeo e dos ésteres de 162 (84%), completou a síntese do (−)-conduritol C (163), um representante de uma família de produtos naturais que apresentam propriedade de inibição de glicosidases.
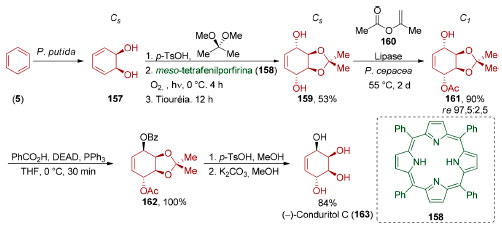 Esquema 35. Síntese do (−)-conduritol C (163), pelo grupo de Johnson137
A dessimetrização enantiosseletiva de compostos aromáticos também pode ser realizada por métodos não-enzimáticos. Em 2017, Sarlah e colaboradores desenvolveram uma dessimetrização do cicloaduto meso 165,138 formado in situ através de uma cicloadição [4+2] fotoquímica entre benzeno (5) e 4-Metil-1,2,4-TriAzolin-3,5-Diona (MTAD, 164, Esquema 36).139 Na presença do ligante 166, foi realizada uma reação de substituição alílica via catálise de níquel utilizando um reagente organomagnésio 167 como nucleófílo (65%, re 98:2). O processo como um todo consiste em uma trans-carboaminação enantiosseletiva.140
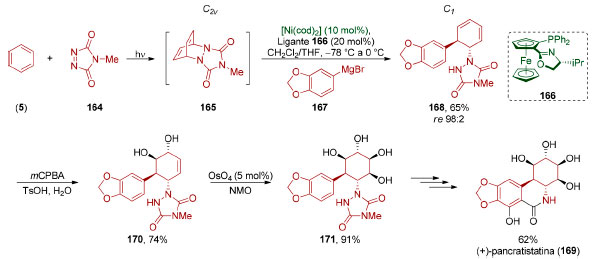 Esquema 36. Síntese da (+)-pancrastistatina (169), por Sarlah e colaboradores138
Esta sequência de dearomatização-dessimetrização foi aplicada pelo grupo de Sarlah na síntese da (+)-pancratistatina (169). Reação de epoxidação de 168, seguida por abertura de epóxido em meio ácido, viabilizou uma reação de trans-di-hidroxilação (74%) seguida de reação de cis-di-hidroxilação (91%), para fornecer o tetraol 171 que foi convertido na (+)-pancratistatina (169). Esta sequência de dearomatização-dessimetrização via catálise metálica pode ser diretamente comparada à via biocatalítica, mesmo considerando-se a diferença de quase duas décadas entre elas. Em 1995, Hudlicky e colaboradores realizaram a primeira síntese total da (+)-pancratistatina (169),141 em 13 etapas a partir do diol obtido por dessimetrização quimioenzimática do bromobenzeno. Em 2008, o grupo de List desenvolveu uma versão assimétrica de reações aldólicas transanulares (Esquema 37).142 Vários dos exemplos estudados por esse grupo se baseiam na ciclização transanular de ciclooctanos formando biciclo[3.3.0]octanos, uma estratégia muito utilizada para a síntese de di-, tri- e tetraquinanos,143 produtos naturais policíclicos que contém anéis de ciclopentano fundidos em sua estrutura.
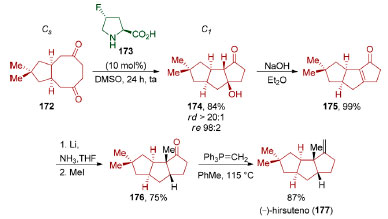 Esquema 37. Dessimetrização aplicada à síntese do (−)-hirsuteno (177), por List e colaboradores142
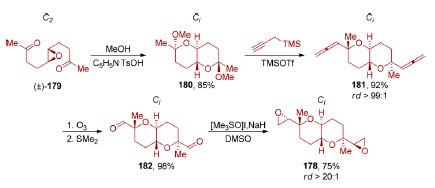
Utilizando o composto meso bicíclico 172, List e colaboradores conseguiram realizar uma dessimetrização via reação aldólica transanular formando o composto 174, contendo um sistema tricíclico presente em triquinanos lineares, em excelente esteoresseletividade (84%, rd >20:1, re 98:2). Reação de eliminação via mecanismo E1cB (99%), redução com lítio em amônia líquida, seguida por uma reação de alquilação (75%) e reação de olefinação de Wittig (87%), levaram ao produto natural (−)-hirsuteno (177). Enquanto compostos meso contendo um plano de simetria foram amplamente estudados, a dessimetrização de compostos contendo um centro de inversão recebeu uma atenção consideravelmente menor por parte da comunidade sintética.144 Em 2001, o grupo de Nelson demonstrou a aplicação da dessimetrização de um bis-epóxido centrossimétrico em uma síntese total (Esquema 38).145 O substrato centrossimétrico 178 pôde ser preparado através de uma síntese bidirecional a partir do epóxido C2-simétrico (±)-179. Uma dupla ciclização através de uma acetalização em meio ácido (85%), seguida por uma adição de propargilsilano na presença de TMSOTf (92%, rd > 99:1), viabilizou a incorporação de dois grupos alenos terminais de 181. Ozonólise do aleno 181 (98%) e epoxidação de carbonila de 182 utilizando ilídeo de enxofre (75% rd > 20:1) completou a preparação do substrato centrossimétrico 178 desejado.
 Esquema 38. Preparação do substrato centrossimétrico 178, pelo grupo de Nelson145
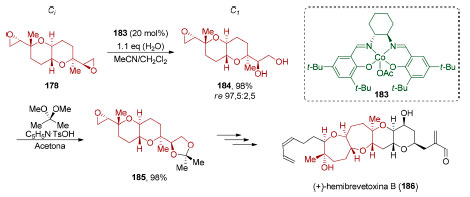
O complexo de cobalto 183 contendo um ligante do tipo Salen foi o ácido de Lewis utilizado pelo grupo de Nelson como catalisador para abertura do epóxido meso centrossimétrico 178 (Esquema 39). A mono-hidratação do substrato centrossimétrico deu-se em excelente enantiosseletividade (98%, re 97,5:2,5), viabilizando a obtenção do composto bicíclico 184 contendo seis centros estereogênicos. A dessimetrização de epóxidos meso contendo um plano de simetria é muito utilizada em Síntese Orgânica,146 mas esse é o primeiro exemplo de uma dessimetrização de um bis-epóxido centrossimétrico. O produto 184 foi então utilizado em uma síntese formal da (+)-hemibrevetoxina B (186).
 Esquema 39. Dessimetrização aplicada à síntese do (+)-hemibrevetoxina (186), por Nelson e colaboradores145
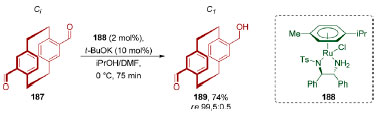
Em 2018, Benedetti, Micouin e colaboradores demonstraram uma dessimetrização de um dialdeído centrossimétrico 187 através de uma transferência de hidrogênio assimétrica utilizando o catalisador de Noyori 188 (Esquema 40).147 As duas unidades formilas estão em posição pseudo-para no [2.2]-paraciclofano 187. Ao realizar uma redução em somente um dos grupos formilas do substrato 187, obteve-se o hidroxi-aldeído 189 de quiralidade planar em excelente enantiosseletividade (74%, re 99,05:0,5).
 Esquema 40. Dessimetrização do dialdeído centrossimétrico 187, pelo grupo de Benedetti e Micouin. O produto 189 apresenta quiralidade planar147
Em 2006, Nelson e colaboradores desenvolveram a dessimetrização de uma piperazina centrossimétrica 190 (Esquema 41).148 A dessimetrização catalítica dessa classe de compostos é desafiadora e, devido a sua relevância em química medicinal,149 é tipicamente realizada via resolução através de formação de um sal com ácido enantiomericamente puro como, por exemplo, ácido canforsulfônico ou ácido tartárico.
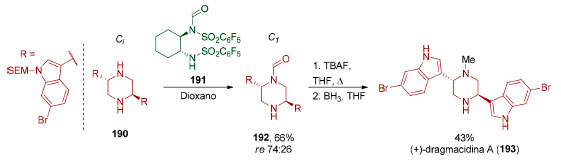 Esquema 41. Dessimetrização da piperazina centrossimétrica 190 aplicada à síntese da (+)-dragmacidina A (193), por Nelson e colaboradores148
O grupo de Nelson utilizou o reagente de formilação quiral 191 para a dessimetrização da piperazina centrossimétrica 190. Infelizmente o produto 192 foi obtido com baixa enantiosseletividade (66%, re 74:26). Uma etapa de desproteção seguida por uma redução do grupo formila viabilizou uma síntese concisa da (+)-dragmacidina A (193). As limitações desse tipo de dessimetrização sugerem que investigações nessa direção ainda se fazem necessárias.
RECONHECIMENTO DE PADRÕES EM SÍNTESE ORGÂNICA Aspectos conceituais O reconhecimento de padrões é um aspecto importante do planejamento sintético na medida em que correlaciona a molécula-alvo a uma subestrutura mais simples ou a uma matéria-prima disponível que seja passível de ser convertida à molécula-alvo.150 Em uma análise retrossintética convencional, realiza-se um processo iterativo sobre o alvo sintético através de sucessivas desconexões de ligações carbono-carbono, carbono-heteroátomo ou carbono-hidrogênio.151 Nesse processo, tem-se como guia reações capazes de formar estas mesmas ligações e, como objetivo, identificar um material de partida e intermediários de estrutura mais simples que a molécula-alvo. Diferentemente da análise retrossintética convencional, no reconhecimento de padrões busca-se a identificação de padrões estruturais presentes tanto no alvo sintético como no material de partida a ser utilizado.152 De formal geral (Figura 10), esses padrões estruturais podem apresentar uma determinada conectividade (ex. o sistema bicíclico do tropano (194), presente em vários alcaloides)153 ou um arranjo estereoquímico específico (ex. a estereotríade 195a-d que pode estar presente em um policetídeo).154
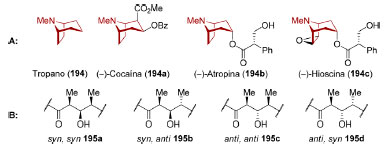 Figura 10. Padrões estruturais identificáveis. A: Alcaloides tropano (194); B: Estereotríades 195a-d presentes em policetídeos
Historicamente, o reconhecimento de determinados padrões estruturais em produtos naturais foi de grande importância para estudos de biossíntese, como a "regra do isopreno (196)" para terpenoides.155 Por exemplo, a origem biossintética do Taxol® (121) apresenta duas sequências reacionais distintas (Esquema 42): as etapas de ciclização (fase das ciclases) em que as ligações carbono-carbono são formadas e a fase de ajuste dos estados de oxidação dos carbonos (fase das oxidases).156
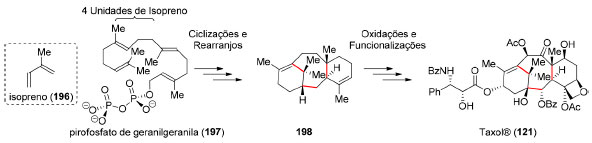 Esquema 42. Biossíntese simplificada do Taxol® (121)
Várias sínteses totais foram realizadas utilizando monoterpenos quirais como material de partida.157 Este tipo de reconhecimento de padrão ainda é amplamente utilizado, principalmente para a síntese total de terpenoides.158 Apesar do advento de metodologias baseadas em catálise enantiosseletiva,159 muitas sínteses que partem de um substrato quiral ainda são vantajosas uma vez que ambos os enantiômeros de muitos monoterpenos podem ser obtidos de forma enantiopura a um custo relativamente baixo. Em 1997, Wender e colaboradores realizaram uma síntese total do Taxol® (121, Esquema 43A).160 Nesta síntese, a (+)-verbenona (199) foi utilizada como material de partida para construção de um anel de seis membros contendo um substituinte gem-dimetila presente no produto natural 121. Em 1967, o (S)-cis-verbenol (200) foi utilizado pelo grupo de Mechoulam161 na síntese total do (−)-Δ9-tetra-hidrocanabinol (202, Esquema 43B),162 o princípio psicoativo presente em C. sativa. Trata-se de um processo em cascata, promovido por ácido de Lewis, que ocorre através de uma reação de Friedel-Crafts, seguida de um rearranjo de carbocátion e interceptação intramolecular por uma hidroxila fenólica. Em 2001, Malkov e Kocovsky, demonstraram a aplicação do (+)-limoneno (202) na síntese do (-)-Δ9-tetra-hidrocanabinol (202, Esquema 43C).163
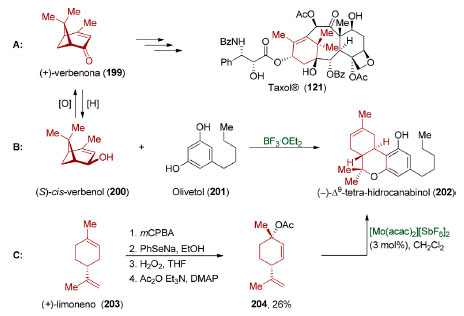 Esquema 43. Reconhecimento de padrão em síntese total. A: (+)-verbenona (199) utilizada na síntese do Taxol® (121). Síntese do (-)-Δ9-tetra-hidrocanabinol (202) a partir do (S)-cis-verbenol (200) (B) e do (+)-limoneno (203) (C)
A (+)-verbenona (199)164 e o (S)-cis-verbenol (200)165 podem ser produzidos através da oxidação biocatalítica do (+)-α-pineno (205, Esquema 44), presente na terebintina, óleo extraído de coníferas abundantes no Brasil como os pinheiros. O mesmo é válido para seus enantiômeros. Dentro da realidade nacional, sínteses utilizando (+)- ou (−)-limoneno (203 ou ent-203) como material de partida são interessantes por ele ser o componente majoritário do óleo extraído da casca de frutas cítricas166 cuja produção, a nível mundial, é liderada pelo Brasil.167
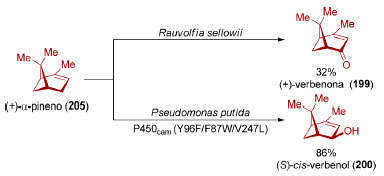 Esquema 44. Conversão biocatalisada de (+)-α-pineno (205) em (+)-verbenona (199)168 e (S)-cis-verbenol (200)161
A síntese total da (±)-adunctina B (206), por Dethe e colaboradores (Esquema 46),168 exemplifica de forma representativa como a simetria molecular permite antecipar limitações no uso de reconhecimento de padrões. Ambos os enantiômeros da carvona (207) foram utilizados em várias sínteses totais.169 Partindo-se de 207, um derivado enantiopuro da (−)-carvona (209), o grupo de Dethe realizou a síntese da (±)-adunctina B (206) em duas etapas: uma substituição eletrofílica aromática (79%, rd 6:1, re 1:1), mediada por ácido de Lewis, seguida de uma ciclização oxidativa do tipo Wacker, catalisada por paládio (Esquema 45A). Apesar de ser uma síntese extremamente concisa, o carbocátion alílico 211 formado na presença do ácido de Lewis apresenta um plano de simetria (Esquema 45B) de modo que a quiralidade presente no material de partida é perdida no processo, dando origem a um produto racêmico.
