
 |
 |
 |
 |
 |
Nota Técnica
| Development and validation of a fast and simple hplc-uv method to determine caffeine in guarana (Paullinia cupana) food supplements |
|
Júlia Cecília Pereira Coura*; Cláudia Aparecida de Oliveira e Silva; Edvane Santos Silva; Sara Araújo Valladão; Maria Gorette Resende Duarte
Divisão de Vigilância Sanitária e Ambiental, Instituto Octávio Magalhães, Laboratório Central de Saúde Pública de Minas Gerais, Fundação Ezequiel Dias, 30510-010 Belo Horizonte - MG, Brasil Recebido em 09/02/2021 *e-mail: julia.coura@funed.mg.gov.br Caffeine and guarana are safe foods according to the American (FDA) and Brazilian (ANVISA) health agencies. However, data regarding the composition, quality, and safety of guarana-based food supplements sold in Brazil are limited. Most of the methods used for quantification of caffeine and other guarana chemical markers are based on complex extraction techniques as well as on gradient elution and do not evaluate the matrix effect nor the uncertainty estimation measurement. A simple and selective method for caffeine detection has been developed and validated using HPLC-UV. It shows linearity between 1 and 10 μg mL-1, has a significant matrix effect (p < 0.05) and its expanded uncertainty varies from 6.9 to 16.7%. Other parameters (selectivity, recovery, precision, robustness, limits of detection, and quantification) were satisfactory. The present study has analyzed 30 commercial samples of guarana-based food supplements (powders and capsules). Powder samples have shown an average caffeine level of 25.27 ± 5.20 mg g-1 while capsules 28.53 ± 13.81 mg g-1. No significant difference between the two types of samples has been observed (p > 0.005). INTRODUCTION Regular physical activity is recommended as one of the necessary measures to preserve as well as enhance human health and quality of life, besides helping prevent non-transmissible chronic diseases, such as diabetes and high blood pressure.1 A significant improvement in body composition is also associated with adequate eating habits, a determining factor to achieve satisfactory performance and adaptation to physical activity. The use of ergogenic resources contributes to the increase in power and physical endurance. Among such resources stand out those of a nutritional character such as caffeine-based food supplements.2-4 Caffeine belongs to the purine alkaloid category and has a stimulant effect, acting in the cardiovascular and central nervous systems, with an increase in its use among athletes since it was removed from the World Anti-Doping Agency list of prohibited substances.5,6 It is present in several widely consumed foods and drinks such as coffee, tea, chocolate, soda, and energy drinks containing guarana (Paullinia cupana), green tea (Camellia sinensis), or mate herb (Ilex paraguariensis).3,4,7-9 In addition to being present in everyday meals, it is widely used as a food supplement in different forms, such as powder and capsules. Caffeine is also found in some drugs such as analgesics, antiflu medications, appetite suppressants, and in some cosmetics.9,10 Among the nutrition claims allowed on food supplement labeling are those citing that "caffeine helps increase alertness and improve concentration" when adults of age 19 or older take a minimum daily dose of 75 mg; and "caffeine helps the increase in resistance capacity and the performance in resistance exercises" when a 200 mg dose is used one hour prior to physical activity.11 Brazil is the largest producer of guarana (Paullinia cupana) in the world, a plant native to the Amazon region which is commercially important.12,13 Guarana presents a complex matrix with active constituents found at distinct concentrations belonging to two chemical classes: methylxanthines and tannins. Caffeine, chemical marker of guarana, is its major component, present at concentrations between 2.5 and 6.0%.12,14 The ergogenic effect of guarana powder was verified in studies showing that people who practice martial arts improved their performance by using this type of product.15 Data on the composition, quality, and safety of Brazilian food supplements are limited, partly because such products are not subject to mandatory registration or safety evaluations before commercialization.3 According to the American health regulatory agency (Food and Drug Administration - FDA) and its Brazilian counterpart (Brazilian Health Regulatory Agency - ANVISA), caffeine and guarana are safe. However, adverse effects after intake have been reported, such as insomnia, nervousness, irritability, seizures, nausea, increased heart rate, and tachycardia. Indiscriminate and excessive use of caffeine (dose higher than 400 mg) may lead to potentially fatal tachydysrythmias, myocardial infarction, and hypertension, which reinforces the importance of quality control of these products.12,13,15 Since the publication of Collegiate Board Resolution - RDC 27/2010, food supplements have been exempt from registration with ANVISA and, consequently, their use and commercialization have increased considerably.16 Such growth was verified not only in Brazil, but also in developed countries, where the use of one or more food supplements was reported by approximately 75% of the population.3,7 In 2018, ANVISA published two important regulations on food supplements: RDC 243/2018 and Normative Instruction - IN 28/2018. Such regulations concern the requirements for composition, quality, safety, and labeling of food supplements, and update the lists of nutrients, bioactive substances, enzymes and probiotics, use limits, claims, and complementary labeling of such products.11,17 Normative Instruction 28/2018 classifies caffeine and guarana powder as bioactive substances present in food supplements.11 According to RDC 243/2018, nutrition labeling of packaged foods must follow RDC 360/2003,18 which approves the technical regulations on such foods. Specificities must be considered regarding the serving presented, which should be determined according to specific population and age groups shown on labels, and nutrition information, which has to include the quantities of all nutrients, bioactive substances, enzymes, and probiotics of the product, as well as the daily value percentage claim.11,17,18 Determination of methylxanthines and tannins by high performance liquid chromatography - HPLC in guarana products is described in several studies that use different chromatographic conditions and mostly complex extraction processes.14,19-22 The Brazilian Pharmacopoeia describes two methods for quantifying caffeine in guarana: one by ultraviolet (UV) spectrophotometry that expresses the result in total methylxanthines, and the other by HPLC-UV. However, the methods do not specify performance parameters, such as detection and quantification limits, linear range, and measurement uncertainty, and their adequacy in the analysis of food supplements containing caffeine has not been proven.23,24 Considering the importance and need for monitoring caffeine contents of guarana-based food supplements, the aim of this study was to develop and validate a simple, fast, and selective method to quantify caffeine in such products by using HPLC-UV.
