
 |
 |
 |
 |
 |
Revisão
|
|
| Atividade anti-Alzheimer de substâncias oriundas de produtos naturais e de origem sintética: alguns exemplos importantes dos últimos 20 anos Anti-Alzheimer activity of substances from natural products and of synthetic origin: some important examples of the last 20 years |
|
Ana Beatriz Silva VellosaI; Valdinei Bueno Lima FilhoII; Breno Almeida SoaresII,*; Nailton M. Nascimento-JúniorI,*
I. Departamento Bioquímica e Química Orgânica, Instituto de Química, Universidade Estadual Paulista,14800-060 - Araraquara - SP, Brasil Recebido em: 22/06/2021 *e-mail: nailton.monteiro@unesp.br Alzheimer's disease has about 9.9 million new cases annually, according to World Health Organization. Due to the increasing incidence of people affected by AD and the absence of an efficient treatment or cure so far, many researchers worldwide are involved in the search for novel promising drugs candidates, through several targets, exploring distinct structural patterns. In this context, we describe herein, focusing the last 20 years, strategic examples of substances with targets related to Alzheimer's disease derived from natural products, such as alkaloids, chalcones, coumarins and flavonoids, in addition to carbamates and heterocyclic compounds. Furthermore, some cases are discussed where computational chemistry is used to better understand protein-ligand interactions. INTRODUÇÃO A doença de Alzheimer (DA) teve seu primeiro relato em 1906, pelo psiquiatra clínico e neuropatologista alemão Alois Alzheimer.1 Porém, apenas a partir da década de 1970, o volume de estudos científicos sobre o tema cresceu significativamente.2 Esse transtorno neurodegenerativo progressivo se manifesta através da diminuição gradual da memória e declínio severo das funções cognitivas.3 A DA é uma das doenças neurodegenerativas mais comuns mundialmente, correspondendo de 50 a 70% dos casos de demência.4 Dados disponibilizados pela Organização Mundial da Saúde (OMS) revelam que aproximadamente 50 milhões de pessoas são acometidas por algum tipo de demência no mundo, com quase 10 milhões de novos casos anuais.5 Estima-se que até 2030, de 70 a 80 milhões de pessoas sejam acometidas de algum tipo de demência no mundo, com esse número podendo atingir aproximadamente 130 milhões de pessoas em 2050, com os impactos econômicos relacionados ao tratamento podendo atingir 2 trilhões de dólares.6,7 No Brasil, há uma carência de dados oficiais, porém estima-se que 1,5 milhão de pessoas sofram de DA, com previsão desse número quadruplicar após 30 anos.8 A DA pode acometer quase 50% dos indivíduos com mais de 85 anos.9 Ademais, homens e mulheres são afetados de forma diferente pela doença. A maior incidência em mulheres pode ser explicada por diversos fatores, como o efeito negativo da apoE (apolipoproteína E) ε4, que é a principal determinante genética da DA. A apoE é um transportador de colesterol, o qual interage com a proteína β-amiloide, podendo contribuir na agregação, no acúmulo e o depósito da proteína β-amiloide no cérebro e na neurodegeneração.10-12 Outro fator determinante na prevalência da DA em mulheres pode estar relacionado à queda rápida na produção dos hormônios sexuais após a menopausa, quando a incidência da DA aumenta em mulheres.13 Em nível celular, a DA é caracterizada pela degradação dos neurônios colinérgicos do sistema nervoso central (SNC), além da diminuição expressiva da acetilcolina (ACh) (1,Figura 1), um importante neurotransmissor relacionado a funções cognitvas como memória, percepção, linguagem, atenção e funções executivas.14
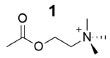 Figura 1. Estrutura química da acetilcolina (ACh)
A hidrólise promovida pelas enzimas colinesterases (AChE e BuChE) sobre a ACh na fenda sináptica, é a principal causa do déficit cognitivo causado pela DA. Em termos estruturais, as sequências primárias das colinesterases apresentam 65% de homologia, além de similaridades relacionadas às suas estruturas tridimensionais,15,16 que apresentam em comum uma cavidade de aproximadamente 20 Å de profundidade (Figura 2(C) e (D)), cuja extremidade inferior localiza-se o sítio catalítico aniônico (CAS), que comporta a tríade catalítica (Ser, Glu, His) (Figura 2(B)), conservada em ambas as enzimas,17 além de alguns subsítios relevantes para o reconhecimento molecular, como a cavidade oxiânion, o sítio de ligação acila e o sítio de ligação colina. Entretanto, na entrada dessa cavidade, encontra-se o sítio aniônico periférico (PAS), região que apresenta diferenças marcantes no número de resíduos aromáticos entre AChE (Trp286, Tyr124, Tyr72, Asp74, Tyr341) e BuChE (Ala277, Gln119, Asn68, Asp70, Tyr332) (Figura 2(A)), levando a diferenças de especificidade do ligante, o que explica a seletividade de alguns inibidores colinesterásicos.18
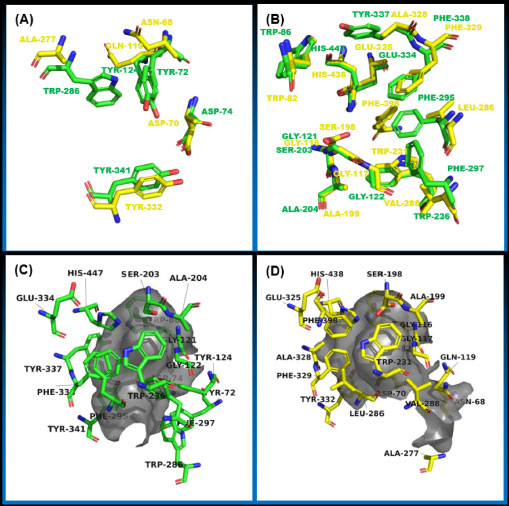 Figura 2. (A) Sobreposição dos resíduos de aminoácidos na região do sítio aniônioco periférico (PAS) nas enzimas hAChE (PDb id: 4EY7) em verde e hBuChE (PDB id: 4BDS) em amarelo. (B) Sobreposição dos resíduos de aminoácidos na região do sítio catalítico (CAS), incluindo a tríade catalítica. (C) Representação da cavidade do sítio ativo da hAChE em cinza. (D) Representação da cavidade do sítio ativo da hBuChE em cinza
Apesar da AChE ser considerada um alvo mais adequado para o planejamento de substâncias devido sua maior incidência nas regiões das sinapses,19 a BuChE também possui relevância em terapias colinérgicas, uma vez que há evidências do aumento dos níveis e da atividade da BuChE com a progressão da DA, levando a mudanças na proporção AChE / BuChE no córtex cerebral de pessoas acometidas pela DA, que podem ir de 0.2 a 11.20 Dessa forma, a BuChE também vem sendo utilizada como um potencial alvo na prospecção de novos anticolinesterásicos.21 Um dos marcadores neuropatológicos da DA é a proteína β-amiloide, que se forma através da clivagem da proteína precursora amiloide (do inglês, APP), por meio das enzimas β- e γ-secretases. Essa proteína insolúvel possui entre 39 e 42 resíduos de aminoácidos e pode ser encontrada em depósitos extracelulares pelo SNC, formando placas senis neurotóxicas (Figura 3).22 Nesse sentido, a busca por inibidores da β-secretase (BACE-1) tem sido uma estratégia adotada por pesquisadores nos últimos anos, gerando grandes expectativas na comunidade científica.23 Contudo, apesar de reduzir a formação de placas β-amiloide, alguns inibidores da BACE-1 que chegaram recentemente até estudos clínicos de fase III, mostraram-se ineficazes em aliviar os sintomas da doença, além de apresentarem efeitos adversos.24,25
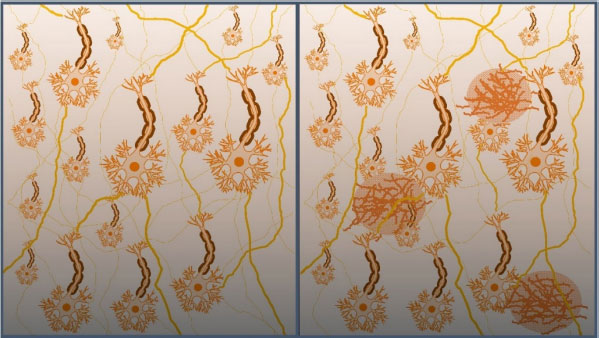 Figura 3. Campo neuronal saudável, à esquerda, e campo neuronal com formação de placas senis, causados pelo acúmulo da proteína β-amiloide, à direita. Adaptada de Alzheimer's Association26
Outro marcador patológico que caracteriza a DA, consiste na formação de emaranhados neurofibrilares (do inglês, NFT), devido ao processo anormal de fosforilação da proteína tau, responsável pelo estabelecimento e manutenção da comunicação interneuronal, além da função de estabilizar os microtúbulos dos axônios, (Figura 4).27 A enzima glicogênio sintase cinase (GSK-3β) possui um papel relevante na fosforilação da proteína tau,28 e vem sendo explorada como um potencial alvo no planejamento de candidatos a protótipos de fármacos multialvo para a DA.29 Além disso, outra isoforma da glicogênio sintase cinase (GSK-3α) está relacionada com a produção do peptídeo β-amiloide via γ-secretase,30 sendo dessa forma, outro alvo terapêutico conveniente na prospecção de novos agentes anti-Alzheimer.
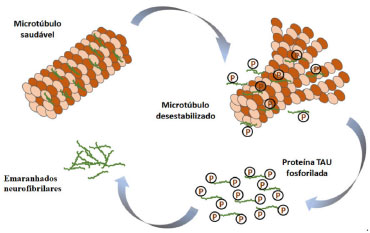 Figura 4. Hiperfosforilação da proteína tau. Adaptado da ref. 31
Atualmente, ainda não há terapias disponíveis para interromper e reverter os danos cerebrais resultantes da DA. Recomenda-se, portanto, tratamentos não-farmacológicos para melhorar a qualidade de vida, incluindo exercícios físicos, terapia ocupacional, musicoterapia e fisioterapia, assim como alguns medicamentos prescritos para amenizar, provisoriamente, os sintomas em indivíduos diagnosticados com DA.32 São conhecidos cinco medicamentos, a partir de 4 fármacos (2-5), aprovados pela Food and Drug Administration, para o tratamento dos pacientes diagnosticados com DA: Aricept® (donepezila (2), 1996), Razadyne® (galantamina (3), 2001), Exelon® (rivastigmina (4), 2000), Namenda® (memantina (5), 2003) e Namzaric® (donepezila (2) + memantina (5), 2014) (Figura 5).33
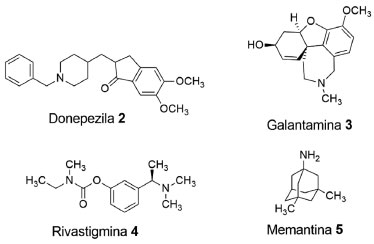 Figura 5. Estrutura química dos fármacos aprovados pela FDA, para o tratamento dos sintomas da doença de Alzheimer33
Os fármacos 2, 3 e 4 são inibidores da acetilcolinesterase (AChE), atuando na diminuição da degradação de ACh, sendo prescritos para os estágios leve e moderado da doença, ajudando a prevenir, retardar e a controlar os sintomas. A rivastigmina (4) pode ser considerada como um inibidor dual, pois seu mecanismo de ação também inclui a inibição da BuChE.34 Já o fármaco 5, é prescrito para tratar casos moderados e graves, atuando como antagonista em receptores de glutamato do tipo N-metil-D-Aspartato (NMDA).35 O medicamento Namzaric®, uma associação de memantina (5) e donepezila (2), também é indicado para casos moderados e graves.36 Por conta do aumento do número de pessoas afetadas pela DA e o fato de não haver tratamento que resulte em cura definitiva até o momento, se torna cada vez mais necessária a busca de novos candidatos a fármacos mais eficazes, tanto por meio da exploração de diferentes alvos (e.g. AChE, BuChE, BACE-1, GSK-3β e α), como de padrões estruturais distintos, sendo estes últimos exemplificados e discutidos a seguir.32
ALCALOIDES E DERIVADOS SINTÉTICOS INIBIDORES DA ACHE E DA BUCHE Alcaloides consistem em uma classe de produtos naturais nitrogenados de baixo peso molecular, os quais participam de processos biológicos de plantas, animais e microrganismos. Cerca de 25% das espécies vegetais apresentam alcaloides em sua composição, sendo especialmente abundantes nas famílias Apocynaceae, Asteraceae, Fabaceae, Papaveraceae, Rubiaceae e Solanaceae.37 Extratos contendo alcaloides são usados pela humanidade desde a antiguidade, entre os quais, destacam-se o ópio (Papaver somniferum), meimendro (Hyoscyamus niger) e coca (Erythroxylum coca).38 É importante destacar que dentre os fármacos aprovados pelo FDA para o tratamento de pessoas portadoras da DA, existe um alcaloide isolado a partir da espécie Galanthus nivalis (Amaryllidaceae) que é a galantamina (3), além da rivastigmina (4), que pode ser considerado um derivado semissintético obtido a partir do alcaloide fisostigmina. Consequentemente, essa classe de substâncias têm sido sistematicamente estudada, visando o desenvolvimento de novos candidatos à fármacos.37 Como exemplo de estudos envolvendo alcaloides, que vêm sendo desenvolvidos, visando a obtenção de novas substâncias com atividade anti-Alzheimer, em 2014, Viayna e colaboradores planejaram uma série de derivados híbridos com o objetivo de inibir alvos relacionados à DA.39 O planejamento estrutural dos candidatos a fármacos foi baseado nas estratégias da química medicinal de: (a) hibridação molecular, entre rhein (6) e hupina (7), e (b) homologação linear, utilizando 5, 6 e 9 espaçadores metileno (-CH2-), para gerar os derivados híbridos 8, 9 e 10, respectivamente (Figura 6).
