
 |
 |
 |
 |
 |
Revisão
|
|
| Métodos analíticos para determinar Lactonas Macrocíclicas en diferentes tipos de matrices. Una revisión Analytical methods for determination of Macrocyclic Lactones in different types of matrices. A review |
|
Patricia Esmeralda Vázquez-QuintalI; Roger Iván Rodríguez-VivasII; David Muñoz-RodríguezI,*
I. Cuerpo Académico de Química Fundamental y Aplicada, Facultad de Ingeniería Química, Universidad Autónoma de Yucatán, 97203, Mérida, Yucatán, México Recebido em: 03/04/2021 *e-mail: david.mr@correo.uady.mx The Macrocyclic Lactones (ML) are drugs widely used to combat endo- and ectoparasites in livestock, pets, crops, etc. Since marketing of ivermectin, the number of available ML increased considerably and, consequently, a wide variety of analytical methodologies for determination of MLs in multiple types of matrices have been published. Most of these methodologies have already been reviewed before so this paper is about those most recent and/or those have not been previously considered. In this review the focus is on biological, alimentary, and environmental matrices emphasizing on the most important techniques employed on sample preparation. Also, the liquid chromatography with of fluorescence and mass spectrometry detectors is discussed as the most used instrumental techniques for identification of MLs. Finally, a brief review is made on the importance of method validation and some strategies for its implementation. INTRODUCCIÓN Las lactonas macrocíclicas (LM) son una familia de fármacos empleados en la medicina humana y veterinaria para el tratamiento contra endo- y ectoparásitos.1 Son muy utilizadas por su actividad de amplio espectro contra nematodos y artrópodos; las LM se pueden encontrar en diversas formulaciones y son fáciles de administrar. Además, son efectivas contra cepas resistentes a otros fármacos.2 Se clasifican en avermectinas y milbemicinas. Ambas se producen directa o indirectamente a partir de la fermentación natural de algunas especies de estreptomicetos del suelo.3 La ivermectina fue la primera LM en desarrollarse en la década de 1970 e inicialmente se aplicó en medicina veterinaria y en la agricultura. En 1981 se comercializó para su uso en ganadería y años después se aplicó en la medicina humana.4 Su exitosa comercialización condujo a enfocar la investigación en la modificación de la estructura química de las avermectinas y milbemicinas para obtener moléculas diferentes con igual o mejor nivel de eficacia que la ivermectina.3 Esto resultó en el desarrollo de varias LM, de las cuales se utilizan principalmente seis avermectinas: ivermectina (IVM), abamectina (ABA), doramectina (DOR), eprinomectina (EPR), emamectina (EMA) y selamectina (SEL); y dos milbemicinas: milbemicina oxima (MIL-O) y moxidectina (MOX).1 La ABA, EMA y MOX se han usado en la agricultura3,5-8 y la ganadería.2,4,9 La IVM, ABA, DOR, EPR y MOX se han aplicado para el tratamiento contra endoparásitos como nematodos gastrointestinales y pulmonares; y ectoparásitos como ácaros, piojos y garrapatas en el ganado bovino, ovino, caprino, equino y porcino.2 Adicionalmente, en la acuacultura se emplea la EMA para el tratamiento del piojo de mar en salmones.1,10 En medicina veterinaria, la MIL-O y la SEL previenen los nematodos cardiacos causantes de filariasis en perros y gatos.1 También ayudan a combatir gusanos redondos, gusanos "con ganchos", ácaros del oído, sarna y pulgas.1,2 Por otro lado, en medicina humana, la IVM y la MOX sirven para combatir el nematodo que causa la enfermedad denominada "ceguera del río" y para tratar la filariasis linfática.2 A pesar de sus beneficios, el uso indiscriminado y la presencia de residuos de LM en el ambiente puede afectar la salud humana2,11,12 y al ecosistema.13 Un ejemplo de esto es el daño a organismos "no blanco", como los escarabajos estercoleros (Coleoptera: Scarabaeidae: Scarabaeinae).14-16 Por lo tanto, es crucial contar con metodologías analíticas para determinar los niveles de las LM en muestras origen agrícola, ganadero y medioambiental. En 20061 y 2012,5 Danaher et al. publicaron dos artículos en los que revisaron los métodos analíticos existentes hasta ese momento. El primero se enfocó en matrices biológicas con énfasis en las técnicas de extracción y detección; además, se consideraron aspectos como el desarrollo y la validación de las metodologías analíticas.1 El segundo artículo se centró en la revisión de los métodos para el análisis de matrices alimenticias (tejidos de animales comestibles, productos lácteos, vegetales) y ambientales (agua, suelo, heces), principalmente con métodos basados en LC-MS/MS.5 En un artículo más reciente, Wang et al. revisaron las metodologías analíticas basadas en inmunoensayos para determinar LM en matrices alimenticias.17 Sin embargo, el desarrollo de nuevas metodologías continúa y han surgido técnicas de pretratamiento analítico que se han diseñado y/o adaptado para aplicarlas en la determinación de LM. Además, el número y variedad de matrices donde estas metodologías pueden aplicarse crece diariamente. En este artículo se revisan las metodologías analíticas para la determinación de LM en matrices biológicas, ambientales y alimenticias. Se enfatiza la etapa del pretratamiento analítico y las técnicas analíticas más utilizadas; además, se exponen las principales estrategias de validación de las metodologías. La revisión aborda los métodos reportados en los últimos 15 años y/o que no se discutieron en revisiones previas.1,5 Además, se incluyeron metodologías que, aunque puede considerarse no recientes, involucran técnicas de extracción y/o matrices inusuales dentro del estado del arte de la determinación de las LM.
ESTRUCTURA QUÍMICA Avermectinas Las avermectinas constituyen una mezcla de ocho compuestos naturales producidos por la fermentación de la bacteria Streptomyces avermitilis.3 Su estructura química consiste en un anillo macrocíclico de 16 miembros formado por un grupo espiroacetal, un anillo de benzofurano y un grupo disacárido o monosacárido (Figura 1).1 Se clasifican según los sustituyentes R5 y R25 y el tipo de enlace entre C22 y C23: la serie A incluye los compuestos que están metoxilados en C5 (R5); y la serie B, los compuestos que tienen un grupo hidroxilo en esa misma posición. Las series A y B se subdividen en dos grupos: en el subgrupo 1 están los compuestos con un enlace doble olefínico entre C22 y C23, mientras que en el subgrupo 2 el C23 está enlazado a un grupo hidroxilo. A su vez, los subgrupos 1 y 2 pueden ser de la clase a si el sustituyente R25 es un radical sec-butil o de la clase b si es un isopropil.2 De las ocho avermectinas naturales se producen en mayor cantidad la B1 y la B2. Además, estas tienen una elevada potencia antiparasitaria, su espectro de actividad es más amplio y son más seguras. La ABA, una mezcla de B1a y B1b, es la avermectina natural más importante y la mayoría de las avermectinas sintéticas o semisintéticas son derivados de ésta (Figura 1).1-3
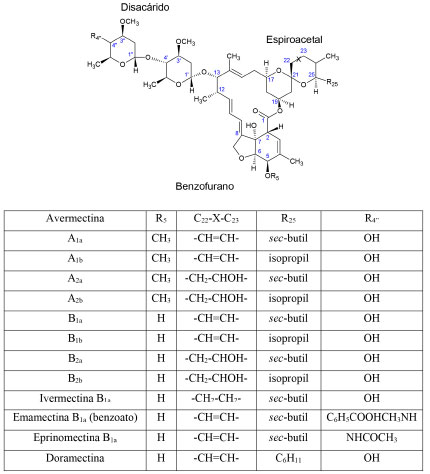 Figura 1. Estructura química de las avermectinas1
La IVM es un derivado semisintético de la ABA (principalmente de la B1a) producido por la hidrogenación selectiva del doble enlace entre C22 y C23. Debido a esta modificación se considera que la IVM es un híbrido entre B1 y B2, e incluso es superior a ambas en términos de eficacia y seguridad.4,18,19 La EMA y la EPR se producen por la aminación de C4" del grupo disacárido de la ABA. En el caso de la EMA se adiciona un grupo metilamino, y el derivado obtenido se cristaliza como benzoato. En la EPR se forma una amina primaria en C4" que luego se somete a una acilación.20 La DOR se obtiene por mutasíntesis o biosíntesis mutacional, un proceso donde S. avermitilis se modifica genéticamente de tal forma que las avermectinas producidas contienen un sustituyente diferente en C25.21 La DOR, por lo tanto, tiene un grupo ciclohexil en C25 y conserva el doble enlace entre C22 y C23, por lo cual en términos de estructura es más cercana a la ABA que a la IVM.22 La SEL (Figura 2) es un derivado semisintético de la DOR. A diferencia de ésta, la SEL tiene un grupo oxima en C5 y un grupo monosacárido en C13. Debido a esta última característica se puede considerar que la SEL es un derivado intermedio entre las avermectinas y las milbemicinas.2,23
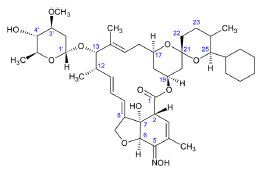 Figura 2. Estructura química de la selamectina13
Milbemicinas Las milbemicinas son producto de la fermentación de bacterias del género Streptomyces, principalmente de las especies S. hygroscopicus y S. cyaneogriseus noncyanogenus.3 Su estructura química es similar a las avermectinas. Sin embargo, las milbemicinas carecen del grupo disacárido en C13 y algunas tienen un anillo abierto de benzofurano.1,4,24 Además, a diferencia de las avermectinas donde el enlace C22-C23 puede ser doble, en las milbemicinas este enlace es simple y el C23 puede ir unido a algún otro sustituyente.3 Las milbemicinas producidas por S. hygroscopicus se dividen en dos series denominadas α y β. Éstas se diferencian por el anillo de benzofurano que está presente en la serie α, pero que en la serie β está parcialmente abierto.24-26 Adicionalmente, cada serie puede subdividirse según los sustituyentes en la fracción espiroacetal. Los compuestos de la serie α, al igual que las avermectinas, tienen un grupo hidroxilo o metoxilo en R5. Sin embargo, a diferencia de éstas, el sustituyente en R25 puede ser un grupo metil o etil (Figura 3). En esta serie, la posición R4 está ocupada por un radical metil o 2-pirrolcarboniloxi y las posiciones R22 y R23, por grupos β-hidroxilo y α-hidroxicarbonil-alquilo, respectivamente.26 Una de las milbemicinas más utilizadas, la MIL-O, se sintetizó a partir de las milbemicinas naturales α1 y α3 adicionando un grupo oxima en R5.27
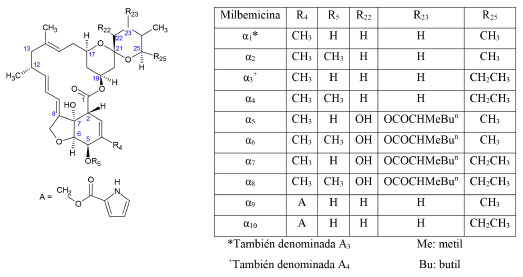 Figura 3. Estructura química de las milbemicinas de la serie α26
La serie β, que se distingue por tener una fracción "incompleta" de benzofurano, está conformada por tres milbemicinas. Las milbemicinas β1 y β2, se caracterizan por la presencia de los grupos β-metoxilo e hidroximetileno en R5 y R8, respectivamente (Figura 4). La diferencia entre estas milbemicinas es el grupo alquilo en R25.26 La milbemicina β3 (Figura 5) es el único miembro que posee un anillo aromático en su estructura, aunque se piensa que éste podría ser un artefacto formado durante el proceso de purificación.25
 Figura 4. Estructura química de las milbemicinas de la serie β26
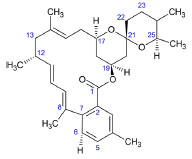 Figura 5. Estructura química de la milbemicina β326
Finalmente, las milbemicinas obtenidas con bacterias S. hygroscopicus genéticamente modificadas se designan con letras mayúsculas. Las milbemicinas D, F, G, J y K (Figura 6) tienen los mismos sustituyentes de la serie α en R4, pero difieren en los otros. Así, en la posición R5 se puede encontrar un grupo β-hidroxilo, β-metoxilo o cetona, mientras que en R25, puede haber un radical alquilo como metil, etil o isopropil. Por otro lado, las milbemicinas E y H (Figura 4) son similares a la serie β; ambas tienen un grupo isopropilo en R25, pero la milbemicina H difiere en los sustituyentes R5 y R8 donde tiene una cetona y un metil, respectivamente.26
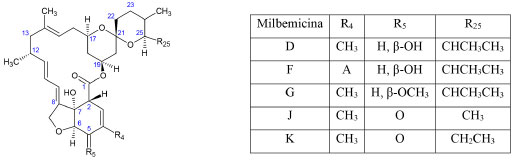 Figura 6. Estructura química de las milbemicinas obtenidas de bacterias genéticamente modificadas26
Las milbemicinas producidas por S. Cyaneogriseus noncyanogenus (Figura 7) presentan cadenas insaturadas en la posición R25 y un grupo -OH en C23 igual que las avermectinas del grupo 2. Las cuatro principales milbemicinas de esta familia se designaron como LL-F 28249 α, λ, β y γ.26,28 La más importante es LL-F 28249α o nemadectina (NEM), que es similar a una milbemicina α, pero con más grupos funcionales en la fracción espiroacetal.2,24
 Figura 7. Estructura química de las milbemicinas de la familia LL-F 2824926
La MOX (Figura 8) es un derivado semisintético de la NEM. Se obtiene introduciendo una fracción de metoxima en lugar del grupo -OH que tiene NEM en R23. La presencia de este grupo y la cadena insaturada en R25 son dos características que no se encuentran en ninguna otra milbemicina o avermectina.2,26,29
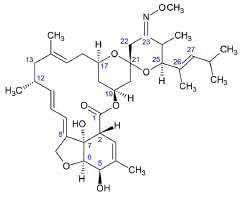 Figura 8. Estructura química de la moxidectina26