EXPERIMENTAL The experiment was carried out at the Product Microscopy Service of the Environmental and Sanitary Division at the Octávio Magalhães Institute (Central Public Health Laboratory of Minas Gerais) of the Ezequiel Dias Foundation (FUNED) in Belo Horizonte, Minas Gerais, Brazil. Standards, reagents, and materials The reference standard, ReagentPlus® Caffeine (purity 100%), was purchased from Sigma-Aldrich (Milwaukee, USA). The guarana seed powder (Paullinia cupana Kunth, Sapindaceae, batch No: 29650, analysis certificate: 1436) was kindly provided by Herboflora (Machado, Brazil), previously evaluated for its microscopic identity, and inserted in the Database of Reference Samples of the Microscopy Service/FUNED under code Fit 67. The following HPLC grade solvents were used: acetonitrile (purchased from Merck, Billerica, USA) and methanol (Sigma-Aldrich, St. Louis, USA). The solvents used in the mobile phase were degassed with helium. For the extraction, phosphoric acid (85% m v-1) from Sigma-Aldrich (Saint Louis, USA) and analytical grade ethyl alcohol from VETEC (Rio de Janeiro, Brazil) were used. Purified water (maximum conductivity = 1.3 μS cm-1 at 25.0 °C and total bacterial count ≤ 100 UFC m L-1)24 was obtained through a system consisting of a Milli-Di Deionizer (DI-PAK cartridge), an A9051 Pump, a Water Prof 230F Filter, and a 0.22 μm Millipak 40 Filter. Analytical conditions A Shimadzu (Kyoto, Japan) high performance liquid chromatography system was used. It was equipped with an SIL-10AF auto injector, an SPD-M10Avp diode-array detector (DAD), an SPD-10Avp UV-VIS detector, two LC-10AD pumps, an SCL-10Avp controller and CLASS VP software, version 6.14 SP2, for data processing. The following materials were also used: a C18 reverse-phase pre-column (4 x 4 mm, 5 μm particles) and a Shim-pack VP-ODS end-capped C18 reverse-phase analytical column (250 x 4 mm I.D., 5 μm particles), Shimadzu (Kyoto, Japan), kept at room temperature (20 ± 2 ºC). The parameters assessed during development of the method included the organic modifier, elution type (isocratic and gradient), mobile phase flow rate, wavelength, and sample injection volume. After optimization, the following parameters were established: isocratic run with mobile phase composed of phosphoric acid (0.1%) (A) and acetonitrile (B), (88:12 v/v), flow rate of 1.5 mL min-1, injection volume of 10 μL, and detection at 272 nm. Extraction optimization Approximately 200 mg of guarana seed powder was weighed in a 15 mL Falcon tube and 10 mL of extracting solution [(8:2 v/v) ethanol-water mixture acidified with a 0.1% phosphoric acid solution at ~3.3 pH] was added.25 Optimization of the caffeine extraction process was performed using an experiment whose outline considered times of 5, 10, and 15 min, and number of extractions (2, 3 and 4 times). For each treatment, three independent replicates were analyzed and three injections were carried out for each of them. The tubes containing guarana powder and extracting solution were immersed in an ultrasound bath (Unique USC 1400; Indaiatuba, SP, Brazil; operating at 40 kHz) for the predetermined time, the solutions were centrifuged at 1,096 x g for 2 min, and the supernatants were collected in 50 mL volumetric flasks. This procedure was repeated 1, 2, or 3 more times and the final volume was completed with extracting solution. Aliquots of 250 μL were transferred to 10 mL volumetric flasks and the volumes were completed with HPLC grade methanol. The solutions were filtered through 0.45 μm membranes into vials and reserved for chromatographic analysis. The results obtained were evaluated for distribution using the Shapiro-Wilk test. Subsequently, the data were submitted to analysis of variance (ANOVA) and the estimated averages for different treatments were discriminated using the Tukey test. The significance level adopted in the hypothesis tests was 5%. R software version 3.6.2 was used for statistical analysis of the data. Standard solutions preparation To prepare the stock solution, approximately 10 mg of caffeine standard was weighed on weighing boats and transferred quantitatively to a 10 mL volumetric flask by using 5 mL HPLC grade methanol. The solution underwent ultrasonification for 10 min, and the final volume was completed with methanol (1 mg mL-1) and kept frozen (-24 to -12 ºC) until it was used. From the stock solution, standard solutions were prepared in solvent (methanol) and in matrix (guarana extract) at concentrations of 1, 2, 4, 6, 8, and 10 μg mL-1. Guarana extract was obtained as described in the subsection above using 3 extractions of 10 min. The solutions were filtered directly into vials through filtrating membranes of 0.45 μm, and 10 μL aliquots were used in the HPLC-UV analyses. Stability of the standard solution and guarana extract The stability of the standard caffeine solution (4μg mL-1) was evaluated in 2 