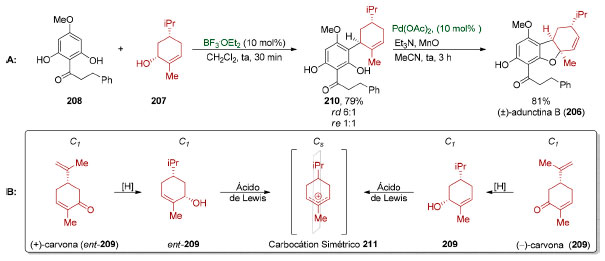 Esquema 45. A: Racemização observada na síntese da (±)-adunctina B 206. B. Mecanismo da racemização168
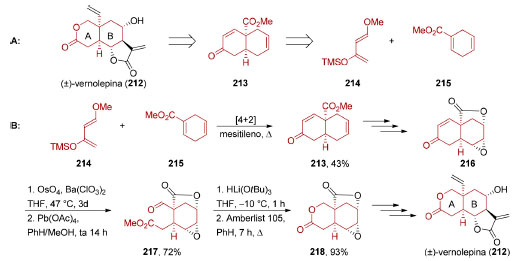 Esquema 46. Reconhecimento de padrões aplicado à (±)-vernolepina (212). A: Análise retrossintética. B: Síntese total, por Danishefsky e colaboradores172
Produtos de reações bem estabelecidas como a reação de Diels-Alder170 e a anelação de Robinson171 apresentam conectividades e arranjos estereoquímicos importantes de serem reconhecidos. Uma aplicação do reconhecimento de padrões cuja visualização exige desconexões adicionais é encontrada na síntese da (±)-vernolepina (212, Esquema 46A), uma bislactona de origem natural que apresentou atividade antitumoral in vitro contra células de carcinoma nasofaringeal. Na síntese da (±)-vernolepina (212) realizada pelo grupo de Danishefsky, a origem dos anéis A e B foi traçada ao cicloaduto 213 resultante da reação de Diels-Alder entre o dieno 214 e o dienófilo cíclico 215.172 Deve-se observar que, para a instalação da lactona presente no anel A da (±)-vernolepina (212), foi necessária uma clivagem oxidativa de 216 (Esquema 46B), seguida por uma redução e uma transesterificação (67%). Ainda que não conheçamos o mecanismo neural pelo qual o reconhecimento de padrões opera, pode-se afirmar que ele se beneficia de um acúmulo de informações prévias sobre possíveis padrões estruturais e do processo cognitivo de superposição de imagens. Representar uma mesma estrutura de vários ângulos diferentes é um recurso simples e acessível que facilita a identificação de tais padrões. Isso permite comparar cada uma das múltiplas perspectivas representadas com estruturas guardadas na memória − ou em anotações pessoais − e eleger o material de partida ou intermediário sintético mais apropriado para a síntese em questão. Sempre se deve ter em mente que a escolha da matéria-prima é um dos aspectos mais importantes de um planejamento sintético, inseparável do custo operacional, eficiência e escalabilidade de uma rota sintética. A morfina (219) é um alcaloide pentacíclico (Figura 11) para o qual mais de 30 sínteses totais já foram descritas.173 Um ícone do esforço da comunidade sintética em sua busca por aperfeiçoar estratégias e aplicar novas reações, constituindo-se em um campo de provas para avaliação de novas metodologias sintéticas. A morfina (219) foi selecionada para ilustrar como diferentes maneiras de representar uma estrutura tridimensional complexa permitem visualizar aspectos que seriam difíceis de ver de outro modo (Figura 11).174
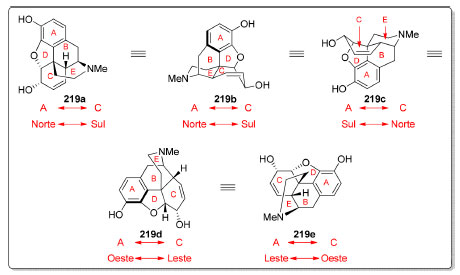 Figura 11. Diferentes perspectivas para a morfina (219, adaptado de Trauner, 2018)182
As duas primeiras representações (219a e 219b) mostram os anéis A e C da morfina (219) na direção Norte−Sul (Figura 11). Enquanto 219a é a representação mais usual, 219b destaca a conformação cadeira do anel E e barco torcido do anel C. Essas mesmas informações estão presentes na representação bidimensional 219c em que os anéis A e C estão orientados nas direções Sul−Norte. As duas últimas representações, 219d e 219e, apresentam os anéis A e C na direção Oeste−Leste e Leste−Oeste, respectivamente, e reforçam a conformação cadeira do anel E. Modelos moleculares, como o de Dreiding, apresentam elevado grau de precisão no que se refere a comprimentos, ângulos de ligação e interações estéricas e oferecem perspectivas que não podem ser facilmente apreciadas com o uso de modelos computacionais. Tais modelos permitem que um arquiteto de moléculas aprecie diferentes vistas de uma obra da natureza, tal qual arquitetos de edifícios e obras de arte apreciam seus objetivos de estudo e trabalho. Com a popularização de técnicas de impressão 3D, os químicos sintéticos em breve poderão apreciar seus objetos de interesse em modelos construídos sob demanda. A relação do reconhecimento de padrão com simetria molecular pode se expressar de várias formas. Em 1996, Overman e colaboradores desenvolveram uma síntese estereosseletiva para o alcaloide bis-pirroloindólico meso-quimonantina (220, Esquema 47A)175 que apresenta um plano de simetria em um de seus confôrmeros. Em meio ácido, este produto natural pode ser convertido na meso-calicantina (221),176,177 um isômero que contém um centro de simetria. Uma maneira de visualizar tal isomerização é considerar uma "hidrólise imaginária" de cada aminal (Esquema 47B), onde as carbonilas do dialdeído 222 estariam diante de grupos amino de aminas primárias e secundárias. Na reação de formação de hemiaminal a partir do intermediário dicarbonílico hipotético 222, tem-se então uma permuta entre os grupos amino que reagem com cada carbonila de 222. Esse não é o mecanismo real dessa isomerização que, provavelmente, deve envolver diferentes hemiaminais e íons imínio, mas apenas uma simplificação daquele. Este tipo de isomerização também está presente nos estereoisômeros quirais de simetria C2 dessa família de alcaloides.178 A síntese de compostos com esse tipo de simetria será discuta a seguir, na sessão 4.2.
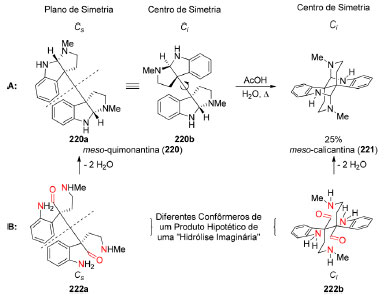 Esquema 47. Isomerização da meso-quimonantina (220) na meso-calicantina (221). A: Condições reacionais. B: Visualização simplificada do processo de isomerização177
Planejamento sintético de moléculas simétricas Em um estudo de 2004, Greer e colaboradores, estimaram que o número de produtos naturais que possuem simetria bilateral, ou seja, compostos que pertencem aos grupos pontuais C2, Cs e C2v, corresponde a 7% dentro do espaço amostral de 3000 artigos analisados (Figura 12).179 Além disso, os autores estimam que cerca de outros 10% dos artigos investigados apresentam moléculas cuja biossíntese passa por intermediários com simetria bilateral que sofreriam modificações estruturais posteriores com perda de simetria.
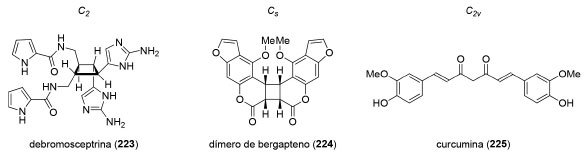 Figura 12. Exemplos selecionados de produtos naturais contendo simetria bilateral
Enquanto os compostos com simetria C2 são quirais, os produtos naturais que pertencem aos grupos pontuais Cs e C2v são aquirais e estão fora do escopo deste artigo. A existência de um eixo de simetria C2 em produtos naturais abre a possibilidade de aumentar a convergência da estratégia sintética ao permitir a preparação simultânea de duas partes iguais da molécula-alvo.180 Uma série de reações como, por exemplo, esterificações, acoplamentos cruzados, alquilações, metátese cruzada de olefinas e cicloadições podem ser empregadas para conectar os dois fragmentos. Uma maneira de se preparar produtos naturais diméricos é a reação de formação de ligação σN−N através de uma oxidação eletroquímica desenvolvida por Baran e colaboradores.181 Nesse trabalho, os autores prepararam o produto natural (+)-xiamicina A (226), em 9 etapas, a partir do álcool 227. A oxidação anódica de 226 levou à formação do alcaloide indólico (+)-dixiamicina B (228), em 28% de rendimento (Esquema 48).