PROPIEDADES FISICOQUÍMICAS Y FARMACOCINÉTICAS Las propiedades fisicoquímicas de las LM dependen de los grupos funcionales en su estructura química. La fracción espiroacetal les proporciona una estructura rígida y diversidad estereoquímica.30 Las LM naturales presentan exclusivamente una configuración R en el C21 de la función espiroacetal. Este tipo de geometría se relaciona con la actividad insecticida pues se han aislado intermediarios producidos por bacterias modificadas genéticamente donde el C21 tiene una configuración S, las cuales presentan un espectro de actividad insecticida menor a los isómeros R.31 La geometría del enlace C22-C23 también influye en las propiedades de las LM. Se ha observado que la saturación de dicho enlace modifica la configuración estereoquímica del anillo de pirano; esto modifica las propiedades de partición de los fármacos en la leche: las LM saturadas en C22-C23 (avermectinas del grupo 2 y milbemicinas) tienen coeficientes de partición leche:plasma superiores a las LM que tienen este enlace insaturado (avermectinas del grupo 1).32,33 Otro ejemplo de esta influencia se encuentra en la IVM, una avermectina B1 que durante su síntesis conserva la conformación de "silla" de la serie B2. En consecuencia, la IVM se caracteriza por tener una elevada potencia parasiticida, propio de las avermectinas B1; pero con un mejor perfil de seguridad que éstas, lo cual la hace similar a las avermectinas B2.3,34 Las LM son moléculas relativamente grandes con pesos moleculares de 640 (MOX) hasta casi 900 (DOR).35 Las LM son escasamente solubles en agua y muy solubles en solventes orgánicos como octanol, n-hexano, benceno, acetona, etanol, metanol, cloroformo, éter dietílico y acetato de etilo.30,36,37 Los coeficientes calculados de partición octanol/agua (log P) de las avermectinas varían de 4.4-5.6, mientras que las milbemicinas tienen valores superiores, aproximadamente de 6. Una excepción notable es la SEL con un log P de 6.4.30 Por su naturaleza lipofílica, las LM tienen un gran volumen de distribución tisular, especialmente en el tejido graso e hígado, y una persistencia prolongada en el organismo hospedador.30,35,38 La alta lipofilicidad de las LM se debe, principalmente, a los anillos de lactona y de benzofurano.30 La presencia de sustituyentes alquílicos, como el ciclohexil, en el C25 de la fracción espiroacetal aumenta el carácter lipofílico de las LM (i.e. DOR, SEL).30 El grupo sacárido (oleandrosa) en C13 también le confiere cierta lipofilicidad a las avermectinas, ya que al tener un solo grupo -OH en C4", su polaridad e hidrofilicidad es menor a la de otros sacáridos.3,30,35 En la EPR, este grupo -OH se sustituye por un grupo amino, lo que disminuye su lipofilicidad (log P = 4.4). Por otro lado, la ausencia de la fracción de oleandrosa en C13 hace que las milbemicinas sean más lipofílicas que las avermectinas.35 En el caso de la MOX, otros factores que contribuyen a su lipofilicidad son la metoxima y la cadena olefínica en el C23 y C25, respectivamente. Las diferencias de lipofilicidad entre las LM se reflejan en algunas de sus características farmacocinéticas. La IVM puede considerarse una LM típica con un volumen de distribución relativamente alto y una baja tasa de eliminación.39,40 Sin embargo, la MOX, que es cien veces más lipofílica se acumula en mayor medida en el tejido adiposo y sus residuos se eliminan más lentamente, lo que resulta en una baja partición grasa:plasma.41 En consecuencia, la MOX se caracteriza por un mayor volumen de distribución, una vida y un tiempo medios de residencia relativamente grandes para una gran variedad de especies.29,30 Por otro lado, la DOR (log P = 5.3) tiene una cinética similar a la MOX, aunque su velocidad de eliminación es ligeramente menor.22,42 Debido a estas características, la leche es una vía importante de eliminación de estos tres fármacos, especialmente MOX y DOR.29,42-44 En contraste, la EPR puede considerarse una LM relativamente polar, por lo que interactúa débilmente con el tejido adiposo.30 Esto se traduce en una mejor distribución tisular, una eliminación más rápida de los residuos y una Cmáx más alta en comparación con otras LM, como la IVM y MOX.34,45 Estas características también influyen en la baja partición leche:plasma de la EPR.35 Por otro lado, la elevada lipofilicidad de la SEL, utilizada como tratamiento tópico en perros y gatos,46 hace que se distribuya eficientemente en la piel de los animales tratados, localizándose principalmente en las glándulas sebáceas.34 La polaridad también influye sobre la actividad biológica de las LM. Se ha reportado que la sustitución del C13 con grupos polares (hidroxi, ciano, amino) o no polares (cloro, fluoro, metoxi) disminuye o aumenta, respectivamente, la actividad de las moléculas resultantes.35,47 Las avermectinas son inestables en medio ácido y básico.48 Bajo condiciones ácidas leves, el grupo disacárido se fragmenta en C4' dando como resultado una especie con un grupo monosacárido. Bajo condiciones ácidas más fuertes, el disacárido se pierde completamente y se forma una aglicona.49 En medio básico, el C2 de las avermectinas pueden sufrir una epimerización.50-52 Por lo tanto, las avermectinas son estables en un rango de pH de 6.2-6.3.53 Por otro lado, las avermectinas y milbemicinas pueden oxidarse en las posiciones C2-C8 del anillo de benzofurano y en los diferentes grupos -OH (C23, C13, C4' y C4") presentes en la molécula.48 Las LM son propensas a la autooxidación, aunque para esto se requiere la presencia de solventes como éteres, alcoholes secundarios y ésteres.54 Por último, las LM pueden degradarse bajo la luz ultravioleta. Debajo de los 280 nm, los enlaces C8-C9 y C10-C11 se isomerizan rápidamente y una irradiación prolongada provoca la aparición de varios productos de descomposición.55
MÉTODOS DE DETERMINACIÓN DE LACTONAS MACROCÍCLICAS A la fecha, se han desarrollado numerosos métodos analíticos para la identificación y cuantificación de LM en matrices biológicas, ambientales y alimenticias. En las matrices biológicas la determinación de LM permite estudiar sus parámetros farmacocinéticos, así como la concentración plasmática y fecal para diseñar mejores planes de tratamiento farmacológico. En cuanto a las matrices alimenticias, las LM suelen permanecer por periodos prolongados y acumularse en los tejidos y órganos de los animales tratados1,38 o excretarse a través de la leche56 lo cual puede ser perjudicial para los consumidores. Además, la presencia de residuos de LM en el medio ambiente puede tener un impacto negativo ya que estos compuestos son tóxicos para organismos acuáticos y terrestres.5 En los últimos años, los métodos desarrollados se han orientado al análisis de matrices biológicas como heces, plasma, suero sanguíneo, y diferentes tejidos animales. También ha habido importantes aportes en el análisis de matrices alimenticias y ambientales. Aunque varios métodos se han desarrollado para determinar LM individuales, la mayoría son métodos multiresiduo donde se determinan dos o más LM simultáneamente (Tabla 1). En el desarrollo de nuevos métodos analíticos se ha buscado mejorar las características analíticas del método, así como el aspecto medioambiental; por lo que muchos de estos métodos se caracterizan por el uso de cantidades relativamente bajas reactivos y generar pocos residuos.
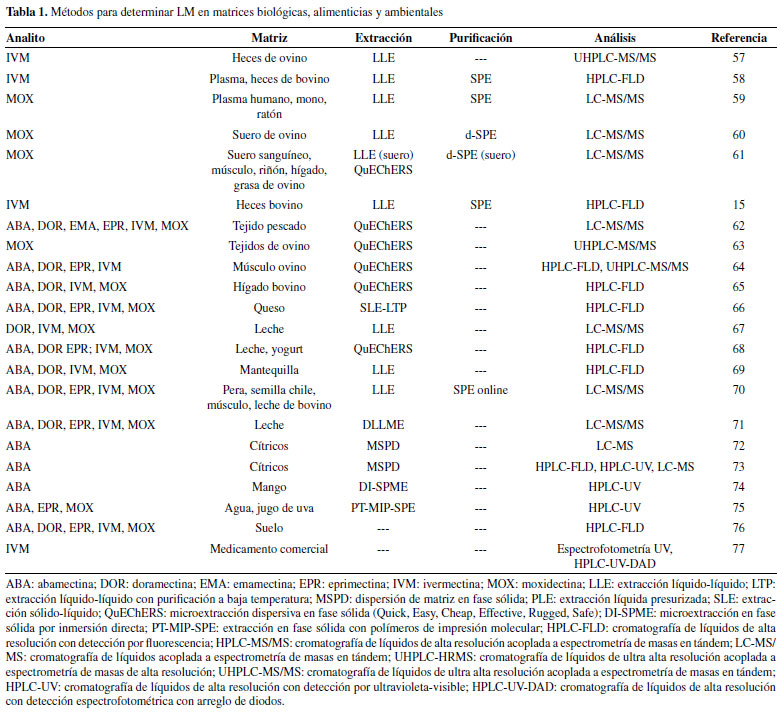
En la Tabla 1 se observa que para el análisis de muestras semisólidas y líquidas (heces y el plasma) la principal técnica de extracción que se utiliza es la LLE seguida de una etapa de limpieza o purificación por SPE. En el caso de los tejidos animales, generalmente con un alto contenido de grasa, predomina el método QuEChERS ya que es eficiente para extraer las LM así como para eliminar interferentes polares y no polares. Por otro lado, en la leche y sus derivados se utilizan tanto la LLE como el método QuEChERS, ya que estas matrices se caracterizan por ser líquidas o semisólidas y también, por su lipofilicidad. Además, se han aplicado otras técnicas como la MSPD, DI-SPME, ASE, LTP, etc. en matrices como frutas, leche, agua, etc. Pretratamiento de la muestra El pretratamiento de la muestra sirve para aislar, preconcentrar y adecuar los analitos para las etapas subsecuentes de separación y detección.70 Es el paso que consume más tiempo, es laborioso y puede introducir errores al procedimiento analítico; además, es posible que durante su desarrollo se pierdan y/o descompongan los analitos y la muestra se contamine.78 Por ello, es una etapa crítica que afecta la calidad de los resultados analíticos, por lo tanto, es crucial seleccionar y optimizar un esquema apropiado.79 En la determinación de LM, se han empleado diferentes procedimientos para el pretratamiento analítico. Típicamente incluyen un paso de extracción y, en algunos casos, un paso adicional de limpieza. La elección del procedimiento adecuado depende del tipo de matriz y la técnica analítica que se utilizará.1 Por su lipofilicidad, las LM son típicamente extraídas con diferentes solventes orgánicos; luego, en la etapa de limpieza del extracto se aplica generalmente la extracción en fase sólida (SPE). Sin embargo, recientemente se han utilizado técnicas de extracción novedosas con las cuales la etapa de limpieza es relativamente más rápida que SPE y en algunos casos no es necesaria. Entre estas técnicas se puede mencionar el método QuEChERS (acrónimo de Quick [rápido], Easy [fácil], Cheap [barato], Effective [efectivo], Rugged [robusto], Safe [seguro]), la extracción sólido-líquido con partición a baja temperatura (SLE-LTP), la dispersión de matriz en fase sólida (MSPD), extracción líquido-líquido con purificación a baja temperatura (LTP), extracción acelerada con solventes (ASE) también llamada extracción líquida presurizada (PLE), extracción por adsorción con barra de agitación (SBSE) y membrana líquida con soporte de fibra hueca (HF-SLM). Plasma y suero sanguíneo El acetonitrilo (ACN) es el solvente más utilizado para la extracción de LM del plasma y suero sanguíneo debido a su polaridad intermedia. Ésta le permite incorporarse fácilmente a las matrices y, al mismo tiempo, disolver eficazmente a las LM. Para la limpieza de los extractos se utilizan comúnmente los cartuchos SPE de octadecilsílica (C18), un adsorbente no polar que retiene fuertemente a las LM, pero no a los interferentes. Estas condiciones se han aplicado para la extracción de IVM,80 MOX81 y DOR82 de plasma de ovino. También, en el análisis de plasma de bovino para extraer IVM,83-85 DOR y MOX86 y para la extracción de MOX de plasma87 y suero sanguíneo88 de equino. Los métodos más recientes aún emplean el ACN y la SPE (C18). Por ejemplo, Cantón et al. utilizaron ACN para extraer IVM de plasma de bovino y cartuchos SPE de C18 para purificar los extractos. La IVM se eluyó con metanol (MeOH).58 El MeOH es un solvente ligeramente más polar que el ACN capaz de desorber la IVM, pero minimizando la coelución de interferentes no polares que pudieran estar presentes en el plasma. Chhonker et al. emplearon MeOH para extraer MOX de plasma humano, de mono y de ratón en presencia de un amortiguador (buffer) de CH3COONa. Al igual que en el estudio anterior, la purificación se realizó con cartuchos de C18, pero el analito se eluyó con isopropanol,59 un solvente menos polar, que puede favorecer la extracción de la MOX, una LM más lipofílica que la IVM. En ambos métodos se obtuvieron recuperaciones superiores al 60%, lo que sugiere que la combinación de solventes fue adecuada. Baptista et al. y Fernandes et al. desarrollaron un método diferente para determinar MOX en suero sanguíneo de cordero. La MOX se extrajo con ACN y la purificación se realizó por SPE dispersiva (dSPE). En esta técnica los adsorbentes utilizados en la SPE tradicional se agregan al extracto. Los autores concluyeron que se obtenían extractos con menos interferentes en dSPE con el uso combinado de los adsorbentes C18 y etilendiamina-N-propil sílica (PSA), en lugar de emplear dichos adsorbentes por separado.60,61Al aplicar la dSPE se evita la necesidad de utilizar cartuchos SPE comerciales, lo que simplifica y abarata el proceso de extracción pues ya no se requiere de etapas de acondicionamiento y lavado, ni de dispositivos especiales para la elución. Además, dSPE permite combinar varios tipos de adsorbentes en diferentes proporciones de acuerdo con los requerimientos de cada análisis. En este caso, los autores combinaron dos adsorbentes de diferente polaridad: C18 (no polar) y PSA (polaridad media). Al combinarlos, los autores lograron abarcar un amplio espectro de polaridades y eliminar así una mayor cantidad de interferentes. Las recuperaciones alcanzadas fueron aceptables (80-107% )y el límite de cuantificación del método estuvo en el orden de los ng mL-1. Estos factores hicieron que esta técnica sea igual de confiable que la SPE tradicional. Heces El procedimiento para extraer LM de heces es similar al empleado en plasma y suero sanguíneo, pues se ha utilizado el ACN para extraer la IVM de heces de bovino83 y la MOX de heces de equino.87 En la extracción también se han usado otros solventes como la acetona e isooctano en la determinación de ABA e IVM en heces de bovino.14,89 En todos estos métodos los extractos se purificaron con cartuchos de C18. La combinación de ACN y C18 se ha reportado incluso en métodos más recientes, sobre todo en la extracción de IVM de heces de bovino.15,58,90 Sin embargo, el uso de la SPE en heces puede presentar inconvenientes ya que los sólidos suspendidos en la matriz pueden obstruir los cartuchos, haciendo más tardado y complejo el pretratamiento de la muestra. En su trabajo, Cantón et al. tuvieron que realizar más etapas de mezclado y centrifugación durante la extracción y purificación de las heces, en comparación con el plasma sanguíneo. No obstante, aunque obtuvieron