
 |
 |
 |
 |
 |
Artigo
|
|
| Infravermelho portátil na região do próximo (NIR) aplicado no controle de qualidade de cafés adulterado por borra Portable near infrared spectroscopy applied to the quality control of coffee adultered by grounds. |
|
Radigya M. CorreiaI; Pedro H. CunhaI; Bárbara Z. AgnolettiI; Lucas L. PereiraII; Fábio L. PartelliIII; Paulo R. FilgueirasI; Valdemar Lacerda Jr.I; Wanderson RomãoI,IV,V,*
I. Departamento de Química, Universidade Federal do Espírito Santo, 29075-910 Vitória - ES, Brasil Recebido em: 01/08/2021 *e-mail: wandersonromao@gmail.com The objective of this work was to control the purity level of canephora coffees (Conilon coffee) from the state of Espírito Santo (Brazil), as a function of the addition of grounds produced by the coffees themselves using a portable microspectrometer in the infrared region. (microNIR), associated with chemometric tools. The PCA model grouped the coffees according to the level of adulterant in the samples and showed that the presence of moisture and polysaccharides is important for the discrimination between pure and adulterated samples. PLS-DA and SIMCA classification models were built with commercial samples and resulted in the discrimination of authentic and non-authentic samples with accuracy values above 90% and 82% for PLS-DA and SIMCA, respectively. The quantification results presented by the PLS model were satisfactory for all models, reaching the best results at 75 °C with RMSEP values of 6.13 wt%, R2p = 0.9917; and LOD in the range of 5.6-8.9 wt%; LOQ = 17-26.8 wt%, according to IUPAC; relative standard deviation for repeatability of 1.0 and 1.7 wt%; and intermediate precision of 3.4 and 1.1 wt% for low (<10 wt%) and high (≈ 90 wt%) levels of adulterants, respectively. Therefore, this work represents the first exploratory study to analyze adulterations with coffee grounds by portable NIR, demonstrating the potential of the equipment against this type of adulteration. INTRODUÇÃO O café é uma das bebidas mais consumidas no mundo, portanto, existe ampla susceptibilidade de adulteração intencional.1 Para o preparo da bebida é realizada uma extração sólido-líquido, que resulta na produção de um subproduto sólido, conhecido como borra do café.2 Há uma grande quantidade de geração de borra no preparo industrial e domiciliar, sendo estimado a geração anual de 6 milhões de toneladas desse resíduo em todo mundo.3 A borra é composta por alta umidade e matéria orgânica e já é comumente aproveitada como biocombustível,4-6 ração para animais,7 fertilizantes,8-10 alimentos,11,12 biossorventes,13,14 dentre outros.15 No entanto, há ainda o uso indevido como adulterante em blends de café, visando obter lucro e reduzindo sua qualidade. Segundo a legislação brasileira (Instrução Normativa 16/2010 - Ministério da Agricultura, Pecuária e Abastecimento - (MAPA)), são definidos como matéria estranha os detritos vegetais não oriundos do cafeeiro, grãos ou sementes de outras espécies, corantes, açúcar e borra de café solúvel ou de infusão, ou seja, os resíduos de cafés. Dentre os adulterantes mais usados encontram-se trigo,16,17 cevada,17,18 arroz,16 soja,16,19 milho,16,19,20 cascas/paus,16,20 e até cafés de variedades e regiões diferentes.21 Nesse contexto, há poucos relatos de adulteração de café por borra.22-24 Logo, o desenvolvimento de um método se faz necessário desde que não exista método normatizado para a detecção do respectivo adulterante. As práticas de adulterações em cafés são frequentes e diversificadas e há vasta variabilidade de técnicas relatadas, conforme citado anteriormente, para suprir a necessidade de identificação de adulterantes em cafés. Segundo a Associação Brasileira da Indústria do Café (ABIC), órgão de fiscalização desse setor, os recursos padrões são as análises visual de lâminas microscópicas e a técnica de reconhecimento de padrões de cores e recursos de fotografias e de processamento digital de imagem por computador. E, mais recentemente, o Sistema Analisador de Alimentos e Café (Ali-C), que tem como base um espectrômetro fotoacústico. Mas de acordo com a literatura, outras metodologias vêm sendo empregadas a fim de contornar obstáculos como, por exemplo, a subjetividade das análises. Reis et al.22 avaliaram o potencial do infravermelho com transformada de Fourier utilizando um sensor de refletância total atenuada (ATR-FTIR) para detectar adulteração de café arábica com milho, cevada, cascas e também a borra do café, sendo que em alguns blends, os compostos estavam presentes simultaneamente. Foram encontrados valores de erro médio de previsão (RMSEP) de 2,00-2,42 wt% e coeficiente de determinação de previsão (R2p) de 0,99 em modelo por regressão por mínimos quadrados parciais (PLS), sem e com eliminação de outliers. Milani et al.16 utilizaram a Ressonância Magnética Nuclear (RMN) para avaliar seis adulterantes diferentes: soja, milho, cevada, cascas de café, arroz e trigo em amostras de cafés arábica, processadas em duas torrefações distintas (média e escura). Os autores consideraram a técnica sensível e seletiva ao construírem 12 curvas de calibração, sendo uma para cada adulterante com duas torras diferentes (média e escura), e encontrarem valores de limites de detecção (LD) abaixo de 0,86 wt%, para uma faixa de trabalho de 0-50 wt% de adulterante. Outras técnicas analíticas empregadas na identificação de adulterantes em cafés são: mass spectrometry (MS),25,26 High Performance Liquid Cromatography (HPLC),17,21,27 capillary electrophoresis-tandem mass spectrometry (CE-MS/MS),28 voltammetric electronic tongue,19 entre outros.20,29 Entre as técnicas existentes, o espectrômetro portátil na região do infravermelho próximo (microNIR), é um equipamento que possui diversas vantagens, destacando tempo de análise, versatilidade (disponibilidade de diversos acessórios para análises de diferentes estados físicos da amostra), baixo custo (técnica verde), portabilidade, seletividade e sensibilidade.30-32 O microNIR tem sido aplicado, também, em análises de explosivos,33 documentos,30 drogas,31,34,35 e alimentos.36-39 Em análise de adulterantes de café, Correia et al.20 identificaram a adição de cascas/paus e milho por microNIR e encontraram, através da construção do modelo PLS, limites de quantificação (LQ) e LD na faixa de 5-8 wt% e 1-3 wt%, respectivamente. A versatilidade da técnica NIR portátil em diferentes aplicações é um indicativo de sua potencialidade, no entanto, devido à grande quantidade de dados gerados, se faz necessário o uso de análises multivariadas a fim de melhor interpretar as informações químicas adquiridas pelo equipamento. Coerente com tais perspectivas, as técnicas quimiométricas como análise de componentes principais (PCA),20,33,39,40 PLS,20,37,39,41 análise discriminante de mínimos quadrados parciais (PLS-DA)35,41,42 e analogia de classe de modelagem independente suave de analogia de classe (SIMCA)38 têm sido amplamente empregadas para o tratamento dos dados de NIR. O microNIR auxilia, portanto, no controle de qualidade realizando análises in situ, com confiabilidade e rapidez, através da resolução de problemas analíticos aliado às ferramentas quimiométricas. O uso de ferramentas estatísticas tornou-se fundamental nas análises químicas devido a necessidade de estudos de diversas propriedades de um determinado analito que continham inúmeras variáveis, como os espectros de infravermelho. Sendo assim, o objetivo deste trabalho foi identificar adulterações de borra de Coffea canephora (Conilon) secas nas temperaturas de 45, 75 e 100 ºC, em diferentes níveis (0 - 100 wt% de adulterante), através da técnica analítica de NIR portátil associada a ferramentas quimiométricas: PCA, PLS-DA, SIMCA e PLS. Nesta pesquisa, as borras de cafés foram secas em três temperaturas diferentes para verificar se este fator dificulta a identificação das mesmas nos blends de cafés.