repetitions and the guarana extract (intrinsic concentration of 3 μg mL-1 of caffeine) in 6 repetitions at times 0, 6, 12, 18, and 24 h at a temperature of 20 ± 2 ºC. The results obtained (concentrations) were analyzed for their distribution by using the Ryan-Joiner test and were subjected to ANOVA, at a significance level of 5%. Method validation System suitability, selectivity, linearity and matrix effect, limits of detection and quantification (LOD and LOQ, respectively), precision, recovery, robustness and measurement uncertainty were the parameters assessed in method validation. The choice of parameters and acceptance criteria were defined as described in the orientation documents published by ANVISA, the General Coordination for Accreditation of the National Institute of Metrology, the European Commission, the FDA, Association of Official Agricultural Chemists (AOAC), EURACHEM, and the studies conducted by Souza, Junqueira, and Ginn; Souza and Junqueira; Souza, Pinto, and Junqueira; and Orozco and Báez.26-35 System suitability and selectivity System suitability was evaluated based on resolution (Rs), retention factor (k), asymmetry (T), number of theoretical plates (N), and relative standard deviation (RSD) concerning retention time and caffeine area, obtained from the CLASS VP software report.36 The results were compared to the limits recommended in the Validation of Chromatographic Methods guide.29 UV spectra (190 to 400 nm) of the caffeine standard and the guarana reference sample were obtained in line to identify the peaks. Peak purity was determined with a diode-array detector (DAD) to assess selectivity. Furthermore, the guarana reference sample and caffeine standard with spiking of theophylline and theobromine were prepared in three independent replicates and separately theophylline and theobromine standards. The results obtained for caffeine (area and retention time) were compared through ANOVA (α = 5%). Linearity and matrix effect Calibration curves of caffeine in solvent (methanol) and matrix (guarana extract) were prepared at six concentration levels (1, 2, 4, 6, 8, and 10 μg mL-1) and in three independent replicates for each level. Linearity was assessed with the ordinary least squares method. The Jackknife technique was used to address outliers. Following the procedure described by Souza and Junqueira,32 it was investigated whether the following premises were met: normality (Ryan-Joiner test), test for homogeneity of variances (Brown-Forsythe test or modified Levene's test), and independence of residuals from the regression (Durbin-Watson test). Matrix effect was evaluated by comparing the slopes and intersections of the calibration curves in solvent and matrix by Student's t-test, after verifying homogeneity of variances (α = 5%).31,33 Limits of detection (LOD) and quantification (LOQ) Theoretical limits of detection and quantification were assessed based on data from the analytical curve using the equations LOD = 3.3s b-1 and LOQ = 10s b-1, respectively, where s = sample standard deviation of the linear coefficient and b = angular coefficient.27 The experimental LOQ of the method was verified by evaluating the lowest analyte concentration level of the curve (n = 6) obtaining acceptable results for the recovery and precision parameters.28,33 Recovery and precision Recovery and precision under repeatability conditions were studied in reference samples with the analyte at low, medium, and high concentration levels (1, 4, and 10 μg mL-1). The samples were prepared by two different analysts, with three replicates for each level (n = 6). Spiking of the standard caffeine solution was performed directly to the guarana powder before the extraction process. Evaluation of the intermediate precision was carried out using solutions of the extracts from the reference sample (n = 12) at 3 μg mL-1 intrinsic concentration of caffeine, on two different days, by two different analysts. All samples were injected in triplicate. Recoveries were estimated and extreme values were investigated for each concentration level using the Grubbs test.33 Average values between 80 and 110% were considered acceptable.27,28,30 Precision under repeatability conditions and intermediate precision were estimated using analysis of variance and expressed as relative standard deviation (RSDr and RSDR, respectively). Premises related to the F-test were previously tested similarly to the linearity studies, and the HorRat ratios were determined for additional assessment. HorRat ratios lower than 2.034 and RSDcalculated ≤ RSDacceptable27 being RSDr ≤ 7% and RSDR ≤ 10%30 were considered acceptance criteria. Robustness Guarana reference samples were analyzed in three independent replicates using the established analytical conditions and varying the following parameters: mobile phase flow rate (1.45 and 1.55 mL min-1), acetonitrile concentration (11.8 and 12.2%), and phosphoric acid concentration (0.09 and 0.11%) at the mobile phase. The concentrations obtained were compared through ANOVA (α = 5%). Estimation of Measurement Uncertainty Uncertainty estimation was determined from the combined standard uncertainty, expanded standard uncertainty, and expanded standard uncertainty expressed in percentage.35,37 The combined uncertainty of measurement was estimated based on the following independent sources: precision under repeatability conditions, calibration curve, uncertainty attributed to sample weighing, the analytical standard, the volumetric flasks, and the automatic pipettes used in the chromatographic analysis. 