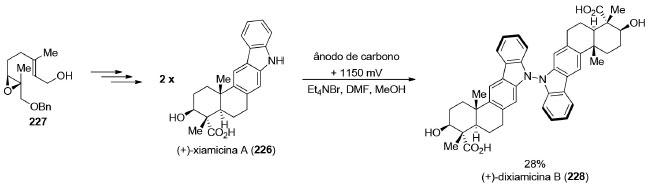 Esquema 48. Síntese total da (+)-dixiamicina B (228) por Baran e colaboradores181
O (+)-testudinariol A (238) é um triterpeno de origem marinha que possui em sua estrutura dimérica um anel ciclopentânico densamente funcionalizado ligado a um 3-tetraidropirano alquilidênico. Cada uma das partes possui quatro centros estereogênicos contíguos. Em 2002, Amarasinghe e Montgomery completaram a síntese total do testudinariol A (229) a partir de uma reação de alquilação bidirecional envolvendo o diânion do composto 230 de simetria C2v. Uma reação de troca de halogênio-metal em 230 permitiu uma reação de dupla alquilação utilizando dois equivalentes do iodeto 231, levando à formação do composto 232 (38%) de simetria C2 (Esquema 49).182 Após algumas transformações bidirecionais, o (+)-testudinariol A (229) foi obtido.
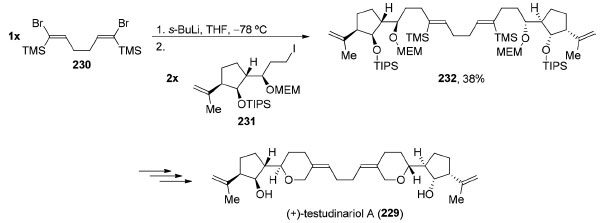 Esquema 49. Síntese total do (+)-testudinariol A (229), por Amarasinghe e Montgomery182
Os cilindrociclofanos são [7,7]-paraciclofanos de 22 membros que possuem atividade citotóxica.183 Alguns deles como, por exemplo, os (−)-cilindrociclofanos A e F (233a e 233b) possuem simetria C2. Em 2000, Smith e colaboradores reportaram a síntese total dos (−)-cilindrociclofanos A e F (233a e 233b) através de uma reação de metátese cruzada de olefinas, seguida por uma reação de metátese de fechamento de anel one-pot (Esquema 50).184 A exposição de dois equivalentes das bisolefinas 234a ou 234b ao catalisador de molibdênio 235 levou à formação dos compostos 236a ou 236b em 77% e 72% de rendimento, respectivamente. Excelentes níveis de seletividade foram observados em favor do produto de acoplamento cabeça-cauda que apresenta simetria C2.
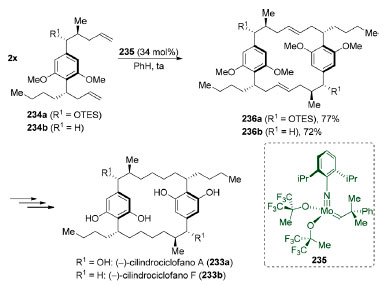 Esquema 50. Síntese total dos (−)-cilindrociclofanos A e F (233a e 233b) por Smith e colaboradores184
O (−)-cianolídeo A (237) é um macrodiolídeo glicosídico de 16 membros contendo 16 centros estereogênicos e que apresenta potente atividade moluscicida. Em 2013, Waldeck e Krische completaram a síntese total deste produto natural utilizando as condições reacionais de Yamaguchi185 para a síntese do macrodiolídeo de simetria C2 (Esquema 51).186
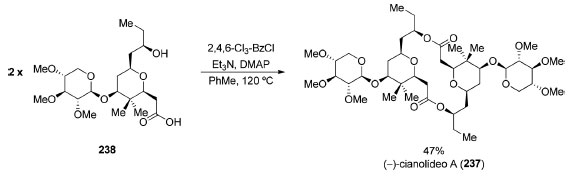 Esquema 51. Síntese total do macrolídeo (−)-cianolídeo A (237) por Waldeck e Krische186
O (−)-clavosolídeo A (239), um macrodiolídeo semelhante ao (−)-cianolídeo A (238), possui 18 centros estereogênicos e estrutura dimérica com simetria C2. Em 2015, Haydl e Breit completaram a síntese total dessa molécula utilizando como etapa chave uma reação de "dimerização"187 do composto 240. Ela se deu a partir de uma reação de adição de ácido carboxílico ao aleno terminal catalisada por ródio levando à formação do macrodiolídeo 242 (72%, rd 92:8, Esquema 52).188 Após algumas transformações bidirecionais, o (−)-clavosolídeo A (239) foi obtido.
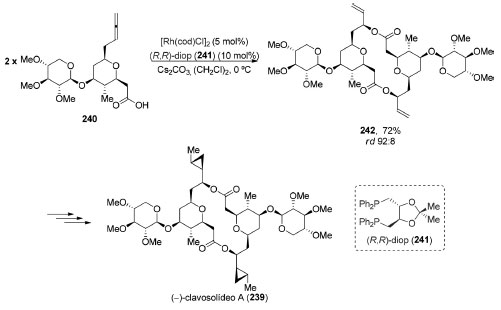 Esquema 52. Síntese total do macrolídeo (−)-clavosolídeo A (239) por Haydl e Breit188
Em situações nas quais encontramos problemas de quimiosseletividade ainda não resolvidos ou a reatividade necessária não foi plenamente desenvolvida, o produto natural dimérico não pode ser preparado a partir de dois fragmentos idênticos. Nesses cenários, pequenas modificações precisam ser feitas em um dos equivalentes para que o acoplamento possa ser realizado. Em 2017, Neumeyer e Brückner completaram a síntese total da (+)-γ-actinorodina (243), uma γ-lactona naftoquinonônica dimérica (Esquema 53).189 Neste trabalho, os autores converteram um equivalente do brometo arílico 244 no correspondente boropinacolato 245. O intermediário formado 245 participa então de um acoplamento de Suzuki-Miyaura in situ com um segundo equivalente do brometo 244 levando à formação do produto dimérico 246, em 52% de rendimento. Após algumas transformações, a (+)-γ-actinorodina (243) foi obtida.
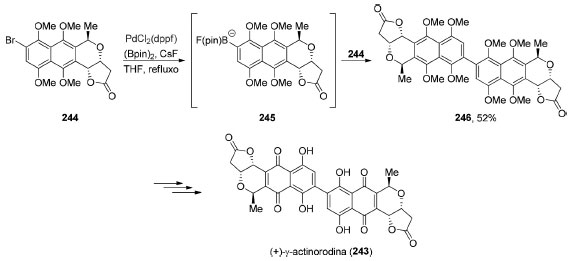 Esquema 53. Síntese total da (+)-γ-actinorodina (243), por Neumeyer e Brückner189
O reconhecimento de padrão faz da "dimerização" de dois fragmentos idênticos a estratégia mais lógica para sintetizar moléculas com eixo de simetria C2. Em muitos casos, não há seletividade envolvida na etapa de acoplamento, em outros é possível utilizar a reação de "dimerização" para criar centros estereogênicos e, por fim, nos casos em que há problemas de seletividade não-solucionados ou reatividade incipiente, os fragmentos a serem acoplados não são exatamente iguais. Reconhecimento de simetria escondida no planejamento sintético Enquanto a simetria presente em um alvo sintético viabiliza diversas estratégias para reduzir o número de etapas da rota sintética, essa não é a única aplicação do reconhecimento de simetria no planejamento sintético. Uma análise retrossintética cuidadosa de um alvo sintético pertencente ao grupo pontual C1 pode viabilizar o reconhecimento de uma relação de simetria ausente no produto final. Os exemplos discutidos a seguir são de sínteses convergentes onde um mesmo intermediário avançado foi utilizado para formar dois ou mais fragmentos do alvo sintético. Esse reconhecimento de padrão pode ser chamado de "simetria escondida", uma vez que a simetria em questão é virtual, resulta exclusivamente da retrossíntese. Em 1971, o grupo de Chapman190 realizou a síntese da (±)-carpanona (247), uma lignana tetracíclica dimérica (benzoxantenonas) contendo cinco estereocentros, isolada na forma de um racemato. Essa síntese foi um dos primeiros exemplos descritos de reconhecimento de simetria escondida (Esquema 54), realizada através de uma dimerização oxidativa biomimética. A isomerização da dupla ligação do grupo alila de 248 (rendimento não reportado) forneceu o monômero 249. Na presença de PdCl2 estequiométrico, Chapman e colaboradores propõem que as hidroxilas fenólicas de 249 se coordenam ao paládio, formando o intermediário chave 250. A dimerização desejada acontece através de um processo redox resultando na formação de Pd(0) e o bis-orto-quinometino (±)-251, de simetria C2, altamente reativo. Através de uma reação de hetero-Diels-Alder intramolecular, o confôrmero 251b, de simetria C1, é convertido na (±)-carpanona (247) em 46% de rendimento. Esse processo biomimético recebeu diversos estudos posteriores visando realizar a dimerização oxidativa de forma catalítica pelos grupos de Kuroda,191 Lindsley,192 Poli,193 e Kozlowski194 para obtenção de benzoxantenonas naturais assim como análogos sintéticos. Os grupos de Ley195 e Shair196 investigaram essa transformação utilizando síntese em fase sólida. Em 2006, Shair e colaboradores utilizaram síntese em fase sólida para o preparo de 10.000 análogos inspirados na estrutura da (±)-carpanona (247).197 Em 2020, Kozlowski194 e colaboradores descreveram a primeira versão enantiosseletiva dessa reação, porém com baixa enantiosseletividade (re 75:25).
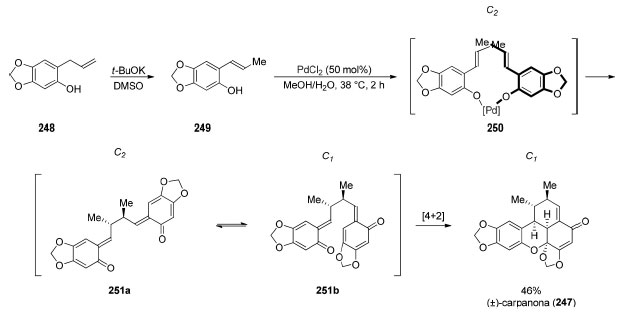 Esquema 54. Síntese total da (±)-carpanona (247) por Chapman e colaboradores190
Em 2017, Liu e colaboradores aplicaram o conceito de simetria escondida na síntese do sesquiterpeno dimérico (−)-sarcandrolídeo J (252)198 cuja estrutura contém oito anéis e 11 estereocentros, três dos quais são tetrassubstituídos e dois quaternários. Eles identificaram que o anel central do sistema octacíclico deste produto natural poderia ser sintetizado através de uma cicloadição [4+2] entre o dieno presente no furano tetracíclico 253 e o γ-alquilideno butenolídeo 254 que age como dienófilo (Esquema 55). Apesar da instabilidade observada para o furano 253, ele pôde ser formado in situ via desidratação da di-hidroxicetona tricíclica 255 em meio ácido. Tanto 254 como 255 puderam ser sintetizados a partir de um mesmo intermediário avançado, o acetal da cetona tricíclica 256.