recuperaciones aceptables (68%), fueron menores que las obtenidas en el plasma sanguíneo (79%).91 Para superar las dificultades que presenta SPE puede utilizarse la LLE sin la necesidad de una etapa de purificación. Vokrál et al. aplicaron esta técnica para extraer IVM de heces de cordero con acetato de etilo, un solvente menos polar que el ACN. La extracción se realizó en presencia de amonio concentrado y no fue necesaria una purificación posterior.57 A pesar de que con esta técnica el pretratamiento analítico se simplificó, la exactitud del método fue pobre pues se obtuvieron recuperaciones del 20-26%.57 Tejidos animales En los primeros métodos para determinar residuos de LM en tejidos animales se aplicaba la extracción con solventes y SPE, principalmente a través de la combinación de ACN y C18. Lifschitz et al. emplearon estas técnicas para extraer MOX de músculo, grasa e hígado de ovino92 y He et al. extrajeron ABA, DOR e IVM de músculo de bovino, porcino, ovino y pollo.91 Wang et al. extrajeron ABA, DOR, EPR e IVM de pescado, y de músculo e hígado de porcino.93 Hou et al. extrajeron estas mismas cuatro LM de hígado de bovino, aunque en este caso fue necesario el uso conjunto de cartuchos de octilsílica (C8) y C18 en la etapa de purificación del extracto.94 Ali et al. y Danaher et al. extrajeron ABA, DOR, EPR, IVM y MOX de hígado de bovino con ACN. Los extractos se pasaron a través de una columna de alúmina (Al) antes de purificarlos con cartuchos SPE de C18.95,96 En otros estudios donde también se ha aplicado el ACN para la extracción se han utilizado otros adsorbentes para la fase de limpieza. Nagata et al. utilizaron cartuchos SPE de aminopropil (NH2) para purificar los extractos en la determinación de ABA, DOR, EPR, IVM, MOX y milbemicina (MIL) en músculo e hígado de bovino.97 Por otro lado, Hernando et al. emplearon cartuchos SPE de Al en el análisis de músculo de salmón para determinar ABA, DOR, EMA e IVM.98 A diferencia del C18, el NH2 y la Al son adsorbentes de fase normal por lo que se utilizan para eliminar interferentes polares presentes en la matriz. En este tipo de aplicaciones, las LM no se retienen y son eluidas más fácilmente que con los adsorbentes no polares. La elución debe realizarse con el solvente adecuado, especialmente en el caso del NH2 donde se logró la elución completa de los analitos con una mezcla de MeOH y acetato de etilo97 a diferencia de la Al donde se utilizó únicamente ACN.98 En ambos estudios las recuperaciones fueron superiores al 60%97 y 70%.98 En estudios como el de Inoue et al. las LM se extrajeron directamente con isooctano, seguidas de una etapa de remoción de lípidos. De esta manera se determinaron ABA, DOR, EMA, EPR, IVM y MOX, entre otros fármacos, en músculo, hígado y grasa de bovino.99 A pesar de que el proceso fue relativamente sencillo, se utilizaron volúmenes relativamente elevados de solvente para la extracción (60 mL) y la deslipidación (40 mL). También se han reportado modificaciones en el proceso de extracción. Por ejemplo, en la ASE el proceso se ha automatizado aplicando temperatura y presión elevadas durante la extracción de los analitos. Xia et al. emplearon este procedimiento para extraer varias LM de músculo e hígado de bovino utilizando una mezcla ACN:agua (40:60) y C18 en el paso de purificación, obteniendo recuperaciones superiores al 85%.100 Tao et al. también emplearon este procedimiento para extraer ABA, DOR e IVM de músculo de porcino; y de músculo, riñón e hígado de bovino con ACN:MeOH (1:1) como solvente sin necesidad de un paso de limpieza adicional. Ellos reportaron recuperaciones por encima del 76%.101 Así, la técnica ASE permite alcanzar buenas recuperaciones de las LM. Sin embargo, es necesario contar con equipos especializados que encarecen el procedimiento. En los métodos desarrollados recientemente las técnicas de extracción se han diversificado. Carro et al. extrajeron ABA, DOR e IVM, junto con otros fármacos, de varias especies de pescado y mariscos por MSPD. En esta técnica, la muestra se dispersa en presencia de un adsorbente sólido donde queda retenido el analito, que luego se eluye con un solvente adecuado. En el estudio mencionado, se utilizó C18 como adsorbente y los analitos se eluyeron con ACN con ácido acético al 0.5%. Adicionalmente se utilizó sílica para la etapa de limpieza.102 La aplicación de la MSPD brinda la posibilidad de utilizar poca muestra (0.2 g) y obtener resultados con buena exactitud (84-112% de recuperación) y precisión (2.7-10.6 % de desviación estándar relativa, RSD). Otra técnica empleada para la etapa de limpieza ha sido la LTP, en combinación con una extracción con solventes. Rübensam et al. extrajeron ABA, DOR EPR, IVM y MOX de músculo de bovino aplicando extracciones sucesivas con ACN en presencia de NaCl. La mezcla se centrifugó y la fase superior se almacenó a-20 °C por 12 horas, tiempo en el que los autores determinaron que la mayoría de los interferentes (i.e. proteínas, grasas, etc.) se congelaron, mientras que los analitos se mantuvieron en fase líquida y se transfirieron fácilmente a otro recipiente.103 La LTP es sencilla y rápida de realizar, además el volumen de ACN empleado para la extracción (7.5 mL) fue menor en comparación con otras técnicas. Además, permitió obtener recuperaciones mayores a 88% y valores de RSD inferiores a 6%, lo que indica una buena exactitud y precisión. Entre las técnicas de extracción de LM de tejidos animales destaca el método QuEChERS. Este consiste en un paso de extracción, una partición líquido-líquido donde se adicionan sales inorgánicas y una etapa de limpieza denominada SPE-dispersiva (dSPE) en la que se agrega al extracto un adsorbente y un agente desecante para eliminar el agua residual. En el método original para cada una de estas etapas se utiliza ACN, MgSO4-NaCl; y PSA con MgSO4, respectivamente.104 Estas condiciones fueron aplicadas por Fernandes et al. para la extracción de MOX de músculo, riñón, hígado y tejido adiposo de ovino.61 En otros estudios se han incluido adsorbentes adicionales para mejorar la fase de limpieza del extracto. Rafidah et al. evaluaron la efectividad de C18, PSA y Al en la determinación de ABA, DOR, EMA, IVM y MOX de tejido muscular de pescado. De los adsorbentes evaluados, la combinación de PSA y C18 tuvo el mejor desempeño.10 Rúbies et al. extrajeron ABA, EMA, EPR, DOR, IVM y MOX de tejido muscular de bovino, ovino, porcino, conejo y ave basándose también en el método QuEChERS original, pero utilizaron ACN con 0.5% de amoniaco en la extracción para un mejor control del pH y C18 en la dispersión.105 Del Bianchi et al. aplicaron el método QuEChERS original sin modificaciones para extraer MOX de músculo, riñón, hígado y tejido adiposo de cordero. Sin embargo, para esta última matriz fue necesario agregar C18.63 El uso del C18 mejora la limpieza de los extractos con alto contenido de interferentes no polares, que son frecuentes en el tejido muscular animal.10,63 Por otro lado, el PSA es útil para eliminar ácidos orgánicos, como los ácidos grasos, que se presentan mayoritariamente en el tejido de pescado.10 Con la combinación de C18 y PSA se recuperaciones y valores de RSD, respectivamente, de 85.8-96.3% y < 11.8%10, 89.2-114.9% y < 17.3%105 y 71.6-111.2% y < 21.8%63. El método QuEChERS también se ha modificado utilizando ACN con 1% de ácido acético como solvente de extracción; y de CH3COONa más MgSO4, para favorecer la partición.106 Por otra parte, Anastassiades et al., además de las dos sales utilizadas originalmente, agregaron citrato tribásico dihidratado y citrato dibásico sesquihidratado de sodio en la partición líquido-líquido.107 Las sales adicionadas funcionan como amortiguadores permitiendo un mejor control del pH (5.5, aproximadamente) durante la extracción. Moschou et al. aplicaron el método QuEChERS amortiguado con acetato para extraer seis diferentes LM de tejidos de pescado y Pimentel-Trapero et al. extrajeron ABA, DOR, IVM y MOX de hígado de bovino empleando el método QuEChERS amortiguado con citrato. En ambos estudios se utilizó C18 en el paso de dSPE.62,65 El método QuEChERS amortiguado con acetato presentó mejores recuperaciones que el método amortiguado con citrato (90-103% y 81.7-93.8%, respectivamente), y requirió de una cantidad de muestra significativamente menor (2.5 g y 10 g, respectivamente). En algunos casos, el método QuEChERS ha sido la base para desarrollar procedimientos más simplificados. Por ejemplo, Lopes et al. extrajeron EMA de pescado aplicando únicamente la fase de extracción y partición con ACN:MeOH (75:25), MgSO4 y CH3COONa.108 Este método se aplicó para extraer EMA junto con 31 fármacos veterinarios más por lo que varias de las condiciones utilizadas (sales de extracción, solvente, etc.) se eligieron con base en la capacidad de extracción de la totalidad de los analitos y no únicamente de la EMA. Esta LM pudo extraerse bajo las condiciones establecidas, aunque en términos generales, su exactitud (78-112%) y precisión (6-20% RSD) fue menor a la de los otros compuestos analizados que alcanzaron recuperaciones superiores al 90% y RSD menores al 10%. Finalmente, Bandeira et al. desarrollaron un método multiresiduo para determinar ABA, DOR, EPR e IVM en tejido muscular de ovino. En este estudio se evaluaron y compararon cuatro metodologías QuEChERS: el método original, los métodos amortiguados con acetato y citrato, y un cuarto método propuesto por los autores. Se utilizó C18 y MgSO4 en la fase dispersiva de los tres primeros métodos. Por su parte, los autores propusieron una metodología utilizando Na2SO4 y NaCl como sales de extracción y la fase dispersiva se realizó únicamente con C18 sin ningún agente desecante. Aunque el desempeño de los cuatro métodos fue bueno, con el método propuesto se obtuvieron extractos más limpios, lo cual fue resultado de la combinación del Na2SO4 y un pH elevado.64 Además, aunque requiere una cantidad de muestra relativamente mayor (5 g), el método es sencillo, rápido y con un menor consumo de solventes. Leche y derivados En la determinación de LM en leche y sus derivados, al igual que con las matrices ya revisadas, uno de los procedimientos más utilizados es la extracción de los analitos con ACN en combinación con la SPE aplicando diversos adsorbentes. El C18 es uno de los adsorbentes más utilizados para extraer LM de leche de diferentes especies animales44,56,109-111 y sus derivados como suero, cuajo y queso.43,112 En otros estudios, se han utilizado cartuchos de C8113-115 y de NH2.116 Li et al. desarrollaron un método para determinar cinco LM en leche, pera, semillas de chile y músculo de bovino. Los analitos se extrajeron con ACN y se aplicó una SPE automatizada, lo cual redujo la manipulación de las muestras, la carga de trabajo y la cantidad de solvente utilizado.70 En la SPE se empleó como adsorbente un polímero hidrofóbico denominado poli(butil metacrilato-co-etileno glicol dimetacrilato), el cual fue desarrollado específicamente para la determinación de las LM. De acuerdo con los autores, el polímero pudo reutilizarse más de 500 veces, a diferencia de los adsorbentes tradicionales que tienen que desecharse. Esto representa una ventaja sobre otros métodos desde el punto de vista económico, práctico y ambiental. Respecto al desempeño, las recuperaciones (71.8-100.1%) y los valores RSD (< 10%) reportados indican que el método fue adecuado para su aplicación. En menor medida se ha aplicado la extracción con solventes. Schenck y Langman extrajeron ABA, DOR, IVM y MOX de leche con extracciones sucesivas usando diferentes solventes orgánicos como etanol, acetato de etilo, isooctano y ACN.117 Giannetti et al. utilizaron ACN para extraer ABA, DOR, EMA, EPR, IVM y MOX de leche, tejido muscular y huevo.118 Dahiya et al. determinaron IVM, DOR y MOX en leche extrayendo los analitos con ACN:MeOH (50:50).67 En estos tres métodos, los analitos fueron reextraídos con hexano, un solvente menos polar, para descartar cualquier interferente que pudo haberse extraído en la primera etapa y no hubo necesidad de aplicar ningún tipo de limpieza adicional. En otro estudio, Macedo et al. extrajeron cuatro LM de mantequilla con una mezcla de ACN, acetato de etilo y agua. La elección de este sistema de solventes se basó en la buena solubilidad de las LM en el ACN, en la eficiencia del acetato de etilo para interactuar con la matriz, y el agua se empleó para favorecer la partición y mejorar la extracción.69 Las recuperaciones alcanzadas en cada método fueron 83.5-113.8%,117 65.4-88.9%,118 92-102%67 y 72.4-106.5% que pueden considerarse adecuadas. Es importante mencionar que la extracción con solventes se caracteriza por su sencillez y rapidez; sin embargo, en ocasiones estas ventajas pueden pasar a segundo plano cuando debido a la complejidad de la muestra el número de extracciones y la cantidad de solvente utilizado aumentan considerablemente. En el caso de los métodos anteriores fue necesario emplear al menos 9 mL de solvente por cada extracción realizada, por lo que al momento de aplicarlos a análisis rutinarios con un elevado número de muestras puede tener