METODOLOGIA Preparo das amostras Amostras de café não comerciais Foram usadas três amostras de café Conilon não comerciais provenientes dos municípios de Iúna (café 1) e São Gabriel da Palha (café 2 e 3) do estado do Espírito Santo, Brasil, fornecidos pela Cooperativa Agrária de Cafeicultores de Iúna (CACIL) e pelo Instituto Federal do Espírito Santo, campus Venda Nova do Imigrante. Para o preparo da borra de café, foi adicionado água quente (0,1 g/mL), com aproximadamente 96-99 ºC, nas amostras torradas e moídas inseridas em papel de filtro comum (n°103) encaixado no suporte de filtro. Posteriormente, com o objetivo de definir as condições de secagem que mantivessem as características mais favoráveis das borras para o estudo, as amostras foram secas em estufa por 60 min, em três temperaturas diferentes (45, 75 e 100 °C). Na segunda etapa, as borras secas foram adicionadas a três tipos de café Conilon não comerciais em pó (nomeados de 1, 2 e 3), resultando em blends com 13 níveis diferentes de adulterações (0, 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 e 100 wt% de adulterante) com três respectivas temperaturas de secagem, totalizando 117 amostras. A escolha da faixa de concentração de adulteração de 0-100 wt% foi baseada nos termos da legislação brasileira, que preconiza baixos níveis de material adicionado, cujo limite estabelecido por lei é de 1 wt% (Instrução Normativa 16/2010 - MAPA). Amostras de cafés comerciais Posteriormente, 31 marcas de cafés foram adquiridas no comércio local do estado do Espírito Santo, Brasil. São elas: Café Falqueto®, Mogiana®, 3 Corações Rituais®, Nº1 Prime®, 3 Corações (Cerrado Mineiro)®, Carnielli® (Café do Nonno),Carnielli® (Cereja Descascada), Café Del Mont Artesanal®, Del Mont 82®, Nº1 Extra Forte®, Nº1 Tradicional®, 3 Corações Tradicional®, Liberdade (Café Especial)®, Liberdade Extra Forte®, Venda Nova Extra Forte®, Grão da Roça®, Café do Ponto Safra Especial®, Cafuso Tradicional®, Cafuso Extra Forte®, 3 Corações Extra Forte®, 3 Corações Tradicional®, 3 Corações Tribos®, Pilão Tradicional®, Caboclo®, Meridiano Extra Forte®, Melita Tradicional®, Melita Extra Forte®, Glória, 3 Corações Forte®, Vista Linda® e Praça Oito®. Todas as marcas foram nomeadas aleatoriamente como amostras A-XE. Para esse conjunto de amostras, borras de café foram produzidas e secas à 75 °C. Posteriormente, todas as amostras comerciais foram divididas em autênticas, que são amostras sem adição de borra (amostras comerciais puras), 0 wt%, Tabela 1S, e seus blends: 25:75, 50:50, 75:25 e 100:0 wt% (Tabela 2S)), totalizando 100 amostras. O outro grupo foi identificado como amostras comerciais não autênticas, ou seja, adulteradas com borra, que são preparadas com adição de 1, 2, 5, 10, 20, 50 e 100 wt% de borra (Tabela 3S), totalizando 112 amostras. Por fim, todas as amostras foram inseridas em vials e submetidas a análise por microNIR. Todas as análises foram triplicadas e feitas de forma randomizada. NIR portátil As análises de NIR portátil foi realizada usando um instrumento modelo miniatura microNIRTM Pro 1700 da Viavi Solutions Inc. Os dados foram obtidos pelo software 2.1 da Viavi Solutions Inc. Espectros de MicroNIR foram adquiridos na faixa de 908-1676 nm, com 100 scans de varredura, e aumento de 6.20 nm de um ponto para outro, resultando num tempo total de medição de aproximadamente 8s por amostra.30,31,39,40,41,43,44 Quimiometria Neste trabalho foram construídas matrizes X com dados de NIR portátil das amostras, originando nove matrizes de tamanho 39x125 cada, para as amostras não comerciais, e uma matriz X(636,125) composta por espectros das amostras comerciais autênticas e não autênticas. O primeiro conjunto foi usado para construção dos modelos PCA, PLS e PLS com seleção de variáveis usando o Algoritmo Genético (GA-PLS) e para os modelos de classificação PLS-DA e SIMCA, foi usado o segundo conjunto citado. Os resultados das amostras não comerciais também foram avaliados pelos modelos PCA e PLS após agrupamentos das nove matrizes mencionadas (X351x125). Anteriormente à construção dos modelos, os espectros NIR foram pré-tratados usando a primeira derivada com suavização pelo algoritmo Savitzky - Golay45 com polinômio de segunda ordem e uma janela de sete pontos com objetivo de reduzir os efeitos da linha de base e assim auxiliar na localização dos máximos de absorção de bandas largas e na resolução de picos sobrepostos obtidas pela NIR. Os espectros médios da amostra em triplicata foram usados para construir os modelos qualitativos e quantitativos. Ademais, antes da construção dos modelos os dados foram centrados na média. Modelos qualitativos O método de reconhecimento de padrões não supervisionado PCA, é uma técnica exploratória,46 e foi usado com objetivo de verificar o comportamento dos dados no espaço multidimensional. Já a modelagem por PLS-DA possui como base a aplicação de um modelo de regressão PLS sobre variáveis que são indicadores dos grupos e o SIMCA a construção de modelos PCA disjuntos utilizados para atribuir uma observação a um ou vários das classes disponíveis,47 dessa maneira, essas ferramentas foram usadas classificar as amostras de acordo com a probabilidade de pertencer a uma determinada classe. Para a construção dos modelos PLS-DA e SIMCA, a seleção das amostras foi realizada dividindo-as entre os conjuntos de treino e teste. Aproximadamente 70% das amostras de cada classe (70 e 78 amostras comerciais autênticas e não autênticas, respectivamente) foram previamente selecionadas para o conjunto de treino, enquanto os 30% restantes foram utilizadas para o conjunto de teste na validação externa, garantindo representatividade das classes. O número ideal de variáveis latentes foi determinado pelo método de validação cruzada k-fold, com k igual a 5 para o modelo PLS-DA e o número de variáveis latentes (nvl) usado foi 6. Para o modelo SIMCA, foram usados 2 componentes para as amostras comerciais autênticas e 5 componentes para as amostras comerciais não autênticas. Os modelos foram avaliados pela exatidão (Equação 1), sensibilidade (Equação 2), especificidade (Equação 3), e taxas de acerto e erro. Por fim, com objetivo de avaliar os modelos construídos em relação a discriminação das amostras foi aplicada a matriz de espectros NIR, X(117,125), amostras não comerciais. 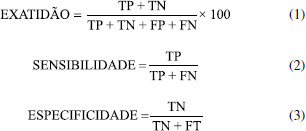 sendo FP e FN o número de amostras falso-positivo e falso-negativo, respectivamente; TP e TN são o número de amostras verdadeiros positivos e negativos verdadeiros, respectivamente. Modelos quantitativos Para verificar a capacidade do modelo e da técnica de NIR portátil em quantificar borra em café Conilon foram construídos modelos PLS.48-50 O número ideal de variáveis latentes foi determinado pelo método análogo ao usado no PLS-DA.51 Na construção do modelo PLS, erro médio de calibração (RMSEC), RMSEP, R2 e viés foram definidos de acordo com os autores.51,52 O LOD e LOQ foram calculados de duas maneiras, usando como parâmetro a metodologia da Agência Nacional de Vigilância sanitária53 e a metodologia preconizada pelo Instituto Nacional de Metrologia, Qualidade e Tecnologia54 - Equação 4 e Equação 5:  em que bk é o vetor com os coeficientes de regressão do modelo PLS para k-variável com base no modelo de regressão e δx corresponde a estimativa do ruído instrumental, determinado como a variância média da matriz de resíduos instrumental.39 De acordo com a União Internacional de Química Pura e Aplicada (IUPAC)55,56 - Equação 6 e Equação 7, o LDmin e LDmax podem ser calculados como: 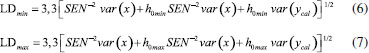 em que h0 é o leverage do branco e var(x) e var(ycal) são as variâncias do sinal do instrumento e concentração de calibração, respectivamente. Para o cálculo do LQ, a fórmula é similar a Equação 6, entretanto, o valor 3,3 é substituído por 10, porque se a concentração do analito é dez vezes a incerteza da previsão, então a última é 10% em relação à concentração prevista.55,56 A IUPAC define o LD como a menor quantidade de uma substância que pode ser distinguida da ausência dessa substância (um valor em branco) dentro de um limite de confiança, e o LQ como a concentração de analito para a qual o erro de predição relativo é no máximo 10%.56 A partir desse conceito estima-se uma faixa de valores ao invés de um valor único, sendo eles valores máximos e mínimos (LDmin, LDmax, LQmin e LQmax). Para realizar a seleção de variáveis, foi realizada otimização dos parâmetros do GA a fim definir os melhores resultados para os cafés secos a 75 °C e 100 °C. A população depende do problema e tem relação direta com o tempo de convergência do algoritmo. Já as n gerações definidas neste trabalho resultaram nos melhores valores para os subconjuntos de variáveis selecionadas. A configuração final do GA foi definida como 1 - tamanho da população, 200, 2 - quantidade de gerações 100, e 3 - probabilidade de mutação, 0.01. O GA foi executado calculando 100 repetições. A precisão intermediária e repetibilidade também foram calculadas usando os espectros de microNIR Café 2 seco a 75 °C (amostras não comerciais), em duas concentrações de adulteração, sendo elas caracterizadas como baixa e alta (10 wt% e 90 wt%), com objetivo de verificar a precisão do método, levando em consideração o desvio padrão relativo (DPR), segundo a International Organization for Standardization (ISO) 5725, conforme Equação 8:  sendo s o desvio padrão e xm a média dos dados do Café 2 com temperatura de secagem à 75 °C.57 De acordo com a ISO 5725, deve haver variação em uma das condições de análise e portanto, foram realizados para cálculo de precisão intermediária, 3 medições (n) por dia, totalizando 7 dias consecutivos. A precisão do método determina as circunstâncias específicas da análise e sua variabilidade, em que a proximidade dos resultados é avaliada em uma série de medições para a mesma amostra. E para a repetitividade, foram realizadas seis medições no mesmo dia, usando um intervalo curto de tempo (30 min.). Todos os cálculos foram realizados usando o MATLAB Software 7.0 (R2013a). Por fim, a estabilidade da borra dos cafés não comerciais foi avaliada levando em conta o armazenamento da mesma na geladeira a 8 °C (caracterizado como média no termostato). Para isso, foi realizado acompanhamento desde o preparo da borra por análise visual durante este período.