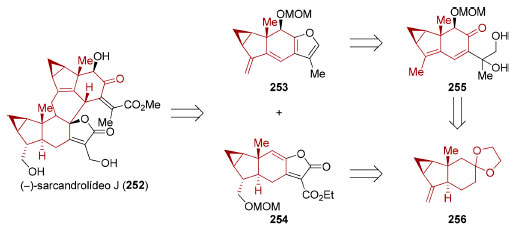 Esquema 55. Análise retrossintética para o (−)-sarcandrolídeo J (252), evidenciando a simetria escondida198
Para a síntese do intermediário avançado 256 (Esquema 56), o grupo de Liu199 utilizou a (+)-verbenona (199) como material de partida. Uma adição 1,4 diastereosseletiva (rd > 20:1) de cuprato de vinila (78%) gerado in situ deu origem ao centro quaternário em 257. A abertura de ciclobutano (75%), seguida pela formação de um acetal (77%) e oxidação alílica (75%), formou o aldeído 260. A conversão deste na hidrazona 261 (77%) viabilizou o preparo do intermediário avançado 256 através de uma reação de ciclopropanação intramolecular catalisada por paládio (94%).200
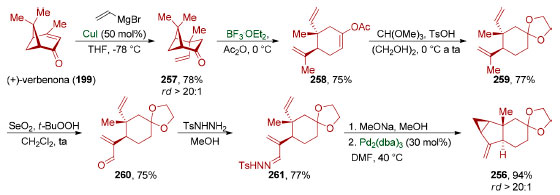 Esquema 56. Preparação de um intermediário avançado utilizado em uma síntese convergente do (−)-sarcandrolídeo J (252), segundo Liu e colaboradores198
O intermediário 256 foi então utilizado na síntese dos fragmentos 254 e 255 (Esquema 57). A reação de cicloadição [4+2] necessária para a formação do anel central pôde ser realizada na presença de excesso de PhCO2H, em um tubo selado a 170 °C (83%). Devido à instabilidade do dieno 253, necessariamente formado in situ, excesso (5 equiv.) do seu precursor, o ceto-diol 255, teve que ser utilizado. Após remoção do grupo de proteção e redução do éster (62%), uma reação de oxidação fotocatalisada do furano, seguida por uma esterificação (47%), forneceu o (−)-sarcandrolídeo J (252).
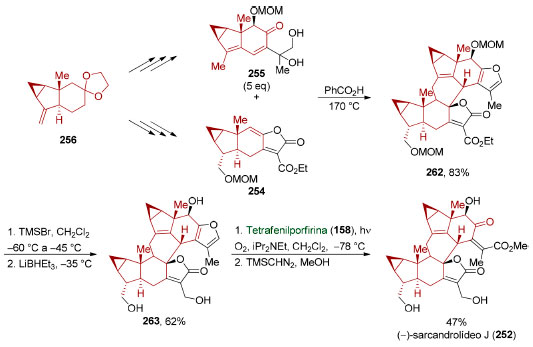 Esquema 57. Síntese convergente do (−)-sarcandrolídeo J (252) através do acoplamento de dois fragmentos oriundos do mesmo intermediário avançado, por Liu e colaboradores198
O reconhecimento de simetria escondida não está limitado à presença de dímeros (ou oligômeros) uma vez que não é necessário que tenha uma interdependência direta com a origem biossintética da molécula-alvo. Em 2013, Carter e colaboradores realizaram a síntese total do (−)-anfidinolídeo F(264),201 um macrolídeo de 25 membros contendo 11 estereocentros e duas unidades tetraidrofurânicas. A análise retrossintética identificou uma simetria escondida ao reconhecer que as unidades tetraidrofurânicas presentes em cada um dos fragmentos (265 e 266) de uma síntese convergente podem ser sintetizadas a partir do mesmo intermediário avançado 267 (Esquema 58).
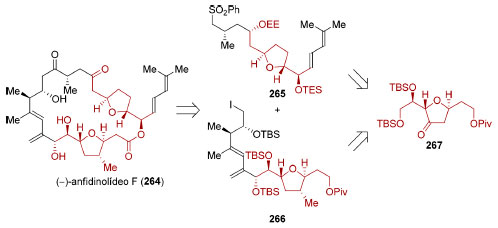 Esquema 58. Análise retrossintética para o (−)-anfidinolídeo F (264), evidenciando a simetria escondida201
Em sua síntese do intermediário avançado 267 (Esquema 59), o grupo de Carter introduziu o primeiro estereocentro através de uma reação de alquinilação enantiosseletiva de 268, seguida de uma reação de benzoilação (35%, re 95:5).202 Uma reação de di-hidroxilação de Sharpless em 271 introduziu outros dois estereocentros (87%, rd > 20:1) e foi seguida pela formação do di-hidrofurano 274, através de uma ciclização catalisada por prata (67%, rd >20:1). Proteção da hidroxila livre de 274 (85%), seguida por uma hidrólise do benzoato de vinila 275 (81%), forneceu o intermediário avançado 267.
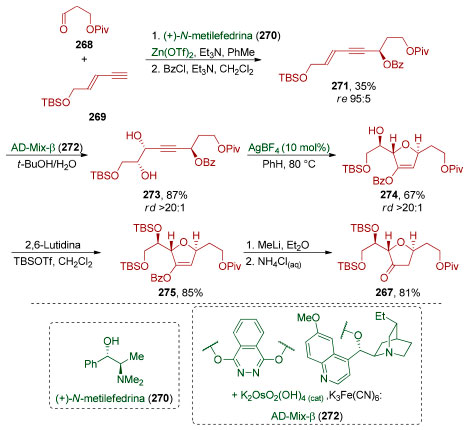 Esquema 59. Preparação de um intermediário avançado utilizado na síntese convergente do (−)-anfidinolídeo F (264), segundo Carter e colaboradores202
O intermediário 267 foi então aplicado na síntese de ambos os fragmentos 265 e 267 (Esquema 60). Uma α-desprotonação da sulfona de 265 viabilizou uma reação de alquilação via substituição nucleofílica em 276 (74%). A α-desprotonação da sulfona em 276 viabilizou a etapa de dessulfurização oxidativa (42%), realizada na presença de um dieno facilmente oxidável. Após uma série de etapas, 277 foi convertido no (−)-anfidinolídeo F (264).
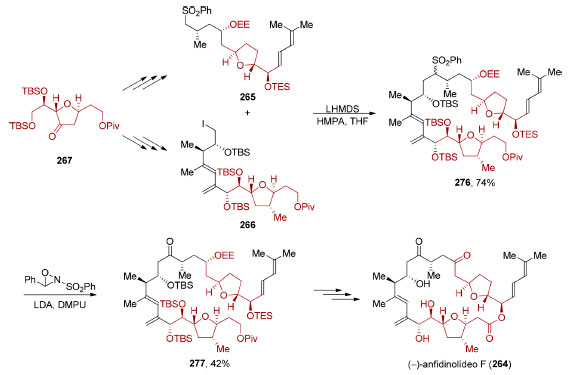 Esquema 60. Síntese convergente do (−)-anfidinolídeo F (264), através do acoplamento de dois fragmentos (265 e 266) sintetizados a partir do mesmo intermediário avançado 267, por Carter e colaboradores202
Em 2005, Ley e colaboradores realizaram uma das sínteses totais da (+)-espongistatina 1 (278),203 um macrolídeo de 42 membros, contendo 24 estereocentros, dois anéis tetraidropirânicos e dois acetais espirocíclicos. A análise retrossintética (Esquema 61) indica que o fragmento contendo os dois acetais espirocíclicos 280 pode ser preparado de forma convergente, a partir de dois intermediários avançados 281 e 282, cada um contendo um acetal espirocíclico. A simetria escondida foi identificada ao se reconhecer que cada espirociclo pode ser preparado a partir de diferentes epímeros do intermediário 283.
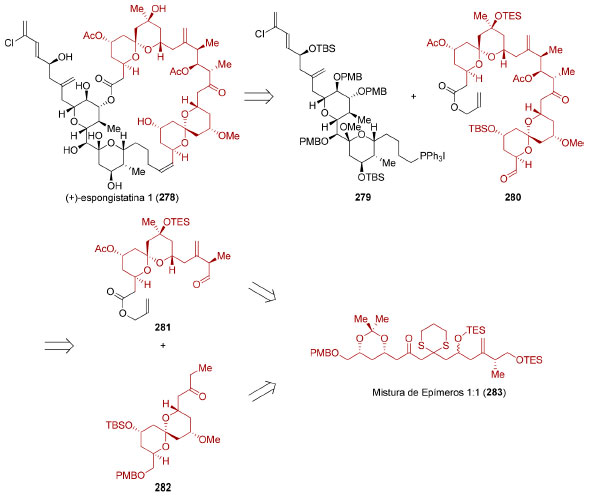 Esquema 61. Análise retrossintética para a (+)-espongistatina 1 (278) evidenciando a simetria escondida, por Ley e colaboradores203
A preparação do intermediário avançado 283 pelo grupo de Ley204 utilizou o éster de Roche ent-70b de configuração S como precursor quiral (Esquema 62). Proteção da hidroxila livre de ent-70b e etapas redox (87%), seguidas por formação de 1,3-ditiana e reproteção (84%), levaram à formação do intermediário 285. Litiação da 1,3-ditiana 285 viabilizou uma abertura de epóxido da epicloridrina racêmica, acompanhada por uma reação SN2 intramolecular, que formou o epóxido terminal 286 (81%). Abertura do epóxido 286 utilizando o ânion de lítio do TIPS-acetileno litiado (287, 87%), etapas de proteção e desproteção e uma olefinação de Tebbe resultaram na formação do enino 290 (56%). Adição do alcino terminal em 290 ao aldeído 291 formando 292 (86%, rd 1:1), seguida de oxidação via periodinana de Dess-Martin e formação de β-ceto-1,3-ditiana205 (87%), completaram a preparação da mistura de epímeros 283.
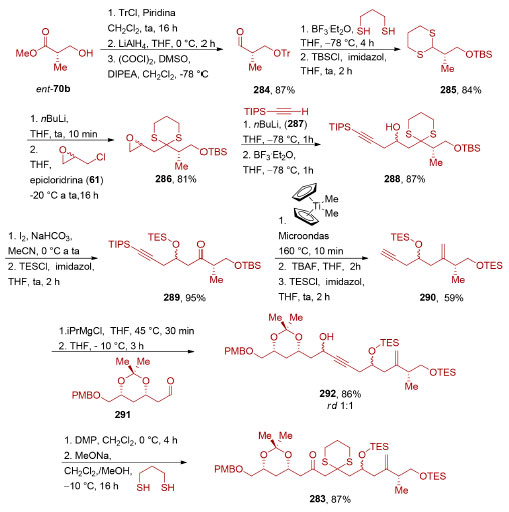 Esquema 62. Preparação do intermediário avançado 283 utilizado na síntese da (+)-espongistatina 1 (278) utilizando simetria escondida, por Ley e colaboradores204
Em meio ácido, 283 foi convertido a uma mistura de três acetais espirocíclicos 293a-c. (86%, rd 4:1:3) que foram separados por CLAE preparativa (Esquema 63). Em meio ácido, o acetal indesejado 293c foi convertido no acetal desejado 293b (rd 3:1, 87% em três ciclos). Após várias etapas, 293a foi convertido no aldeído 281 e 293b na etilcetona 282.
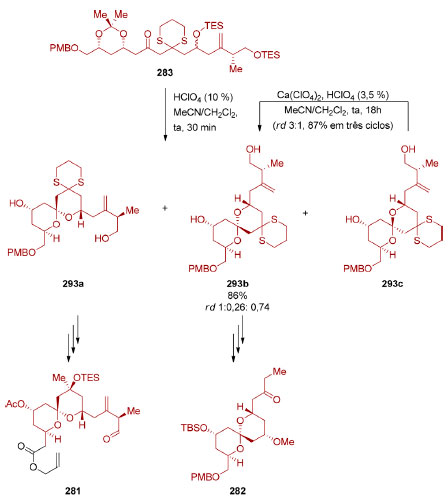 Esquema 63. Preparação de dois fragmentos da (+)-espongistatina 1 (278) a partir do mesmo intermediário avançado, por Ley e colaboradores203
Os dois fragmentos espirocíclicos 281 e 282 (Esquema 64) foram combinados através de uma reação aldólica anti-seletiva mediada por enolato de boro (rd 5:1), seguida por acetilação e desproteção (51%). O fragmento 280 foi então conectado ao fragmento altamente funcionalizado206 279 através de uma reação de olefinação de Wittig Z-seletiva (50%). Para completar a síntese, foram necessárias as desproteções dos éteres p-metoxibenzila (65%) e do éster alílico, seguida por macrolactonização de Yamaguchi (56%). A (+)-espongistatina 1 (278) foi finalmente obtida após a remoção dos grupos de proteção de silício em 57% de rendimento.