un impacto negativo en términos prácticos, económicos y medioambientales. El método QuEChERS se ha aplicado a este tipo de muestras. Whelan et al. emplearon el método original para extraer ABA, DOR, EMA, EPR, IVM y MOX de leche, aunque debido a la composición de la matriz emplearon C18 en la etapa de dSPE.119 Furlani et al. aplicaron el mismo método en la determinación de cinco LM en leche y yogurt, con PSA y MgSO4 en el paso de dSPE.68 En el primer estudio se reportaron recuperaciones superiores al 90% y valores de RSD por debajo de 20%; mientras que en el segundo los valores reportados fueron de 83% y < 15%, respectivamente. La exactitud y precisión de ambos métodos fueron similares, pero se pueden destacar algunas diferencias: el método desarrollado por Whelan et al. no sólo es aplicable a las LM sino que se desarrolló para determinar otros 34 antihelmínticos más, lo cual indica que se trata de un método multiresiduo. Por otro lado, el método de Furlani et al. es adecuado para determinar únicamente las LM, pero puede aplicarse en dos matrices diferentes (leche y yogurt), lo que le confiere una mayor versatilidad. En otros estudios se desarrollaron procedimientos similares a QuEChERS. Aguilera-Luiz et al. y Gómez et al. determinaron ABA, EMA e IVM en leche y queso, respectivamente, mediante una extracción con ACN acidificado en combinación con EDTA disódico utilizando MgSO4 y CH3COONa para promover el proceso de partición, y sin la necesidad de un paso adicional de limpieza.120,121 Al igual que en otros métodos, la eliminación del paso de limpieza simplifica y agiliza notablemente el pretratamiento de la muestra: los autores reportaron que es posible realizar la extracción en menos de cuatro120,121 y nueve120,121 minutos. Por otro lado, las recuperaciones de ambos métodos resultaron adecuadas; sin embargo, las obtenidas en el análisis de queso (84.5-109.8%) fueron superiores a las del análisis de leche (70.6-106.4%). Rübensam et al. aplicaron la extracción con solventes y la LTP para determinar ABA, DOR, EPR, IVM y MOX de leche empleando varias adiciones de ACN con NaCl en la fase de extracción para después congelar los extractos durante 12 h y recuperar los analitos de la fase líquida.122 De Souza Santos Cheibub et al. desarrollaron un método multiresidual con estas mismas técnicas para determinar cinco diferentes LM en queso. En este caso, se empleó ACN y agua en la extracción. Como ya se mencionó, la LTP tiene la ventaja de ser una técnica rápida y, aunque la etapa de congelamiento podría parecer tardada, es un proceso que no requiere supervisión y los extractos quedan prácticamente libres de agua residual después de la extracción.66 Otro aspecto importante es el relativo bajo consumo de solventes y que se requiere de poca muestra para realizar la extracción. En particular, de Souza Santos Cheibub obtuvieron mejores recuperaciones con muestras pequeñas (0.5 y 1 g) en comparación con muestras de mayor tamaño (2.5 g).66 Las recuperaciones de ambos métodos fueron mayores a 90%, aunque el segundo tuvo mejor precisión (RSD < 5%). La MSPD fue empleada por García-Mayor et al. para extraer IVM, junto con otros fármacos, de leche de oveja. Para adsorber los analitos se utilizó arena de mar comercial lavada y Na2SO4. La elución se realizó con una mezcla de MeOH y acetato de etilo después de un lavado con hexano para extraer las grasas interferentes.123 A diferencia de otros métodos basados en MSPD, donde se ha empleado C18 como adsorbente,102 en el descrito anteriormente se optó por utilizar arena de mar ya que con el C18 se obtuvieron bajas recuperaciones además de que la mezcla formada durante la dispersión dificultó la elución del extracto. Es importante mencionar que, al igual que en otros métodos multiresiduales, en éste se determinaron otros tipos de macrólidos además de la IVM. Las recuperaciones para este analito estuvieron en el rango de 89.4-98% con una excelente precisión (RSD < 3%). Por otra parte, Campillo et al. extrajeron cinco LM de diferentes tipos de leche utilizando la microextracción dispersiva líquido-líquido (DLLME) en la cual las LM son extraídas con un solvente orgánico que es dispersado en gotas pequeñas en un solvente diferente. En este estudio, las LM fueron extraídas con cloroformo, el cual se dispersó en ACN. Éste último se dejó sedimentar para descartarlo. Esta técnica es igual de rápida que el método QuEChERS, pero tiene la ventaja de utilizar una menor cantidad de solventes.71 En este método sólo fue necesario utilizar 2 mL de ACN más 200 µL de cloroformo en comparación con los 10 mL de ACN que se utilizan típicamente en QuEChERS. Adicionalmente, las recuperaciones obtenidas con DLLME fueron mayores al 90%, este valor es igual o mayor a las recuperaciones obtenida por QuEChERS. Alimentos grasos La determinación de LM en alimentos con alto contenido de grasas es menos frecuente e involucra algunas técnicas de extracción no reportadas para otras matrices. En dos diferentes estudios Hernández-Borges et al. determinaron ABA en pulpa de aguacate. En el primero, la ABA se extrajo con una mezcla ACN:agua (4:1) aplicando ultrasonido, y el extracto se purificó con cartuchos SPE de C18.124 En su siguiente trabajo, utilizaron la misma mezcla, pero usaron una extracción asistida con microondas (MAE) y cartuchos de C18. La aplicación del ultrasonido o las microondas resultan en un proceso mejor controlado, más rápido y con un menor consumo de reactivos.125 Aunque en ambos métodos se obtuvieron recuperaciones superiores al 87% (RSD < 12%), éstas fueron ligeramente mejores cuando se utilizó la MAE, lo que indica que esta técnica es capaz de alcanzar una mayor exactitud y precisión en comparación con el ultrasonido. Huang et al. determinaron ABA e IVM en diferentes tipos de aceite vegetal y en manteca de cerdo. Los analitos se extrajeron con ACN saturado de hexano y se aplicó la LTP para limpiar los extractos a-30 °C durante 16 h. Este procedimiento se comparó con una SPE con Al y se obtuvieron resultados similares: recuperaciones superiores al 71% y 80%; y valores de RSD inferiores a 11 y 12% para LTP y SPE, respectivamente. Sin embargo, la LTP tiene la ventaja de ser un proceso más rápido y utilizar una menor cantidad de solvente que la SPE.126 Frutas y vegetales Filho et al. aplicaron la microextracción en fase sólida por inmersión directa (DI-SPME) para extraer ABA de pulpa de mango junto con otros plaguicidas. En esta técnica, la muestra se pone en contacto con una fibra cubierta de un material adsorbente (polidimetilsiloxano) donde el analito queda retenido. Posteriormente, esta fibra se coloca directamente en el equipo, donde el analito es desorbido utilizando un solvente adecuado (ACN).74 Aunque es una técnica sencilla de realizar, presenta algunas desventajas prácticas como el tiempo requerido para llevar a cabo la extracción (alrededor de 1 h) y la necesidad de contar con una interfase que permita conectar el sistema de extracción con el instrumento de medición. La recuperación de ABA fue aceptable (76-82.4%) aunque inferior a la obtenida con otros métodos. Valenzuela et al. extrajeron ABA de naranja aplicando la MSPD con C18 como adsorbente y diclorometano para la elución. Las principales ventajas de MSPD es su sencillez y que puede aplicarse con material típico de laboratorio. Además, requiere cantidades relativamente pequeñas de muestra y solventes, lo que hace a MSPD más económica en comparación con otras técnicas. Los métodos desarrollados con esta técnica presentaron buena exactitud y precisión (R > 90% y RSD < 5%).72,73 Muestras de interés ambiental El análisis de muestras ambientales para determinar LM se ha enfocado, principalmente, en agua y suelo. Raich-Montiu et al. determinaron IVM y dos de sus metabolitos en agua corriente y residual.127 La extracción se realizó con una membrana hueca con soporte líquido (HF-SLM). Este tipo de dispositivos están conformados por un polímero hidrofóbico que funciona como soporte, un agente extractor, por lo general una molécula anfifilica; y un solvente orgánico.128 El analito es transferido a través del solvente orgánico desde la fase acuosa original a otra fase acuosa conformada por el agente extractor donde finalmente queda retenido.129 Raich-Montiu et al. utilizaron una membrana con soporte de polipropileno, y bicarbonato de amonio y undecano, como agente extractor y solvente orgánico, respectivamente. Los autores compararon la selectividad y la precisión de la HF-SLM y la SPE. Determinaron que estadísticamente se obtenían los mismos resultados con ambas técnicas; sin embargo, la HF-SLM fue un método más rápido, barato y que generaba menos residuos.127 En otro estudio, Giordano et al. extrajeron ABA de agua superficial utilizando la extracción por sorción con barras de agitación (SBSE). Esta técnica es similar a la SPME, pero con un mayor volumen de fase adsorbente, la cual es colocada como recubrimiento a una barra de agitación magnética y se pone en contacto con la matriz a analizar. Después de un tiempo, el analito se desorbe de la barra con un solvente adecuado. La fase utilizada en este estudio fue polidimetilsiloxano y el analito se desorbió con MeOH.130 En el método desarrollado se extrajeron 15 plaguicidas además de la ABA y se optimizaron algunos parámetros críticos como el volumen de la muestra, los tiempos de agitación y de desorción, adición de sales, etc. En general, se reportaron recuperaciones por debajo de 70%, pero en el caso específico de la ABA las recuperaciones no superaron el 8%. Sin embargo, los autores afirman que su principal objetivo fue desarrollar un método selectivo y práctico. En ese aspecto, el método fue capaz de identificar correctamente los analitos. En otro método reportado, Krogh et al. extrajeron ABA, DOR, EMA, EPR, IVM, MOX y SEL de muestras de agua, sedimentos y suelo. En las muestras sólidas se aplicó la ASE con MeOH:agua (1:1) y los extractos se purificaron con SPE. Las muestras acuosas solo se pasaron a través de cartuchos de SPE.131 En este trabajo se evaluaron varios parámetros importantes en la extracción como el adsorbente, el pH, el solvente de elución, etc. Se probaron varios adsorbentes SPE (C18, Al, HLB, etc.) con diferentes características estructurales y de adsorción. El HLB, un adsorbente polimérico de fase reversa, mostró el mejor desempeño. En este sentido, resulta interesante que con el C18 se obtuvieran bajas recuperaciones a pesar de ser uno de los adsorbentes más utilizados para extraer LM. También se evaluaron diferentes valores de pH, donde resultó que con el HLB y un pH de 7 se obtuvieron las recuperaciones más altas. En la validación final se obtuvieron recuperaciones de 70-88% para la mayoría de los analitos, con una precisión adecuada (RSD 8-23%). Recientemente, Rath et al. desarrollaron un método multiresiduo para determinar LM y otros antimicrobianos veterinarios en diferentes tipos de suelo. En este estudio se hicieron pruebas de adsorción-desorción mediante un procedimiento de equilibrio donde se establecieron diversas condiciones experimentales para cada analito. En este análisis, no se aplicó ningún pretratamiento, únicamente fue necesario filtrar la muestra antes del análisis instrumental.76 Finalmente, también se han analizado algas marinas. Lorenzo et al. determinaron ABA, DOR e IVM en algas marinas aplicando la ASE con ACN como eluyente y carbón negro grafitizado (GCB), Florisil y arena como adsorbentes; no se aplicó ningún paso de limpieza adicional.132 El GCB ayudó a eliminar los pigmentos presentes en las muestras, y en combinación con el Florisil permitió obtener recuperaciones aceptables (92-120%) para las avermectinas analizadas. Análisis instrumental de lactonas macrocíclicas Las LM tienen poca volatilidad y un elevado peso molecular. Así, la técnica analítica predominante para su separación, detección y cuantificación es la cromatografía de líquidos de alta y ultra alta resolución (HPLC y UHPLC, respectivamente) seguida de diferentes detectores cuya elección depende del nivel de concentración de las LM en la muestra. Si la concentración está en el orden de mg kg-1, la detección por ultravioleta (UV) es la más adecuada; mientras que si está en el orden de μg kg-1 se prefiere la detección por fluorescencia (FLD) o por espectrometría de masas (MS y MS/MS).1 En los análisis de LM por HPLC/UHPLC se emplean columnas de fase inversa como C8 o C18 con longitud de 50 a 300 mm, diámetro interno de 2.1 a 4.6 mm y tamaño de partícula de 1.7 a 5.0 μm; en ocasiones, se utilizan pre-columnas,59,60,62,69,93,102,103,116,122,124,125,127,130,132 que contienen la misma fase estacionaria que la columna. En la mayoría de los estudios, la temperatura de la columna se mantiene constante (25-60 °C) durante la separación. La composición de la fase móvil varía y depende del detector. Se suele emplear una mezcla de solvente orgánico y agua. El solvente orgánico, que se utiliza en una proporción de 70% o mayor, puede ser ACN, MeOH o una mezcla de ambos, aunque también se han empleado otros solventes. Kolberg y Bandeira et al. utilizaron una fase móvil compuesta