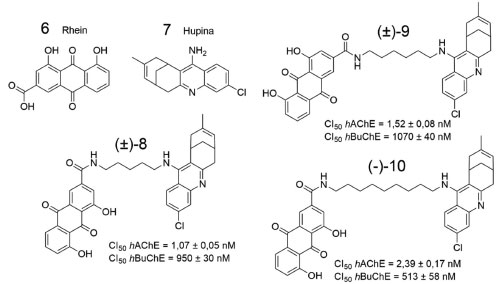 Figura 6. Estruturas químicas de Rhein (6) e Hupina (7), além de três alcaloides sintéticos híbridos, sintetizados por Viayna e colaboradores39
Os híbridos (8-10) foram sintetizados e avaliados frente a AChE, a butirilcolinesterase (BuChE) e em relação à atividade antiagregante da proteína tau. Ensaios in vitro, realizados em células de Escherichia coli, mostraram que os híbridos 8, 9 e 10 exibiram atividade inibitória frente ambas colinesterases humanas (hChE's), e atividade antiagregante tau, além de apresentarem resultados adequados de permeabilidade cerebral.39 Estudos com membranas artificiais revelaram que as substâncias sintetizadas podem ser capazes de atravessar a barreira hematoencefálica. O híbrido 8 foi o mais potente inibidor da hAChE. Paralelamente, o derivado 10 foi o mais potente inibidor da β-secretase 1 (BACE-1) (80 ± 10 nmol L-1) e da agregação da tau (34,3 ± 0,1%). Dessa forma, 10 foi submetida a ensaios in vivo que confirmaram sua capacidade de diminuição dos níveis de agregação da proteína β-amiloide, além de ação protetora contra efeitos de diminuição das sinapses, que são induzidas pelos oligômeros da mesma proteína. Todos os híbridos sintetizados apresentaram seletividade para hAChE em relação a hBuChE, com índices de seletividade entre 28 e 888.39 Em outro trabalho, publicado um ano depois, Yang e colaboradores isolaram constituintes químicos de sementes de Peganum harmala, os quais tiveram a atividade inibitória avaliada frente a AChE e a BuChE.40 A seguir, são representadas as seis substâncias que apresentaram melhor atividade inibitória frente a AChE (Figura 7).
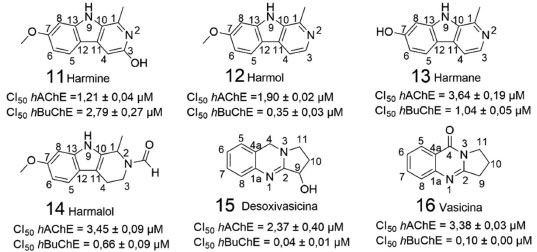 Figura 7. Alcaloides isolados por Yang e colaboradores40
As substâncias 11-16 apresentaram destacada inibição da AChE e da BuChE, com valores de concentração inibitória (CI50) nas faixas de 1,21-3,64 µmol L-1 e 0,04-2,79 µmol L-1, respectivamente. A presença de substituintes nos carbonos 1, 3 e 7, assim como no nitrogênio 2 (14), foram importantes para a inibição de ambas as enzimas. Cabe destacar que a presença dos grupos metoxila ou hidroxila no carbono 7 das β-carbolinas levou ao aumento da inibição frente a AChE. Já em 15 e 16, foram relevantes para a atividade inibitória: o anel de cinco membros abrangendo o carbono 2 e o nitrogênio 3, assim como a carbonila presente no carbono 4 ou a hidroxila presente no carbono 9. Outro estudo que merece destaque, envolve oito alcaloides naturais de padrão estrutural isoquinolínico, isolados por Gonzalez e colaboradores, a partir da raiz de Zanthoxylum rigidum. As substâncias obtidas foram avaliadas quanto a atividade em diversos alvos, envolvidos nos principais mecanismos associados à patogênese da DA.41 A seguir, são mostrados os resultados mais relevantes de atividade inibitória frente a AChE da enguia elétrica (EeAChE) e recombinante humano (HrAChE) e BuChE de soro equino (EqBuChE), permitindo a identificação de dois alcaloides benzofenantridínicos (17 e 18), com resultados mais promissores (Figura 8). Cabe ressaltar que as formas oligoméricas das AChE's, oriundas de diferentes espécies de animais vertebrados, possuem alta similaridade estrutural e funcional, indicando uma conservação ao longo do processo evolucionário.42 Essa semelhança estrutural, principalmente na região do sítio catalítico, permite a realização de ensaios de inibição enzimática utilizando AChE's de ocorrência em diferentes organismos, com a ressalva de que uma análise comparativa só terá validade em caso de valores de inibição obtidos em ensaios realizados com AChE's oriundas do mesmo organismo.
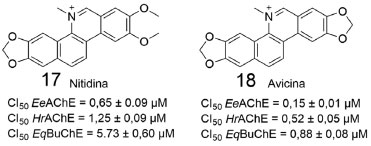 Figura 8. Alcaloides benzofenantridínicos estudados por Gonzalez e colaboradores41
Ambos alcaloides benzofenantridínicos apresentaram inibição da AChE e da BuChE, sendo mais potentes frente a AChE, com valores de CI50 na faixa submicromolar. Além disso, esses alcaloides apresentaram moderada atividade antiagregante da proteína β-amiloide.
DERIVADOS SINTÉTICOS DE CHALCONAS INIBIDORES DA ACHE As chalconas são substâncias precursoras na biossíntese de flavonóides, e podem ser encontradas em diversas espécies vegetais. Sua estrutura básica (1,3-difenil-2-propen-1-ona) é composta por dois anéis aromáticos conectados por uma carbonila α,β-insaturada, em um sistema de elétrons π completamente deslocalizado, conectando ambos anéis aromáticos (Figura 9).43,44
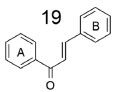 Figura 9. Estrutura química da 1,3-difenil-2-propen-1-ona (19)43,44
As chalconas apresentam um amplo espectro de atividades biológicas, tais como: antioxidante, antibacteriana, anti-helmíntica, amebicida, antiúlcera, antiviral, inseticida, antiprotozoária, antitumoral (citotóxica) e imunossupressora.45 Em vista desse histórico, pesquisadores vem utilizando as chalconas no desenvolvimento de novos candidatos a fármacos.44 Um estudo envolvendo a síntese e avaliação de uma série de novos derivados de chalconas, numa abordagem multialvo, visando atividade anti-Alzheimer, foi publicado por Zhang e colaboradores.46 A maioria das substâncias apresentaram inibição seletiva da AChE, inibição significativa da monoamina oxidase (MAO-B), atividade antioxidante adequada, além de atividade inibitória moderada para agregação da proteína β-amiloide (auto-induzida ou com indução por Cu2+). As seis substâncias mais promissoras (Figura 10) exibiram melhor atividade inibitória frente a EeAChE em relação a BuChE de soro de rato, sendo 20 o mais potente da série (CI50 = 0,07 µmol L-1). Pode ser observado que a atividade inibitória da AChE está diretamente relacionada com a presença dos grupos (N,N-dietilamino)metila (anéis A e B) e metoxila (anel A) da subunidade chalcona.46
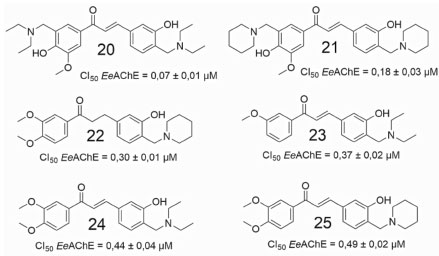 Figura 10. Derivados de chalconas estudados por Zhang e colaboradores46
A substância 24 apresentou potência moderada frente a AChE (CI50 = 0,44 µmol L-1), além de inibição da agregação do peptídeo β-amiloide auto-induzida (55%, a 25 µmol L-1), propriedade quelante de biometal e atividade antioxidante moderada. Num outro trabalho, envolvendo as estratégias de hibridação molecular e homologação linear, Chandrika e colaboradores sintetizaram doze derivados chalcona-donepezila, divididos em duas séries (1,3 e 1,4-chalcona-donepezila). Os derivados obtidos foram avaliados quanto a inibição da EeAChE e da EfBuChE, assim como a prevenção e/ou interrupção da oligomerização da proteína β-amiloide.47 As quatro substâncias que apresentaram melhor inibição da AChE estão representadas na Figura 11.
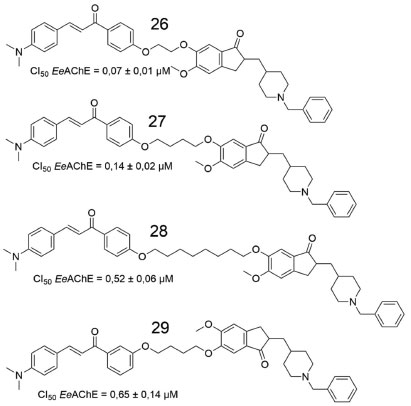 Figura 11. Derivados de chalconas sintetizados a avaliados por Chandrika e colaboradores47
Comparando-se os híbridos das duas séries, foi possível observar que quase todos os derivados 1,4-chalcona-donepezila foram mais eficazes na inibição da EeAChE. Porém, quando comparados à donepezila (CI50 = 0,12 ± 0,01), a maioria exibiu valores de CI50 de 1 a 15 vezes mais elevados, com a exceção de 26 e 27 (0,07 ± 0,01 µmol L-1, 0,14 ± 0,02 µmol L-1, respectivamente). Concluiu-se, portanto, que o aumento do comprimento da cadeia que conecta as subunidades 1,4-chalcona e donepezila causa diminuição gradual da atividade inibitória frente a AChE. Os estudos computacionais mostraram que, na proteína β-amiloide, os resíduos de glutamato (Glu22) e aspartato (Asp23) formaram uma região de superfície carregada negativamente, que poderia interagir favoravelmente com o grupo N,N-dimetilamino, carregado positivamente. Dessa forma, o canal amplamente hidrofóbico, que se mostrou alinhado à cadeia lateral, incluindo os resíduos Phe19, Ala21 e Val24, se encaixa e interage favoravelmente com as regiões hidrofóbicas dos derivados de chalcona. Além disso, oxigênio carbonílico dos derivados de chalcona pode formar ligações de hidrogênio com átomos de nitrogênio amídicos, de regiões específicas da proteína β-amiloide, devido à distorção das folhas β. Num outro estudo envolvendo derivados de chalcona, Reeta e colaboradores avaliaram uma série de etil-acetohidroxamato-chalconas quanto às atividades inibitórias frente a hAChE,48 onde a maioria dos derivados exibiram efeito inibitório seletivo e potente. Estudos de reversibilidade e cinética mostraram que o derivado fluorado 33 (Figura 12) exibiu atividade inibitória destacada para a AChE.