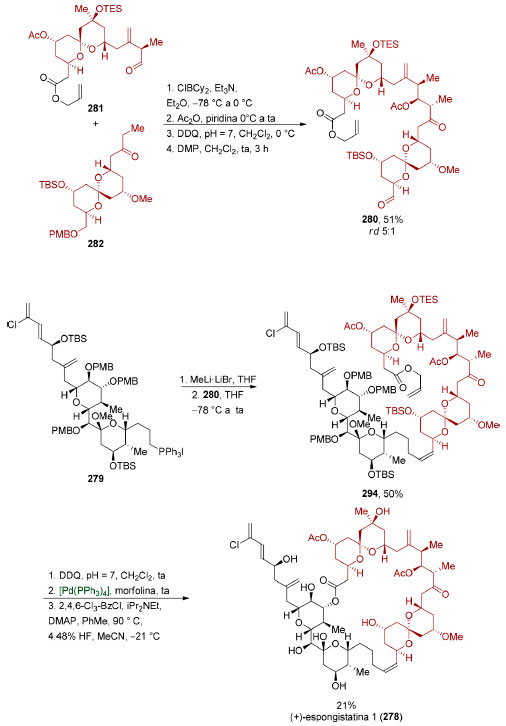 Esquema 64. Síntese convergente da (+)-espongistatina 1 (278), através do acoplamento de dois fragmentos sintetizados do mesmo intermediário avançado, por Ley e colaboradores203
Para fins de consistência conceitual, é preciso mencionar que Ley e colaboradores se referem à identificação de um pseudo-eixo C2 na estrutura da (+)-espongistatina 1 (278). Entretanto, na literatura de síntese orgânica o uso desse termo é tipicamente associado a um arranjo estereoquímico diferente.207 De acordo com nossa compreensão,208 uma estrutura de simetria pseudo-C2 se refere a um composto quiral que possui um centro proquiral ligado a dois grupos enantiotópicos (Figura 12).209,210 Essa disposição espacial211 foi utilizada não só em estratégias sintéticas212,213 como no planejamento e síntese de catalisadores.214 Para facilitar a compreensão desta disposição espacial, compostos contendo um eixo C2 (Figura 13A), um centro pseudo-assimétrico (Figura 13B) e um pseudo-eixo C2 (Figura 13C) estão representados lado a lado. Por definição, uma rotação em torno de um eixo C2 verdadeiro resulta em uma disposição espacial equivalente à original (Figura 13A). Diferente de um centro assimétrico, um composto contendo um carbono pseudo-assimétrico é meso devido a presença de um plano de simetria (Figura 13B). Ao se comparar com as estruturas das figuras 13A e 13B, o pseudo-eixo C2pode ser compreendido como o eixo que coincide com um eixo C2 hipotético que estaria presente caso o carbono sp3 proquiral não estivesse presente ou não fosse proquiral. Diferente de um eixo C2 verdadeiro, uma rotação em torno de um pseudo-eixo C2 resulta em uma disposição espacial não-equivalente à original, o que é perceptível ao se observar a posição dos substituintes no carbono proquiral.
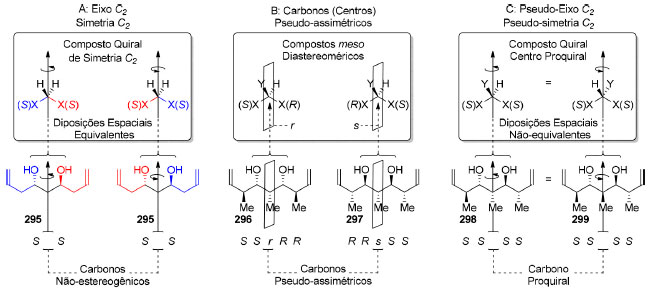 Figura 13. Comparação entre compostos contendo eixos C2 (A), centros pseudo-assimétricos (B) e pseudo-eixo C2 (C)
CONSIDERAÇÕES FINAIS O conceito de simetria encontra uma definição precisa no âmbito da Matemática e refere-se à propriedade de objetos que permanecem invariantes mediante uma transformação como reflexão em relação a um plano ou rotação em torno de um eixo. Essa definição refere-se tanto a objetos reais, como as moléculas orgânicas abordadas neste trabalho, como a objetos imaginários como as letras de nosso alfabeto (por exemplo, a letra "A" apresenta eixo de simetria C2 e planos de simetria (σv e σh) pertencendo ao grupo pontual C2v enquanto a letra "J" não tem eixo de simetria mas apenas um plano de simetria, podendo ser classificada como pertencente ao grupo pontual Cs). No âmbito da Química Orgânica, a relação entre simetria e quiralidade assume várias possibilidades distintas: 1) a molécula apresenta um único átomo de carbono sp3 com quatro substituintes diferentes (grupo pontual C1) e é quiral; 2) a molécula é simétrica como o trans-1,2-dimetilciclopropano (grupo pontual C2) e é quiral; e 3) a molécula é simétrica como o cis-1,2-dimetilciclopropano (grupo pontual Cs) e é aquiral. Um processo de dessimetrização estereosseletivo envolve a perda de um ou mais elementos de simetria de uma molécula (plano de simetria, centro de inversão ou eixo de rotação impróprio) e viabiliza a conversão de um composto proquiral ou meso em quiral. A incorporação de tais processos ao planejamento sintético constitui uma estratégia valiosa para a construção de compostos orgânicos enantiomericamente puros ou enriquecidos. Em compostos meso, um processo de dessimetrização permite estabelecer a estereoquímica de vários centros estereogênicos simultaneamente, mesmo que em sítios distantes do sítio reativo da molécula, aumentando assim a brevidade e a eficiência de uma rota sintética. A dessimetrização de um composto meso inverte a ordem tradicional de controle de centros estereogênicos, de modo que as etapas diastereosseletivas ocorrem no início da rota sintética. Em certo sentido, uma dessimetrização é um processo que se assemelha a uma resolução clássica que emprega um reagente quiral para converter uma mistura racêmica em uma mistura de diastereoisômeros para então viabilizar sua separação. No entanto, ao contrário de uma resolução clássica, o rendimento máximo teórico de uma dessimetrização é de 100%. Neste trabalho procuramos destacar a utilidade desses processos na síntese de produtos naturais e insumos farmacêuticos ativos de diferentes graus de complexidade. Vários desafios e oportunidades associados à dessimetrização são descritos, como o aproveitamento do glicerol (59) e do ácido isobutírico (71) e a dificuldade da dessimetrização de meso-piperazinas (190). Aplicações de produtos de dessimetrização são descritas para as sínteses totais de produtos naturais com estruturas relativamente complexas, como o Taxol® (121), a (-)-agelastatina (143), o (+)-neopeltolídeo (152) e a (+)-hemibrevetoxina (186) . A aplicação de reconhecimento de padrões em análise retrossintética foi exemplificada com as sínteses do Taxol® (121) e do (-)-Δ9-tetra-hidrocanabinol (202) partindo de monoterpenos acessíveis como materiais de partida. A constatação de simetria C2 na estrutura de produtos naturais permite realizar sínteses convergentes ou bidirecionais, como representado pelas sínteses totais do (+)-dixiamicina B (228), do (+)-testudinariol A (229) e do clavosolídeo A (239). Enquanto a desconexão de unidades monoméricas presentes em estruturas simétricas é uma estratégia auto-evidente de simplificação do planejamento sintético, o reconhecimento de elementos de simetria escondidos em moléculas com simetria C1 requer um nível mais elaborado de abstração cognitiva. Ao procurar reconhecer características comuns (ou sinteticamente equivalentes) em partes distintas de um alvo sintético, essa estratégia viabiliza sínteses convergentes nas quais dois fragmentos podem ser preparados a partir de um mesmo intermediário. Tal estratégia foi empregada nas sínteses totais da (±)-carpanona (247),do (−)-sarcandrolídeo J (252), do (−)-anfidinolídeo F (264) e da (+)-espongistatina 1 (278). Considerados em conjunto, os conceitos expostos neste trabalho têm a finalidade de evidenciar o potencial do reconhecimento de simetria e do uso de processos de dessimetrização a fim de agregar convergência e eficiência ao planejamento e à execução da síntese de moléculas orgânicas, ao mesmo tempo que procura expor desafios e oportunidades para expandir o emprego dessas estratégias na área de síntese de produtos naturais e interesse da indústria farmacêutica.
AGRADECIMENTOS Os autores expressam agradecimento à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) pelo apoio financeiro. (Processos Nº 2018/05742-9, 2018/04837-6, 2019/13104-5)
REFERÊNCIAS E NOTAS 1. Tavakoli, A.; Gisin, N.; Quantum 2020, 4, 293. 2. Lloyd, D. R.; Symmetry 2010, 2, 455. 3. Dunitz, J. D.; Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 14260. 4. Consoli, M.; Pluchino, A.; Michelson-Morley Experiments: An Enigma for Physics and the History of Science, 1st ed., World Scientific Publishing: Singapore, 2018. 5. Silvi, B.; Theoretical Chemistry Accounts (2017), doi:10.1007/s00214-017-2146-3. 6. Pauling, L.; The Nature of the Chemical Bond and the Structure of Molecules and Crystals, 1st ed., Cornell University Press: Cornell, 1939; Gillespie, R. J.; Robinson, E. A.; Chem. Soc. Rev. 2005, 34, 396. 7. Senn, H. M.; Thiel, W.; Angew. Chem., Int. Ed. 2009, 48, 1198. 8. Schwarzenbach, D.; Acta Crystallogr., Sect. A 2012, A68, 57. 9. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Introduction to Spectroscopy, 5th ed., Cengage Learning: Stamford, 2014. 10. Em 2017, Bizzini, Mayor e colaboradores sintetizaram o tetraciclo[5.2.2.01,6.04,9]-undecano, o "trinorbornano", de simetria C2. Durante anos, este esteve entre os três hidrocarbonetos saturados hipotéticos contendo 11 carbonos que ainda não haviam sido sintetizados: Bizzini, L. D.; Müntener, T.; Häussinger, D.; Neuburger, M.; Mayor, M.; Chem. Commun. 2017, 53, 11399. 11. Hoffmann, R.; Journal of Aestetics and Art Criticism 1990, 48, 191. 12. Ternansky, R. J.; Balogh, D. W.; Paquette, L. A.; J. Am. Chem. Soc. 1982, 104, 4503. 13. Doyle, M.; Parker, W.; Gunn, P.A.; Martin, J.; Macnicol, D. D.; Tetrahedron Lett. 1970, 42, 3619. 14. Sekine, Y.; Brown, M.; Boekelheide, V.; J. Am. Chem. Soc. 1979, 101, 3126. 15. Rohlfing, E. A; Cox, D. M.; Kaldor, A.; J. Chem. Phys. 1984, 81, 3322. 16. Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E.; Nature 1985, 318, 162. 