por THF, ACN y MeOH para separar dos y cuatro LM, respectivamente;64,111 de Souza Santos Cheibub et al. separaron cinco LM con una combinación de ACN e isopropanol.66 La cantidad de agua en la fase móvil depende del número de LM y del detector. Si se utiliza FLD se debe aumentar el carácter no polar de la fase móvil debido a que los productos derivatizados son menos polares que las LM sin derivatizar.1 En cuanto a la elución, se ha aplicado tanto la elución isocrática15,59,60,64,67,68,71,100,101,111,113,118,122,133 como la elución con gradiente.10,57,61-66,69,74,76,93,102,103,105,108,116,121-123,127,130,132,134 La primera se utiliza principalmente en FLD, mientras que la segunda se prefiere con MS. Esta última, es más selectiva y proporciona separaciones robustas con picos bien definidos.1 Cromatografía de líquidos con detección espectrofotométrica Las LM absorben fuertemente la luz debajo de los 300 nm, con máximos entre 238-266 nm37,48,135 por lo que pueden determinarse midiendo su absorbancia en la región UV. Macedo et al. aplicaron esta propiedad para determinar IVM en medicamentos comerciales. Compararon un método espectrofotométrico y un método cromatográfico (HPLC-UV-DAD) cuyos límites de detección (LOD) (0.093 y 0.310 mg L-1) y de cuantificación (LOQ) (0.016 y 0.055 mg L-1), respectivamente; fueron adecuados para determinar la IVM en los medicamentos. Sin embargo, cuando se analizaron muestras reales, el método espectrofotométrico sobreestimó las concentraciones hasta casi tres veces. Los autores concluyeron que el método HPLC-UV-DAD tuvo mejor desempeño, ya que la separación cromatográfica mejoró la selectividad y cuantificación del fármaco.134 Teixeira et al. desarrollaron un método para determinar ABA, EPR y MOX en agua y jugo de uva basado en una SPE donde el adsorbente es colocado en una punta de pipeta en vez de un cartucho convencional. Este dispositivo de extracción se acopló directamente al HPLC-UV lo que hizo al método rápido, simple, sensible y confiable.75 El límite de detección reportado fue de 25 ng mL-1 que se consideró adecuado para aplicar el método a muestras reales. Sin embargo, es importante tener cuidado en este aspecto, pues como los autores señalan, se ha reportado que la detección por UV no es adecuada para cuantificar algunas LM, como la ABA, ya que a bajas concentraciones los componentes de la matriz pueden interferir en el análisis.73 Cromatografía de líquidos con detección de fluorescencia La elección de HPLC-FLD para la determinación de residuos de LM se debe a su bajo costo, buena selectividad y sensibilidad, entre otras características. Además, algunos organismos oficiales, como la Comisión Europea, lo consideran un detector adecuado de confirmación para este tipo de fármacos según lo establecido en el Anexo 1 de la Directiva 96/23/EC.136 Debido a que las LM no son intrínsecamente fluorescentes, se requiere una derivatización antes del análisis cromatográfico para producir una molécula que presente fluorescencia. Tolan et al. desarrollaron el primer procedimiento de derivatización para determinar ABA e IVM en plasma sanguíneo. Utilizaron anhídrido acético y piridina como catalizador para producir la pérdida de dos grupos hidroxilo y dos protones en las posiciones 5 y 7 del anillo de benzofurano. Esta deshidratación produce un anillo aromático en conjugación con un grupo funcional dieno lo que da como resultado la fluorescencia (Figura 9). Estas condiciones también producen la acetilación de los grupos hidroxilo en R4" y R23 cuando están presentes. La reacción se completó de 22-24 horas a una temperatura de 105-110 °C y los derivados fluorescentes se purificaron con sílica antes del análisis.137
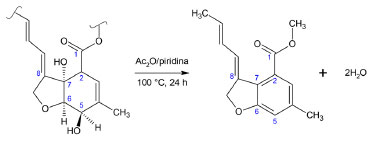 Figura 9. Reacción de derivatización de la avermectina (estructuras parciales)137
Tway et al. sustituyeron la piridina por dimetilformamida y N-metilimidazol (N-MImdz), un catalizador nucleofílico más eficaz, y produjeron el mismo derivado fluorescente en 60 min a 90 °C y, al igual que Tolan et al. antes de la determinación purificaron los derivados en cartuchos SPE de sílica.138 Por otro lado, de Montigny et al. redujeron notablemente el tiempo de reacción a 30 s reemplazando el anhídrido acético por anhídrido trifluoroacético (TFAA) para producir un mejor grupo saliente y utilizaron N-MImdz como catalizador a temperatura ambiente, sin aplicar una limpieza adicional.139 En estos primeros estudios se logró disminuir el tiempo de la derivatización sin la necesidad de aplicar condiciones extremas y sin un paso de limpieza post-derivatización. Sin embargo, algunos de los derivados fluorescentes, como el de la EPR, son relativamente inestables por lo que se han desarrollado métodos para aumentar su estabilidad. Payne et al. emplearon una derivatización pre-columna automatizada140 y Ali et al. incluyeron una etapa de calentamiento a 65 °C y fue necesario aumentar el tiempo de reacción de 30 s a 90 min.96 Danaher et al. modificaron este último procedimiento adicionando una mínima cantidad de ácido acético glacial. La reacción tomó 30 min a 65 °C y se obtuvieron derivados igualmente estables tanto para la EPR como para las demás LM involucradas en el estudio.95 Rupp et al. reportaron que durante la derivatización de la IVM se formaron dos especies: una hidroxilada y una esterificada, ésta última provoca una disminución en la señal y en la estabilidad del analito. Para corregir este problema adicionaron una pequeña cantidad de acetato de amonio con calentamiento a 55 °C para favorecer la formación del derivado hidroxilado.13 En contraparte, Berendsen et al. reportaron que con los derivados esterificados se obtuvieron mejores resultados en la determinación de IVM, DOR, ABA, EPR y MOX. Para la producción de estos derivados utilizaron trietilamina y ácido trifluoroacético además de los reactivos tradicionales (TFAA y N-MImdz).113 Felici et al. derivatizaron la IVM mediante una oxidación catalizada con dióxido de manganeso para producir un derivado 5-cetónico, el cual se tautomerizó a un fenol fluorescente agregando acetato de amonio. Aunque los resultados obtenidos en este estudio fueron similares a otros, la estabilidad del derivado fue relativamente baja.141 En resumen, aunque se han desarrollado varios procedimientos para mejorar la derivatización de las LM en términos de estabilidad, tiempo y sencillez; en la mayoría de los métodos se sigue aplicando la reacción de derivatización clásica donde se utilizan el TFAA y N-Mimdz como agentes derivatizantes, pues ha demostrado ser simple, rápida, ocurre a temperatura ambiente, no requiere condiciones especiales y produce compuestos estables. Cromatografía de líquidos con detección por espectrometría de masas La LC con detección por MS se utilizó inicialmente para la elucidación estructural de las LM y sus metabolitos28,37,88,142,143 ya que se prefería la LC-FLD para la cuantificación y confirmación de las LM por su alta selectividad. Por ejemplo, Valenzuela et al. compararon el desempeño de FLD y MS en el análisis de ABA en cítricos y, aunque ambos detectores mostraron resultados similares, concluyeron que la FLD es preferible para los análisis rutinarios por su selectividad y bajo costo. Sin embargo, algunos autores han reportado que la FLD es poco eficiente para la confirmación de las LM ya que puede producir falsos positivos, sobre todo si el análisis es a nivel de trazas144 por lo que en los últimos años se ha preferido MS. En algunos estudios se ha empleado FLD junto con MS para la cuantificación y la confirmación de LM, respectivamente. Rübensam et al. aplicaron esta estrategia en el análisis de cinco LM en leche122 y músculo de bovino.103 Bandeira et al. desarrollaron dos métodos empleando FLD y MS para determinar LM en músculo de ovino; sin embargo, concluyeron que la MS es más recomendable ya que incrementa la selectividad y disminuye la probabilidad de obtener falsos positivos.64 Cuando se utiliza la LC acoplada a MS para la confirmación y cuantificación de LM se utiliza predominantemente la configuración en tándem (LC-MS/MS) con diferentes tipos de ionización, entre los que destacan la ionización por electrospray (ESI) en modo positivo (ESI+)10,57,60-64,67,93,101-103,108,109,119-121,126,130,132 y negativo (ESI-),59,60,105,109,110,119 la ionización química a presión atmosférica (APCI) en modo positivo (APCI+)94,127,131 y negativo (APCI-),70,71 y el turboionspray.98 Cuando se utilizan ESI+ y APCI+, los iones que se monitorean son [M + Na]+, [M + K]+ y [M + H]+. La abundancia de dichos iones depende en gran medida de la composición de la fase móvil y de la estructura química de los analitos. Los iones [M + Na]+ se forman frecuentemente sin la necesidad de agregar aditivos debido a la presencia de Na+ en los solventes y en el material de laboratorio.145 Durden reportó que cuando se aplica ESI+ junto con una fase móvil compuesta por MeOH o ACN, agua y ácido fórmico o acético, el ion [M + Na]+ predomina en las LM-CHO, es decir, las que contienen únicamente carbono, hidrógeno y oxígeno (ABA, DOR e IVM), mientras que en las LM que contienen nitrógeno (EMA, EPR, SEL y MOX) predomina el ion [M + H]+.109 Por otro lado, Moschou et al. encontraron que el uso exclusivo de MeOH en la fase móvil producía mayor abundancia de los iones [M + Na]+ en comparación con el uso de ACN o una mezcla de ambos solventes utilizando ESI+.62 En otro estudio se reportó la presencia de iones [M + Li]+ cuya formación se favoreció con la adición de acetato de litio en la solución de inyección.145 En general, los iones [M + Na]+ tienen una intensidad elevada por lo que se utilizan para el análisis cuantitativo. Sin embargo, en la determinación de LM por ESI+ se ha observado que los iones [M + Na]+ son muy estables, difíciles de fragmentar y la abundancia de los iones resultantes es baja, lo que causa una disminución en la sensibilidad. Además, las curvas de calibración construidas con los iones [M + Na]+ son no lineales y poco reproducibles, y por lo tanto se pueden obtener resultados incorrectos.60,62,101,109,145,146 Una estrategia para evitar los problemas relacionados con los iones [M + Na]+ es reemplazarlos por iones amonio [M + NH4]+. Estos, junto con los iones monoprotonados [M + H]+ se fragmentan fácilmente y las intensidades de los iones resultantes son mayores que las obtenidas de la fragmentación de los iones [M + Na]+. Además, a diferencia de estos, los iones [M + NH4]+ y [M + H]+ pueden generarse tanto en ESI como en APCI. La formación de los iones [M + NH4]+ se favorece con la adición de formiato o acetato de amonio a la fase móvil.101,109,145,147,148 Durden reportó que, con una cantidad baja de formiato, la señal de los iones [M + NH4]+ en las LM-CHO es mayor que la señal de [M + Na]+, en tanto que en las LM nitrogenadas, predomina el ion [M + H]+ y es necesario agregar cantidades mayores de formiato para que la intensidad del ion [M + NH4]+ sea significativa.109 Esta estrategia ha sido aplicada en diversos métodos.57,61,62,64,67 Por otro lado, la formación de los iones protonados [M + H]+ puede favorecerse por la adición de ácido fórmico en el modo ESI+, aunque con estas condiciones también pueden formarse iones con carga doble ([M + 2H]2+) y triple ([M + 3H]3+), lo que puede provocar pérdida de sensibilidad en el análisis.62 En otros estudios se han utilizado las fuentes de ionización en modo negativo ya que es más sensible o reproducible que el modo positivo.59,60,109 Turnipseed et al. emplearon APCI- para confirmar la presencia de IVM, DOR, EPR y MOX en diferentes matrices alimenticias y encontraron que el ion desprotonado [M - H]- predominó para todos los analitos.149 Li et al. reportaron que una fase móvil compuesta de ACN al 10% y acetato de amonio mejora la ionización y la resolución con APCI-.70 También se ha reportado el uso de ESI-, donde la formación de los iones desprotonados de los analitos se ha favorecido por adición de trietilamina a la fase móvil.60,105,109 En la gran mayoría de los métodos desarrollados recientemente, el triple cuadrupolo (QqQ) se ha posicionado como el analizador de masas más utilizado en la identificación y cuantificación de LM ya que, en comparación con los instrumentos de un cuadrupolo, son más sensibles debido a que la fragmentación extra y las etapas de selección aumentan la selectividad y disminuyen el ruido. Además, son menos propensos al efecto de matriz y su rango dinámico es más amplio.1 La trampa de iones (IT) también se ha utilizado como analizador de masas en la determinación de LM.71,150-153 Algunas de las ventajas de la IT es que se puede generar un espectro completo sin perder sensibilidad; además, ciertos iones (i.e. [M + Na]+) son más fáciles de fragmentar en comparación con el QqQ. Sin embargo, el rango de masas es limitado y puede haber interferencia isobárica, lo cual afecta la sensibilidad y repetibilidad del método.1 A pesar de estas limitaciones, los métodos para determinar LM basados en el uso de IT han mostrado ser igual de sensibles, específicos y exactos que otros métodos que aplican FLD151, UV71 y con QqQ.150 Otro de los