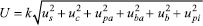 where U is the expanded measurement uncertainty of the analyte (μg mL-1); k, the coverage factor; us, the measurement precision uncertainty in terms of precision under repeatability conditions; uc, the uncertainty corresponding to the calibration curve; upa, the uncertainty for the standard; uba, the uncertainty associated with the analytical balance; ub, the uncertainty attributed to the volumetric flasks; and upi, the uncertainty related to the automatic pipettes used. Method application To carry out an evaluation of the content and variability of the caffeine content in guarana-based food supplements, the developed and validated method was applied to 30 samples (15 of which were marketed in the form of capsules and 15 in powder) purchased from establishments in Belo Horizonte, Minas Gerais, Brazil. Each sample was analyzed in three independent replicates and the profile of caffeine levels was characterized by measures of central tendency (mean and median) and dispersion (minimum, maximum, and standard deviation). The percentiles of data distribution were also estimated. The means obtained for the different forms of presentation of the supplements were compared by t test, after the evaluation of the homogeneity of the variances by F test (both with a 5% significance level).
RESULTS AND DISCUSSION Defining the analytical conditions Composition of the mobile phase and elution type were previously tested based on the studies conducted by Machado25 and Klein, Longhini and De Mello.21 Machado observed a retention time of approximately 11.5 min for caffeine under chromatographic conditions established using mobile phase consisting of linear gradient of 0.1% phosphoric acid solution (A) and acetonitrile at concentrations from 0 to 20% (B).25 In the present study, multiple tests altering the proposed gradient were performed. However, the alterations did not result in a considerable decrease in retention time nor in total analysis time. Then, isocratic elution was tested by employing 10% methanol-acetonitrile as modifier in different proportions.21 It was observed that the caffeine peaks were dissipated and asymmetrical. In addition, the resolution was < 2 between the posterior peak and the analyte. The Brazilian Pharmacopoeia uses as a mobile phase the mixture of water, methyl alcohol, and trifluoroacetic acid (70:30:0.005 v/v/v).24 As it is a toxic, corrosive composition and with 30% organic solvent, it was decided not to test this condition. To optimize the analytical conditions, the isocratic elution was evaluated, using higher concentrations of acetonitrile (10 to 18%), to obtain a faster rebalancing of the column to the initial analysis conditions, generating less waste. The flow rate was tested in the range of 1.2 to 1.7 mL min-1 to obtain a more symmetrical peak, without a tail, which was faster for the analysis. The mobile phase composed of 0.1% (A) (pH ~2.1) and acetonitrile (B) (88:12 v/v), and flow rate of 1.5 mL min-1 provided the best results. In the chromatogram obtained, caffeine was the major peak, with a retention time of 8 min and a tail / asymmetry factor of 1.09. The resolution in relation to the posterior (unknown) peak was 2.89 and the total running time was 18 min due to the presence of an unknown peak in 16 min. (Figure 1). Maximum caffeine absorption was verified at 272 nm based on the DAD spectral profile assessment, which was the wavelength defined for the proposed method.
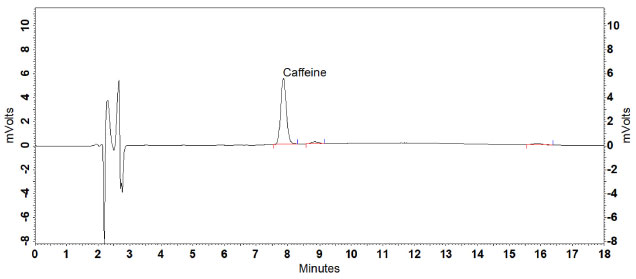 Figure 1. Chromatogram of caffeine determination in a guarana powder reference sample (3 μg mL-1) under the chromatographic conditions established in the HPLC-UV method
Extraction optimization and caffeine stability Data normality (ρ > 0.05) and significant difference between the evaluated treatments (ρ > 0.05) were observed in the extraction process optimization. The treatment in which three 10-min extractions were used presented significantly higher mean yield than the others; thus, it was defined as the extraction process of the method. There was no difference among the 5-min extraction treatments, and the treatment with two 15-min extractions presented considerably lower yield than the others (Table 1).