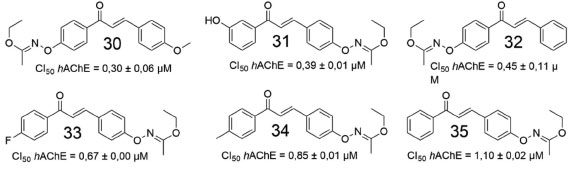 Figura 12. Derivados de chalconas sintetizados e avaliados por Reeta e colaboradores48
Ensaios farmacológicos complementares demonstraram que a maioria das substâncias inibiu seletivamente a hAChE. O efeito inibitório de 35 e 32 frente a AChE tem uma diferença notável, com valores de CI50 de 1,10 e 0,45 µmol L-1, respectivamente. Portanto, a mudança da posição do grupo etil-acetohidroxamato (EAH), em cada um dos anéis aromáticos, pode resultar em alterações na inibição da AChE.
DERIVADOS DE CUMARINAS INIBIDORES DA ACHE E DA BUCHE As cumarinas constituem uma classe de compostos que apresentam em sua estrutura dois anéis de 6 membros condensados, sendo um aromático e outro uma lactona α,β-insaturada, tendo como estrutura mais simples a 1,2-benzopirona (36), representada pela Figura 13.49 As principais fontes de cumarinas são plantas de diferentes espécies, frutas, bactérias, fungos e mel, sendo responsável pelo seu aroma.50
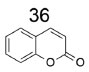 Figura 13. Estrutura química da 1,2 - benzopirona (36)50
A maioria das cumarinas comercializadas são sintetizadas a partir do aminoácido fenilalanina, salicilaldeído ou isoladas da espécie vegetal Dipteryx odorata. Suas utilizações são amplas, transitando entre aromatizantes de alimentos, fixador de perfumes, aditivo em tintas em spray, produtos de limpeza, até a utilização como fármacos, considerando as propriedades antibiótica, broncodilatadora, fungicida, anticoagulante, analgésica e antitumoral dessa classe de substâncias.51 Explorando o potencial farmacológico das cumarinas, Zhou e colaboradores (2008) planejaram e sintetizaram 3 séries de derivados, com base em características estruturais da ensaculina (37), para serem testadas na inibição da AChE.52 Os testes in vivo para obtenção dos valores médios de inibição seguiram a metodologia de Ellman et al. (1961) e foram realizados em peixes da espécie Electrophorus electricus (poraquê).53 As 8 estruturas com melhores valores de CI50 seguem representadas na (Figura 14).
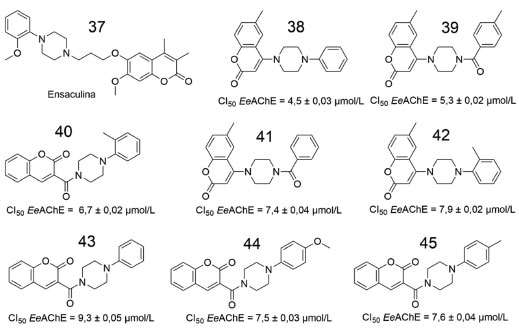 Figura 14. Estruturas de derivados de cumarinas estudadas por Zhou e colaboradores52
Com base nos resultados obtidos, os autores concluíram que os derivados substituídos com a subunidade N-fenilpiperazina nas posições 3 e/ou 4 do anel da cumarina apresentaram melhor atividade inibitória (variando entre 4,5 e 9,3 g/mL). Outro ponto observado foi a influência da carbonila, entre subunidade cumarina e a N-fenilpiperazina, na inibição da EeAChE. Além disso, a distância entre o átomo de carbono carbonílico da cumarina e a piperazina mostrou-se importante para a atividade inibitória.52 Um estudo realizado por Shen et al., envolvendo três séries de derivados contendo a subunidade benzofurano condensada ao anel aromático cumarínico, revelou o potencial anticolinesterásico (hAChE e hBuChE) de diversas substâncias, estando sete delas apresentadas na (Figura 15).54
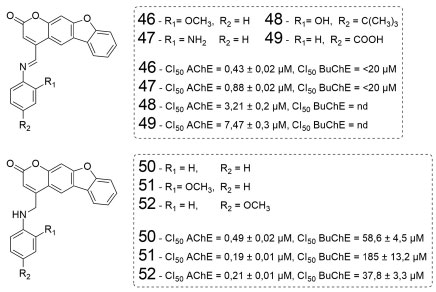 Figura 15. Estruturas dos derivados cumarínicos estudados por Shen e colaboradores54
O composto 51, com a subunidade metilanilino metoxilada em orto, foi o mais potente da série, com CI50 = 0,19 ± 0,01 µmol L-1. Os derivados 50, 51 e 52 apresentaram maior seletividade para a AChE, em comparação com a BuChE. Já a estrutura 46, metoxilada na mesma posição que 51, exibiu a maior atividade contra a AChE entre seus análogos 47-49, indicando que essa característica pode ter influência em alguma interação relevante com algum resíduo de aminoácido chave do sítio ativo da AChE. Outro aspecto importante relatado pelos autores é que os compostos com grupos substituintes mais volumosos, como 48 e 49, resultaram na diminuição da atividade quando comparados com os que possuem substituintes menores. Isso sugere que o impedimento estérico pode influenciar no acesso das estruturas ao sítio ortostérico da enzima. Pisani e colaboradores (2010) analisaram os valores de inibição da AChE bovina e da BuChE de soro equino de diversos derivados cumarínicos.55 Dentre eles, a cumarina dimetoxilada 53 (Figura 16) foi a que apresentou melhor perfil de inibição, com índice de seletividade BoAChE/EqBuChE > 300.000.
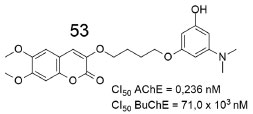 Figura 16. Derivado cumarínico dimetoxilado estudado por Pisani e colaboradores55
A cinética da inibição da AChE bovina pelo composto 53 demonstrou uma inibição do tipo competitiva, com valor de Ki = 2,1 ± 0,4 nmol L-1. Segundo os autores, esse mecanismo difere da donepezila e de outros inibidores da literatura, que geralmente se ligam nos sítios catalítico e periférico da AChE. Para melhor compreensão dos requisitos moleculares responsáveis pela alta afinidade e seletividade de ligação de 53 com AChE, foi realizado um estudo de modelagem molecular, sugerindo que a presença dos dois grupos metoxilas estabilizam a subunidade cumarínica dentro do sítio aniônico periférico (PAS), em uma área com ampla exposição ao solvente. Dessa forma, foi possível a inserção dessa subunidade na fenda aberta, entre os anéis aromáticos presentes no Trp286 e na Tyr341, respectivamente. Outros derivados substituídos na posição 7 da subunidade cumarina também foram avaliados pelo mesmo grupo de pesquisa, todavia nenhum apresentou resultado adequado de inibição da AChE,56 confirmando 53 como a substância analisada com comprimento e flexibilidade conformacional ideais, dentro da série. Em publicação recente, Abdshahzadeh e colaboradores (2019) sintetizaram e analisaram duas séries de derivados de cumarinas.57 O efeito dos substituintes N,N-dietilamina e o comprimento do espaçador foram investigados na inibição da AChE e da BuChE. Os cinco derivados mais potentes são apresentados na Figura 17.
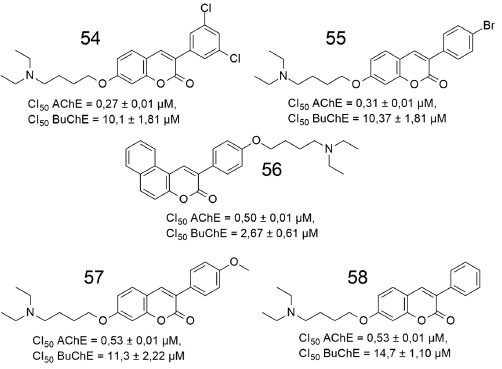 Figura 17. Estruturas de derivados de cumarinas estudadas de Abdshahzadeh e colaboradores57
O derivado contendo uma subunidade 3,5-diclorofenila (54) foi o mais potente frente a AChE, apresentando CI50 = 0,27 ± 0,01 µmol L-1. Estudos de modelagem molecular e cinética revelaram que 54 atua de forma mista, interagindo com o sítio catalítico e com o sítio aniônico periférico da enzima. Adicionalmente, 54 bloqueou a auto-agregação de β-amiloide em 44,11% a 100 µmol L-1, além de proteger significativamente as células PC12 contra o estresse oxidativo induzido por H2O2. Os derivados cumarínicos 54-58 apresentaram melhores resultados do que os demais análogos modificados na posição 3 que também fizeram parte dos estudos, provavelmente, devido à interação hidrofóbica da subunidade fenila na posição 3 da cumarina com o sítio ativo da enzima, sugerindo que cumarinas substituídas podem ser exploradas como inibidores da AChE.
DERIVADOS DE FLAVONÓIDES INIBIDORES DA ACHE E DA BUCHE Os flavonóides constituem uma classe de substâncias polifenólicas de origem vegetal, cuja estrutura básica é composta por 15 átomos de carbonos, distribuídos em dois anéis benzênicos e um pirano central, conforme mostra a (Figura 18).58 Devido a possibilidade de variação das estruturas fenólicas, esses compostos apresentam diversas propriedades farmacológicas, se destacando pelas propriedades antioxidantes, anti-inflamatórias e inibição da amiloidogênese.59
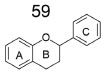 Figura 18. Estrutura química básica dos flavonóides (59)58
Em relação a DA, alguns flavonóides podem interferir na produção e agregação das proteínas β-amiloide e tau,60 promovendo a depuração da proteína β-amiloide e inibindo a fosforilação da tau pela via de sinalização mTOR/autofagia. Além disso, podem atuar como inibidores da AChE e da BuChE, enzimas diretamente relacionadas com a patogênese da DA.61 Em 2015, o potencial anticolinesterásico de 4 flavonoides isolados de Achillea millefolium L. foi avaliado por Sevindik e colaboradores (Figura 19).62 Os estudos de ancoragem molecular revelaram o composto 62 como o mais promissor, interagindo com os sítios da AChE e da BuChE. Ademais, de acordo com o mesmo protocolo in silico, 63 apresentou interações nos sítios catalítico e periférico de AChE e da BuChE, sendo possível observar interações relevantes com resíduos de aminoácidos chave de ambas as colinesterases, reforçando a capacidade inibitória de 63. Adicionalmente, ensaios in vivo confirmaram o potencial de inibição de 63 (CI50 AChE = 1.65 ± 0.122 µmol L-1 e CI50 BuChE = 1.97 ± 0.220 µmol L-1). Por outro lado, 60 apresentou o menor valor de CI50 para ambas as enzimas.
 Figura 19. Estruturas de derivados de flavonoides estudadas por Sevindik e colaboradores62
No mesmo ano, Balkis e colaboradores avaliaram a potência (CI50) de diversos flavonóides,63 conforme pode ser observado na (Figura 20), onde são destacadas as quatro substâncias mais ativas.