RESULTADOS E DISCUSSÃO Os espectros de NIR portátil das amostras não comerciais dos blends de café Conilon/borra de café após pré-tratamento de derivada primeira e centrar na média (café 1 - Figura 1, café 2 e 3 - Figura 1S) foram mostrados em função da temperatura de secagem da borra (45 °C, 75 °C e 100 °C), totalizando 13 espectros sobrepostos para cada conjunto. Os espectros foram em degradê de cor, onde o maior teor da borra no blend corresponde a tonalidade do espectro mais escuro. A presença das bandas nas regiões de 1100-1200 nm, 1350 1450 nm e 1500-1600 nm foram identificadas em todos os conjuntos de amostras (Figura 1 e Figura 1S), no entanto, nota-se que a intensidade das bandas varia conforme a adição de adulterante no blend.
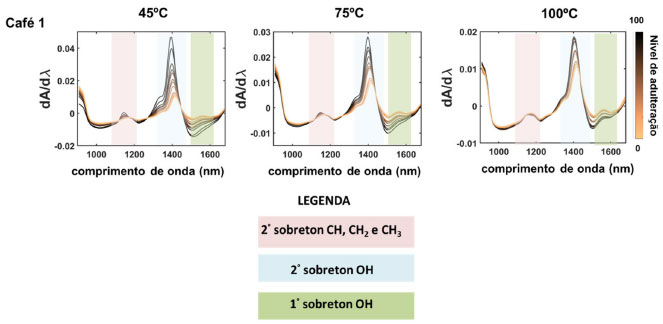 Figura 1. Espectros NIR portátil depois do pré-tratamento com primeira derivada de blends de café Conilon 1/borra de café secas a 45, 75 e 100oC (amostras não comerciais)
A região de 1100-1200 nm, que caracteriza o segundo sobretom dos grupos CH, CH2 e CH3,39 apresentou maior intensidade para amostras com maior teor de adulterante. Santos et al.39 citaram que essa região é atribuída aos ácidos graxos insaturados e apesar de amostras de cafés serem normalmente adulteradas com outros componentes como milho, cevada, dentre outros,16,20,22 a borra é derivada da própria amostra e, portanto, possui teores significativos de ácidos que são compostos majoritários nos grãos deste fruto. Isso ocorre devido a relação da polaridade e o tamanho das cadeias carbônicas dos principais ácidos, como palmítico, oléico, esteárico, dentre outros,47 pois os mesmos não são solubilizados no processo de extração com água (isso é, solubilidade (~20 °C) do ácido esteárico = 0,05 mg/100 mL, ácido palmítico e oleico = insolúvel),58 sendo, portanto, concentrados na composição química da borra. Independentemente da temperatura e da origem das amostras, houve maior intensidade das bandas na região de 1350-1500 nm para amostras com maiores níveis de borra de café. Essa região caracteriza o grupo funcional OH (segundo sobretom) e está relacionada a dois fatores: 1) a presença da molécula de H2O,39 devido ao teor de umidade da borra e 2) a presença de outros compostos, como os polissacarídeos (exemplo a galactose e arabinose) e proteínas,59 que por não serem solúveis tem sua maior concentração no sólido residual, afetam a intensidade desta banda na absorção. Mussatto et al.60 caracterizaram quimicamente a borra de café, e relataram a alta presença de açúcares hemicelulósicos nesse subproduto, alcançando níveis de 45 wt% da composição. A região de 1500-1600 nm, refere-se ao primeiro sobretom de OH e mostra que quanto menor o nível de adulteração maior intensidade da banda. Segundo Barbin et al.,59 existe uma relação desta faixa de comprimento de onda com os açúcares, sendo eles os carboidratos solúveis como frutose, glicose, sacarose, dentre outros (isto é: solubilidade (~20 °C) da frutose = 79 g/100mL, sacarose = 197g/mL e glicose = 47g/mL),58 assim durante o processo de extração com água, uma alta parcela destes componentes são extraídos, fazendo com que haja menor teor destes na borra de café. No que diz respeito à temperatura de secagem da borra (45, 75 e 100 °C), os espectros de microNIR obtidos (Figura 1 e 1S) sugerem que cafés secos a 100 °C possuem menor variação na intensidade das bandas, independente do teor de borra, em todas as regiões do espectro ao compararmos as secagens a 45 °C e 75 °C. Esse resultado mostra que a alta temperatura de secagem influencia no perfil espectral dos espectros NIR, dificultando a identificação dos cafés puros e adulterados. Isso pode ser claramente observado na região de 1350-1500 nm (Figura 1). Mei e Oliveira61 realizaram estudos de degradação térmica da borra de café e constataram, através da análise termogravimétrica (TGA), que ocorre perda de massa após aumento da temperatura de secagem desse resíduo. Os autores afirmaram que essa perda está relacionada a compostos voláteis presentes na amostra, tais como moléculas de água que evaporam nessa faixa de temperatura. Outros compostos que caracterizam esta região são as proteínas, que podem sofrer degradação na faixa de 40 a 80 °C.62 PCA Modelos PCA foram construídos e os gráficos dos scores para os blends dos três conjuntos de amostras não comerciais de café Conilon (1, 2 e 3) e suas respectivas borras em função das temperaturas de secagem são apresentados nas Figura 2a-c. A média da soma das variâncias explicadas das componentes PC1 e PC2 de cada café em função da temperatura de secagem das borras, foram iguais a 99,73 (45 °C), 99,79 (75 °C), e 97,70 (100oC) com desvio padrão de ±0,14, ±0,04 e ±1,07, respectivamente. Esses resultados demonstram que os modelos contêm elevado nível de informação usando PC1xPC2, pois são superiores a 99%, exceto à temperatura de secagem de 100 °C.