analizadores de masas que se ha aplicado en el análisis de LM es el tiempo de vuelo (TOF) el cual separa los iones aprovechando las diferentes velocidades de estos cuando se les aplica el mismo potencial.154 Los métodos desarrollados que hacen uso del TOF han sido lo suficientemente exactos, precisos y sensibles. La mayoría de estos métodos son multiresiduales que se han utilizado para la determinación exclusiva de avermectinas146 o en conjunto con otros fármacos veterinarios.155 Sin embargo, en algunos casos, se han presentado problemas en la cuantificación de las avermectinas por una baja señal analítica de los iones seleccionados y por interferencia de la matriz.156 Los instrumentos más recientes operan con analizadores de masas híbridos como la trampa de iones lineal cuadrupolar (QqQLIT) y el cuadrupolo con tiempo de vuelo (Qq-TOF), los cuales funcionan de manera similar al QqQ, pero el último cuadrupolo es sustituido por una IT o un TOF, respectivamente. Estos analizadores se han aplicado en la determinación de LM con menos frecuencia que el QqQ; sin embargo, se han obtenido buenos resultados. Hernando et al. desarrollaron un método LC-MS/MS con QqQLIT para identificar y cuantificar ABA, DOR, EMA e IVM en muestras de salmón y pimiento. La selectividad del QqQLIT en combinación con el modo de monitoreo de reacciones múltiples permitió confirmar y cuantificar las avermectinas a nivel de trazas.98 En un estudio más reciente, Zrnčić et al. desarrollaron y validaron un método UHPLC-QqLIT para determinar 10 diferentes antihelmínticos, entre ellos la MOX, en agua superficial obteniendo una separación rápida de los analitos y límites de detección en el orden de ng L-1.153 El analizador Qq-TOF se ha utilizado para la detección y cuantificación de IVM y algunos de sus metabolitos en muestras ambientales127 y biológicas.157 Kang et al. aplicaron el sistema híbrido (IT)-TOF/MS para el monitoreo de más de 100 fármacos veterinarios. Este sistema facilita la identificación y elucidación de los compuestos en muestras complejas; sin embargo, en el caso de las avermectinas no mostró la sensibilidad suficiente para estimarlas correctamente.158 Una de las técnicas de detección más novedosas que se acoplan a LC es la espectrometría de masas de alta resolución (HRMS), que se basa en el uso de analizadores de masas híbridos como el cuadrupolo-Orbitrap (Q-Orbitrap). Kaufmann et al. analizaron el desempeño de un instrumento Orbitrap de etapa simple para determinar IVM, ABA y otros antihelmínticos en leche y tejido muscular. La alta resolución (50000 FWHM) del equipo permitió una detección selectiva y sensible de los analitos sin la necesidad de fragmentar los iones precursores, lo que además hace que la cuantificación sea más fácil que en LC-MS/MS. Esto fue especialmente beneficioso para el análisis de las avermectinas, que forman iones poco intensos y/o muy resistentes a la fragmentación ([M-Na]+).159 Rúbies et al. exploraron la aplicación del Q-Orbitrap acoplado a UHPLC en la determinación de varias LM. Evaluaron el desempeño de los modos de operación escaneo (full scan-all ion fragmentation, aif) completo y tSIM-dd MS/MS, y de la ionización por electrospray calentado en modo positivo (HESI+) y negativo (HESI-). En el escaneo completo no se utiliza el cuadrupolo y todos los iones son fragmentados, en el modo tSIM-dd MS/MS sólo se monitorean y fragmentan los iones precursores. En general, la técnica mostró un desempeño excelente. Cuando se operó en AIF, el modo HESI+ fue más sensible que el modo negativo. En contraste, cuando se trabajó con tSIM-dd MS/MS, ambos tipos de ionización tuvieron un desempeño similar, y se detectaron las LM a concentraciones de 0.1-0.5 µg kg-1.105 Wang et al. desarrollaron y validaron un método basado en la misma técnica para determinar residuos de 105 fármacos veterinarios en leche, incluyendo seis diferentes LM. El método mostró buena exactitud, repetibilidad y sensibilidad para la cuantificación, además de que fue posible identificar los fármacos mediante sus espectros.160
VALIDACIÓN DE MÉTODOS La mayoría de los métodos desarrollados para determinar LM son validados siguiendo los criterios establecidos en la Directiva 2002/657/CE, documento emitido por la Comisión Europea161 donde se propone un enfoque sistemático para validar los métodos analíticos. Según este documento, las características analíticas que deben determinarse para validar los métodos analíticos cuantitativos son, entre otros, la veracidad o recuperación, precisión, especificidad, robustez, linealidad, límite de decisión (CCα) y capacidad de detección (CCβ). La robustez es otra característica que se recomienda evaluar. La Directiva 2002/657/CE establece dos tipos de robustez: la robustez por cambios menores y la robustez por cambios importantes, dependiendo de la magnitud de las modificaciones que se realicen.161 Esta característica es especialmente complicada porque implica la realización de numerosas pruebas y en general, es difícil establecer cuáles son los factores que deben ser modificados.162 En los métodos de determinación de LM, la robustez no se evalúa frecuentemente. Sin embargo, Galarini et al. evaluaron la robustez haciendo cambios importantes en su metodología, los cuales consistieron en analizar tejidos, leche y alimento de especies animales diferentes a las analizadas en la metodología original.116 Cerkvenik-Flajs et al. aplicaron una estrategia similar y evaluaron el impacto de utilizar leche de bovinos y ovinos en el desempeño de su método.115 Durden y Wostke realizaron una evaluación más profunda de la robustez. Modificaron ocho factores (volumen del solvente de extracción, tiempos de mezclado y centrifugación, pH del buffer de extracción, marca de los cartuchos de SPE, composición y volumen de las soluciones de lavado y volumen de elución) y midieron el efecto combinado e individual de estos factores sobre las recuperaciones, áreas de pico, concentraciones, etc., para lo cual aplicaron conjuntamente el método de Youden, el diseño Plackett-Burmann y la prueba Vander Heyden-Waters-Dovletoglou.110 De acuerdo con la Directiva 2002/657/CE la cuantificación en los métodos cromatográficos debe realizarse con el método del patrón interno.161 Para la determinación de LM suele adicionarse una avermectina o milbemicina diferente a las que se están determinando, aunque también se han empleado otros compuestos como benzo(k)fluoranteno.65 La Directiva 2002/657/CE también recomienda utilizar análogos con etiquetado isotópico especialmente adecuados para la MS,161 por lo que algunos estudios los han empleado.62,153 Otro aspecto que considerar en la determinación de las LM es la complejidad de las matrices analizadas, ya que pueden causar el aumento o la supresión de la señal analítica (efecto matriz). Debido a la ionización, el efecto matriz es más común cuando se utiliza la detección por MS, a diferencia de los detectores FLD y UV donde este proceso no ocurre.163 Una manera de verificar la presencia del efecto matriz es comparar las señales analíticas de las curvas de calibración preparadas en solvente y en presencia de la matriz. La magnitud y la naturaleza del efecto dependen del tipo de matriz y del analito. Por ejemplo, en el análisis de tejido muscular, la ABA, DOR e IVM mostraron un aumento similar de la señal analítica.91 En otro estudio, se encontró que el incremento de la señal por efecto de la matriz (leche) era menor en la DOR que en la IVM y MOX.67 En contraste, cuando se analizó agua, el efecto de la matriz sobre la MOX fue negativo, es decir, la señal analítica disminuyó.153 Moschou et al. encontraron efectos de supresión (EPR, ABA, IVM) y aumento (DOR y MOX) de la señal en el análisis de tejido de pescado.62 Una evaluación más profunda del efecto matriz la realizaron de Souza Santos Cheibub et al. Compararon estadísticamente las pendientes de las curvas de calibración en solvente y en matriz (queso). En este último caso se emplearon curvas preparadas con muestras individuales y agrupadas. En ninguno de los casos se observó algún efecto de la matriz, excepto en la EPR, que mostró un efecto leve en la muestra agrupada.66 Además de la adición de un patrón interno, el uso de curvas de calibración preparadas en presencia de la matriz y el método de adición estándar62,150,156,164 son otras estrategias para contrarrestar el efecto matriz. En la Directiva 2002/657/CE se establece que las curvas de calibración que se utilicen para la cuantificación deben contener, al menos, cinco niveles de concentración. Además, es necesario calcular el rango de trabajo, función de calibración, bondad de ajuste, entre otros parámetros estadísticos.161 Macedo et al. construyeron una curva de calibración con muestras de mantequilla fortificadas y calcularon el CCα y CCβ de la ABA, DOR, IVM y MOX a partir del valor del intercepto de la curva.69 Por otra parte, Rübensam et al. calcularon el CCα y CCβ de la ABA, DOR, EPR, EMA, IVM y MOX analizando 21 muestras de tejido de bovino fortificadas.103 A pesar de que la Directiva 2002/657/CE no contempla el cálculo de los límites de detección (LOD) y cuantificación (LOQ), estos parámetros son importantes, sobre todo para analitos que no tienen un valor de límite máximo permitido establecido.165 Por eso, la gran mayoría de los métodos revisados reportan el LOD y LOQ junto con el CCα y CCβ. Finalmente, otra característica de funcionamiento, según la Directiva 2002/657/CE, es la estabilidad del analito.161 Del Bianchi evaluaron la estabilidad a corto plazo de la MOX en muestras de tejido animal bajo diferentes condiciones de temperatura, luminosidad y humedad. También evaluaron el efecto de los ciclos de congelamiento/descongelamiento sobre la estabilidad del analito. Los autores concluyeron que la MOX es estable bajo condiciones estándar de laboratorio y después de ser congelada y descongelada en periodos de 24, 48 y 96 horas.63
CONCLUSIONES El desarrollo de nuevos métodos ha crecido notablemente en las últimas dos décadas y su alcance se ha hecho cada vez mayor, llegando a matrices poco convencionales (i.e. agua, tierra, algas). También, se han adaptado y desarrollado técnicas de extracción y análisis que permitan que la determinación sea más rápida y confiable. La mayoría de los métodos revisados se enfocan en el análisis de LM en heces y plasma sanguíneo con el fin de caracterizar su farmacocinética. El análisis de matrices biológicas y ambientales tiene como objetivo confirmar la presencia de LM y determinar si su concentración rebasa los límites permitidos. La extracción con ACN es el principal método de aislamiento de las LM, aunque se han utilizado otros solventes como el isooctano y la acetona. La principal técnica de purificación de los extractos sigue siendo la extracción en fase sólida (SPE). Sin embargo, en los últimos años se han aplicado el método QuEChERS, la dispersión de la matriz en fase sólida (MSPD), la extracción con purificación a baja temperatura (LTP), extracción acelerada con solventes (ASE), entre otras. La cromatografía de líquidos de alta resolución (HPLC) es la técnica más empleada, aunque en la actualidad la cromatografía líquida de ultra alta resolución (UHPLC) ha ganado terreno. La separación de los analitos se realiza aplicando sistemas de elución isocráticos y con gradiente, empleando columnas cromatográficas de fase reversa relativamente cortas. En algunos casos, es posible hacer que los extractos pasen directamente a la columna cromatográfica lo que agiliza los análisis y mejora el desempeño del método. La fase móvil es otro aspecto crítico y su composición depende de los analitos y el detector empleado. La detección de LM puede llevarse a cabo por UV/Vis, FLD, y MS. La fluorescencia se ha preferido por su selectividad y sensibilidad, además de que es una técnica de bajo costo en comparación con MS, pero para utilizarse con las LM es necesario realizar una derivatización. Sin embargo, recientemente se ha cuestionado las ventajas de esta técnica de detección y se ha sustituido paulatinamente por MS. La espectrometría de masas en tándem es la más utilizada, con diferentes modos de ionización y analizadores de masas. Recientemente se han desarrollado métodos basados en el uso de analizadores de masas como la trampa de iones lineal, el tiempo de vuelo y el Orbitrap que han mejorado considerablemente la sensibilidad y selectividad de los métodos, permitiendo, además, el análisis de más de 100 fármacos simultáneamente. También se revisaron brevemente las características analíticas más importantes que se evalúan en la validación de métodos de acuerdo con la Directiva 2002/657/CE, haciendo énfasis en su aplicación en los métodos de determinación de LM.
AGRADECIMIENTOS Patricia Esmeralda Vázquez-Quintal es estudiante del Doctorado Institucional en Ciencias Agropecuarias y Manejo de Recursos Naturales Tropicales de la Universidad Autónoma de Yucatán y recibió una beca del Consejo Nacional de Ciencia y Tecnología (Conacyt). Agradecemos a los Doctores J. A. Barrón, E. Gutiérrez y J. A. Rosado por sus valiosos comentarios y correcciones a este artículo.