17. Castro, E.; Garcia, A. H.; Zavala, G.; Echegoyen, L.; J. Mater. Chem. B 2017, 5, 6523. 18. Trauner, D.; Angew. Chem., Int. Ed. 2018, 57, 4177. 19. Benfrey, O. T.; Morris, P. J. T.; Robert Burns Woodward: Architect and Artist in the World of Molecules, 1st ed., Chemical Heritage Foundation, 2001. 20. Bishop, D. M.; Group Theory and Chemistry, 2nd ed., Dover Publications: Mineola, 1993; Kettle, S. A.; Symmetry and Structure: Readable Group Theory for Chemists, 3rd ed., John Wiley & Sons: West Sussex, 2007. 21. Lesk, A. M.; Introduction to Symmetry and Group Theory for Chemists, 1st ed., Kluwer Academic Publishers: Norwell, 2004; Wolfram, T.; Ellialttioglu, S.; Applications of Group Theory to Atoms, Molecules and Solids, 1st ed., Cambridge University Press: Cambridge, 2014. 22. Bunker, P. R.; Jensen, P.; Fundamentals of Molecular Symmetry, 1st ed., IOP Publishing: Bristol, 2005. 23. Housecroft, C. E.; Sharpe, A. G.; Inorganic Chemistry, 4th ed., Pearson: Harlow, 2012. 24. McGlinchey M. J.; Symmetry 2014, 6, 622. 25. Molloy, K. C.; Group Theory for Chemists: Fundamental Theory an Applications, 2nd ed., Woodhead Publishing: Sawston, 2011. 26. Fukui, K.; Yonezawa, T., Shingu, H.; J. Chem. Phys. 1952, 20, 722; Fukui, K.; Science 1982, 218, 747. 27. Woodward, R. B.; Hoffmann, R.; J. Am. Chem. Soc. 1965, 87, 395; Longuet-Higgins, H. C.; Abrahamson, E. W.; J. Am. Chem. Soc. 1965, 87, 2045; Woodward, R. B.; Hoffmann, R.; J. Am. Chem. Soc. 1965, 87, 2046; Hoffmann, R.; Woodward, R. B.; Acc. Chem. Res. 1968, 1, 17; Halevi, E. A.; Orbital Symmetry and Reaction Mechanism: The OCAMS View, 3rd ed., Springer-Verlag: Berlin, 1992. 28. Woodward, R. B.; Hoffmann, R.; Angew. Chem., Int. Ed. Engl. 1969, 8, 781; Geerlings, P.; Ayers, P. W.; Toro-Labbé, A.; Chattaraj, P. K.; De Proft, F.; Acc. Chem. Res. 2012, 45, 683. 29. Maier, G.; Pfriem, S.; Schäfer, U.; Matusch, R.; Angew. Chem., Int. Ed. Engl. 1978, 17, 520. 30. Maier, G.; Neudert, J.; Wolf, O.; Pappusch, D.; Sekiguchi, A.; Tanaka, M.; Matsuo, T.; J. Am. Chem. Soc. 2002, 124, 13819. 31. Eaton, P. E.; Cole, T. W.; J. Am. Chem. Soc. 1964, 86, 3157. 32. Reekie, T. A.; Williams, C. M.; Rendina, L. M.; Kassiou, M.; J. Med. Chem. 2019, 62, 1078. 33. Biegasiewicz, K. F.; Griffiths, J. R.; Savage, G. P.; Tsanaktsidis, J.; Priefer, R.; Chem. Rev. 2015, 115, 6719. 34. Cahn, R. S.; Ingold, C.; Prelog, V.; Angew. Chem., Int. Ed. Engl. 1966, 5, 385; Eliel, E. L.; Wilen, S. H.; Stereochemistry of Organic Compounds, 1st ed., Wiley: New York, 1994. 35. North, M.; Principles and Applications of Stereochemistry, 1st ed., CRC Press: Boca Raton, 2017. 36. Kelvin, W. T.; The Molecular Tactics of a Crystal, 1st ed., Claredon Press: Oxford, 1894. 37. Gal, J.; Helv. Chim. Acta 2013, 96, 1617. 38. Flack, H. D.; Acta Cryst. 2009, A65, 371. 39. Tobe, Y.; Mendeleev Commun. 2003, 13, 93. 40. Lewis, D. E.; Angew. Chem., Int. Ed. 2019, 58, 3694. 41. Kekulé, A.; Justus Liebigs Ann. Chem. 1857, 104, 129; Kekulé, A.; Justus Liebigs Ann. Chem. 1858, 106, 129. 42. Couper, A. S.; Justus Liebigs Ann. Chem. 1859, 110, 46. 43. Butlerov, A.; Justus Liebigs Ann. Chem. 1859, 110, 51; Hargittai, I.; Struct. Chem. 2011, 22, 243. 44. van't Hoff, J. H.; Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445. 45. Le Bel, J. A.; Bull. Chim. Soc. Fr. 1874, 22, 337. 46. Fletcher S. P.; Nat. Chem. 2009, 1, 692. 47. Bada, J. L.; Nature 1995, 374, 594; Wagner, A. J.; Blackmond D. G.; ACS Cent. Sci. 2016 2, 775; Hawbaker, N. A.; Blackmond, D. G.; Nat. Chem. 2019, 11, 957. 48. Mo, Y.; Wires Comput. Mol. Sci. 2011, 1, 164. 49. Lesot, P.; Luz, Z.; Aroulanda, C.; Zimmermann, H.; Magn. Reson. Chem. 2014, 52, 581; Lesot, P.; Aroulanda, C.; Zimmermann, H.; Luz, Z.; Chem. Soc. Rev. 2015, 44, 2330. 50. Longuet-Higgins H. C.; Mol. Phys. 1963, 6, 445; Altmann, S. L.; Proc. R. Soc. London, Ser. A 1967, 298, 184. 51. Gilson, M. K.; Irikura, K. K.; J. Phys. Chem. B 2010, 114, 16304. 52. de Oliveira, G. M.; Simetria de Moléculas e Cristais: Fundamentos da Espectroscopia Vibracional, 1I ed., Bookman: Porto Alegre, 2009. 53. Ercolani, G.; J. Chem. Educ. 2005, 82, 1703. 54. Liu, S.; J. Phys. Chem. A 2013, 117, 962. 55. Pophristic, V.; Goodman, L.; Nature 2001, 411, 565. 56. Schreiner, P. R.; Angew. Chem., Int. Ed. 2002, 41, 3579. 57. Pendás, A. M.; Blanco, M. A.; Francisco, E.; J. Comp. Chem. 2009, 30, 98. 58. Dantus, M.; Bowman, R. M.; Zewail, A. H.; Nature 1990, 343, 737. 59. Liu, S.; Schauer, C. K.; J. Chem. Phys. 2015, 142, 054107. 60. Yang, M. T.; Woerpel, K. A.; J. Org. Chem. 2009, 74, 545; Siebler, C.; Maryasian, B.; Kuemin, M.; Erdmann, R. S.; Rigling, C.; Grünenfelder, C.; Ochsenfeld, C.; Wennemers, H.; Chem. Sci. 2015, 6, 6725. 61. Villa, M.; Senent, M. L.; Carvajal, M.; Phys. Chem. Chem. Phys. 2013, 15, 10258; Perrin, A.; Flaud, J. M.; Kwabia-Tchana, F.; Manceron, L.; Groner, P.; Mol. Phys. 2019, 117, 323; Drouin, B. J.; Pearson, J. C.; Walters, A.; Lattanzi, V.; J. Mol. Spectrosc. 2006, 240, 227. 62. Testa, B.; Vistoli. G.; Pedretti, A.; Helv. Chim. Acta 2013, 96, 564. 63. Murcko, M. A.; Castejon, H.; Wiberg, K. B.; J. Phys. Chem. 1996, 100, 16162. 64. Stojanović, M.; Aleksić, J.; Baranac-Stojanović, M.; Tetrahedron 2015, 71, 5119. 65. Lüttschwager, N. O. B.; Wassermann, T. N.; Mata, R. A.; Suhm, M. A.; Angew. Chem., Int. Ed. 2013, 52, 463. 66. Tóbiás, R.; Császár, A. G.; Gyevi-Nagy, L.; Tasi, G.; J. Comput. Chem. 2018, 39, 424. 67. Balabin, R. M.; J. Phys. Chem. A 2009, 113, 1012. 68. Blake, T. A.; Xantheas, S. S.; J. Phys. Chem. A 2006, 110, 10487. 69. Li, H.; Miller, C. C.; Philips, L. A.; J. Chem. Phys. 1994, 100, 8590. 70. Kummli, D. S.; Frey, H. M.; Leutwyler, S.; J. Phys. Chem. A 2007, 111, 11936. 71. Kowalewski, P.; Frey, H.-M.; Infanger, D.; Leutwyler, S.; J. Phys. Chem. A 2015, 119, 11215. 72. Ocola, E. J.; Bauman, L. E.; Laane, J.; J. Phys. Chem. A 2011, 115, 6531. 73. Brügger, G.; Frey, H.-M.; Steinegger, P.; Balmer, F.; Leutwyler, S.; J. Phys. Chem. A 2011, 115, 9567. 74. Kang, Y. K.; Park, H. S.; Chem. Phys. Lett. 2018, 702, 82. 75. Hasha, D. L.; Eguchi, T.; Jonas, J.; J. Am. Chem. Soc. 1982, 104, 2290. 76. Nelson, D. J.; Brammer, C. N.; J. Chem. Educ. 2011, 88, 292. 77. O'Leary, B. M.; Grotzfeld, R. M.; Rebek, J.; J. Am. Chem. Soc. 1997, 119, 11701. 78. Alabugin, I. V.; J. Org. Chem. 2000, 65, 3910; Weldon, A. J.; Vickrey, T. L.; Tschumper, G. S.; J. Phys. Chem. A 2005, 109, 11073. 79. IUPAC; Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"); McNaught, A. D., Wilkinson, A., comp.; Blackwell Scientific Publications: Oxford, 1997. Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. 80. McClymont, K. S.; Wang, F.-Y.; Minakar, A.; Baran, P. S.; J. Am. Chem. Soc. 2020, 142, 8608. 81. Sharpe, R. J.; Johnson, J. S.; J. Am. Chem. Soc. 2015, 137, 4968. 82. Nilewski, C.; Deprez, N. R.; Fessard, T. C.; Li, D. B.; Geisser, R. W.; Carreira, E. M.; Angew. Chem., Int. Ed. 2011, 50, 7940. 83. Poss, C. S.; Schreiber, S. L.; Acc. Chem. Res. 1994, 27, 9. 84. Lu, Y.; Woo, S. K.; Krische, M. J.; J. Am. Chem. Soc. 2011, 133, 13876. 85. Novaes, L. F. T.; Sarotti, A. M.; Pilli, R. A.; J. Org. Chem. 2015, 80, 12027. 86. Merad, J.; Candy, M.; Pons, J.-M.; Bressy, C.; Synthesis 2017, 49, 1938. 87. Historicamente, o conceito de excesso enantiomérico (ee) foi designado para estabelecer a pureza ótica de um determinado composto, diretamente relacionado às quantidades relativas entre os enantiômeros presentes na amostra. Entretanto, esta medida apresenta inconveniências quando aplicada à estereosseletividade. Do ponto de vista analítico, as quantidades relativas entre dois enantiômeros não são mais determinadas utilizando-se polarímetros. O método moderno de determinação consiste na separação analítica dos enantiômeros em uma coluna cromatográfica (fase estacionária quiral) que permite obter as razões entre os enantiômeros; Gawley, R. E.; J. Org. Chem. 2006, 71, 2411. 88. Rosen, T.; Heathcock, C. H.; J. Am. Chem. Soc. 1985, 107, 3731; Heathcock, C. H.; Hadley, C. R.; Rosen, T.; Theisen, P. D.; Hecker, S. J.; J. Med. Chem. 1987, 30, 1858. 89. Brown, A. G.; Smale, T. C.; King, T. J.; Hasenkamp, R.; Thompson, R. H.; J. Chem. Soc., Perkin Trans. 1. 1976, 1165; Endo, A.; Kuroda, M.; Tsujita, Y.; J. Antibiot. 1976, 29, 1346. 90. Alberts, A. W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; Patchett, A.; Monaghan, R.; Currie, S.; Stapley, E.; Albers-Schonberg, G.; Hensens, O.; Hinshfield, J.; Hoogsteen, K.; Liesch, J.; Springer, J.; Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 3957. 