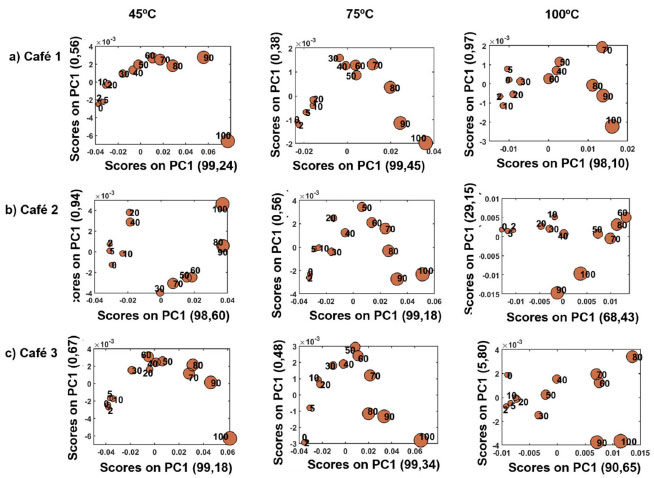 Figura 2. Gráficos dos scores dos modelos de PCA para os blends (amostras não comerciais) de café Conilon a) café 1, b) café 2 e c) café 3, e borra em função dos diferentes níveis de adulteração
Ao observar a Figura 2a-c, verificamos que houve um melhor gradiente de separação na componente PC1 em função da concentração do adulterante presente nas blends de café Conilon/borra de café. Logo, as amostras seguem um aumento do teor de adulterante que é linear ao longo de uma única componente, isso é, a PC1. Dessa forma, em todos os modelos PCA, os cafés puros (0 wt%) e/ou baixos níveis de adulteração foram perfeitamente discriminados no quadrante negativo da PC1 (PC1<0), enquanto as amostras adulteradas com maiores teores de borra, a partir de 40 50 wt%, se encontraram na região da PC1>0. A partir do gráfico de loadings (Figura 3) de PC1, a região de maior influência nas amostras para todos os modelos construídos é a de 1350-1500 nm. Esse resultado está correlacionado à diferença na concentração de H2O, mostrando que os espectros de microNIR são sensíveis à presença da umidade nos blends proveniente das borras adicionadas e a outros compostos característicos que apresentam a ligação O-H.
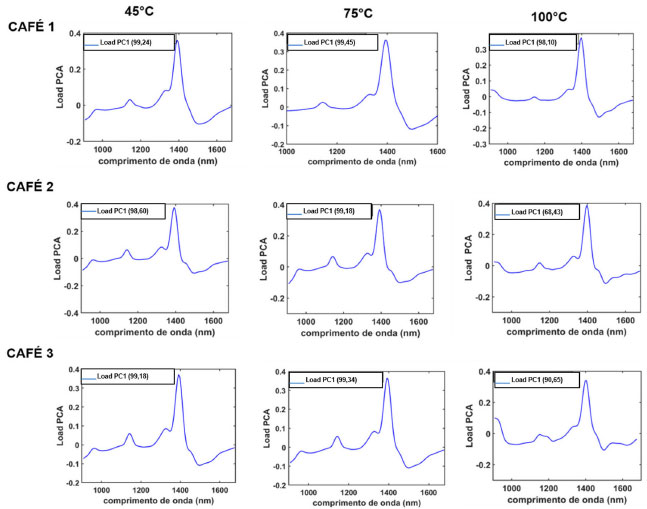 Figura 3. Gráfico dos loadings de PC1 do modelo PCA para as amostras de cafés (amostras não comerciais) adulteradas com diferentes concentrações de borra de café, de acordo com a temperatura de secagem das borras
Ao estudar as blendas de café/borra produzidas em função de distintas condições de temperatura de secagem e, consequentemente umidade,63,64 nota-se que essas propriedades influenciaram nas intensidades das bandas de absorção, principalmente, aquelas relacionadas ao grupo funcional OH. Portanto, é possível afirmar que o conjunto de amostras que proporcionou maiores informações para a construção do modelo PCA foram os blends preparados com as borras secas à 75 °C. Cardoso65 analisou também três temperaturas de secagem da borra de café expresso a fim de verificar o comportamento higroscópico (capacidade de absorver a água) das amostras e assim ajustar o melhor processo de conservação para posterior transporte, armazenamento e uso do resíduo. Os autores constataram que o nível mínimo ideal de secagem foi a partir de 45 °C, conforme utilizado neste estudo. Benicá et al.66 construíram modelos estatísticos relacionando o processo de torrefação dos cafés, a temperatura de secagem da borra e seu reaproveitamento em muffins e concluíram que a faixa de 70-80 °C foi ideal para cafés classificados como torra clara e escura. Um novo modelo PCA foi construído contendo as amostras não comerciais de blends de Conilon (cafés 1-3) e suas respectivas borras obtidas na temperatura de secagem de 75oC. A Figura 4a mostra que o gráfico de scores apresenta uma variância explicada pela PC1 e PC2 de 99,35%, onde novamente a tendência de adulteração aumenta em função da PC1. Já ambas as PCs, PC1 vs. PC2 conseguem separar as amostras entre Café 1 - Iúna e Cafés 2-3, ambos de São Gabriel da Palha. Esse resultado mostra que é possível reconhecer os municípios de origem.
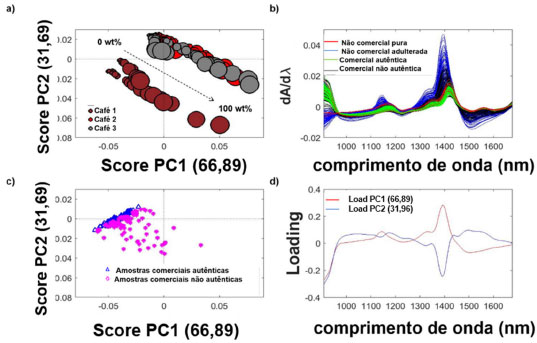 Figura 4. a) gráfico dos scores do modelo PCA para as amostras não comerciais. O tamanho do marcador é proporcional ao teor de adulterante nas amostras; b) espectros microNIR, após pré-tratamento com a primeira derivada, de cafés não comerciais puros e adulterados e amostras comerciais autênticas e não autênticas; c) gráfico dos scores para amostras comerciais (amostras comerciais autênticas (n = 100) são representadas pelo símbolo (Δ) e as amostras comerciais não autênticas (n = 112) são representadas pelo símbolo (*)); d) gráfico dos loadings do modelo PCA para os blends de café
Com objetivo de avaliar a qualidade das amostras dos cafés comerciais, espectros NIR dessas amostras e seus blends foram adquiridos (Figura 4b) e um novo modelo de PCA foi construído. Os gráficos de scores e loadings são apresentados nas Figuras 4c to 4d, respectivamente. Analisando os espectros NIR, as amostras adulteradas (não comerciais e comerciais), apresentam uma maior intensidade de absorção na região de1350-1500 nm (Figura 4b). O gráfico de scores da PC1 x PC2, Figura 4c, mostra que as amostras comerciais autênticas (sem adição de borra), estão bem agrupadas e distribuídas na região da PC1<0 e PC2 ~ 0. Próximo dessa região, há a presença de um conjunto de amostras comerciais não autênticas contendo menores níveis de adulterante, isso é, concentração de borra na faixa de 1 a 20 wt%. As demais amostras comerciais não autênticas com concentração de borra de 50 a 100 wt%, estão dispersas ao longo da PC1. O gráfico de loadings (Figura 4d) mostra que a região de maior importância na construção do modelo foi em 1350-1500 nm nas duas PCs, mas em direções opostas da PCs (PC1 >0 e PC2 <0). PLS-DA e SIMCA A partir dos espectros de NIR portátil dos cafés comerciais, foram construídos modelos de classificação (autênticas e não autênticas), sendo PLS-DA e SIMCA, a fim de comparação dos resultados. As matrizes de confusão dos modelos PLS-DA e SIMCA são representadas na Tabela 1 e mostram a probabilidade de uma determinada amostra pertencer a cada classe.40,41 A classe alvo para a construção do modelo é o conjunto de amostras autênticas, ou seja, sem a presença de borra.
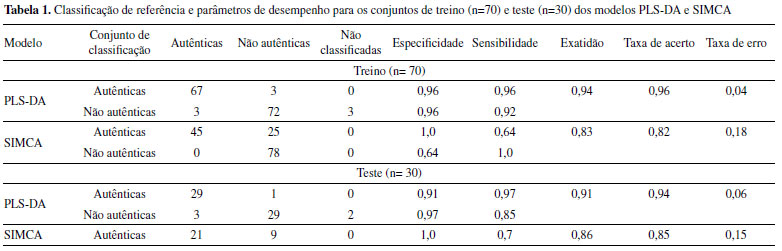
Ao observar os resultados do conjunto de treino dos modelos PLS-DA (Tabela 1 e Figura 5), verifica-se que das 70 amostras caracterizadas como autênticas, somente três foram classificadas incorretamente e entre as 78 amostras não pertencentes a classe alvo, ou seja, não autênticas, o modelo classificou 72 corretamente, três incorretamente, não sendo capaz de classificar apenas três amostras. Os resultados para o conjunto de teste também foram excelentes com, por exemplo, somente uma amostra classificada incorretamente no grupo das autênticas.