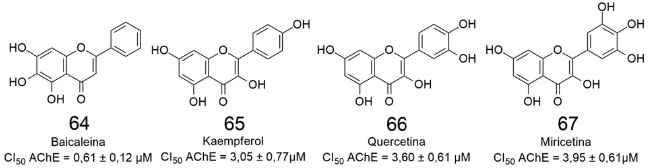 Figura 20. Estruturas de derivados de flavonoides estudadas por Balkis e colaboradores (2015)63
A (Figura 21) revela que a baicaleina (64) é a inibidora mais potente da EeAChE, apresentando CI50 = 0,61 µmol L-1. As três hidroxilas ligadas aos átomos 5, 6 e 7 do anel A de 64 se mostram importantes para a atividade inibitória destacada, assim como a ausência de hidroxilas no anel C, ao compararmos os derivados 64-67.63
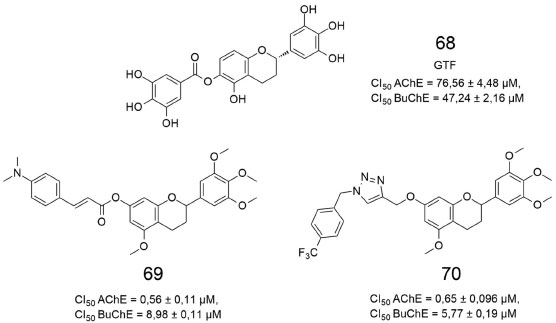 Figura 21. Estruturas de derivados de flavonoides estudadas por Shi e colaboradores64
Em outro estudo recente, desenvolvido por Shi, três séries oriundas do flavonoide 7-O-galoiltricetiflavona (GTF) (68) foram sintetizadas e avaliadas quanto a inibição da agregação da proteína β-amiloide e inibição frente as enzimas AChE e BuChE.64 Os derivados 69 e 70 se apresentaram como os mais potentes (Figura 21). Comparando-se os resultados de todas as substâncias analisadas, os derivados cinamato (e.g. 69) e 1,2,3-triazol substituídos (e.g. 70) apresentaram maiores atividades inibitórias frente à AChE em detrimento dos derivados benzoato e benzenossulfonato substituídos.64 Além disso, os autores também avaliaram a capacidade das substâncias na inibição da agregação de β-amiloide, usando um ensaio fluorométrico baseado em tioflavina-T. Comparando todas as estruturas analisadas, constatou-se que os híbridos de cinamato possuem atividades de inibição mais altas do que as substâncias substituídas por benzoato e benzenossulfonato. Um estudo cinético revelou que as substâncias 69 e 70 são inibidoras de tipo misto, enquanto que estudos computacionais indicaram ligações de hidrogênio entre o grupo carbonila de 69 com a Arg289 e a Ile287 da AChE. Já a estrutura 70 posicionou-se na bolsa de ligação da BuChE, realizando ligação de hidrogênio com a Ser198. Ensaios in vitro revelaram que 69 e 70 possuem atividade neuroprotetora contra danos às células SH-SY5Y do neuroblastoma humano e quase nenhuma toxicidade nas mesmas.64
DERIVADOS PIPERAZÍNICOS COMO INIBIDORES DA ACHE E DA BUCHE A subunidade estrutural piperazina (71) (Figura 22) consiste em um anel de seis membros, contendo dois átomos de nitrogênio nas posições 1 e 4, os quais podem apresentar diversos substituintes, em um ou em ambos os átomos de nitrogênio. Nesse contexto, derivados piperazínicos são descritos como estruturas privilegiadas, por apresentarem um amplo grupo de atividades biológicas, por exemplo: antipsicótica, antidepressiva, anticonvulsivante, anti-histamínica, anticâncer, anti-HIV e anticolinesterásica,65 sendo essa última explorada a seguir.
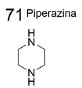 Figura 22. Formula estrutural da piperazina (71)65
Sergeant e colaboradores sintetizaram dois derivados piperazínicos, contendo subunidades oriundas da tacrina e da rivastigmina, gerando 72 e 73, respectivamente (Figura 23). Ambos apresentaram inibição em escala subnanomolar frente a AChE, além de apresentarem potencial de redução da agregação da β-amiloide. Estudos in vitro e in vivo indicaram que o composto 72 também reduziu a formação de emaranhados fibrilares causados pela fosforilação da proteína tau, prevenindo lesões cerebrais e deficiências associadas à memória.66
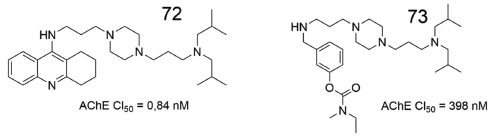 Figura 23. Derivados piperazínicos, 72 e 73, com dados de atividade frente a AChE66
Um estudo recente, envolvendo a síntese de derivados pirazolopiridínicos, contendo a subunidade piperazina, revelou que os derivados 74, 75 e 76 são capazes de inibir a agregação β-amiloide, interferindo na formação de folhas β, de forma mais expressiva do que a curcumina (52,70%) (Figura 24). Nenhum dos três derivados destacados apresentou atividade inibitória frente a BuChE, entretanto 76 foi o único capaz de inibir a AChE, de forma potente e seletiva (CI50 = 0,045 ± 0,010 µmol L-1). Estudos computacionais, visando explicar a atividade inibitória de 76 frente a AChE demonstraram que ambas as subunidades pirazolopiridina estão envolvidas em interações do tipo empilhamento π com o Trp84 e com o Trp279, a distâncias de 3,3 Å e 5,1 Å, respectivamente. Para o mesmo derivado, foi possível observar uma ligação de hidrogênio (2,3 Å) entre o carboxilato da cadeia lateral do Asp72 e o hidrogênio (NH) de uma das subunidades pirazolopiridina.67
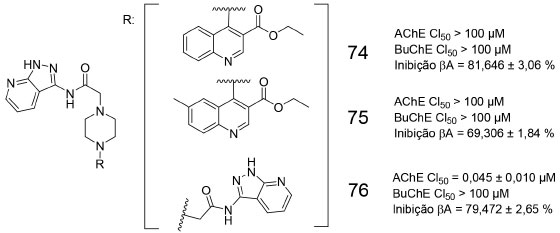 Figura 24. Derivados pirazolopiridínicos (74-76), avaliados frente a AChE, BuChE e inibição da proteína β-amiloide67
A donepezila (2), um os quatro fármacos já aprovados para tratamento de pacientes com DA, é comumente utilizada como padrão de comparação, com possíveis candidatos a inibidores da AChE. Nesse contexto, Tripathi e colaboradores desenvolveram uma série de N-benzil piperazinas e os resultados dos estudos in vitro mostraram que o composto 77 foi o mais ativo da série de candidatos a inibidores, apresentando um resultado de inibição da AChE na mesma escala que os valores apresentados para a donepezila, no mesmo estudo (CI50 = 0,1 ± 0,01 µmol L-1) (Figura 25). Estudos comportamentais in vivo indicaram melhora significativa nas disfunções cognitivas, através do modelo de camundongos com amnésia induzida por escopolamina. Adicionalmente, estudos de ancoragem molecular evidenciaram que 77 apresenta interações com a His440 e com a Ser200, ambos pertencentes à tríade catalítica do sítio ativo da AChE.68
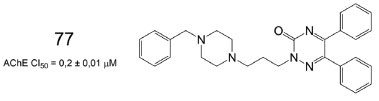 Figura 25. Estrutura química de 77 e valor de CI50 frente a AChE68
Em outro estudo visando a obtenção de substâncias com perfil multialvo frente a doença de Alzheimer, uma série de derivados da donepezila (2) foram sintetizados e apresentaram atividade inibitória frente a EeAChE e a EqBuChE, além de inibirem a agregação de β-amiloide e exibirem propriedades antioxidantes. Os derivados 79 e 80 mostraram-se mais eficazes que o fármaco padrão donepezila (CI50 = 0,039 ± 0,002 µmol L-1) na inibição da AChE. Adicionalmente, 78, 79 e 80 diminuíram de forma efetiva o estresse oxidativo induzido por H2O2 em células SH-SY5Y e apresentaram inibição da agregação de β-amiloide melhores do que a curcumina (55,7 ± 0,56%), exibindo atividade neuroprotetora (Figura 26).69
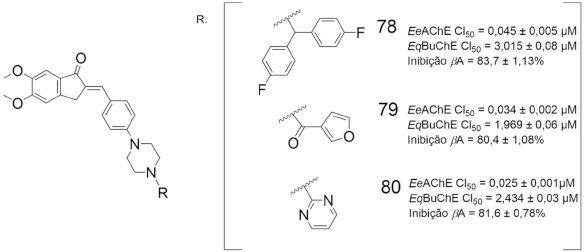 Figura 26. Estruturas químicas de 78-80 e atividade inibitória frente a AChE, BuChE e inibição da βA69
Os resultados globais indicam que a maioria dos compostos testados apresentaram potência inibitória comparativamente menor contra frente à BuChE, com valores de CI50 variando entre 0,214 a 12,22 µmol L-1, indicando maior seletividade em relação à AChE.69
DERIVADOS CARBAMATOS COMO INIBIDORES DA ACHE E DA BUCHE Considerando as características multifatoriais da DA, substâncias híbridas vêm sendo desenvolvidas para serem testadas frente aos diversos alvos envolvidos. Nesse contexto, Sang e colaboradores planejaram derivados híbridos baseados no fármaco rivastigmina (4) e na apigenina (81), a qual ainda passa por ensaios clínicos.70 O derivado bicarbamato (82) (Figura 27) se mostrou um potente antioxidante, com capacidade de absorvância do radical oxigênio (ORAC, oxygen radical absorbance capacity) igual a 1,3 eq, além de inibir ambas colinesterases em ensaios realizados em córtex homogeneizado de rato (CI50 = 6,80 ± 0,31 µmol L-1) e BuChE de soro humano (CI50 = 16,1 ± 0,52 µmol L-1). Adicionalmente, 82 apresentou potencial como agente quelante seletivo, inibindo de forma significativa a agregação de oligômeros β-amiloide.70
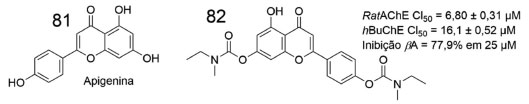 Figura 27. Estrutura da Apigenina (81) e do dicarbamato 8270
Os autores ainda realizaram estudos in vivo, utilizando modelo em peixe-zebra (zebrafish) para a DA e os resultados indicaram a baixa toxicidade de 82, mesmo quando administrado em doses maiores que 2000 mg kg-1, além de efeitos protetores nos danos vasculares mediados por β-amiloide. Ademais, os ensaios in vivo também apontaram que 82 pode melhorar déficits de memória induzidos por escopolamina, complementando os estudos que evidenciam 82 como um promissor candidato a agente anti-Alzheimer com potencial ação multialvo.70 Estudos de ancoragem molecular no sítio ativo da AChE, indicaram que o espaçador metileno de uma das subunidades N-etil-N-metilcarbamato de 82 interage com o resíduo Trp86, pertencente ao sítio catalítico aniônico, por meio de interações sigma-π. A carbonila central realiza ligação de hidrogênio com a Phe295, enquanto subunidade bicíclica central interage com os resíduos Trp286 e Tyr341, via interações π-π. No que diz respeito às interações de 82 no sítio ativo da BuChE, foi possível observar uma ligação de hidrogênio entre o grupo hidroxila e a Tyr128, além de interações do tipo empilhamento π entre o anel aromático fenólico e o Trp82. Um dos átomos de oxigênio de uma das subunidades carbamato interage com o Asp70, como aceptor de ligação hidrogênio (Figura 28).70
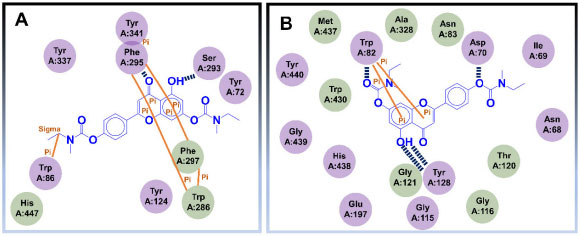 Figura 28. Interações do derivado bicarbamato 82 com a AChE (A) e com a BuChE (B)70