91. Scandinavian Simvastatin Survival Group; Lancet 1994, 344, 1383. 92. Bergeron, S.; Chaplin, D. A.; Edwards, J. H.; Ellis, B. S. W.; Hill, C. L.; Holt-Tiffin, K.; Knight, J. R.; Mahoney, T.; Osborne, A. P.; Ruecroft, G.; Org. Process Res. Dev. 2006, 10, 661. 93. Lessons from Lipitor and the broken blockbuster drug model, Editorial, Lancet 2011, 378, 1976. 94. Umpierre, A.; Machado, F.; Rev. Virtual Quim. 2013, 5, 106. 95. Beatriz, A.; Araújo, Y. J. K., de Lima, D. P.; Quim. Nova 2011, 34, 306. 96. Mandai, H.; Ashihara, K.; Mitsudo, K.; Suga, S.; Chem. Lett. 2018, 47, 1360. 97. You, Z.; Hoveyda, A. H.; Snapper, M. L.; Angew. Chem., Int. Ed. 2009, 48, 547. 98. Sakakura, A.; Umemura, S.; Ishihara, K.; Adv. Synth. Catal. 2011, 353, 1938; Lewis, C. A.; Sculimbrene, B. R.; Xu, Y.; Miller, S. J.; Org. Lett. 2005, 7, 3021. 99. Batovska, D. I.; Tsubota, S.; Kato, Y.; Asano, Y.; Ubukata, M.; Tetrahedron Asymmetry 2004, 15, 3551. 100. Giustra, Z. X.; Tan, K. L.; Chem. Commun. 2013, 49, 4370. 101. Hasegawa, J.; Ogura, M.; Hamaguchi, S.; Shimazaki, M.; Kawaharada, H.; Watanabe, K.; J. Ferment. Technol. 1981, 59, 203. 102. Shimazaki, M.; Hasegawa, J.; Kan, K.; Nomura, K.; Nose, Y.; Kondo, H.; Ohashi, T.; Watanabe, K.; Chem. Pharm. Bull. 1982, 30, 3139. 103. Ng, K. K. F.; Vane, J. R.; Nature 1967, 216, 762. 104. Ferreira, S. H.; Brit. J. Pharmacol. 1965, 24, 163. 105. Goodhue, C. T.; Schaeffer, J. R.; Biotechnol. Bioeng. 1971, 13, 203. 106. Mickel, S. J.; Sedelmeier, G. H.; Niederer, D.; Schuerch, F.; Grimler, D.; Koch, G.; Daeffler, R.; Osmani, A.; Hirni, A.; Schaer, K.; Gamboni, R.; Bach, A.; Chaudhary, A.; Chen, S.; Chen, W.; Hu, B.; Jagoe, C. T.; Kim, H.-Y.; Kinder, F. R., Jr.; Liu, Y.; Lu, Y.; McKenna, J.; Prashad, M.; Ramsey, T. M.; Repic, O.; Rogers, L.; Shieh, W.-C.; Wang, R.-M.; Waykole, L.; Org. Proc. Res. Dev. 2004, 8, 101. 107. Wu, Q.-F.; Shen, P.-X.; He, J.; Wang, X.-B.; Zhang, F.; Shao, Q.; Zhu, R.-Y.; Mapelli, C.; Qiao, J. X.; Poss, M. A.; Yu, J,-Q.; Science 2017, 355, 499. 108. Oliveira, J. M.; Angnes, R. A.; Khan, I. U.; Polo, E. C.; Heerdt, G.; Servilha, B. M.; da Silva, V. H. M.; Braga, A. A. C.; Correia, C. R. D.; Chem. Eur. J. 2018, 24, 11738. 109. Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H.; Nature 2008, 456, 933. 110. Aggarwal, V. K.; Olofsson, B.; Angew. Chem., Int. Ed. 2005, 44, 5516. 111. Yamagata, A. D. G.; Datta, S.; Jackson, K. E.; Stegbauer, L.; Paton, R. S.; Dixon, D. J.; Angew.Chem. Int. Ed. 2015, 54, 4899. 112. Gallier, F.; Martel, A.; Dujardin, G.; Angew. Chem., Int. Ed. 2017, 56, 12424. 113. Bui, T.; Barbas, C. F., III; Tetrahedron Lett. 2000, 41, 6951. 114. Eder, U.; Sauer, G.; Wiechert, R.; Angew. Chem., Int. Ed. Engl. 1971, 10, 496; Hajos, Z. G.; Parrish D. R.; J. Org. Chem. 1974, 39, 1615. 115. Lee, H. M.; Nieto-Oberhuber, C.; Shair M. D.; J. Am. Chem. Soc. 2008, 130, 16864. 116. Tang, Y.; Liu, J.-t.; Chen, P.; Lv, M.-C.; Wang, Z.-Z.; Huang, Y.-K.; J. Org. Chem. 2014, 79, 11729. 117. Hog, D. T.; Huber, F. M. E.; Jiménez-Osés, G.; Mayer P.; Houk, K. N.; Trauner, D.; Chem. Eur. J. 2015, 21, 13646. 118. Reddy, T. J.; Bordeau, G.; Trimble, L.; Org. Lett. 2006, 8, 5585. 119. Para mais exemplos, ver: Eagan, J. M.; Hori, M.; Wu, J.; Kanyiva, K. S.; Snyder, S. A.; Angew. Chem., Int. Ed. 2015, 54, 7842. 120. List, B. Hoang, L.; Martin, H. J.; Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5839. 121. List, B.; Angew. Chem., Int. Ed. 2010, 49, 1730. 122. Bahmanyar, S.; Houk, K. N.; J. Am. Chem. Soc. 2001, 123, 12911; Clemente, F. R.; Houk, K. N.; Angew. Chem., Int. Ed. 2004, 43, 576. 123. Zhu, H.; Clemente, F. R.; Houk, K. N.; Meyer, M. P.; J. Am. Chem. Soc. 2009, 131, 1632. 124. Smith, A. B., III; Kanoh, N.; Ishiyama, H.; Minakawa, N.; Rainier, J. D.; Hartz, R. A.; Cho, Y. S.; Cui, H.; Moser, W. H.; J. Am. Chem. Soc. 2003, 125, 8228. 125. Harada, N.; Sugioka, T.; Ando, Y.; Uda, H.; Kuriki, T.; J. Am. Chem. Soc. 1988, 110, 8483. 126. Grieco, P. A.; Collins, J. L.; Moher, E. D.; Fleck, T. J.; Gross, R. S.; J. Am. Chem. Soc. 1993, 115, 6078. 127. Waters, S. P.; Tian, Y.; Li, Y.-M; Danishefsky, S. J.; J. Am. Chem. Soc. 2005, 127, 13514. 128. Masters, J. J.; Link, J. T.; Snyder, L. B.; Young, W. B.; Danishefsky, S. J.; Angew. Chem., Int. Ed. Engl. 1995, 34, 1723. 129. Danishefsky, S. J.; Masters, J. J.; Young, W. B.; Link, J. T.; Snyder, L. B.; Magee, T. V.; Jung, D. K.; Isaacs, R. C. A.; Bornmann, W. G.; Alaimo, C. A.; Coburn, C. A.; Di Grandi, M. J.; J. Am. Chem. Soc. 1996, 118, 2843. 130. Bolm, C.; Schiffers, I.; Dinter, C. L.; Gerlach, A.; J. Org. Chem. 2000, 65, 6984. 131. Seebach, D., Jaeschke, G., Wang, Y. M.; Angew. Chem., Int. Ed. Engl. 1995, 34, 2395; Jaeschke, G. Seebach, D.; J. Org. Chem. 1998, 63, 1190. 132. Chen, T-G.; Barton, L. M.; Lin, Y.; Tsien, J.; Kossler, D.; Bastida, I.; Asai, S.; Bi, C.; Chen, J. S.; Shan, M.; Fang, H.; Fang, F. G.; Choi, H-w.; Hawkins, L.; Qin, T.; Baran, P. S.; Nature 2018, 560, 350. 133. Anugu, R. R.; Mainkar, P. S.; Sridhar, B.; Chandrasekhar, S.; Org. Biomol. Chem. 2016, 14, 1332. 134. Trost, B. M.; Dong, G.; J. Am. Chem. Soc. 2006, 128, 6054. 135. Trost, B. M.; Dong, G.; Chem. Eur. J. 2009, 15, 6910. 136. Yu, M.; Schrock, R. R.; Hoveyda, A. H.; Angew. Chem., Int. Ed. 2015, 54, 215. 137. Johnson, C. R.; Plé, P. A.; Adams, J. P.; J. Chem. Soc., Chem. Commun. 1991, 1006. 138. Hamrock, S. J.; Sheridan, R. S.; J. Am. Chem. Soc. 1989, 111, 9247. 139. Hernandez, L. W.; Pospech, J.; Klöckner, U.; Bingham, T. W.; Sarlah, D.; J. Am. Chem. Soc. 2017, 139, 15656. 140. Apesar desse processo ser altamente estereosseletivo e escalonável, as condições reacionais foram otimizadas para poder utilizar um precursor de níquel estável ao ar e diminuir a carga de catalisador: Hernandez, L. W.; Klöckner, U.; Pospech, J.; Hauss, L.; Sarlah, D.; J. Am. Chem. Soc. 2018, 140, 4503. 141. Tian, X.; Hudlicky, T.; Koenigsberger, K.; J. Am. Chem. Soc. 1995, 117, 3643. 142. Chandler, C. L.; List, B.; J. Am. Chem. Soc. 2008, 130, 6737. 143. Wender, P. A.; Correia, C. R. D.; J. Am. Chem. Soc. 1987, 109, 2523; Mehta, G.; Rao, K. S.; J. Am. Chem. Soc. 1986, 108, 8015; Whitesell, J. K.; Matthews, R. S.; Solomon, P. A.; Tetrahedron Lett. 1976, 17, 1549. 144. Anstiss, M.; Holland, J. M.; Nelson, A.; Titchmarsh, J. R.; Synlett 2003, 1213. 145. Holland, J. M.; Lewis, M.; Nelson, A.; Angew. Chem., Int. Ed. 2001, 40, 4082; Holland, J. M.; Lewis, M.; Nelson, A.; J. Org. Chem. 2003, 68, 747. 146. Schneider, C.; Synthesis 2006, 3919. 147. Delcourt, M.-L.; Felder, S.; Benedetti, E.; Micouin, L.; ACS Catal. 2018, 8, 6612. 148. Anstiss, M.; Nelson, A.; Org. Biomol. Chem. 2006, 4, 4135. 149. Calderon, S. N.; Rice, K. C.; Rothman, R. B.; Porreca, F.; Flippen-Anderson, J. L.; Xu, H.; Becketts, K.; Smith, L. E.; Bilsky, E. J.; Davis, P.; Horvath, R.; J. Med. Chem. 1997, 40, 695; Katsura, Y.; Zhang, X.; Homma, K.; Rice, K. C.; Calderon, S. N.; Rothman, R. B.; Yamamura, H. I. Davis, P.; Flippen-Anderson, J. L.; Xu, H.; Becketts, K.; Foltz, E. J.; Porreca, F.; J. Med. Chem. 1997, 40, 2936. 150. Wilson, R. M.; Danishefsky, S. J.; J. Org. Chem. 2007, 72, 4293; Gerlinger, C. K. G.; Gaich, T.; Chem. Eur. J. 2019, 25, 10782. 151. A síntese do (±)-longifoleno por Corey e colaboradores em 1961 é considerada o primeiro exemplo de aplicação explícita da análise retrossintética na síntese de produtos naturais: Corey, E. J.; Ohno, M.; Vatakencherry, P. A.; Mitra, R. B.; J. Am. Chem. Soc. 1961, 83, 1251; Em 1917, em sua síntese total da tropinona, Robinson introduziu o conceito de "hidrólise imaginária". Este pode ser visto como um precedente para o conceito de desconexão: Robinson, R.; J. Chem. Soc., Trans. 1917, 111, 762. 152. Corey, E. J.; Cheng, X.-M.; The Logic of Chemical Synthesis,1st ed., John Wiley: New York, 1989. De acordo com a filosofia de Síntese desenvolvida por Corey, a análise retrossintética convencional à qual nos referimos se encaixaria na lógica associada ao "target-goal". Já a estratégia de reconhecimento de padrões, descrita por Danishefsky, e parcialmente adotada desde os tempos da semissíntese, seria a estratégia que Corey definiu como "substrate-goal". 153. Afewerki, S.; Wang, J.-X.; Liao, W.-W.; Córdova, A.; The Alkaloids: Chemistry and Biology 2019, 81, 151. 154. Koskinen, A. M. P.; Karisalmi, K.; Chem. Soc. Rev. 2005, 34, 677. 155. Ruzicka, L.; Experientia 1953, 9, 357. 156. Croteau, R.; Ketchum, R. E. B.; Long, R. M.; Kaspera, R.; Wildung, M. R.; Phytochem. Rev. 2006, 5, 75. 