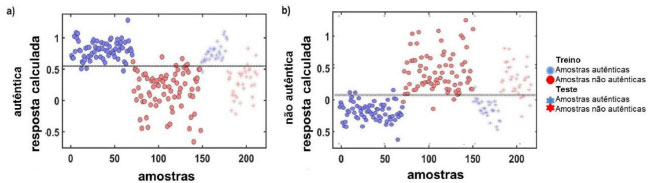 Figura 5. Resposta calculada para o modelo PLS-DA para a probabilidade das amostras pertencerem a determinada classe. a) autêntica e b) não autêntica
Para o modelo SIMCA, observa-se a partir do gráfico dos scores (Figura 2S) da classe alvo, ou seja, amostras autênticas, que a variância total explicada com duas componentes principais foi de 99,91% e que houve agrupamento das amostras de treino e teste em torno de PC1. O modelo classificou corretamente todas as amostras não autênticas do conjunto de treino e teste (Tabela 1), mostrando resultados ao nível de significância acima de 99% para esse grupo. Para o grupo das amostras autênticas, foram classificadas incorretamente 25 e 9 amostras de blends nos conjuntos de treino e teste, respectivamente. Os resultados mostram que a modelagem por PLS-DA classificou corretamente maior quantidade de amostras em relação ao modelo SIMCA. Após a observação dos parâmetros de desempenho (especificidade, sensibilidade e exatidão) dos modelos e da classificação correta das amostras autênticas, é possível concluir que o PLS-DA é mais eficiente do que o modelo SIMCA. Os parâmetros de desempenho do modelo PLS-DA e SIMCA são mostrados na Tabela 1. Foram realizados testes de exclusão de algumas amostras para detecção de outliers, no entanto, não houve ganho nos parâmetros de desempenho e, portanto, as amostras não são prejudiciais à qualidade dos modelos. A nível de comparação, é possível observar que o modelo PLS-DA apresentou valores de exatidão e taxa de acerto acima de 93% para o conjunto de treino e maior que 90% para o conjunto de teste. As taxas de erro (na faixa de 4-6%) mostram a consistência da técnica em classificar amostras de cafés autênticos e não autênticos pelo modelo PLS-DA. Núñez et al.21 usaram a técnica de HPLC acoplada ao ultravioleta (UV) e construíram modelos PLS-DA para discriminar amostras de cafés de diferentes regiões e torras para fins de autenticação de fraudes, e encontraram valores de taxa de acerto a partir de 89.3% para o conjunto de treinamento. Baqueta et al.40 analisaram misturas de cafés com torras e classificações diferentes (duro e rio) usando NIR portátil, na mesma faixa de comprimento de onda (906-1676 nm), e encontraram taxa de erros entre 1,9 - 8%, através da construção do modelo PLS-DA. Com relação a especificidade e sensibilidade do conjunto de treino, os autores encontraram sensibilidade: café duro/rio torra média (classe 1) - 0,98; café duro/rio torra escura (classe 2) - 1.0 e café rio/rio torra média/escura (classe 3) - 0,91; especificidade: classe 1 - 0,97; classe 2 - 0,97 e classe 3 - 0,92,42 valores similares aos obtidos neste trabalho, conforme Tabela 1. O modelo SIMCA apresenta valores acima de 82% de exatidão e taxa de acerto para os conjuntos de treino e teste. Apesar desse último ter apresentado valores inferiores aos obtidos no modelo PLS-DA, os resultados foram satisfatórios, alcançando alta sensibilidade para amostras não autênticas (Tabela 1), mostrando-se eficiente em identificar amostras com a presença da borra. Segundo a Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA) os métodos tradicionais de análise, como a análise visual de lâminas microscópicas, possuem dificuldade ou não conseguem detectar este subproduto, pois o café é uma matriz complexa e a borra é um dos resíduos do próprio café. Outro parâmetro que comprova a eficácia do modelo SIMCA é especificidade para as amostras autênticas, ou seja, os negativos verdadeiros, mostrando a capacidade do método em identificar corretamente as amostras que não apresentam a presença do adulterante. A diferença entre os resultados do PLS-DA e SIMCA deste estudo, está principalmente nos resultados de sensibilidade das amostras autênticas e na especificidade das amostras não autênticas de ambos os conjuntos, além da menor exatidão e taxa de acerto do modelo SIMCA, uma vez que a classe alvo são as amostras autênticas. Bylesjö et al.47 afirma que em relação à taxa de classificação, desde que a variabilidade dentro da classe seja baixa, modelos PLS-DA superam a modelagem SIMCA. A partir dos resultados dos modelos construídos, é possível mostrar o bom desempenho da técnica de microNIR associada a modelos de classificação para a discriminação entre amostras autênticas de cafés e amostras que não pertencem a essa classe (não autênticas) com baixo erro de resposta calculado. Neste sentido, o conjunto de amostras não comerciais (n= 117) foram aplicadas nos modelos SIMCA e PLS-DA construídos. Os melhores resultados foram obtidos pelo modelo SIMCA, que classificou 92,3% das amostras corretamente. Este resultado está relacionado ao conjunto de amostras aplicadas ao modelo, uma vez que, dos 117 cafés, 98 não pertencem à classe alvo, ou seja, não são autênticas, e como visto anteriormente, o modelo SIMCA obteve maior taxa de acerto para a classificação deste conjunto. Concentrações baixas (2 wt%) de borra nos blends foram classificadas corretamente. Já o modelo PLS-DA classificou corretamente 81,2% do conjunto de amostras não comerciais, identificando teores acima de 10 wt% de borra. Por fim, pode-se afirmar que o modelo PLS-DA foi melhor para a classificação de amostras autênticas e não autênticas e o modelo SIMCA foi melhor na classificação das amostras não autênticas e do conjunto aplicado (amostras não comerciais) ao modelo. PLS Os resultados médios de RMSEP, R2p, LD, LQ, e erro sistemático (viés) e número de variáveis latentes (nvl) dos nove modelos PLS construídos a partir das amostras não comerciais foram apresentados na Tabela 2. Uma variável latente foi utilizada para a construção do modelo PLS, sendo estabelecida a melhor relação de RMSEC x RMSEP para que não houvesse super ajuste do modelo. Outra justificativa sobre o uso de apenas uma variável latente está relacionada a boa separação em PC1 (Figura 2).