Um trabalho recente envolveu a obtenção de carbamatos derivados do bambuterol (83, Figura 30), um potente inibidor da BuChE.71 Os ensaios de inibição enzimática com EeAChE e a EqBuChE revelaram que 11 das 16 substâncias da série apresentaram potencial inibitório dual maior do que a rivastigmina (4,Figura 5). Dentre essas, destacam-se os carbamatos BMC-3 (84) e BMC-16 (85), os quais obtiveram os melhores valores de CI50 para ambas as colinesterases (EeAChE e a EqBuChE) (Figura 29). Esses resultados incentivaram os autores a realizarem novos ensaios de 84 e 85, utilizando ChE's de plasma humano, já que a atividade e a suscetibilidade de ChE's à inibidores aparenta ser diferente entre humanos e outros organismos.72 Os resultados confirmaram a atividade inibitória dual desses derivados, que também apresentaram potencial de permeação através da barreira hematoencefálica (BHE).69
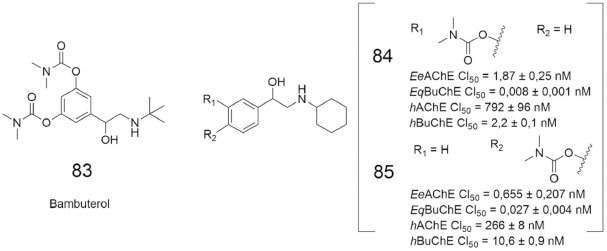 Figura 29. Estruturas químicas do inibidor seletivo da BuChE bambuterol (83) e dos carbamatos 84 e 8569
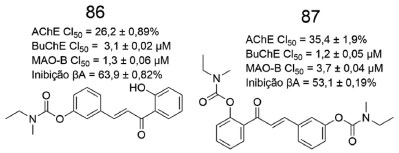 Figura 30. Estruturas químicas dos híbridos chalcona-carbamato 86 e 87, assim como os respectivos dados de avaliação farmacológica73
Sang e colaboradores sintetizaram uma série de híbridos chalcona- carbamato e avaliaram o potencial multialvo em ensaios de inibição enzimática para a EeAChE, EqBuChE e as monoaminooxidases (MAO-A e MAO-B), além de avaliarem o potencial antioxidante e de inibição da agregação de β-amiloide. Os derivados 86 e 87 apresentaram atividade inibitória frente a BuChE, além de inibirem seletivamente a MAO-B e serem capazes de inibir a agregação de β-amiloide (Figura 30).73 Estudos de ancoragem molecular, envolvendo o sítio ativo de BuChE, revelaram que 86 apresenta uma ligação de hidrogênio intramolecular entre sua carbonila α,β-insaturada e a hidroxila fenólica, estando essa útima também envolvida em uma ligação de hidrogênio com a carbonila da espinha dorsal do Trp82, enquanto que o oxigênio da carbonila α,β-insaturada de 86 também faz outra ligação de hidrogênio com o hidrogênio (NH) da Gly121. Além disso, a fenila ligada à C=C de 86 faz uma interação do tipo empilhamento π com o Trp82 (Figura 31, (A)). Já o derivado bicarbamato 87, ao ser analisado no sítio ativo de BuChE, apresentou ligação de hidrogênio entre o oxigênio da carbonila α,β-insaturada e a hidroxila fenólica da Tyr332, a qual tem sua cadeia lateral aromática realizando empilhamento π com a fenila orto-substituída de 87. Ademais, também são observados: um empilhamento π entre a fenila meta-substituída de 87 e o Trp82, além da ligação de hidrogênio entre o oxigênio da subunidade carbamato próxima e a hidroxila da Thr120 (Figura 31, (B)).73
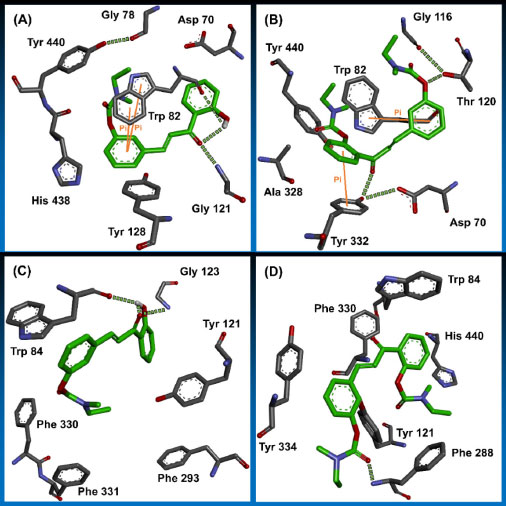 Figura 31. Modos de ligação dos derivados híbridos chalcona-carbamato 86 (A) e 87 (B), no sítio ativo da BuChE. Em (C) e (D), são mostrados os modos de ligação de 86 e 87 no sítio ativo da AChE, respectivamente73
Paralelamente, ambos os derivados híbridos carbamato-chalcona foram avaliados in silico no sítio ativo de AChE. Nesse contexto, 86 também apresenta uma ligação de hidrogênio intramolecular entre sua carbonila α,β-insaturada e a hidroxila fenólica, estando essa útima também envolvida em uma ligação de hidrogênio com a carbonila da espinha dorsal do Trp84, enquanto que o oxigênio da carbonila α,β-insaturada de 86 também faz outra ligação de hidrogênio com o hidrogênio (NH) da Gly123 (Figura 31, (C)). Por fim, ao analisarmos 87 no sítio da AChE, podemos destacar uma ligação de hidrogênio entre a carbonila da subunidade carbamato, ligada à fenila meta-substituída, e o hidrogênio (NH) da espinha dorsal da Phe288 como a mais relevante (Figura 31, (D)).73 Num trabalho envolvendo salicilanilidas N-fenil-para-substituídas, com a hidroxila fenólica substituída por uma subunidade carbamato N-substituída, Krátký e colaboradores sintetizaram vinte desses derivados, os quais foram avaliados contra AChE de enguia elétrica e BuChE do soro equino. As substâncias 88 e 89 apresentaram o melhor resultado de inibição da AChE, com CI50 melhores que a rivastigmina, a qual foi utilizada como referência nos ensaios. Em relação a inibição da BuChE, 90 e 91 se destacaram na série avaliada, apresentando potencial inibitório superior a rivastigmina (CI50 = 38,40 ± 1,97 µmol L-1) (Figura 32).74
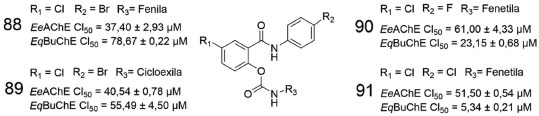 Figura 32. Salicilanilidas N-fenil-para-substituídas (88-91), avaliadas frente a AChE e a BuChE74
Em um estudo otimização estrutural do composto líder 92 (Figura 33), através de modificações estruturais que forneceram 15 derivados, Roy e colaboradores obtiveram êxito ao identificar o carbamato N-(meta-metilfenil)substituído 93, o qual apresentou inibição da AChE de rato melhor do que a rivastigmina (CI50 = 1,11 ± 0,05 µmol L-1). Os autores sugeriram que a substituição da benzila pela 2,4-diclorobenzila (Sítio 1), resultando em 93, foi relevante para melhor inibição da AChE. Adicionalmente, simulações de ancoragem molecular revelaram similaridade entre o padrão de interação dos carbamatos mais potentes, incluindo 93, e a ganstigmina (PDB id: 2BAG). Estudos de dinâmica molecular no complexo TcAChE-93 apontaram que a subunidade m-toluila apresenta interações hidrofóbicas com Trp84, Phe330 e Phe331.75
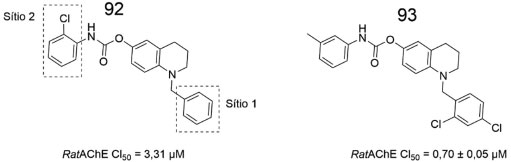 Figura 33. Otimização estrutural de 92, para fornecer o carbamato N-(meta-metilfenil) substituído 9375
DERIVADOS TRIAZÓLICOS COMO INIBIDORES DA BACE-1, ACHE E BUCHE Dentre as subunidades heterocíclicas aromáticas empregadas na construção de substâncias bioativas, os triazóis, os quais são constituídos por anéis de cinco membros contendo três átomos de nitrogênio, se apresentando como 1,2,3- e 1,2,4-triazóis, em duas formas tautoméricas aromáticas cada (Figura 34), merecem destaque na busca de novos candidatos a fármacos para o tratamento de pacientes com a DA.76
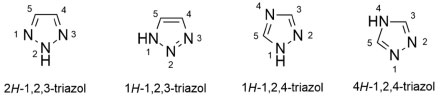 Figura 34. Estruturas dos 1,2,3- e 1,2,4-triazóis e suas respectivas formas tautoméricas76
Nesse contexto, Rastegari e colaboradores obtiveram uma série de derivados 1,2,3-triazólicos, dentre os quais 94 apresentou CI50 = 1,80 ± 0,09 µmol L-1 frente a AChE, não apresentou inibição da BuChE e foi capaz de inibir a BACE-1 com CI50 = 21,13 µmol L-1 (Figura 35). Já o derivado clorado 95 apresentou atividade inibitória tanto em relação a AChE (CI50 = 5,03 ± 0,35 µmol L-1), como em relação a BuChE (CI50 = 1,71 ± 0,21 µmol L-1). Estudos complementares demonstraram que 94 apresenta efeito neuroprotetor contra a morte celular induzida por H2O2 em neurônios PC12 a 50 µmol L-1, bem como a capacidade quelante de alguns metais, como o Fe2+, Cu2+ e Zn2+.77
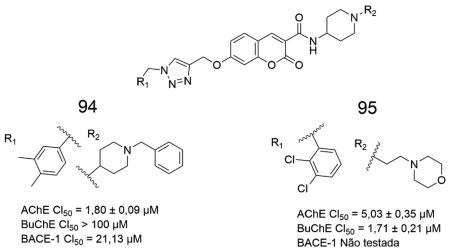 Figura 35. Estruturas dos derivados 1,2,3-triazólicos 94 e 9577
Num trabalho envolvendo inibidores da beta-secretase 1 (BACE 1) com padrão estrutural 1,2,3-triazólico, a subunidade ftalimida ligada ao nitrogênio triazólico, por meio de espaçadores metileno, resultou na obtenção de 96 e 97, os quais apresentaram os melhores valores de inibição da BACE-1 (Figura 36), juntamente com o derivado no qual a ftalimida foi substituída pela para-bromobenzila (98), sem espaçadores, na mesma posição. Paralelamente, o percentual de proteção das células PC12, desafiadas com β-amiloide25-35 e 10 µmol L-1 dos compostos 97 e 98, foram de 83,5 e 55,7%, respectivamente. Ademais, esses mesmos derivados não apresentaram citotoxicidade contra células PC12 até a concentração de 100 µmol L-1.78
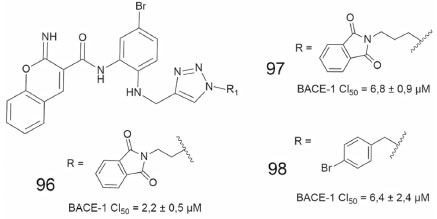 Figura 36. Derivados 1,2,3-triazólicos (96-98) e os dados de potência frente a BACE-178
Em outro estudo envolvendo inibidores da BACE-1, dez derivados 1,2,3-triazólicos foram sintetizados e avaliados, revelando 99 e 100 como os mais potentes na inibição da BACE-1 (Figura 37), além de apresentarem efeito neuroprotetor em linhagens de células neuronais PC12 e proteção moderada contra a toxicidade causada pela proteína β-amiloide.79
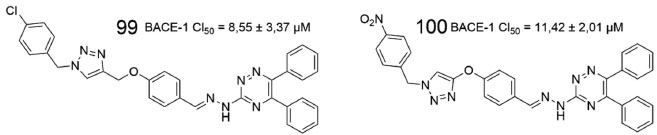 Figura 37. Estruturas químicas de 99 e 10079
A. Kaur e colaboradores obtiveram o derivado 101, o qual possui duas subunidades 1,2,3-triazólicas (Figura 38) e inibe da agregação de β-amiloide42, além de demonstrar excelente taxa de inibição. Ademais, 101 age como agente quelante, inibindo a agregação de β-amiloide 42 induzida por Cu+2 e é capaz de controlar a geração de espécies reativas de oxigênio (EROs), prevenindo o ciclo redox de cobre. Os estudos de ancoragem molecular indicaram que a energia de ligação de-5,52 kcal mol-1 evidencia uma ligação favorável entre o composto 101 e β-amiloide. Nesse contexto, foi possível identificar uma ligação de hidrogênio entre um nitrogênio triazolico e o hidrogênio (NH) da Ala42, além de contatos hidrofóbicos entre 101 e os resíduos de Ala30, Ile31, Leu34, Met35, Gly38, Val39, Val40, Ile41 e Ala42, do monômero β-amiloide42.80
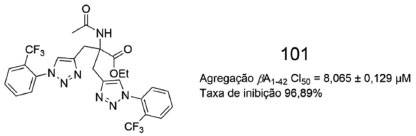 Figura 38. Estrutura química do derivado 1,2,3-triazólico 10180
Um potente inibidor da BuChE, contendo a subunidade 1,2,3-triazola ligada ao nitrogênio isatínico, por meio de um espaçador metileno (102), foi desenvolvido por S. Marques e colaboradores. As inibições contra as BuChE equina (CI50 = 0,46 µmol L-1) e humana (CI50 = 0,51 µmol L-1) foram significativas, mas 102 não inibiu a AChE, apesar de ter sido capaz de inibir de agregação β-amiloide42, com 11,5 ± 4,2% (Figura 39). Estudos de ancoragem molecular evidenciaram interações relevantes de 102 no sítio ativo de BuChE, dentre as quais podemos destacar uma ligação de hidrogênio entre o oxigênio carbonílico da isatina e a hidroxila presente na cadeia lateral da Thr120, além de uma interação π-π entre a subunidade triazólica de 102 e a subunidade indola, presente na cadeia lateral do Trp82.81