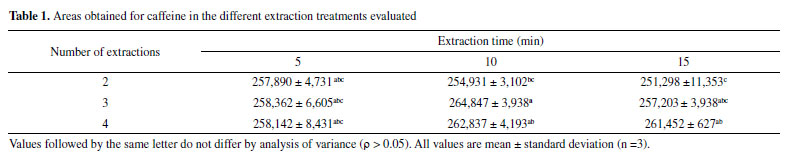
Table 1. Areas obtained for caffeine in the different extraction treatments evaluated Regarding caffeine stability, no significant degradation was evidenced from the extract of the reference guarana samples and from standard solutions at temperature of 20 ± 2 ºC for 6, 12, 18, and 24 h (ρ > 0.05). Therefore, it was confirmed that the standard and guarana extract solutions remain stable for enough time to perform the analyses (Table 1S, Supplementary material). Validation System suitability and selectivity The results obtained for the parameters resolution (3.12), retention factor (2.64), asymmetry (1.09), number of theoretical plates (9,342), and RSD of retention time (0.56%) and caffeine area (0.59%) fell within the recommended limits.29 Method selectivity was verified by obtaining satisfactory results for the resolution of the posterior caffeine peak (Rs > 2.0) and spectral purity (> 99.9%). Evaluation of the peak purity was carried out by building a ratiogram on the CLASS VP Software, where the ratio between absorbances in two wavelengths as a function of run time was plotted (Figure1S, Supplementary material). The result was a rectangular graph, showing that the chromatographic ratio is constant, lower than 1.0 and higher than the method noise, as recommended by Snyder, Kirkland, and Glajch.38 The section in which the purity curve is above zero indicates the absence of impurities (Figure 2S, Supplementary material).36 Adequate results were also obtained for selectivity assessment in the presence of interferents inherent in guarana: theophylline and theobromine, which are methylxanthines of structures similar to caffeine (Figure 2). No significant differences were observed for the area and retention time of caffeine in the presence of such substances (ρ > 0.05).
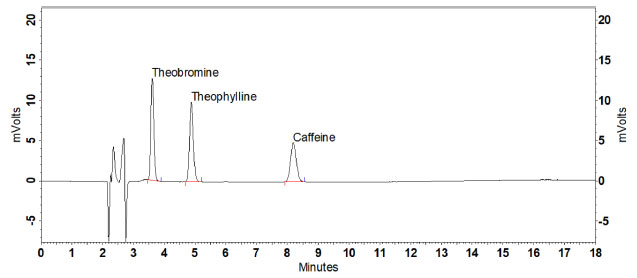 Figure 2. Chromatogram of the pool of theobromine, theophylline, and caffeine standards under the chromatographic conditions established in the method
Linearity and matrix effect After the treatment of dispersed values, 3 outliers were identified and removed only from the matrix curve. The premises related to the simple linear regression analysis (normality, homogeneity of variances, and independence of residuals from the regression) were met regarding the established concentration range, both for the calibration curve of caffeine in methanol and in matrix. Coefficients of determination (R2) of 0.9994 and 0.9981 were observed for the curve in solvent and in matrix, respectively. The matrix effect was proven, and a significant difference (ρ < 0.05) was observed between the slopes and intersections of the calibration curves developed. Thus, it was demonstrated that the chromatographic response of caffeine analysis is affected by the guarana powder matrix, and the preparation of calibration curves by dissolving the standard in the solvent is not recommended (Figure 3).