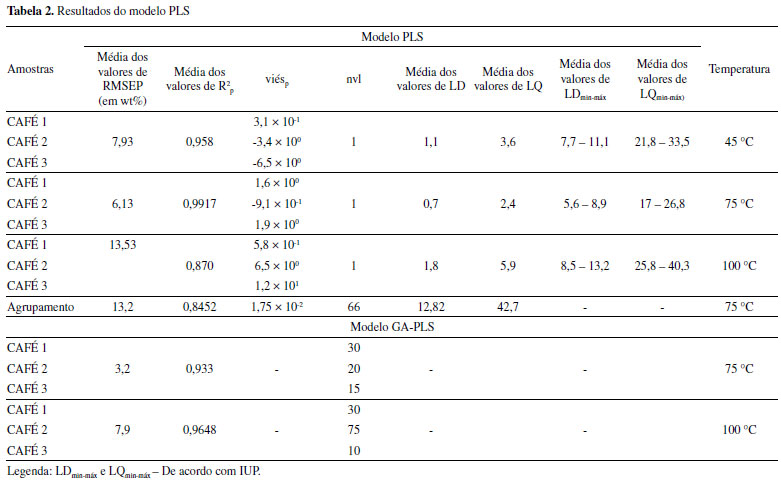
A partir da Tabela 2, observa-se também que o RMSEP foi de 7,93 wt%, 6,13 wt% e 13,53 wt% para as temperaturas de secagem da borra em 45 °C, 75 °C e 100 °C, respectivamente. E o menor valor médio de RMSEP foi para as amostras secas à 75 °C, provando que este resultado mostra que todas as amostras (Cafés 1, 2 e 3) nessa temperatura apresentaram RMSEP abaixo de 7 wt%, e assim o conjunto de dados que exibiu excelentes valores para a modelagem em torno da exatidão foram os cafés secos à 75 °C. Reis et al.22 usaram dados obtidos por ATR-FTIR para construção de modelos PLS a fim de identificar adulterantes simultâneos em amostras de café, sendo um deles a borra. Os autores usaram uma faixa de trabalho de 0 a 66% de adulteração, faixa menos ampla do que usada no presente estudo (0-100 wt%) e aplicaram seleção de variáveis ao conjunto de dados, encontrando valores de RMSEP entre 2,4-3,6 wt%. Em estudo anterior, os autores encontraram por espectroscopia na região do infravermelho por reflectância difusa (DRIFT-IV), RMSEP de 3,7 wt% usando conjunto de amostras e parâmetros similares.23 O R2p também foi avaliado e este parâmetro permite uma estimativa da qualidade da curva obtida de acordo com o percentual de variação total do sinal analítico explicado pela variação da concentração do analito.67 A ANVISA recomenda um coeficiente de correlação igual ou maior que 0,99, portanto, os dados espectrais fornecidos por NIR portátil para as amostras secas a temperatura de 75 °C alcançaram um ajuste ideal dos dados para a linha de regressão. Analisando alguns dados da literatura provenientes de análises de café por microNIR é possível perceber que os resultados de R2 obtidos foram excelentes. Em 2018, Correia et al.20 obtiveram valores de 0,97 para blends de café arábica e milho. Baqueta et al.,37 em 2019, encontraram valores de R2 na faixa de 0,73 a 0,84 ao prever parâmetros de controle da qualidade de cafés como granulometria, umidade, cor e tempo de infusão dos grãos torrados. Já no estudo de Tugnolo et al.68 aplicaram a ferramenta quimiométrica PLS e obtiveram R2 na faixa de 0,18 a 0,84 ao prever o teor de umidade, densidade compactada e granulometria de pó no café moído e para estimar a umidade no grão torrado com visualização de medições em tempo real. A viés, ou erro sistemático foi calculado, para o conjunto de previsão (viésp). Observa-se que foram encontrados para todos os cafés (75 °C e 45 °C), exceto à 100 °C, valores que indicam a ausência destes erros para o modelo PLS ao nível de significância de 5%. O LD e LQ são indicadores numéricos simples, confiáveis e de fácil interpretação para verificar a qualidade do modelo de calibração.56 De acordo com Allegrini e Olivieri55 ainda não há estimador destes limites geralmente aceito para estudos PLS, portanto, neste estudo foram realizados dois cálculos diferentes para identificar o LD e LQ das amostras de cafés Conilon adulteradas com a borra, Tabela 2. Seguindo a metodologia da ANVISA53/INMETRO54 foram encontrados resultados a partir de 0.7 wt% e 2,4 wt% de LD e LQ, respectivamente, para os cafés secos à 75 °C. Ao comparar estes resultados aos valores de exatidão pode-se afirmar que estão superestimados, mas pode-se levar em consideração o fato de serem amostras sólidas que podem sofrer efeito de espalhamento, devido a possível não uniformidade de amostras nesse estado físico. Outros autores encontraram, LD e LQ, usando NIR portátil em análises de cafés, de 4,15 e 12,59 wt%, respectivamente, ao prever a granulometria.37 Os valores de LDmin-máx e LQmin-máx (IUPAC) foram superiores aos definidos pela ANVISA53/INMETRO54, conforme Tabela 2. Esse resultado possivelmente tem relação com os valores de h0 (leverage), que é caracterizado como a distância da amostra até o centro do conjunto de dados medido no subespaço das variáveis latentes, ou seja, quando a concentração do analito é zero. Nessa investigação o branco usado nos cálculos foram as amostras caracterizadas como 0%, sem a presença de adulterante. No entanto, o café é uma matriz complexa, que possui muitos componentes contribuindo para seu espectro que podem provocar efeito de matriz, pois são analisados os espectros inteiros. O LDmin-máx e LQmin-máx levam em consideração dois possíveis erros, tipo I e tipo II, enquanto os resultados obtidos por cálculos de LD e LQ, segundo a ANVISA e INMETRO, usam somente a possibilidade do erro tipo I. O erro tipo I caracteriza-se probabilidade de rejeitar H0 uma vez que ela é verdadeira e o tipo II probabilidade de aceitar H0 uma vez que ela é falsa. Os resultados obtidos conforme cálculo da IUPAC, onde foram encontrados valores médios entre 5,6-8,9 wt% de LD e 17-26,8 wt% para LQ na temperatura de 75 °C, por exemplo, vão de acordo com os valores encontrados de RMSEP, mostrando ser promissora a metodologia da IUPAC. Após avaliação dos resultados de exatidão, LD, LQ e R2p é possível inferir que a secagem da borra afeta a qualidade do modelo e que há uma tendência na excelência dos resultados na ordem crescente de 100 °C, 45 °C e 75 °C, além de que os resultados do modelo PCA corroboram com essas afirmações. Apesar da exatidão à 100 °C ser maior (13,53 wt%) e o limite de detecção de borra em café Conilon ser na faixa de 8,5 - 13,2 (IUPAC), conforme Tabela 2, foram encontrados bons resultados para todos os conjuntos de amostras. Para verificar a exatidão dos modelos quimiométricos foi construído também um modelo PLS agrupando os espectros de microNIR dos três cafés e as três temperaturas de secagem da borra. Os resultados de RMSEP e R2p do modelo foram 13,2 wt% e 0,8452, respectivamente. Posteriormente, foi avaliado o conjunto de amostras comerciais autênticas e não autênticas (n = 212) preparadas a partir das 31 marcas adquiridas no comércio local. Os valores previstos obtidos para as amostras comerciais autênticas, Tabela 4S, mostram que as amostras obtiveram, predominantemente, resultados negativos. Esse resultado está dentro da faixa de erro do modelo e comprova a ausência de adulterante nas amostras. Já os resultados para as amostras comerciais não autênticas Tabela 5S, apresentaram variabilidade (> desvio padrão = 17 wt%) quanto aos valores de previsão sendo alguns iguais e/ou próximos aos valores de referência, levando em consideração a faixa de erro do modelo PLS, como por exemplo, os blends V, W e Y. Em contrapartida, outros conjuntos apresentaram maior variabilidade nos valores de referência versus previsão. Estes resultados podem estar relacionados a 3 fatores sendo a) o LQ de 42,7 wt%, segundo a ANVISA; b) à origem e variedade das amostras, conforme apresentado pelos modelos PCA e c) a influência da temperatura de secagem das borras, observado nos demais modelos PLS construídos. O próximo passo foi avaliar se a seleção de uma faixa específica do comprimento de onda auxiliaria na melhora dos resultados. Alguns trabalhos usam a seleção de variáveis para melhorar a capacidade preditiva dos modelos PLS.69-73 Dentre os métodos disponíveis, este trabalho usou o Algoritmo Genético (GA), proposto por John H. Holland na década de 60.74 A Tabela 2 também mostra os valores médios de RMSEP e R2p para os Cafés 1, 2 e 3, bem como a quantidade de variáveis selecionadas pelo modelo para os cafés secos à 75 °C e 100 °C. Levando em consideração os fatores RMSEP, tempo de processamento e R2 observa-se excelentes valores para todas as amostras, sendo os resultados abaixo de RMSEP <8 wt% do GA-PLS. Observa-se também que os resultados para borras secas à 100 °C, prevalecem maiores que a temperatura de 75 °C, portanto, há um limite de detecção de adulteração dos cafés nesta temperatura, apresentado no modelo através da menor exatidão. Ao comparar os resultados de RMSEP obtidos pelos modelos anteriores (Tabela 2) é possível constatar que houve a promoção de melhorias em relação ao método full scan e após a aplicação da seleção de variáveis por GA, sendo que para o último foram encontrados, aproximadamente metade dos valores em relação a primeira metodologia para as duas temperaturas. Contudo, independentemente do método utilizado os valores de predição foram excelentes. A precisão do método foi definida através dos parâmetros de repetibilidade e precisão intermediária. Os valores de RDS para repetibilidade foram de 1.0 para o nível de 90 wt% e 1,7 para 10 wt% de adulterante. Já para a precisão intermediária os valores encontrados foram 3,4 e 1,1 wt% para baixa e alta concentração, respectivamente. Correia et al.48 encontraram valores de repetibilidade e precisão intermediária de 4 e 5 wt%, respectivamente, ao analisarem blends de café arábica e Conilon por espectrometria de massas com ionização por eletrospray (ESI(-)FT-ICR MS) e 1,7 wt%, em ambas figuras de mérito, por ATR-FTIR. Outros autores analisaram amostras de cafés e encontraram valores de repetibilidade 7-27,9% usando um microscópio estereoscópico,75 1,65% por HPLC,76 1,7 - 3,3% também por HPLC77 e 1,6-3,5% por RMN. Segundo as agências responsáveis por verificarem as competências dos ensaios de laboratório, o valor limite a ser alcançado é 5%. Dessa maneira, os resultados obtidos neste estudo demonstram a confiabilidade da técnica de microNIR em análises de adulterações de café Conilon com borra de café, sendo mínima a variabilidade observada em intervalos de tempos diferentes. Por fim, foi realizada uma análise qualitativa, a fim de identificar o tempo de conservação dos blends sem que haja interferências de microrganismos, devido ao alto teor de umidade presente nas borras de café. O estudo foi delineado através do tempo (em dias) versus análise visual, levando em consideração que ao armazenar as misturas o indivíduo só poderá usar estes parâmetros. Observou-se que o tempo estimado de conservação foi de 137 dias. A EMBRAPA considera que esta prática além de lesar o consumidor, pode proporcionar o crescimento de fungos e bactérias nocivas à saúde. Ribani et al.67 cita que em certos tipos de amostras, faz-se necessário avaliar a estabilidade da substância para determinar o tempo de estocagem das amostras. Os autores exemplificam ao discutir que tempos longos de estocagem de amostras biológicas aumentam a probabilidade de degradação dos compostos de interesse, com subsequente formação de metabólitos. Portanto, identificar a estabilidade ajuda no processo de análises, pois as mesmas podem ser completadas antes de ocorrer a degradação.