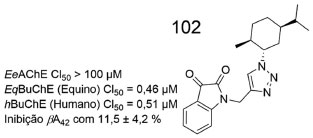 Figura 39. Estrutura química do derivado 1,2,3-triazólico 102 e seus respectivos dados de atividade81
DERIVADOS OXADIAZÓLICOS COMO INIBIDORES DA GSK-3 (Α E Β), BACE-1, ACHE E BUCHE Oxadiazóis são constituídos por anéis de cinco membros, contendo dois átomos de nitrogênio e um átomo de oxigênio, apresentando-se como 1,3,4-, 1,2,4-, 1,2,3- e 1,2,5-oxadiazóis (Figura 40). Essas subunidades são amplamente encontradas em substâncias bioativas, especialmente como equivalentes bioisostéricos de amidas e ésteres, o que evidencia a importância desses heterociclos aromáticos na química medicinal.82
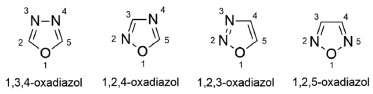 Figura 40. Isômeros dos núcleos oxadiazólicos82
Nesse contexto, Onishi e colaboradores avaliaram um derivado 1,3,4-oxadiazólico, contendo uma subunidade benzofurano central (103, Figura 41), como inibidor as duas isoformas da GSK-3 (α e β), o qual exibiu alta potência para ambas, com CI50 = 37 nmol L-1 e CI50 = 53 nmol L-1, respectivamente.83
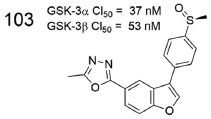 Figura 41. Derivado 1,3,4-oxadiazólico 103 e valores de inibição da GSK-3 (α e β)83
Derivados 1,3,4-oxadiazólicos, contendo a subunidade 4-piridinila, foram sintetizados e avaliados quanto ao potencial inibidor contra as formas humanas de AChE e BuChE. Dentre os derivados testados, o composto 104, contendo a subunidade fenol para-substituída, foi o mais ativo frente a AChE (CI50 = 1,098 ± 0,140 µmol L-1), enquanto que 105, contendo a meta-toluíla, apresentou seletividade frente a BuChE, com valor de CI50 = 8,322 ± 0,231 µmol L-1 (Figura 42). Dados farmacológicos complementares indicaram propriedades antiagregantes de proteína β-amiloide do composto 104 (38,2-65,9%), comparáveis à donepezila (2) (39,6-68,9%). Paralelamente, estudos de ancoragem molecular para 104, no sítio ativo da AChE, demonstraram modo de ligação similar à donepezila (2). Em relação às interações de 104 com resíduos de aminoácidos do sítio ativo, podemos destacar: (i) interações polares com dois resíduos da tríade catalítica (Ser200 e His440), através de interações polares, (ii) empilhamento π entre a subunidade 1,3,4-oxadiazola e os resíduos de Trp84 e Phe330, (iii) além de uma ligação de hidrogênio entre a subunidade piridinila protonada de 104 e a cadeia lateral do resíduo de Glu199, presente no subsítio aniônico.84
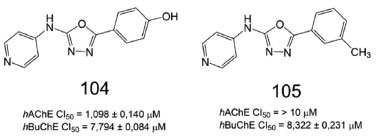 Figura 42. Derivados 1,3,4-oxadiazólicos (104 e 105) e inibição da AChE e BuChE84
Tripathi e colaboradores sintetizaram uma série de derivados 1,3,4-oxadiazólicos conectados à subunidade 2-piridinila, por meio de um espaçador piperazínico, e avaliaram inibição frente a BACE 1, AChE e BuChE. Dentre os melhores derivados, 106 e 107 se destacaram com resultados de inibição em ambas as colinestarases, além de significante inibição da BACE-1 (Figura 43). Em simulações de ancoragem molecular no sítio ativo da AChE, o núcleo 1,3,4-oxadiazólico de 107 realiza interação do tipo empilhamento π com a Tyr341, assim como interação eletrostática com o resíduo de Asp74, ambos localizados no sítio periférico aniônico (PAS) da AChE. Adicionalmente, a subunidade 2-piridinilpiperazina de 107 realiza interações hidrofóbicas com os resíduos de Tyr124 e Trp286, também localizados no SPA. Outra interação de destaque envolve uma ligação de hidrogênio entre o nitrogênio piridinico de 107 e a espinha dorsal da Phe295, na região de ligação da acila, na AChE.85
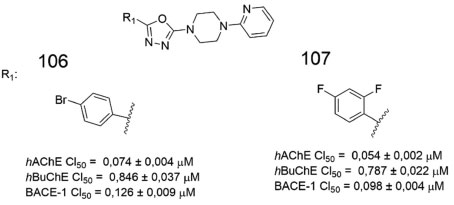 Figura 43. Derivados 1,3,4-oxadiazólicos (106 e 107) e potência frente a AChE, BuChE e BACE-185
Em outro estudo, desenvolvido pelo mesmo grupo de pesquisa, derivados 1,3,4-oxadiazólicos, contendo a subunidade pirimidina, foram sintetizados e avaliados, revelando 108 como o melhor inibidor para as colinestareses AChE e BuChE (Figura 44). O potencial antioxidante de 108 (60,0%) foi comparável ao do ácido ascórbico (64,6%), utilizado como padrão, nesse ensaio.86
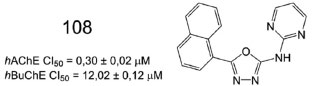 Figura 44. Derivado 1,3,4-oxadiazólico 108 e valores de inibição da AChE e BuChE86
Num trabalho envolvendo 1,3,4-oxadiazóis análogos do ácido ferúlico (antioxidante e anticolinesterásico), dentre os derivados sintetizados e avaliados, 109 foi o mais promissor, com resultados de inibição submicromolares para a AChE, BuChE e BACE-1 (Figura 45). Simulações de ancoragem molecular no sítio ativo da AChE revelaram que 109 interagiu com diversos resíduos do sítio periférico aniônico (Tyr72, Tyr124, Trp286 e Tyr341), através de interações hidrofóbicas. O grupo para-trifluorometilfenila de 109 estabeleceu interações polares com resíduos da tríade catalítica (Ser203 e His447).87
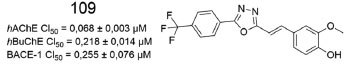 Figura 45. Derivado oxadiazólico 109 e valores de inibição frente a AChE, BuChE e BACE-187
DERIVADOS TIADIAZÓLICOS COMO INIBIDORES DA BACE-1, ACHE E BUCHE O núcleo tiadiazólico possui quatro formas isoméricas e consiste em um sistema heterocíclico aromático de cinco membros, contendo um átomo de enxofre e dois átomos de nitrogênio (Figura 46). Assim como os oxadiazóis, substâncias contendo a subunidade tiadiazólica também estão presentes em diversas substâncias bioativas88
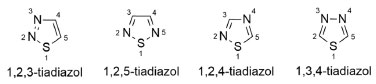 Figura 46. Formas isoméricas do núcleo tiadiazólico
Uma série de quatro derivados (110-113), contendo a subunidade tacrina e substituídos na posição 5 da 1,2,4-tiadiazola por um grupo anilina para-substituído, foram sintetizados e avaliados frente a AChE humana e a BuChE de soro bovino, apresentando melhores resultados para essa última, com potência em faixa submicromolar, conforme pode ser destacado para o derivado fluorado 111 na BuChE (CI50 = 0,073 ± 0,002 µmol L-1) (Figura 47).89
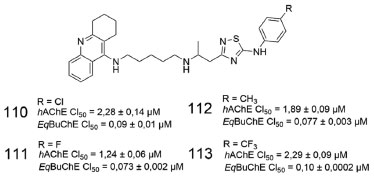 Figura 47. Derivados tacrina/1,2,4-tiadiazola (110-113) e atividade frente a AChE e BuChE89
Uma triagem virtual com base na BACE-1 foi realizada com o objetivo de selecionar potenciais inibidores não-peptídicos da classe dos 1,2,4-tiadiazóis. Os resultados dos estudos de ancoragem indicaram que algumas das substâncias da série interagiram com os ácidos aspárticos catalíticos (Asp32 e/ou Asp228), que desempenham um papel crucial na catálise enzimática e na ligação do inibidor. Especificamente, o grupo 5-amino de algumas das substâncias investigadas interagiu por meio da ligação de hidrogênio com a díade catalítica e via interações eletrostáticas, com os dois resíduos do subsítio S2 (Gly230 e Thr231). Além disso, foi possível observar que o átomo de nitrogênio, localizado na posição 2 do anel heterocíclico, formou ligações de hidrogênio, direta ou intermediada por água, com os resíduos Thr72 e Gln73 (Figura 48). Os resultados experimentais apontaram 114 como o mais potente da série.90
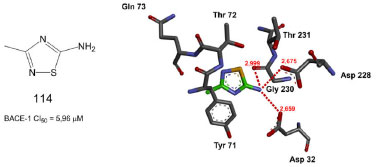 Figura 48. Estrutura química do composto 114 e sua pose no sítio ativo da BACE-190
Em estudo recente, investigou-se a atividade anticolinesterásica de uma série de derivados 1,3,4-tiadiazólicos, contendo a subunidade piperazina. Os resultados de inibição da AChE indicaram valores na escala nanomolar, com destaque para o composto 115 (CI50 = 0,0181 ± 0,0009 µmol L-1) (Figura 49). Estudos cinéticos revelaram um mecanismo de inibição do tipo misto para 115 e o valor de Ki indicou alta afinidade de ligação, mesmo em baixas concentrações (Ki = 0,0031 µmol L-1). Adicionalmente, estudos de ancoragem molecular revelaram ligação de hidrogênio (2,34 Å) entre o grupo amino de 115 e a hidroxila da Tyr124 da AChE, além de interação do tipo empilhamento π entre a subunidade 1,3,4-tiadiazola de 115 e a subunidade aromática da Tyr341. Também vale destacar uma interação hidrofóbica entre a subunidade morfolina de 115 e o Trp286.91
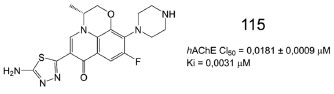 Figura 49. Estrutura química e valores de atividade para o derivado 1,3,4-tiadiazólico 11591
Outra série de derivados 1,3,4-tiadiazol foi sintetizada e avaliada como inibidores de EeAChE e EqBuChE. Dentre os compostos obtidos, os que apresentaram os melhores resultados de CI50 foram 116, 117 e 118 (Figura 50), com destaque para a alta seletividade de 117 e 118 em relação a AChE.92
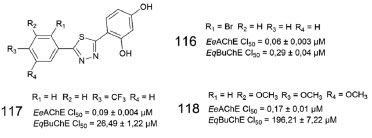 Figura 50. Derivados 1,3,4-tiadiazólicos (116-118) e valores de CI50 para AChE e BuChE92
Visando o entendimento do modo de ligação do composto mais ativo no sítio da AChE, estudos de ancoragem molecular com o derivado bromado 116 indicaram que sua subunidade 1,3,4-tiadiazólica realiza interação do tipo empilhamento π com a subunidade indola do Trp84. Também foi possível observar que as hidroxilas fenólicas de 116 formam ligações de hidrogênio com os resíduos Glu199, Gly117 e Tyr130. Por fim, podemos destacar a ligação de hidrogênio não-clássica entre um dos hidrogênios da subunidade orto-bromofenila de 116 e a hidroxila fenolíca da Tyr334 (Figura 51).