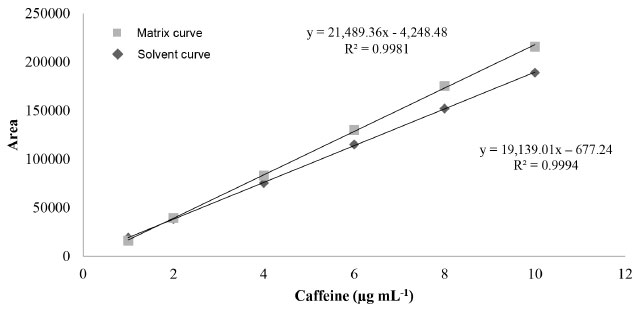 Figure 3. Calibration curves of caffeine in methanol and in guarana powder extract
Evaluation of the matrix effect on quantification of endogenous or exogenous compounds is an important assessment parameter in the development and validation of analytical methods, including chromatographic ones. Substances intrinsic to the biological matrix may co-elute with the analyte of interest, thus interfering in different analytical parameters, such as selectivity, recovery, and precision.39 Nonetheless, the matrix effect is still scarcely explored in studies about the development and validation of methodologies, both for the determination of different analytes in guarana and for caffeine analysis in foods and food supplements.14,20,40 To analyze catechins, procyanidins, and caffeine in guarana extract, Yonekura and Tamura proposed an isocratic method by HPLC-UV .22 Teixeira et al. also developed a method using CLAE-UV to determine caffeine in soft drinks and energy drinks.8 Both studies used a calibration curve with standards in solvent. Work developed by Yousefi et al. describes an analytical method by HPLC-UV to determine caffeine in teas and energy drinks, in which an analytical curve developed by dissolving the standard in deionized water was used.41 However, in none of these studies was the matrix effect evaluated. Limits, recovery, precision, robustness, and estimation measurement of uncertainty Table 2 shows results for limits of detection and quantification, recovery, precision under repeatability conditions, and intermediate precision and measurement of uncertainty. The experimental limit of quantification was confirmed as the low level of the linear range (1.0 μg mL-1), for which satisfactory results were found regarding recovery and precision evaluation. Teixeira et al. found the same value of LOQ.8
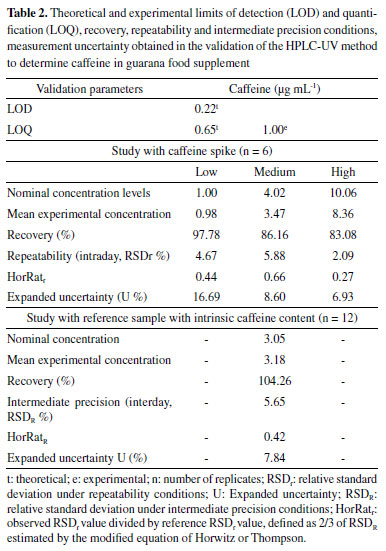
When developing a method for simultaneous analysis of caffeine and five soda additives through HPLC-UV, Agçj, Zor, and Dönmez observed similar results for the LOD and LOQ theoretical calculation (LOD = 0.19 μg mL-1 and LOQ = 0.63 μg mL-1) by using the intersection and slope of the calibration curve.42 On the other hand, Klein, Longhini, and De Mello found slightly lower values (LOD = 0.13 μg mL-1 and LOQ = 0.39 μg mL-1) in the validation of a methodology through HPLC-PDA for the analysis of the same analyte and matrix assessed in the present study.21 Satisfactory results were obtained for all concentration levels studied considering the recommended recovery range of 80 to 110%.27,28,30 Machado et al. reported more exact and less wide-ranging results in a study of caffeine recovery in guarana powder (94.9 to 95.6 % and RSD < 1.0%);14 however, a lower number of replicates was used for such estimate compared to this study. The premises related to the F-test (normality of residuals and homogeneity of variances) were met in the repeatability and intermediate precision evaluation. Satisfactory results for RSDr, RSDR, HorRatr, and HorRatR confirmed the precision of the method for both conditions. The developed method was robust in relation to the predetermined variations in the concentrations of acetonitrile and phosphoric acid in the composition of the mobile phase. There were no significant differences (p > 0.05) in the concentration of caffeine due to the subtle changes in these parameters. However, for the variation of the flow rate, a significant difference was observed (p < 0.05), this parameter being a critical control point (Table 2S, Supplementary Material). The expanded uncertainty varied between 6.9 and 16.7%, over the entire concentration range studied, with a smaller difference being observed between the medium and high levels of concentration evaluated. An asymmetric distribution of the reported values was observed, making it necessary to adopt two interval ranges for the association of uncertainty.35 Pipetters, standard solution, and calibration curve were the most relevant sources in the composition of the combined uncertainty. Caffeine content in commercial samples of food supplements based on guarana The developed method was applied in the determination of caffeine in commercial samples of food supplements based on guarana, produced by different manufacturers, including 15 samples marketed in the form of capsules and 15 in powder. Average caffeine levels of 25.27 ± 5.20 mg g-1 were found for powdered products and 28.53 ± 13.81 mg g-1 for those sold in capsules, with no significant difference between the two forms of presentation (p > 0.005) (Table 3S, Supplementary Material). The medians were also similar and approached the estimated average levels. However, a greater variability in caffeine content was observed for the samples in capsules, whose contents found varied between 6.29 ± 0.05 and 67.64 ± 4.45 mg g-1 (Table 3).