CONCLUSÕES Neste trabalho foi desenvolvida a primeira metodologia usando microNIR, como ferramenta analítica, associado a quimiometria (PCA, PLS, PLS-DA e SIMCA) para determinação de adulteração de café Conilon com borras, com excelentes resultados. O modelo PCA mostrou a sensibilidade da técnica à presença de borra como adulterante em amostras de cafés, e através dos loadings, podemos concluir que a região de 1350-1500 nm, caracterizada pelo grupo funcional OH, é importante nessa discriminação. A partir do modelo PLS e das figuras de mérito obteve-se maior e menor exatidão para as borras secas a 75 e 100 °C, respectivamente. Os modelos construídos com os blends de cafés secos à 75ºC obtiveram RMSEP médio de 6,1 wt%. No entanto, mesmo com valores superiores às demais temperaturas, as amostras secas à 100 °C podem ser detectadas na faixa de 1,8-8,5 wt% de adulterante, com erro de predição médio na faixa de 13,5 wt%, sendo menores ao utilizar seleção de variáveis (7,9 wt%), o que mostra a eficácia da técnica. Os modelos obtidos por PLS-DA e SIMCA tiveram boa performance de discriminação entre os cafés autênticos e não autênticos, resultando na obtenção de valores de taxa de acerto e exatidão acima de 90% e 82%, respectivamente. O modelo SIMCA classificou corretamente 92,3% do conjunto de amostras externas e a partir de níveis mínimos (2 wt%) de adulterante no blend. O DPR (<4 wt%) calculado para precisão intermediária e repetibilidade, corroborou com os demais resultados confirmando a confiabilidade da técnica de microNIR. Observou-se também que o tempo máximo de armazenamento na geladeira das borras para análise é de 137 dias sem que haja degradação das amostras.
MATERIAL SUPLEMENTAR As Figuras 1S-2S e as Tabelas 1S-5S estão disponíveis em http://quimicanova.sbq.org.br em formato PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem a Cooperativa Agrária dos Cafeicultores de Iúna - CACIL e o Instituto Federal do Espirito Santo - Venda Nova do Imigrante pelo fornecimento das amostras. Os autores também agradecem à CAPES, à FAPES (Pronem 596/2018 e PPE Agro 76459934/16) e ao CNPq (310057/2020-5) pelo suporte financeiro.
REFERÊNCIAS 1. Wang, X.; Lim, L.; Fu, Y.; J. AOAC Int. 2020, 103, 1. 2. Angeloni, G.; Guerrini, L.; Masella, P.; Bellumori, M.; Daluiso, S.; Parenti, A.; Innocrnti, M.; Food Res. Int. 2019, 116, 1327. 3. Silva, M. A.; Nebra, S. A.; Silva, M. J. M.; Sanchez, C. G.; Biomass Bioenergy 1998, 14, 457. 4. Bispo, M.; Barros, J.; Tomasini, D.; Primaz, C.; Caramão, E.; Dariva, C.; Krause, L.; J. Earth Sci Eng. 2016, 6, 235. 5. Primaz, C.; Schena, T.; Lazzari, E.; Caramão, E.; Jacques, R.; Fuel 2018, 232, 572. 6. Rocha, M. V. P.; de Matos, L.; de Lima, L. P.; Figueiredo, P. M. D.; Lucena, I. L.; Fernandes, F. A. N.; Goncalves, L. R. B.; Bioresour. Technol. 2014, 167, 343. 7. Givens, D. I.; Barber, W. P.; Agric. Wastes 1986, 18, 69. 8. Cruz, R.; Mendes, E.; Torrinha, Á.; Morais, S.; Pereira, J.; Baptista, P.; Casal, S.; Food Res. Int. 2015, 73, 190. 9. Gomes, T.; Pereira, J. A.; Ramalhosa, E.; Casal, S.; Baptista, P.; VII Congreso Ibérico de Agroingeniería y Ciencias Horticolas, Madrid, 2013. 10. Ribeiro, J. P.; Vicente, E. D.; Gomes, A. P.; Nunes, M. I.; Alves, C.; Tarelho, L. A. C.; Environ. Sci. Pollut. Res. 2017, 24, 15270. 11. Ballesteros, L. F.; Ramirez, M. J.; Orrego, C. E.; Teixeira, J. A.; Mussatto, S. I.; Food Chem. 2017, 237, 623. 12. Martinez-saez, N.; García, A. T.; Pérez, I. D.; Rebollo-Hernanz, M.; Mesías, M.; Morales, F. J.; Martín-Cabrejas, M. A.; Del Castillo, M. D.; Food Chem. 2017, 216, 114. 13. Safarik, I.; Horska, K.; Svobodova, B.; Safarikova, M.; Eur. Food Res. Technol. 2012, 234, 345. 14. Yeung, P. T.; Chung, P. Y.; Tsang, H. C.; Tang, J. C. O.; Cheng, G. Y. M.; Gambari, R.; Chui, C. H.; Lam, K. H.; RSC Adv. 2014, 4, 38839. 15. Janissen, B.; Huynh, T.; Resour., Conserv. Recycl. 2018, 128, 110. 16. Milani, M. I.; Rossini, E. L.; Catelani., T. A.; Pezza, L.; Toci, A. T.; Pezza, H. R.; Food Control 2020, 112. 17. Song, H. Y.; Jang, H. W.; Debnat, T.; Lee, K.; Int. J. Food Sci. Technol. 2018, 54, 256-262. 18. Ebrahimi-najafabadi, H.; Leardi, R.; Oliveri, P.; Casolino, M. C.; Jalali-heravi, M.; Lanteri, S.; Talanta 2012, 99, 175. 19. Arrieta, A. A.; Arrieta, P. L.; Mendonza, J. M.; Acta Sci. Pol., Technol. Aliment. 2019, 18, 35. 20. Correia, R. M.; Tosato, F.; Domingos, E.; Rodrigues, R. R. T.; Aquino, L. F. M.; Filgueiras, P. R.; Lacerda Jr., V.; Romão, W.; Talanta 2018a, 176, 59. 21. Núñez, N.; Collado, X.; Martínez, C.; Saurina, J.; Núñez, O.; Foods 2020, 9, 378. 22. Reis, N.; Franca, A.; Oliveira, L.; J. Spectrosc. 2016, 1. 23. Reis, N.; Franca, A. S.; Oliveira, L. S.; Talanta 2013, 115, 563. 24. Reis, N.; Botelho, B. G.; Franca, A. S.; Oliveira, L. S.; Food Anal. Methods 2017, 10, 2700. 25. Garrett, R.; Vaz, B. G.; Hovell, A. M. C.; Eberlin, M. N.; Rezende, C. M.; J. Agric. Food Chem. 2012, 60, 4253. 26. Assis, C.; Pereira, H. V.; Amador, V.S.; Augusti, R.; de Oliveira, L. S.; Sena, M. M.; Food Chem. 2019, 281, 71. 27. Domingues, D. S.; Pauli, E. D.; de Abreu, J. E. M.; Massura, F. W.; Cristiano, V.; Santos, M. J.; Nixdorf, S. L.; Food Chem. 2014, 146, 353. 28. Daniel, D.; Lopes, F. S.; dos Santos, V. B.; Lago, C. L.; Food Chem. 2017, 243, 305. 29. Sezer, B.; Apaydin, H.; Bilge, G.; Boyaci, I. H.; Food Chem. 2018, 264, 142. 30. Correia, R.; Domingos, E.; Tosato, F.; Aquino, F.; Fontes, A.; Cáo, V.; Filgueiras, P.; Romão, W.; Forensic Chem. 2018b, 8, 57. 