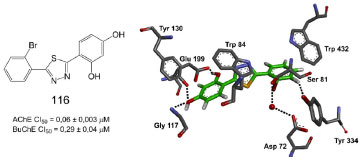 Figura 51. Modo de ligação de 116 no sítio ativo da AChE92
DERIVADOS CARBAZÓLICOS COMO INIBIDORES DA ACHE E DA BUCHE Os carbazóis são substâncias que apresentam uma subunidade tricíclica, contendo um anel pirrólico central fundido com duas fenilas (Figura 52). A subunidade carbazola (119) está presente em diversas substâncias bioativas, conforme será exemplificado a seguir.93
 Figura 52. Formula estrutural do carbazol93
Ghobadian e colaboradores sintetizaram e avaliaram derivados N-benzilpiridínicos, contendo a subunidade carbazola, como inibidores de colinesterases (EeAChE e EqBuChE) e na agregação de proteína β-amiloide. Ensaios in vitro revelaram que todos os derivados se mostraram mais potentes frente a BuChE, com destaque para 120, que apresentou o melhor resultado (BuChE CI50 = 0,073 ± 0,003 µmol L-1). Estudos de ancoragem molecular no sítio ativo da BuChE revelaram que a subunidade carbazólica de 120 realiza interações do tipo empilhamento π com o Trp82. Ademais, a subunidade piridina de 120 realiza empilhamento π com a Tyr332, além de interação do tipo π-alquil com a Leu286, ambas pertencentes ao sítio periférico (Figura 53).94
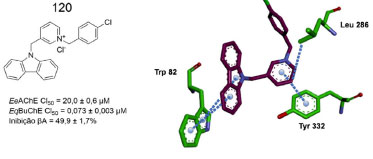 Figura 53. Estrutura do derivado carbazólico 120 e seu modo de ligação no sítio ativo da BuChE94
Outros derivados carbazólicos foram sintetizados e avaliados como inibidores da EeAChE, da EqBuChE e na agregação de βA. Entre as substâncias investigadas, 121 e 122 exibiram os melhores resultados, com inibição seletiva frente a AChE, além inibição da agregação de β-amiloide, com valores superiores a 60% (Figura 54).95
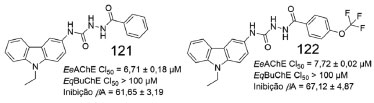 Figura 54. Derivados carbazólicos (121 e 122) e valores de inibição da AChE, da BuChE e da agregação de proteína β-amiloide95
Em trabalho envolvendo híbridos ácido ferúlico-carbazol, visando novos agentes anti-Alzheimer, foram sintetizadas e avaliadas diversas substâncias, dentre as quais 123 e 124 obtiveram os melhores resultados para as colinesterases (EeAChE e EqBuChE) (Figura 55), mostrando a importância da subunidade carbazólica em 123, que exibiu atividade relevante, mesmo com a ausência da subunidade oriunda do ácido felúrico.96
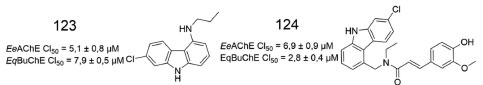 Figura 55. Derivados carbazólicos (123 e 124) e valores de atividade frente a AChE e BuChE96
Choubdar e colaboradores sintetizaram uma série de derivados carbazólicos, conectados a subunidades heterocíclicas, por meio de espaçadores alifáticos. Após avaliação do potencial de inibição frente a EeAChE e EqBuChE, observou-se valores de CI50 variando entre 0,11-36,5 µmol L-1 e 0,02-98,6 µmol L-1, respectivamente. O derivado 125 (Figura 56) foi o inibidor mais potente para ambas as colinesterases e também apresentou inibição da agregação de βA (autoagregação e induzida por AChE).97
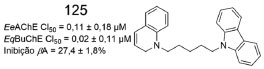 Figura 56. Derivado carzazólico 125 e dados de inibição da AChE, BuChE e agregação de βA97
Em um estudo recente, uma série de híbridos foi desenhada com base na fusão do núcleo carbazólico com o estilbeno. Os compostos planejados foram sintetizados e avaliados quanto a inibição das colinesterases e da agregação de β-amiloide. Dentre os compostos avaliados, 126 foi o que apresentou os melhores resultados para inibição da EqBuChE e da agregação de β-amiloide, além de apresentar valores adequados para inibição da hAChE (Figura 57). Além disso, 127 apresentou melhor índice de inibição frente a AChE, com CI50 = 1,84 ± 0,27 µmol L-1.98
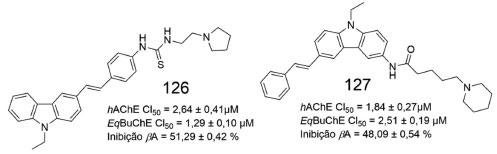 Figura 57. Compostos carbazólicos (125 e 126) e inibição da AChE, BuChE e agregação de βA98
Estudos de ancoragem molecular no sítio ativo da AChE mostraram que 126 apresenta modo de ligação semelhante ao da donepezila (2). Ademais, a subunidade pirrolidínica protonada de 126, posicionada no sítio catalítico aniônico e no sítio periférico aniônico, realiza interação cátion-π com o Trp84, além de ligação de hidrogênio e interações de natureza iônica com o Glu199. A subunidade carbazólica de 126 exibe interações π-π com os resíduos de Tyr70 e Tyr334.98 Já o modo de interação de 126 no sítio ativo da BuChE mostrou que o ligante ocupou a cavidade catalítica da enzima através da subunidade carbazola, na bolsa hidrofóbica, contendo os resíduos de aminoácidos não polares Ala277, Val280, Pro285 e Leu286. O subunidade fenila de 126 formou uma interação π-π com o resíduo Tyr332 e o nitrogênio protonado da pirrolidina de 126 realizou interação cátion-π com o Trp82.98
CONCLUSÕES A atividade anti-Alzheimer foi abordada para diferentes classes de substâncias de origem natural (e. g. alcaloides, chalconas, cumarinas e flavonoides), e sintética, tais como: derivados de piperazinas, carbamatos e subunidades heterocíclicas aromáticas (e.g. triazóis, oxazóis, tiadiazóis e carbazóis). Nesse sentido, foram descritos alguns estudos realizados nos últimos 20 anos, envolvendo estratégias de desenvolvimento bem como avaliação farmacológica dessas substâncias, em alvos terapêuticos relacionados com a etiologia da DA, com foco nas colinesterases (AChE e BuChE), sem deixar de abordar outros alvos relevantes como BACE-1 e GSK-3β e α. Alguns exemplos de interações ligante-biomacromolécula (e.g. inibidor-AChE), são apresentados, por meio de estudos in silico, visando fornecer um conhecimento complementar relevante para o planejamento de novos candidatos à fármacos baseados na estrutura da biomacromolécula. De forma complementar, os vários exemplos reportados, também podem fornecer material útil para o planejamento de novos candidatos à fármacos baseados na estrutura do ligante. Analisando a diversidade de alvos terapêuticos relevantes, assim como os mais variados padrões estruturais de substâncias bioativas da literatura, visando terapias mais eficazes para o tratamento de pessoas portadoras da DA, é possível afirmar que a AChE ainda se destaca dentre os alvos mais clássicos, sendo inibida por 3 dos fármacos atualmente aprovados para uso terapêutico. Nesse contexto, alguns estudos citados nesta revisão que apontam substâncias com maior seletividade e potência para a AChE em relação a BuChE, além de potencial de inibição da agregação de proteína β-amiloide, merecem posição de destaque e devem ser considerados para estudos farmacológicos complementares, como por exemplo, verificação do potencial de penetração através da barreira hematoencefálica (BHE), característica indispensável para um fármaco com ação no SNC. A diversidade estrutural das substâncias bioativas descritas ao longo do texto, evidencia a importância de explorar a variedade de padrões estruturais dos produtos naturais, em conjunto com a quimiodiversidade proveniente da química orgânica sintética, em programas de prospecção de novos candidatos a protótipos de fármacos. Por fim, a inexistência de uma cura definitiva para a DA, a tendência de progressão no número de pessoas acometidas e o fato dos fármacos disponíveis para tratamento serem incapazes de impedir a progressão da demência explicam o tamanho do desafio que vem sendo enfrentado por pesquisadores da área pelo mundo. Como consequência, um grande volume de estudos reportando novas substâncias que atingiram fases II/III de testes clínicos, tem sido reportados, especialmente nos últimos anos.99-101 Em geral, esses estudos abordam o desenvolvimento não somente de inibidores colinesterásicos, mas de substâncias que possam apresentar ação multialvo (e. g. BACE-1, GSK-3β e α), explorando, dessa forma, o conceito da polifarmacologia no caráter multifatorial da DA.102,103
AGRADECIMENTOS O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Código de Financiamento 001. Adicionalmente, agradecemos à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Projeto Regular, Processo 2018/00187-7, pelo apoio financeiro ao LaQMedSOMM, que foi fundamental para o desenvolvimento deste trabalho.