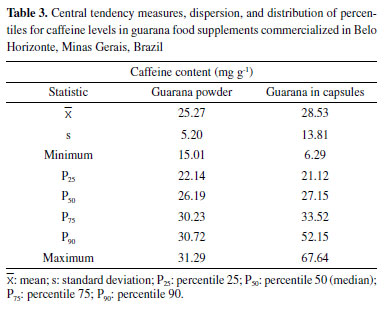
High variability of the caffeine content between the samples was also observed when considering the dose of use recommended by the manufacturer. The estimated levels of caffeine ranged from 6.9 to 224.6 mg day-1 for capsules and 34.2 to 245.7 mg day-1 for powdered supplements. Such values are in disagreement with the minimum and maximum limits recommended by IN 28/2018 from 75 to 200 mg day-1. A high variability was also observed by Viana et al. when analyzing by HPLC-DAD about 100 samples of dietary supplements sold on Brazilian websites. The authors found levels of caffeine in supplements between 25.0 and 1,476.7 mg day-1.43
CONCLUSIONS The established extraction procedure is simple, fast, and low cost, using less aggressive solvents and minimizing the generation of waste, compared to similar studies. In the present study, it was found that the method developed is adequate for the purpose, since satisfactory results were obtained for all evaluated parameters. The proof of the matrix effect and the calculation of the measurement uncertainty estimate reinforce the need for a complete study of the performance parameters to guarantee the reliability of the results. Therefore, the proposed method may be used both in health surveillance and monitoring, and in quality control of food supplements, especially concerning the confirmation of caffeine content, which must be shown on the labels of products available on the market.
SUPPLEMENTARY MATERIAL In the supplementary material, available at http://quimicanova.sbq.org.br in pdf format, with free access, there are the gross results of the commercial samples analyzed, as well as tables on the stability and robustness tests studied, as well as complementary data on purity, in the DAD of the validation of methodology by HPLC-UV for determination of caffeine in food supplements based on guarana (Paullinia cupana).
ACKNOWLEDGEMENTS The authors would like to thank the Ezequiel Dias Foundation for funding the research, in particular the Product Microscopy Service for the availability and support for carrying out the chromatographic tests and Priscilla R. V. Campana for the initial statistical treatment of extraction data.
REFERENCES 1. Freire, R. S.; Lélis, F. L. O.; Filho, J. A. F.; Nepomuceno, M. O. N.; Silveira, M. F.; Revista Brasileira de Medicina do Esporte 2014, 20, 345. 2. Almeida, C.; Sangiovanni, D; Liberali, R.; Revista Brasileira de Nutrição Esportiva 2009, 3, 198. 3. Bessada, S. M. F.; Alves, R. C.; Oliveira, M. B. P. P.; Food Res. Int. 2018, 109, 310. 4. Soares, E. M. K. V. K.; Garcia G. L.; Molina G. E.; Fontana K. E.; Revista Brasileira de Medicina do Esporte 2019, 25, 168. 5. World Anti-Doping Agency (WADA); The world anti-doping code international standard, Prohibited list January 2020, Montreal, 2020. 6. Southward, K.; Rutherfurd-Markwick, K.; Badenhorst, C.; Ali A.; Nutrients 2018, 10, 1. 7. Gurley, B. J.; Steelman, S. C.; Thomas, S. L.; Clin. Ther. 2015, 37, 275. 8. Teixeira, L. S.; Silva, C. F.; Oliveira, H. L.; Dinali L. A. F.; Júnior, C. S. N.; Borges, K. B.; Microchem. J. 2020, 158, 105252. 9. De Maria, C. A. B.; Moreira, R. F. A.; Quim. Nova 2007, 30, 99. 10. Funasaki, M.; Barroso, H. S.; Fernandes, V. L. A.; Menezes, I. S.; Quim. Nova 2016, 39, 194. 11. Agência Nacional de Vigilância Sanitária (ANVISA); Estabelece as listas de constituintes, de limites de uso, de alegações e de rotulagem complementar dos suplementos alimentares, Instrução Normativa - IN n° 28, de 26/07/2018. 12. Santana, A. L.; Macedo, G. A.; J. Funct. Foods 2018, 47, 457. 13. Marques, L. L. M.; Ferreira, E. D. F.; de Paula, M. N.; Klein, T.; De Mello, J. C. P.; Rev. Bras. Farmacogn. 2019, 29, 77. 14. Machado, K. N.; Freitas, A. A.; Cunha, L. H.; Faraco, A. A. G.; Pádua, R. M.; Braga, F. C.; Vianna-Soares, C. D.; Castilho, R. O.; Food Chem. 2018, 239, 180. 