31. Correia, R.; Domingos, E.; Tosato, F.; Araujo, N.; Almeida, J.; Silva, M.; Marcelo, M.; Ortiz, R.; Filgueiras, P.; Romão, W.; Anal. Methods 2018c, 10, 593. 32. Paiva, E. M.; Rohwedder, J. J. R.; Pasquini, C.; Pimentel, M. F.; Pereira, C. F.; Fuel 2015, 160, 57. 33. Risoluti R.; Gregori A.; Schiavone S.; Materazzi S.; Analyst 2018, 90, 4288. 34. Risoluti, R.; Gullifa, G.; Battistini, A.; Materazzi, S.; Anal. Chem. (2019), doi: 10.20944/preprints201907.0235.v1. DOI: http://dx.doi.org/10.20944/preprints201907.0235.v1 35. Risoluti, R.; Gullifa, G.; Battistini, A.; Materazzi, S.; Talanta 2020, 211, 120672. 36. Basri, K. N.; Hussain, M. N.; Bakar, J.; Sharif, Z.; Khir, M. F. A.; Zoolfakar, A.; S. Spectrochim. Acta, Part A 2017, 173, 335. 37. Baqueta, M. R.; Coqueiro, A.; Março, P. H.; Valderrama, P.; Food Anal. Methods 2019, 13, 50. 38. Neves, M. D. G.; Poppi, R. J.; Siesler, H. W.; Molecules 2019, 24, 3. 39. Santos, P. C.; Tosato, F.; Cesconetto, M.; Corrêa, T.; Santos, F.; Lacerda Jr., V.; Pires, A.; Ribeiro, A.; Filgueiras, P.; Romão, W.; Quim. Nova 2020, 43, 891. 40. Borghi, F. T.; Santos, P. C.; Santos, F. D.; Nascimento, M. H. C.; Corrêa, T.; Cesconetto, M.; Pires, A. A.; Ribeiro, A. V. F. N.; Lacerda Jr., V.; Romão, W.; Filgueiras, P. R.; Microchem. J. 2020, 159, 105544. 41. Santos. F. D.; Santos, L. P.; Cunha, P. H. P.; Borghi, F. T.; Romão, W.; de Castro, E. V. R. de Oliveira, E. C., Filgueiras, P. R.; Fuel 2021, 283, 118854. 42. Baqueta, M. R.; Coqueiro, A.; Março, P. H.; Valderrama, P.; Talanta 2021, 222, 121526. 43. O'Brien, N. A.; Hulsea, C. A.; Friedricha, D. M.; Van Milligena, F. J.; von Guntena, M. K.; Pfeifer F.; Siesler, H. W.; Proc. SPIE 2012, 8374, 837404. 44. Silva, L. C. R.; Folli, G. S.; Santos, L. P.; Barros, I. H. A. S.; Oliveira, B. G.; Borghi, F. T.; Dos Santos, F. D.; Filgueiras, P. R.; Romão, W.; Vib. Spectrosc. 2020, 111, 103158. 45. Savitzky, A.; Golay, M. J. E.; Anal. Chem. 1964, 36, 1627. 46. Wold, S.; Chemom. Intell. Lab. Syst. 1987, 2, 37. 47. Bylesjo, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J. K.; Holmes, E.; Trygg, J.; J. Chemom. 2006, 20, 341. 48. Correia, R. M.; Loureiro, L. B.; Rodrigues, R. R. T.; Costa, H. B.; Oliveira, B. G.; Filgueiras, P. R.; Thompson, C. J.; Lacerda Jr. V.; Romão, W.; Anal. Methods, 2016, 8, 76. 49. Ribeiro, J. S.; Ferreira, M. M. C.; Salva, T. J. G.; Talanta, 2011, 83, 1352. 50. Geladi, P.; Kowalski, B. R.; Anal. Chim. Acta 1986, 185, 1. 51. De Souza, A. M.; Filgueiras, P. R.; Coelho, M. R.; Fontana, A.; Winkler, T. C. B.; Valderrama, P.; Poppi, R. J.; J. Infrared Spectrosc. 2016, 24, 293. 52. Portela, N. A.; Oliveira, E. C. S.; Cunha Neto, A.; Rodrigues, R. R. T.; Silva, S. R. C.; Castro, E. V. R.; Filgueiras, P. R.; Fuel 2016, 166, 12. 53. Agência Nacional de Vigilância Sanitária (ANVISA), RE no 899, de 29 de maio de 2003, Guia para validação de métodos analíticos e bioanalíticos. 54. Instituto Nacional de Metrologia, Normalização e Qualidade Industrial (INMETRO), Orientações sobre Validação de Métodos de Ensaios Químicos, DOQ-CGCRE-008, 2003. 55. Allegrini, F.; Olivieri, A. C.; Anal. Chem. 2014, 86, 7858. 56. Olivieri, A. C.; Introduction to Multivariate Calibration 2018, 159. 57. Folli; G. S.; Souza, L. M.; Araújo, B. Q.; Romão, W.; Filgueiras P.R.; Rapid Commun. Mass Spectrom. (2020), doi:10.1002/rcm.8861. DOI: http://dx.doi.org/10.1002/rcm.8861 58. PubChem, disponível em: https://pubchem.ncbi.nlm.nih.gov/ acessado em novembro 2021. 59. Barbin, D. F.; Felicio, A. L. de S. M.; Sunb, D.; Nixdorf, S. L.; Hirooka, E. Y.; Food Res. Int. 2014, 61, 23. 60. Mussatto, S. I.; Machado; E. M. S.; Martins, S.; Teixeira. J. A.; Food Bioprocess Technol. 2011, 4, 661. 61. Mei, L. H.; Oliveira, N.; Polímeros 2017, 27, 99. 62. Toda, T. A.; Dissertação de mestrado, Universidade de São Paulo, Brasil, 2016. 63. Ribas, A. I.; Cánovas, G. V. B.; Garza S. G.; Añó, V. G.; Métodos Experimentales en la Ingeniería Alimentaria, Acribia: Zaragoza, 2000. 64. Oetterer, M.; D'arce, M. A. B. R.; Spoto, M.; Fundamentos de ciência e tecnologia de alimentos, 1a ed., Manole: Barueri, 2006, 632p. 65. Cardoso, B. T.; Dissertação de mestrado, Universidade Tiradentes, Brasil, 2013. 66. Benicá, D. B.; Dissertação de mestrado, Universidade Federal do Espírito Santo, Brasil, 2019. 67. Ribani, M.; Bottoli, C. B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.; Quim. Nova 2004, 27, 771. 68. Tugnolo, A.; Beghi, R.; Giovenzana, V.; Guidetti. R.; J. Near Infrared Spectrosc. 2019, 27, 1. 69. Gomes. A. A.; Dissertação de mestrado, Universidade Federal da Paraíba, Brasil, 2012. 70. Zhang, C.; Jiang, H.; Liu, F.; He, Y.; Food Bioprocess Technol. 2016, 10. 71. Assis, C.; Oliveira, L. S.; Sena, M. M.; Food Anal. Methods 2017, 11, 578. 72. De Assis, M. W.; De Fusco, D. O.; Costa, R. C.; de Lima, K. M.; Cunha Jr, L. C.; Teixeira, G. H. de A.; J. Sci. Food Agric. 2018, 98, 5750. 73. Zareef, M.; Hassan, M. M.; Arslan, M.; Ahmad, W.; Ali, S.; Ouyang, Q.; Li, H.; Wu, X.; Chen. Q.; Microchem. J. 2020, 159, 105431. 74. Costa Filho, C. A.; Poppi, R. J.; Quím. Nova 1999, 22, 405. 75. Mendes, L. C.; Santos, J. C. F.; Corrêa, J. A.; de Alkimim Filho, J. F.; da Silva, D. V. R.; de Jesus, L. N.; Dibai, W. L. S.; Brazilian J. Food Technol. 2016, 19, 1. 76. de Lima, V. B.; Salva, T. de J. G.; Barboza, F. R.; Zago, C. de M. C.; Zanini B. S.; X Simpósio de pesquisa dos Cafés do Brasil, Vitória, 2019. 77. Macheiner, L.; Schmidt, A.; Schreiner, M.; Mayer, H. K.; J. Food Compos. Anal. 2019, 103307. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access