REFERENCIAS 1. Danaher, M.; Howells, L. C.; Crooks, S. R. H.; Cerkvenik-Flajs, V.; O'Keeffe, M.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2006, 844, 175. 2. Prichard, R.; Ménez, C.; Lespine, A.; Int. J. Parasitol.: Drugs Drug Resist. 2012, 2, 134. 3. Shoop, W. L.; Mrozik, H.; Fisher, M. H.; Vet. Parasitol. 1995, 59, 139. 4. Campbell, W. C.; Curr. Pharm. Biotechnol. 2012, 13, 853. 5. Danaher, M.; Radeck, W.; Kolar, L.; Keegan, J.; Cerkvenik-Flajs, V.; Curr. Pharm. Biotechnol. 2012, 13, 936. 6. Humeres, E. C.; Morse, J. G.; Exp. Appl. Acarol. 2005, 36, 51. 7. Pitterna, T.; Cassayre, J.; Hüter, O. F.; Jung, P. M. J.; Maienfisch, P.; Kessabi, F. M.; Quaranta, L.; Tobler, H.; Bioorg. Med. Chem. 2009, 17, 4085. 8. Yoshii, K.; Kaihara, A.; Tsumura, Y.; Ishimitsu, S.; Tonogai, Y.; J. Chromatogr. A 2000, 896, 75. 9. Fisher, M. H.; Mrozik, H.; Annu. Rev. Pharmacol. Toxicol. 1992, 32, 537. 10. Rafidah, I.; Ghanthimathi, S.; Fatimah, A. B.; Mahyudin, N. A.; Anal. Methods 2013, 5, 4172. 11. Macdonald, N.; Gledhill, A.; Arch. Toxicol. 2007, 81, 553. 12. Sung, Y. F.; Huang, C. T.; Fan, C. K.; Lin, C. H.; Lin, S. P.; Clin. Toxicol. 2009, 47, 686. 13. Rupp, H. S.; Turnipseed, S. B.; Walker, C. C.; Roybal, J. E.; Long, A. R.; J. AOAC Int. 1998, 81, 549. 14. Iwasa, M.; Maruo, T.; Ueda, M.; Yamashita, N.; Bull. Entomol. Res. 2007, 97, 619. 15. Pérez-Cogollo, L. C.; Rodríguez-Vivas, R. I.; Reyes-Novelo, E.; Delfín-González, H.; Muñoz-Rodríguez, D.; Bull. Entomol. Res. 2017, 107, 118. 16. Rodríguez-Vivas, R. I.; Basto-Estrella, G. del S.; Reyes-Novelo, E.; Arcila-Fuentes, W.; Ojeda-Chi, M.; Trinidad-Martínez, I.; Martínez-M, I.; J. Asia-Pac. Entomol. 2019, 22, 239. 17. Wang, Z.; Beier, R. C.; Shen, J.; TrAC - Trends Anal. Chem. 2017, 92, 42. 18. Campbell, W. C.; Fisher, M. H.; Stapley, E. O.; Albers-Schönberg, G.; Jacob, T. A.; Science 1983, 221, 823. 19. Campbell, W. C.; In Ivermectin and Abamectin; Campbell, W. C., Ed.; 1989; pp. 234. 20. Cvetovich, R. J.; Kelly, D. H.; DiMichele, L. M.; Shuman, R. F.; Grabowski, E. J. J.; J. Org. Chem. 1994, 59, 7704. 21. Dutton, C. J.; Gibson, S. P.; Goudie, A. C.; Holdom, K. S.; Pacey, M. S.; Ruddock, J. C.; Bu'Lock, J. D.; Richards, M. K.; J. Antibiot. 1991, 44, 357. 22. Goudie, A. C.; Evans, N. A.; Gration, K. A. F.; Bishop, B. F.; Gibson, S. P.; Holdom, K. S.; Kaye, B.; Wicks, S. R.; Lewis, D.; Weatherley, A. J.; Bruce, C. I.; Herbert, A.; Seymour, D. J.; Vet. Parasitol. 1993, 49, 5. 23. Bishop, B. F.; Bruce, C. I.; Evans, N. A.; Goudie, A. C.; Gration, K. A. F.; Gibson, S. P.; Pacey, M. S.; Perry, D. A.; Walshe, N. D. A.; Witty, M. J.; Vet. Parasitol. 2000, 91, 163. 24. Blizzard, T. A.; Org. Prep. Proced. Int. 1994, 26, 617. 25. Davies, H. G.; Green, R. H.; Chem. Soc. Rev. 1991, 20, 211. 26. Kornis, G. I.; In Agrochemicals from Natural Products; Godfrey, C. R. A., ed.; Marcel Dekker: New York, 1995; pp. 215. 27. Jung, M.; Saito, A.; Buescher, M.; Maurer, M.; Graf, J.-F.; In Macrocyclic lactones in antiparasitic therapy; Vercruysse, J., Rew, R. S., eds.; CABI Publishing: Wallingford, 2002; pp. 51. 28. Carter, G. T.; Nietsche, J. A.; Borders, D. B.; J. Chem. Soc., Chem. Commun. 1987, 6, 402. 29. Rock, D. W.; DeLay, R. L.; Gliddon, M. J.; In Macrocyclic lactones in antiparasitic therapy; Vercruysse, J., Rew, R. S., eds.; CABI Publishing: Wallingford, 2002; pp. 75. 30. Lespine, A.; Expert Opin. Drug Metab. Toxicol. 2013, 9, 1581. 31. Sun, P.; Zhao, Q.; Zhang, H.; Wu, J.; Liu, W.; ChemBioChem 2014, 15, 660. 32. Shoop, W. L.; Demontigny, P.; Fink, D. W.; Williams, J. B.; Egerton, J. R.; Mrozik, H.; Fisher, M. H.; Skelly, B. J.; Turner, M. J.; Int. J. Parasitol. 1996, 26, 1227. 33. Shoop, W. L.; Egerton, J. R.; Eary, C. H.; Haines, H. W.; Michael, B. F.; Mrozik, H.; Eskola, P.; Fisher, M. H.; Slayton, L.; Ostlind, D. A.; Skelly, B. J.; Fulton, R. K.; Barth, D.; Costa, S.; Gregory, L. M.; Campbell, W. C.; Seward, R. L.; Turner, M. J.; Int. J. Parasitol. 1996, 26, 1237. 34. Shoop, W.; Soll, M.; In Macrocyclic lactones in antiparasitic therapy; Vercruysse, J., Rew, R. S., eds.; 2002; pp. 1. 35. McKellar, Q. A.; Gokbulut, C.; Curr. Pharm. Biotechnol. 2012, 13, 888. 36. El-Saber Batiha, G.; Alqahtani, A.; Ilesanmi, O. B.; Saati, A. A.; El-Mleeh, A.; Hetta, H. F.; Magdy Beshbishy, A.; Pharmaceuticals 2020, 13, 196. 37. Takiguchi, Y.; Muramatsu, S.; Ide, J.; Mishima, H.; Terao, M.; J. Antibiot. 1983, 502. 38. Rodríguez-Vivas, R. I.; Arieta-Román, R. J.; Pérez-Cogollo, L. C.; Rosado-Aguilar, J. A.; Ramírez-Cruz, G. T.; Basto-Estrella, G.; Arch. Med. Vet. 2010, 42, 115. 39. González Canga, A.; Sahagún Prieto, A. M.; José Diez Liébana, M.; Martínez, N. F.; Vega, M. S.; Vieitez, J. J. G.; Vet. J. 2009, 179, 25. 40. Mckellar, Q. A.; Benchaoui, H. A.; J. Vet. Pharmacol. Ther. 1996, 19, 331. 41. Hayes, P. W.; In Australian Veterinary Association Congress; Canberra, Australia, 1994. 42. Conder, G. A.; Baker, W. J.; In Macrocyclic lactones in antiparasitic therapy; Vercruysse, J., Rew, R. S., eds.; CABI Publishing: Wallingford, 2002; pp. 30. 43. Imperiale, F.; Lifschitz, A.; Sallovitz, J.; Virkel, G.; Lanusse, C.; J. Dairy Res. 2004, 71, 427. 44. Imperiale, F. A.; Busetti, M. R.; Suárez, V. H.; Lanusse, C. E.; J. Agric. Food Chem. 2004, 52, 6205. 45. Alvinerie, M.; Sutra, J. F.; Galtier, P.; Mage, C.; Res. Vet. Sci. 2006, 67, 229. 46. Banks, B. J.; Bishop, B. F.; Evans, N. A.; Gibson, S. P.; Goudie, A. C.; Gration, K. A. F.; Pacey, M. S.; Perry, D. A.; Witty, M. J.; Bioorg. Med. Chem. 2000, 8, 2017. 47. Mrozik, H.; Linn, B. O.; Eskola, P.; Lusi, A.; Matzuk, A.; Preiser, F. A.; Ostlind, D. A.; Schaeffer, J. M.; Fisher, M. H.; J. Med. Chem. 1989, 32, 375. 48. Awasthi, A.; Razzak, M.; Al-Kassas, R.; Harvey, J.; Garg, S.; Chem. Pharm. Bull. 2012, 60, 931. 49. Mrozik, H.; Eskola, P.; Arison, B. H.; Albers-Schönberg, G.; Fisher, M. H.; J. Org. Chem. 1982, 47, 489. 50. Fraser-Reid, B.; Wolleb, H.; Faghih, R.; Barchi, J.; J. Am. Chem. Soc. 1987, 109, 933. 51. Hanessian, S.; Dube, D.; Hodges, P. J.; J. Am. Chem. Soc. 1987, 109, 7063. 52. Pivnichny, J. v.; Arison, B. H.; Preiser, F. A.; Shim, J. S. K.; Mrozik, H.; J. Agric. Food Chem. 1988, 36, 826. 53. Fink, D. W.; Anal. Profiles Drug Subst. Excipients 1988, 17, 155. 54. Stong, J. D.; Pivnichny, J. v.; ozik, H.; Waksmunski, F. S.; J. Pharm. Sci. (Philadelphia, PA, U. S.) 1992, 81, 1000. 55. Mrozik, H.; Eskola, P.; Reynolds, G. F.; Arison, B. H.; Smith, G. M.; Fisher, M. H.; J. Org. Chem. 1988, 53, 1820. 56. Imperiale, F. A.; Farias, C.; Pis, A.; Sallovitz, J. M.; Lifschitz, A.; Lanusse, C.; Food Addit. Contam., Part A 2009, 26, 57. 57. Vokřál, I.; Michaela, S.; Radka, P.; Jiří, L.; Lukás, P.; Dominika, S.; Kateřina, L.; Barbora, S.; Lenka, S.; Ecotoxicol. Environ. Saf. 2019, 169, 944. 58. Canton, C.; Canton, L.; Domínguez, M. P.; Moreno, L.; Lanusse, C.; Alvarez, L.; Ceballos, L.; Vet. Parasitol. 2018, 256, 43. 59. Chhonker, Y. S.; Sleightholm, R. L.; Murry, D. J.; Biomed. Chromatogr. 2019, 33, 1. 60. Baptista, R. C.; Fernandes, M. A. M.; Gilaverte, S.; Queiroz, S. C. N.; Assalin, M. R.; Ferracini, V. L.; Monteiro, A. L. G.; Reyes, F. G. R.; J. Braz. Chem. Soc. 2017, 28, 250. 61. Fernandes, M. A. M.; Gilaverte, S.; Bianchi, M. D.; da Silva, C. J. A.; Molento, M. B.; Reyes, F. G. R.; Monteiro, A. L. G.; Res. Vet. Sci. 2017, 114, 406. 62. Moschou, I. C.; Dasenaki, M. E.; Thomaidis, N. S.; J. Chromatogr. B 2019, 1104, 134. 63. del Bianchi A. Cruz, M.; Fernandes, M. A. M.; Patricia, P. A.; Monteiro, A. L. G.; Daniel, D.; Reyes, F. G. R.; J. Chromatogr. B 2018, 1072, 390. 64. Bandeira, N. M. G.; Ribeiro, L. C.; Rizzetti, T. M.; Martins, M. L.; Adaime, M. B.; Zanella, R.; Prestes, O. D.; J. Braz. Chem. Soc. 2016, 28, 878. 65. Pimentel-Trapero, D.; Sonseca-Yepes, A.; Moreira-Romero, S.; Hernández-Carrasquilla, M.; J. Chromatogr. B 2016, 1015-1016, 166. 66. de Souza Santos Cheibub, A. M.; Silva Bahiense de Lyra, E.; Pereira Netto, A. D.; Food Chem. 2019, 272, 148. 67. Dahiya, M.; Dubey, N.; Singh, P.; Singh, G. N.; Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2013, 52, 1313. 68. Furlani, R. P. Z.; Dias, F. F. G.; Nogueira, P. M.; Gomes, F. M. L.; Tfouni, S. A. V.; Camargo, M. C. R.; Food Control 2015, 48, 43. 69. Macedo, F.; Marsico, E. T.; Conte-Júnior, C. A.; de Resende, M. F.; Brasil, T. F.; Pereira Netto, A. D.; Food Chem. 2015, 179, 239. 70. Li, X.; Wang, M.-M.; Zheng, G.-Y.; Ai, L.-F.; Wang, X.-S.; J. Agric. Food Chem. 2015, 63, 4096. 71. Campillo, N.; Viñas, P.; Férez-Melgarejo, G.; Hernández-Córdoba, M.; J. Chromatogr. A 2013, 1282, 20. 72. Valenzuela, A. I.; Redondo, M. J.; Pico, Y.; Font, G.; J. Chromatogr. A 2000, 871, 57. 73. Valenzuela, A. I.; Popa, D. S.; Redondo, M. J.; Mañes, J.; J. Chromatogr. A 2001, 918, 59. 74. Filho, A. M.; dos Santos, F. N.; de Paula Pereira, P. A.; J. Sep. Sci. 2011, 34, 2960. 75. Teixeira, R. A.; Flores, D. H. Â.; da Silva, R. C. S.; Dutra, F. V. A.; Borges, K. B.; Food Chem. 2018, 262, 86. 76. Rath, S.; Fostier, A. H.; Pereira, L. A.; Dioniso, A. C.; de Oliveira Ferreira, F.; Doretto, K. M.; Maniero Peruchi, L.; Viera, A.; de Oliveira Neto, O. F.; Dal Bosco, S. M.; Martínez-Mejía, M. J.; Chemosphere 2019, 214, 111. 77. Pacheco-Silva, É.; de Souza, J. R.; Caldas, E. D.; Quim. Nova 2014, 37, 111. 78. de Koning, S.; Janssen, H. G.; Brinkman, U. A. T.; Chromatographia 2009, 69, 33. 