15. Silveira, J. Q.; Amorim, L. L.; Burian, J. P.; Revista Brasileira de Nutrição Esportiva 2018, 12, 246. 16. Abe-Matsumoto, L. T.; Sampaio, G. R.; Bastos, D. H. M.; Rev. Inst. Adolfo Lutz 2016, 75, 1. 17. Agência Nacional de Vigilância Sanitária (ANVISA); Dispõe sobre os requisitos sanitários dos suplementos alimentares, Resolução da Diretoria Colegiada RDC nº 243, de 26/07/2018. 18. Agência Nacional de Vigilância Sanitária (ANVISA); Regulamento técnico sobre rotulagem nutricional de alimentos embalados, Resolução da Diretoria Colegiada RDC n° 360 de 23/12/2003. 19. Sousa, S. A.; Alves, S. F.; de Paula, J. A. M.; Fiuza, T. S., Paula, J. R.; Bara, M. T. F.; Rev. Bras. Farmacogn. 2010, 20, 866. 20. Sousa, S. A.; Pascoa, H.; Conceição, E. C.; Alves, S. F.; Diniz, D. G. A; Paula, J. R.; Bara, M. T. F.; Braz. J. Pharm. Sci. 2011, 47, 269. 21. Klein, T.; Longhini, R.; De Mello, J. C. P.; Talanta 2012, 88, 502. 22. Yonekura, L.; Tamura, H.; MethodsX 2019, 6, 850. 23. Farmacopeia Brasileira, 5ª ed., Agência Nacional de Vigilância Sanitária Editora: Brasília, Brasil, 2017, Segundo Suplemento. 24. Farmacopeia Brasileira, 6ª ed., Agência Nacional de Vigilância Sanitária Editora: Brasília, Brasil, 2019. 25. Machado, K. N. Dissertação de mestrado, Universidade Federal de Minas Gerais, Brasil, 2015. 26. Agência Nacional de Vigilância Sanitária (ANVISA); Guia para Validação de Métodos Analíticos e Bioanalíticos, Resolução RE n° 899 de 29/05/2003. 27. Coordenação Geral de Acreditação (CGCRE); DOQ-CGCRE-008 Orientação sobre validação de métodos analíticos, Rev.05, Rio de Janeiro, 2016. 28. Official Journal of the European Communities (OJEC); Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, 08/2002. 29. Food and Drug Administration (FDA). Center for Drug Evaluation and Research (CDER). Reviewer guidance-Validation of chromatographic methods, 1994. 30. Zhang, Y.; Betz, J. M.; Griffiths, J.; Hildreth, J. B.; Jennens, M.; Ji, D.; Joseph, G.; Kennedy, D. C.; Mudge, E.; Ofitserova, M.; Phillips, M. M.; Phillips, T.; Richards, L. D.; Rimmer, C. A.; Royce, S.; Schaneberg, B. T.; Solyom, A. M.; Sullivan, D. M.; Szpylka, J.; Traub, J.; Wood, L.; Yang, J.; Yoo, S.-J.; Zhou, J.; Zhu, J.; Zielinski, G.; Coates, S. G.; J. AOAC Int. 2016, 99, 314. 31. Souza, S. V. C.; Junqueira, R. G.; Ginn, R.; J. Chromatogr. A 2005, 1077, 151. 32. Souza, S. V. C.; Junqueira, R. G.; Anal. Chim. Acta 2005, 552, 25. 33. Souza, S. V. C., Pinto, C. T.; Junqueira, R. G.; J. Food Compos. Anal. 2007, 20, 241. 34. Orozco, C. A. R.; Báez, M. R. R.; Uso de la ecuación de Horwitz em laboratorios de ensayos NMX-EC-17025-IMNC-2006, Simposio de Metrología, Centro Nacional de Metrología SM2010-S5C-3, Chihuahua, Mexico, 10/2010. 35. Co-operation on International Traceability in Analytical Chemistry (CITAC)/ EURACHEM Guide CG 4.; Quantifying Uncertainty in Analytical Measurement 3th ed., Teddington, 2012. 36. Shimadzu Corporation. CLASS-VP Instruction Manual, Chromatography Data System. Kyoto, 1997, Ch. 9. 37. Joint Committee for Guides in Metrology (JCGM); JCGM 100:2008. Evaluation of measurement data - Guide to the expression of uncertainty in measurement, 1st ed., 2008. 38. Snyder, L. R.; Kirkland, J. J.; Glajch, J. L. Practical HPLC method development, 2nd ed.; John Wiley & Sons: United States of America, 1997, ch. 3. 39. Cassiano, N. M.; Barreiro, J. C.; Martins, L. R. R.; Oliveira, R. V.; Cass, Q. B.; Quim. Nova 2009, 32, 1021. 40. Al-Othman, Z. A.; Aqel, A.; Alharbi, M. K. E.; Badjah-Hadj-Ahmed A. Y.; Al-Warthan A. A.; Food Chem. 2012, 132, 2217. 41. Yousefi, S.; Kamankesh, M., Jazaeri, S.; Attaran, A.; Mohammadi, A.; Anal. Methods 2017, 10, 1665. 42. Agçj, B.; Zor, F. D.; Dönmez, O. A.; Int. J. Anal. Chem. 2016, 2016, 1. 43. Viana, C.; Zemolin G. M.; Müller, L. S.; Molin, T. R.; Seiffertb, H; Carvalho, L. M.; Food Addit. Contam., Part A (2015), doi:10.1080/19440049.2015.1112040. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access