157. Brill, Z. G.; Condakes, M. L.; Ting, C. P.; Maimone, T. J.; Chem. Rev. 2017, 117, 11753. 158. Harmange, C. S.; Thach, D. Q.; Haelsig, K. T.; Maimone, T.; J.; Acc. Chem. Res. 2020, 53, 949. 159. Caprio, V.; Williams, J. M. J.; Catalysis in Asymmetric Synthesis, 2nd ed., Wiley: Hoboken, 2009. 160. Wender, P. A.; Badham, N. F.; Conway, S. P.; Floreancig, P. E.; Glass, T. E.; Houze, J. B.; Krauss, N. E.; Lee, D.; Marquess, D. G.; McGrane, P. L.; Meng, W.; Natchus, M. G.; Shuker, A. J.; Sutton, J. C.; Taylor, R. E.; J. Am. Chem. Soc. 1997, 119, 2757. 161. Mechoulam, R.; Braun, P.; Gaoni, Y.; J. Am. Chem. Soc. 1967, 89, 4552. 162. Em 2020, a Anvisa concedeu autorização para a comercialização de fitofármaco contendo até 0,2% de THC, além de outros canabinoides como o canabidiol e que pode ser prescrito para o tratamento de algumas doenças neurodegenerativas depois de esgotadas outras opções terapêuticas: https://www.gov.br/anvisa/pt-br/assuntos/noticias-anvisa/2020/anvisa-autoriza-primeiro-produto-a-base-de-cannabis, acessada em outubro 2020. 163. Malkov, A. V.; Kocovsky, P.; Collect. Czech. Chem. Commun. 2001, 66, 1257. 164. Limberger, R. P.; Aleixo, A. M.; Fett-Neto, A. G.; Henriques, A. T.; Electron. J. Biotechnol. (2007), doi: 10.2225/vol10-issue4-fulltext-8. 165. Bell, S. G.; Chen, X.; Sowden, R. J.; Xu, F.; Williams, J. N.; Wong, L.-L.; Rao, Z.; J. Am. Chem. Soc. 2003, 125, 705. 166. Pires, T. C. M.; Ribeiro, M. G. T. C.; Machado, A. A. S. C.; Quim. Nova 2018, 41, 355. 167. Erpen, L.; Muniz, F. R.; Moraes, T. S.; Tavano, E. C. R.; Rev. iPecege 2018, 4, 33. 168. Dethe, D. H.; Dherange, B. D.; J. Org. Chem. 2018, 83, 3392. 169. Hagiwara, H.; Nat. Prod. Commun. 2016, 11, 1391. 170. Peng, F.; Grote, R. E.; Wilson, R. M.; Danishefsky, S. J.; Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10904. 171. Peng, F.; Dai, M.; Angeles, A. R.; Danishefsky, S.; J.; Chem. Sci. 2012, 3, 3076. 172. Danishefsky, S.; Kitahara, T.; Schuda, P. F.; Etheredge, S. J.; J. Am. Chem. Soc. 1976, 98, 3028; Danishefsky, S.; Schuda, P. F.; Kitahara, T.; Etheredge, S. J.; J. Am. Chem. Soc. 1977, 99, 6066. 173. Reed, J. W.; Hudlicky, T.; Acc. Chem. Res. 2015, 48, 674. 174. Trauner, D.; Angew. Chem., Int. Ed. 2018, 57, 4177. 175. Link, J. T.; Overman, L. E.; J. Am. Chem. Soc. 1996, 118, 8166. 176. Hendrickson, J. B.; Göschke, R.; Rees, R.; Tetrahedron 1964, 20, 565. 177. Hall, E. S.; McCapra, F.; Scott, A. I.; Tetrahedron 1967, 23, 4131. 178. Movassaghi, M.; Schmidt, M. A.; Angew. Chem., Int. Ed. 2007, 46, 3725. 179. Voloshchuk, T.; Farina, N. S.; Wauchope, O. R.; Kiprowska, M.; Haberfield, P.; Greer, A.; J. Nat. Prod. 2004, 67, 1141. 180. Vrettou, M.; Gray, A. A.; Brewer, A. R. E.; Barrett, A. G. M.; Tetrahedron 2007, 63, 1487. 181. Rosen, B. R.; Werner, E. W.; O'Brien, A. G.; Baran, P. S.; J. Am. Chem. Soc. 2014, 136, 5571. 182. Para o isolamento, ver: Spinella, A.; Mollo, E.; Trivellone, E.; Cimino, G.; Tetrahedron 1997, 53, 16891; Para a síntese total, ver: Amarasinghe, K. K. D.; Montgomery, J.; J. Am. Chem. Soc. 2002, 124, 9366. 183. Para o isolamento, ver: Moore, B. S.; Chen, J. L.; Patterson, G. M. L.; Moore, R. E.; Brinen, L. S.; Kato, Y.; Clardy, J.; J. Am. Chem. Soc. 1990, 112, 4061; Moore, B. S.; Chen, J.-L.; Patterson, G. M. L.; Moore, R. E.; Tetrahedron 1992, 48, 3001; Bobzin, S. C.; Moore, R. E.; Tetrahedron 1993, 49, 7615. 184. Para a síntese total, ver: Smith, A. B., III; Kozmin, S. A.; Adams, C. M.; Paone, D. V.; J. Am. Chem. Soc. 2000, 122, 4984. 185. Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M.; Bull. Chem. Soc. Jpn. 1979, 52, 1989. 186. Para o isolamento, ver: Pereira, A. R.; McCue, C. F.; Gerwick, W. H.; J. Nat. Prod. 2010, 73, 217; Para a síntese total, ver: Waldeck, A. R.; Krische, M. J.; Angew. Chem., Int. Ed. 2013, 52, 4470. 187. Segundo a IUPAC, dimerização é uma transformação de uma entidade molecular A para fornecer a entidade molecular A2, ou seja, uma reação de adição aonde todos os átomos do material de partida devem ser encontrados no produto. Como, neste caso, nem todos os átomos são conservados no produto final, mas este conserva uma boa semelhança com o material de partida, optamos por colocar a expressão dimerização entre aspas. https://goldbook.iupac.org/terms/view/D01744, acessada em abril 2021. 188. Para o isolamento, ver: Rao, M. R.; Faulkner, D. J.; J. Nat. Prod. 2002, 65, 386; Erickson, K. L.; Gustafson, K. R.; Pannell, L. K.; Beutler, J. A.; Boyd, M. R.; J. Nat. Prod. 2002, 65, 1303; Para a síntese total, ver: Haydl, A. M.; Breit, B.; Angew. Chem. Int. Ed. 2015, 54, 15530. 189. Neumeyer, M.; Brückner, R.; Angew. Chem., Int. Ed. 2017, 56, 3383. 190. Chapman, O. L.; Engel, M. R.; Springer, J. P.; Clardyy, J. C.; J. Am. Chem. Soc. 1971, 93, 6696. 191. Matsumoto, M.; Kuroda, K.; Tetrahedron Lett. 1981, 22, 4437. 192. Daniels, R. N.; Fadeyi, O. O.; Lindsley, C. W.; Org. Lett. 2008, 10, 4097. 193. Liron, F.; Fontana, F.; Zirimwabagabo, J.-O.; Prestat, G.; Rajabi, J.; Rosa, C. L.; Poli, G.; Org. Lett. 2009, 11, 4378. 194. Neuhaus, W. C.; Kozlowski M. C.; Chem. Asian J. 2020, 15, 1039. 195. Baxendale, I. R.; Lee, A.-L.; Ley, S. V.; J. Chem. Soc. Perkin Trans. 1 2002, 1850. 196. Lindsley, C. W.; Chan, L. K.; Goess, B. C.; Joseph, R.; Shair, M. D.; J. Am. Chem. Soc. 2000, 122, 422. 197. Goess, B. C.; Hannoush, R. N.; Chan, L. K.; Kirchhausen, T.; Shair, M. D.; J. Am, Chem. Soc. Rev. 2006, 128, 5391. 198. Yuan, C.; Du, B.; Deng, H.; Man, Y.; Liu, B.; Angew. Chem., Int. Ed. 2017, 56, 637. 199. Yuan, C.; Du, B.; Yang, L.; Liu, B.; J. Am. Chem. Soc. 2013, 135, 9291. 200. Yang, Y.; Li, J.; Du, B.; Yuan, C.; Liu, B.; Qin, S.; Chem. Commun. 2015, 51, 6179. 201. Mahapatra, S.; Carter, R. G.; Angew. Chem., Int. Ed. 2012, 51, 7948. 202. Mahapatra, S.; Carter, R. G.; J. Am. Chem. Soc. 2013, 135, 10792. 203. Ball, M.; Gaunt, M. J.; Hook, D. F.; Jessiman, A. S.; Kawahara, S.; Orsini, P.; Scolaro, A.; Talbot, A. C.; Tanner, H. R.; Yamanoi, S.; Ley, S. V.; Angew. Chem., Int. Ed. 2005, 44, 5433. 204. Gaunt, M. J.; Jessiman, A. S.; Orsini, P.; Tanner, H. R.; Hook, D. F.; Ley, S. V.; Org. Lett. 2003, 5, 4819. 205. Gaunt, M. J.; Sneddon, H. F.; Hewitt, P. R.; Orsini, P.; Hook, D. F.; Ley, S. V.; Org. Biomol. Chem. 2003, 1, 15. 206. Kraus, H.; Français, A.; O'Brien, M.; Frost, J.; Diéguez-Vázquez, A.; Polara, A.; Baricordi, N.; Horan, R.; Hsu, D.-S.; Tsunoda, T.; Ley, S. V.; Chem. Sci. 2013, 4, 1989. 207. Para uma perspectiva mais ampla de pseudosimetria, ver: Glaser, R.; Symmetry,Spectroscopy and Crystallography: The Structural Nexus, 1st ed., Wiley: Hoboken, 2015. 208. O termo simetria pseudo-C2 tem sido amplamente utilizado na literatura de forma inconsistente. Para exemplos onde simetria pseudo-C2é chamada de simetria C2, ver: Erickson, J.; Neidhart, D. J.; VanDrie, J.; Kempf, D. J.; Wang, X. C.; Norbeck, D. W.; Plattner, J. J.; Rittenhouse, J. W.; Turon, M.; Wideburg, N.; Kohlbrenner, W. E.; Simmer, R.; Helfrich, R.; Paul, D. A.; Knigge, M.; Science 1990, 249, 527; Randolph, J. T.; Voight, E. A.; Greszler, S. N.; Uno, B. E.; Newton, J. N.; Gleason, K. M.; Stolarik, D.; Handel, C. V.; Bow, D. A. J.; DeGoey, D. A.; J. Med. Chem. 2020, 63, 11034. Para referências onde simetria pseudo-C2 foi atribuída a compostos sem um pseudoeixo-C2, ver: Perron, Q.; Alexakis, A.; Tetrahedron Asymmetry 2007, 18, 2503; Bouhrara, M.; Jeanneau, E.; Veyre, L.; Copéret, C.; Thieuleux, C.; Dalton Trans. 2011, 40, 2995; Ma, D.-Y.; Xiao, Z.-Y.; Etxabe, J.; Wärnmark, K.; ChemCatChem 2012, 4, 1321; Borrego, L. G.; Recio, R.; Álvarez, E.; Sánchez-Coronilla, A.; Khiar, N.; Fernández, I.; Org. Lett. 2019, 21, 6513. 209. Gao, X.; Han, H.; Krische, M. J.; J. Am. Chem. Soc. 2011, 133, 12795. 210. Kasun, Z. A.; Gao, X.; Lipinski, R. M.; Krische, M. J.; J. Am. Chem. Soc. 2015, 137, 8900. 211. Yamazaki e colaboradores se referem a pseudo-simetria C2 ao descrever um composto que nesta revisão seria classificado como proquiral sp3: Inoue, Y.; Hatayama, T.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T.; Yamazaki, T.; Chem. Commun. 2018, 54, 9913. 212. Tanis, P. S.; Infantine, J. R.; Leighton, J. L.; Org. Lett. 2013, 15, 5464. 213. Ishikawa, T.; Kawasaki-Takasuka, T.; Kubota, T.; Yamazaki, T.; Beilstein J. Org. Chem. 2017, 13, 2473. 214. Whitesell, J. K.; Chem. Rev. 1989, 89, 1581.
#Dedicamos esse artigo à memória do prof. José Augusto Rosário Rodrigues |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access