79. Hyötyläinen, T.; Anal. Bioanal. Chem. 2009, 394, 743. 80. Alvinerie, M.; Sutra, J. F.; Galtier, P.; Vet. Res. 1993, 24, 417. 81. Alvinerie, M.; Escudero, E.; Sutra, J.-F.; Eeckhoutte, C.; Galtier, P.; Vet. Res. 1998, 29, 113. 82. Escudero, E.; Carceles, C. M.; Diaz, M. S.; Sutra, J. F.; Galtier, P.; Alvinerie, M.; Res. Vet. Sci. 1999, 67, 177. 83. Alvinerie, M.; Sutra, J. F.; Galtier, P.; Lifschitz, A.; Virkel, G.; Sallovitz, J.; Lanusse, C.; Res. Vet. Sci. 1999, 66. 84. Anastasio, A.; Esposito, M.; Amorena, M.; Catellani, P.; Serpe, L.; Cortesi, M. L.; J. Agric. Food Chem. 2002, 50, 5241. 85. Lifschitz, A.; Virkel, G.; Pis, A.; Imperiale, F.; Sanchez, S.; Alvarez, L.; Kujanek, R.; Lanusse, C.; Vet. Parasitol. 1999, 86, 203. 86. Lanusse, C.; Lifschitz, A.; Virkel, G.; Alvarez, L.; Sánchez, S.; Sutra, J. F.; Galtier, P.; Alvinerie, M.; J. Vet. Pharmacol. Ther. 1997, 20, 91. 87. Pérez, R.; Cabezas, I.; Godoy, C.; Rubilar, L.; Díaz, L.; Muñoz, L.; Arboix, M.; Alvinerie, M.; Arch. Med. Vet. 2001, 33, 77. 88. Afzal, J.; Burke, A. B.; Batten, P. L.; DeLay, R. L.; Miller, P.; J. Agric. Food Chem. 1997, 45, 3627. 89. Payne, L. D.; Hicks, M. B.; Wehner, T. A.; J. Agric. Food Chem. 1995, 43, 1233. 90. Fernandez, C.; San Andrés, M.; Porcel, M. A.; Rodriguez, C.; Alonso, A.; Tarazona, J. V.; Soil Sediment Contam. 2009, 18, 564. 91. He, L.; Zhao, D.; Su, Y.; Liu, Y.; Nie, J.; Lian, J.; J. AOAC Int. 2009, 92, 348. 92. Lifschitz, A.; Imperiale, F.; Virkel, G.; Cobeñas, M. M.; Scherling, N.; Delay, R.; Lanusse, C.; J. Agric. Food Chem. 2000, 48, 6011. 93. Wang, F.; Chen, J.; Cheng, H.; Tang, Z.; Zhang, G.; Niu, Z.; Pang, S.; Wang, X.; Lee, F. S. C.; Food Addit. Contam., Part A 2011, 28, 627. 94. Hou, X.; Jiang, H.; Ding, S.; Zhang, S.; Li, X.; Shen, J.; J. AOAC Int. 2006, 89, 1110. 95. Danaher, M.; O'Keeffe, M.; Glennon, J. D.; Howells, L.; Analyst (Cambridge, U. K.) 2001, 126, 576. 96. Ali, M. S.; Sun, T.; McLeroy, G. E.; Phillippo, E. T.; J. AOAC Int. 2000, 83, 31. 97. Nagata, T.; Miyamoto, F.; Hasegawa, Y.; Ashizawa, E.; J. AOAC Int. 2003, 86, 490. 98. Hernando, M. D.; Suárez-Barcena, J. M.; Bueno, M. J. M.; Garcia-Reyes, J. F.; Fernández-Alba, A. R.; J. Chromatogr. A 2007, 1155, 62. 99. Inoue, K.; Yoshimi, Y.; Hino, T.; Oka, H.; J. Sep. Sci. 2009, 32, 3596. 100. Xia, X.; Xiao, Z.; Huang, Q.; Xia, L.; Zhu, K.; Wang, X.; Shen, J.; Ding, S.; Chromatographia 2010, 72, 1089. 101. Tao, Y.; Yu, G.; Chen, D.; Pan, Y.; Liu, Z.; Wei, H.; Peng, D.; Huang, L.; Wang, Y.; Yuan, Z.; J. Chromatogr. B 2012, 897, 64. 102. Carro, A. M.; García-Rodríguez, D.; Gonzalez-Siso, P.; Lorenzo, R. A.; J. Sep. Sci. 2012, 35, 2866. 103. Rübensam, G.; Barreto, F.; Hoff, R. B.; Pizzolato, T. M.; Food Control 2013, 29, 55. 104. Anastassiades, M.; Lehotay, S. J.; Stajnbaher, D.; Schenck, F. J.; J. AOAC Int. 2003, 86, 412. 105. Rúbies, A.; Antkowiak, S.; Granados, M.; Companyó, R.; Centrich, F.; Food Chem. 2015, 181, 57. 106. Lehotay, S. J.; J. AOAC Int. 2007, 90, 485. 107. British Standard; British Standard 2008, 24, 1. 108. Lopes, R. P.; Reyes, R. C.; Romero-González, R.; Vidal, J. L. M.; Frenich, A. G.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 895-896, 39. 109. Durden, D. A.; J. Chromatogr. B 2007, 850, 134. 110. Durden, D. A.; Wotske, J.; J. AOAC Int. 2009, 92, 580. 111. Kolberg, D.; J. Braz. Chem. Soc. 2009, 20, 1220. 112. Imperiale, F.; Pis, A.; Sallovitz, J.; Lisfchitz, A.; Busetti, M.; Suárez, V.; Lanusse, C.; J. Food Prot. 2006, 69, 2424. 113. Berendsen, B. J. A.; Mulder, P. P. J.; van Rhijn, H. (J)A; Anal. Chim. Acta 2007, 585, 126. 114. Cerkvenik, V.; Doganoc, D. Z.; Skubic, V.; Beek, W. M. J.; Keukens, H. J.; Eur. Food Res. Technol. 2001, 213, 72. 115. Cerkvenik-Flajs, V.; Milčinski, L.; Süssinger, A.; Hodosček, L.; Danaher, M.; Antonić, J.; Anal. Chim. Acta 2010, 663, 165. 116. Galarini, R.; Saluti, G.; Moretti, S.; Giusepponi, D.; Dusi, G.; Food Addit. Contam., Part A 2013, 30, 1068. 117. Schenck, F. J.; Lagman, L. H.; J. AOAC Int. 1999, 82, 1340. 118. Giannetti, L.; Giorgi, A.; Necci, F.; Ferretti, G.; Buiarelli, F.; Neri, B.; Anal. Chim. Acta 2011, 700, 11. 119. Whelan, M.; Kinsella, B.; Furey, A.; Moloney, M.; Cantwell, H.; Lehotay, S. J.; Danaher, M.; J. Chromatogr. A 2010, 1217, 4612. 120. Aguilera-Luiz, M. M.; Martínes Vidal, J. L.; Romero-González, R.; Garrido Frenich, A.; J. Chromatogr. A 2008, 1205, 10. 121. Pérez, M. L. G.; Romero-González, R.; Vidal, J. L. M.; Frenich, A. G.; J. Sep. Sci. 2013, 36, 1223. 122. Rübensam, G.; Barreto, F.; Hoff, R. B.; Kist, T. L.; Pizzolato, T. M.; Anal. Chim. Acta 2011, 705, 24. 123. García-Mayor, M. A.; Gallego-Picó, A.; Garcinuño, R. M.; Fernández-Hernando, P.; Durand-Alegría, J. S.; Food Chem. 2012, 134, 553. 124. Hernández-Borges, J.; Ravelo-Pérez, L. M.; Hernández-Suárez, E. M.; Carnero, A.; Rodríguez-Delgado, M. A.; J. Chromatogr. A 2007, 1165, 52. 125. Hernández Borges, J.; Ravelo-Pérez, L. M.; Hernández-Suárez, E. M.; Carnero, A.; Rodríguez-Delgado, M. Á.; Chromatographia 2008, 67, 69. 126. Huang, J. X.; Lu, D. H.; Wan, K.; Wang, F. H.; Chin. Chem. Lett. 2014, 25, 635. 127. Raich-Montiu, J.; Krogh, K. A.; Granados, M.; Jönsson, J. A.; Halling-Sørensen, B.; J. Chromatogr. A 2008, 1187, 275. 128. Eljaddi, T.; Lebrun, L.; Hlaibi, M.; J. Membr. Sci. Res. 2017, 3, 199. 129. Lezamiz, J.; Jönsson, J. Å.; J. Chromatogr. A 2007, 1152, 226. 130. Giordano, A.; Fernández-Franzón, M.; Ruiz, M. J.; Font, G.; Picó, Y.; Anal. Bioanal. Chem. 2009, 393, 1733. 131. Krogh, K. A.; Björklund, E.; Loeffler, D.; Fink, G.; Halling-Sørensen, B.; Ternes, T. A.; J. Chromatogr. A 2008, 1211, 60. 132. Lorenzo, R. A.; Pais, S.; Racamonde, I.; García-Rodríguez, D.; Carro, A. M.; Anal. Bioanal. Chem. 2012, 404, 173. 133. Pérez, R.; Núñez, M. J.; Palma, C.; Riquelme, J.; Arboix, M.; J. Vet. Pharmacol. Ther. 2014, 37, 550. 134. Macedo da Costa, F.; Pereira Netto, A. D.; Quim. Nova 2012, 35, 616. 135. Mishima, H.; Milbemycin: A Family of Macrolide Antibiotics With Insecticidal Activity; International Union of Pure and Applied Chemistry, 1983. 136. The Commission of the European Communities; Off. J. Eur. Communities: Legis. 1996, 1. 137. Tolan, J. W.; Eskola, P.; Fink, D. W.; Mrozik, H.; Zimmerman, L. A.; J. Chromatogr. A 1980, 367. 138. Tway, P. C.; Wood, J. S.; Downing, G. v.; J. Agric. Food Chem. 1981, 29, 1059. 139. de Montigny, P.; Shim, J. S. K.; Pivnichny, J. v.; J. Pharm. Biomed. Anal. 1990, 8, 507. 140. Payne, L. D.; Mayo, V. R.; Morneweck, L. A.; Hicks, M. B.; Wehner, T. A.; J. Agric. Food Chem. 1997, 220, 3501. 141. Felici, E.; Wang, C. C.; Casado, C.; Vicario, A.; Pereyra, V.; Gómez, M. R.; Heliyon 2019, 5, 1. 142. Afzal, J.; Stout, S. J.; DaCunha, A. R.; Miller, P.; J. Agric. Food Chem. 1994, 42, 1767. 143. Stout, S. J.; DaCunha, A. R.; Wu, S.-S.; Zulalian, J.; Afzal, J.; J. Agric. Food Chem. 1994, 42, 388. 144. Zuidema, T.; Mulder, P. P. J.; Stappers, S.; Lasaroms, J.; van Rhijn, H.; Food Addit. Contam. 2006, 23, 1149. 145. Grimalt, S.; Pozo, Ó. J.; Marín, J. M.; Sancho, J. v.; Hernández, F.; J. Am. Soc. Mass Spectrom. 2005, 16, 1619. 146. Wu, Z.; Li, J.; Zhu, L.; Luo, H.; Xu, X.; J. Chromatogr. B 2001, 755, 361. 147. Ali, M. S.; Sun, T.; McLeroy, G. E.; Phillippo, E. T.; J. AOAC Int. 2000, 83, 39. 148. Stout, S. J.; Wickremesinhe, E.; DaCunha, A. R.; Khunachak, A.; J. AOAC Int. 2000, 83, 1446. 149. Turnipseed, S. B.; Roybal, J. E.; Rupp, H. S.; Gonzales, S. A.; Pfenning, A. P.; Hurlbut, J. A.; Rapid Commun. Mass Spectrom. 1999, 13, 493. 150. Cheng, C.; Liu, L. C.; Anal. Methods 2014, 6, 1581. 151. Noppe, H.; Verheyden, K.; Bussche, J. vanden; Wille, K.; de Brabander, H.; Food Addit. Contam., Part A 2009, 26, 1232. 152. Ortiz, A. J.; Cortez, V.; Azzouz, A.; Verdú, J. R.; PLoS ONE 2017, 12, 1. 153. Zrnčić, M.; Gros, M.; Babić, S.; Kastelan-Macan, M.; Barcelo, D.; Petrović, M.; Chemosphere 2014, 99, 224. 154. Rubinson, K. A.; Rubinson, J. F.; Análisis instrumental; Pearson Educación, S. A.: Madrid, 2001. 155. Hernando, M. D.; Mezcua, M.; Suárez-Barcena, J. M.; Fernández-Alba, A. R.; Anal. Chim. Acta 2006, 562, 176. 156. Ortelli, D.; Cognard, E.; Jan, P.; Edder, P.; J. Chromatogr. B 2009, 877, 2363. 157. Vokřál, I.; Jedličková, V.; Jirásko, R.; Stuchlíková, L.; Bártíková, H.; Skálová, L.; Lamka, J.; Holčapek, M.; Szotáková, B.; Parasitology 2013, 140, 361. 158. Kang, J. W.; Park, S. J.; Park, H. C.; Hossain, M. A.; Kim, M. A.; Son, S. W.; Lim, C. M.; Kim, T. W.; Cho, B. H.; Appl. Biochem. Biotechnol. 2017, 182, 635. 159. Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M.; Talanta 2011, 85, 991. 160. Wang, J.; Leung, D.; Chow, W.; Chang, J.; Wong, J. W.; J. Agric. Food Chem. 2015, 63, 9175. 161. The Commission of the European Communities; Off. J. Eur. Communities: Legis. 2002, L 221, 8. 162. Kaufmann, A.; Anal. Chim. Acta 2009, 637, 144. 163. Wagil, M.; Białk-Bielińska, A.; Maszkowska, J.; Stepnowski, P.; Kumirska, J.; Chemosphere 2015, 119, S9. 164. Gentili, A.; Perret, D.; Marchese, S.; TrAC - Trends Anal. Chem. 2005, 24, 704. 165. Codex Alimentarius; Directrices para el diseño y la implementación de programas reglamentarios de aseguramiento de inocuidad alimentaria relacionados con el uso de medicamentos veterinarios en los animales destinados a la producción de alimentos CAC/GL 71-2009; 2009. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access