REFERÊNCIAS 1. Zeitschrift, A.; Alzheimer Dis. Assoc. Disord. 1987, 1, 3. 2. Zhagn, L.; Li, Z.; Zhonghua Yi Shi Za Zhi 2014, 44, 288. 3. Viegas, F. P. D.; Simões, M. C. R.; da Rocha, M. D.; Castelli, M. R.; Moreira, M. S.; Viegas Junior, C.; Rev. Virtual Quim. 2011, 3, 286. 4. Alzheimer's Dement. 2020, 16, 391. 5. https://www.who.int/health-topics/dementia#tab=tab_2 acessada em novembro 2021. 6. Nichols, E.; Szoeke, C. E. I.; Vollset, S. E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M. T. E.; Akinyemi, R. O.; Alahdab, F.; Asgedom, S. W.; Awasthi, A.; Barker-Collo, S. L.; Baune, B. T.; Béjot, Y.; Belachew, A. B.; Bennett, D. A.; Biadgo, B.; Bijani, A.; Bin Sayeed, M. S.; Brayne, C.; Carpenter, D. O.; Carvalho, F.; Catalá-López, F.; Cerin, E.; Choi, J.-Y. J.; Dang, A. K.; Degefa, M. G.; Djalalinia, S.; Dubey, M.; Duken, E. E.; Edvardsson, D.; Endres, M.; Eskandarieh, S.; Faro, A.; Farzadfar, F.; Fereshtehnejad, S.-M.; Fernandes, E.; Filip, I.; Fischer, F.; Gebre, A. K.; Geremew, D.; Ghasemi-Kasman, M.; Gnedovskaya, E. V; Gupta, R.; Hachinski, V.; Hagos, T. B.; Hamidi, S.; Hankey, G. J.; Haro, J. M.; Hay, S. I.; Irvani, S. S. N.; Jha, R. P.; Jonas, J. B.; Kalani, R.; Karch, A.; Kasaeian, A.; Khader, Y. S.; Khalil, I. A.; Khan, E. A.; Khanna, T.; Khoja, T. A. M.; Khubchandani, J.; Kisa, A.; Kissimova-Skarbek, K.; Kivimäki, M.; Koyanagi, A.; Krohn, K. J.; Logroscino, G.; Lorkowski, S.; Majdan, M.; Malekzadeh, R.; März, W.; Massano, J.; Mengistu, G.; Meretoja, A.; Mohammadi, M.; Mohammadi-Khanaposhtani, M.; Mokdad, A. H.; Mondello, S.; Moradi, G.; Nagel, G.; Naghavi, M.; Naik, G.; Nguyen, L. H.; Nguyen, T. H.; Nirayo, Y. L.; Nixon, M. R.; Ofori-Asenso, R.; Ogbo, F. A.; Olagunju, A. T.; Owolabi, M. O.; Panda-Jonas, S.; Passos, V. M. de A.; Pereira, D. M.; Pinilla-Monsalve, G. D.; Piradov, M. A.; Pond, C. D.; Poustchi, H.; Qorbani, M.; Radfar, A.; Reiner Jr., R. C.; Robinson, S. R.; Roshandel, G.; Rostami, A.; Russ, T. C.; Sachdev, P. S.; Safari, H.; Safiri, S.; Sahathevan, R.; Salimi, Y.; Satpathy, M.; Sawhney, M.; Saylan, M.; Sepanlou, S. G.; Shafieesabet, A.; Shaikh, M. A.; Sahraian, M. A.; Shigematsu, M.; Shiri, R.; Shiue, I.; Silva, J. P.; Smith, M.; Sobhani, S.; Stein, D. J.; Tabarés-Seisdedos, R.; Tovani-Palone, M. R.; Tran, B. X.; Tran, T. T.; Tsegay, A. T.; Ullah, I.; Venketasubramanian, N.; Vlassov, V.; Wang, Y.-P.; Weiss, J.; Westerman, R.; Wijeratne, T.; Wyper, G. M. A.; Yano, Y.; Yimer, E. M.; Yonemoto, N.; Yousefifard, M.; Zaidi, Z.; Zare, Z.; Vos, T.; Feigin, V. L.; Murray, C. J. L.; Lancet Neurol. 2019, 18, 88. 7. Alzheimer's Association; Alzheimers. Dement. 2016, 12, 459. 8. https://portalhospitaisbrasil.com.br/casos-de-alzheimer-no-brasil-podem-quadruplicar-sem-plano-nacional-de-cuidado-as-demencias/ acessada em novembro 2021. 9. Gutierrez, B. A. O.; da Silva, H. S.; Guimarães, C.; Campino, A. C.; Cienc. e Saude Coletiva 2014, 19, 4479. 10. Wei, W.; Qiu, Q.; Ph, D.; Lai, A.; Mon, T.; Mwamburi, M.; Rosenzweig, J.; Kowall, N.; Stern, R.; Zhu, H.; Steffens, D. C.; American Journal of Geriatric Psychiatry 2014, 22, 177. 11. Liu, C. C.; Kanekiyo, T.; Xu, H.; Bu, G.; Nat. Rev. Neurosci. 2013, 9, 106. 12. Hoyer, F. F.; Steinmetz, M.; Zimmer, S.; Becker, A.; Lütjohann, D.; Buchalla, R.; Zimmer, A.; Nickenig, G.; J. Mol. Cell. Cardiol. 2011, 51, 1007. 13. Hall, J. R.; Wiechmann, A. R.; Cunningham, R. L.; Johnson, L. A.; Edwards, M.; Barber, R. C.; Singh, M.; Winter, S.; O'Bryant, S. E.; Alzheimer's Res. Ther. 2015, 7, 1. 14. Korolev, I. O.; Med. Student Res. J. 2014, 04, 24. 15. Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J. C.; Nachon, F.; J. Biol. Chem. 2003, 278, 41141. 16. Cheung, J.; Rudolph, M. J.; Burshteyn, F.; Cassidy, M. S.; Gary, E. N.; Love, J.; Franklin, M. C.; Height, J. J.; J. Med. Chem. 2012, 55, 10282. 17. Shafferman, A.; Kronman, C.; Flashner, Y.; Leitner, M.; Grosfeld, H.; Ordentlich, A.; Gozes, Y.; Cohen, S.; Ariel, N.; Barak, D.; J. Biol. Chem. 1992, 267, 17640. 18. Rosenberry, T. L.; Brazzolotto, X.; Macdonald, I. R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F.; Molecules 2017, 22. 19. Barreiro, E. J.; Camara, C. A.; Verli, H.; Brazil-Más, L.; Castro, N. G.; Cintra, W. M.; Aracava, Y.; Rodrigues, C. R.; Fraga, C. A. M.; J. Med. Chem. 2003, 46, 1144. 20. Giacobini, E.; Int. J. Geriatr. Psychiatry 2003, 18, S1. 21. Do Goulart, P. N.; Caruso, L.; Nadur, N. F.; Franco, D. P.; Kümmerle, A. E.; Lacerda, R. B.; Rev. Virtual Quim. 2021, 13, 90. 22. Serrano-Pozo, A.; Frosch, M. P.; Masliah, E.; Hyman, B. T.; Cold Spring Harb. Perspect. Med. 2011, 1, a006189. 23. Coimbra, J. R. M.; Marques, D. F. F.; Baptista, S. J.; Pereira, C. M. F.; Moreira, P. I.; Dinis, T. C. P.; Santos, A. E.; Salvador, J. A. R.; Front. Chem. 2018, 6, 178. 24. ClinicalTrials.gov; A 24-Month Study to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Participants With Early Alzheimer's Disease. 25. ClinicalTrials.gov; An Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer's Dementia. 26. https://www.alz.org/brain_portuguese/10.asp acessada em novembro 2021. 27. García-Ayllón, M. S.; Small, D. H.; Avila, J.; Sáez-Valero, J.; Front. Mol. Neurosci. 2011, 4, 1. 28. Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J.; Front. Mol. Neurosci. 2014, 7, 46. 29. De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A.; J. Med. Chem. 2021, 64, 26. 30. Phiel, C. J.; Wilson, C. A.; Lee, V. M. Y.; Klein, P. S.; Nature 2003, 423, 435. 31. Paula, V. J. R.; Guimarães, F. M.; Diniz, B. S.; Forlenza, O. V.; Dement. Neuropsychol. 2009, 3, 188. 32. Fang, R.; Ye, S.; Huangfu, J.; Calimag, D. P.; Transl. Neurodegener. 2017, 6, 1. 33. Cummings, J. L.; Isaacson, R. S.; Schmitt, F. A.; Velting, D. M.; Ann. Clin. Transl. Neurol. 2015, 2, 307. 34. Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K. W.; Karanam, A. K.; Christopher, S.; Clin. Interv. Aging 2017, 12, 697. 35. Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y.; Front. Neurosci. 2019, 13, 43. 36. Molino, I.; Colucci, L.; Fasanaro, A. M.; Traini, E.; Amenta, F.; Sci. World J. 2013, 2013. 37. Lima, J. A.; Hamerski, L. Em Studies in Natural Products Chemistry; Atta-ur-Rahman, ed.; Elsevier: Amsterdam, 2018, Vol. 61. 38. Brownstein, M. J.; Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5391. 39. Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F. G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F. J.; Andrisano, V.; Clos, M. V.; Inestrosa, N. C.; Muñoz-Torrero, D.; J. Med. Chem. 2014, 57, 2549. 40. Yang, Y.; Cheng, X.; Liu, W.; Chou, G.; Wang, Z.; Wang, C.; J. Ethnopharmacol. 2015, 168, 279. 41. Plazas, E.; Hagenow, S.; Avila Murillo, M.; Stark, H.; Cuca, L. E.; Bioorg. Chem. 2020, 98, 103722. 42. Wiesner, J.; Kříz, Z.; Kuča, K.; Jun, D.; Koča, J.; J. Enzyme Inhib. Med. Chem. 2007, 22, 417. 43. Yerragunta, V.; Kumaraswamy, T.; Suman, D.; Anusha, V.; Patil, P.; Samhitha, T.; PharmaTutor 2013, 1, 54. 44. Rampa, A.; Bartolini, M.; Pruccoli, L.; Naldi, M.; Iriepa, I.; Moraleda, I.; Belluti, F.; Gobbi, S.; Tarozzi, A.; Bisi, A.; Molecules 2018, 23, 1. 45. Iwashina, T.; The structure and distribution of the flavonoids in plants. J. Plant Res. 2000, 113, 287. 46. Zhang, X.; Song, Q.; Cao, Z.; Li, Y.; Tian, C.; Yang, Z.; Zhang, H.; Deng, Y.; Bioorg. Chem. 2019, 87, 395. 47. Chandrika, N. T.; Fosso, M. Y.; Tsodikov, O. V.; LeVine, H.; Garneau-Tsodikova, S.; Molecules 2020, 25. 48. Reeta; Baek, S. C.; Lee, J. P.; Rangarajan, T. M.; Ayushee; Singh, R. P.; Singh, M.; Mangiatordi, G. F.; Nicolotti, O.; Kim, H.; Mathew, B.; CNS & Neurological Disorders - Drug Targets 2019, 18, 643. 49. Silva, C. R. da; Gomes, V. S.; Kulkamp, I. C.; Kanis, L. A.; Rev. Bras. Farmacogn. 2008, 18, 594. 50. Celeghini, R. M. S.; Vilegas, J. H. Y.; Lanças, F. M.; J. Braz. Chem. Soc. 2001, 12, 706. 51. Rodrigues, R. F.; Dissertação de Mestrado, Universidade Estadual de Campinas, Brasil, 2005. 52. Zhou, X.; Wang, X. B.; Wang, T.; Kong, L. Y.; Bioorg. Med. Chem. 2008, 16, 8011. 53. Ellman, G. L.; Courtney, K. D.; Andres, V.; Featherstone, R. M.; Biochem. Pharmacol. 1961, 7, 88. 54. Shen, Q.; Peng, Q.; Shao, J.; Liu, X.; Huang, Z.; Pu, X.; Ma, L.; Li, Y. M.; Chan, A. S. C.; Gu, L.; Eur. J. Med. Chem. 2005, 40, 1307. 55. Pisani, L.; Catto, M.; Giangreco, I.; Leonetti, F.; Nicolotti, O.; Stefanachi, A.; Cellamare, S.; Carotti, A.; ChemMedChem 2010, 5, 1616. 56. Leonetti, F.; Catto, M.; Nicolotti, O.; Pisani, L.; Cappa, A.; Stefanachi, A.; Carotti, A.; Bioorg. Med. Chem. 2008, 16, 7450. 57. Abdshahzadeh, H.; Golshani, M.; Nadri, H.; Saberi Kia, I.; Abdolahi, Z.; Forootanfar, H.; Ameri, A.; Tüylü Küçükkılınç, T.; Ayazgok, B.; Jalili-Baleh, L.; Sadat Ebrahimi, S. E.; Moghimi, S.; Haririan, I.; Khoobi, M.; Foroumadi, A.; Chem. Biodivers. 2019, 16. 58. Dornas, W. C.; Oliveira, T. T.; Rodrigues-Das-Dores, R. G.; Santos, A. F.; Nagem, T. J.; Rev. Ciênc. Farm. Básica Apl. 2007, 28, 241. 59. Silva, R.; Vale, L.; Calou, B. F.; de Deus, M.; Socorro, M.; Pinheiro, P. M.; Paula, A.; Acta Toxicol. Argent. 2015, 23, 36. 60. Uddin, M. S.; Kabir, M. T.; Front. Cell Dev. Biol. 2019, 7, 1. 61. Uddin, M. S.; Kabir, M. T.; Niaz, K.; Jeandet, P.; Clément, C.; Mathew, B.; Rauf, A.; Rengasamy, K. R. R.; Sobarzo-Sánchez, E.; Ashraf, G. M.; Aleya, L.; Molecules 2020, 25. 62. Sevindik, H. G.; Güvenalp, Z.; Yerdelen, K. Ö.; Yuca, H.; Demirezer, L. Ö.; Ind. Crops Prod. 2015, 76, 873. 63. Balkis, A.; Tran, K.; Lee, Y. Z.; Ng, K.; J. Agric. Sci. 2015, 7, 26. 64. Shi, S.; Wang, H.; Wang, J.; Wang, Y.; Xue, X.; Hou, Z.; Yao, G. D.; Huang, X. X.; Zhao, H.; Liu, Q.; Song, S. J.; Bioorg. Chem. 2020, 100, 103917. 65. Al-Ghorbani, M.; Bushra Begum, A.; Zabiulla, Z.; Mamatha, S. V.; Khanum, S. A.; Res. J. Pharm. Technol. 2015, 8, 611. 66. Sergeant, N.; Vingtdeux, V.; Eddarkaoui, S.; Gay, M.; Evrard, C.; Le Fur, N.; Laurent, C.; Caillierez, R.; Obriot, H.; Larchanché, P. E.; Farce, A.; Coevoet, M.; Carato, P.; Kouach, M.; Descat, A.; Dallemagne, P.; Buée-Scherrer, V.; Blum, D.; Hamdane, M.; Buée, L.; Melnyk, P.; Neurobiol. Dis. 2019, 129, 217. 67. Umar, T.; Shalini, S.; Raza, M. K.; Gusain, S.; Kumar, J.; Seth, P.; Tiwari, M.; Hoda, N.; Eur. J. Med. Chem. 2019, 175, 2. 68. Tripathi, P. N.; Srivastava, P.; Sharma, P.; Tripathi, M. K.; Seth, A.; Tripathi, A.; Rai, S. N.; Singh, S. P.; Shrivastava, S. K.; Bioorg. Chem. 2019, 85, 82. 69. Mishra, C. B.; Kumari, S.; Manral, A.; Prakash, A.; Saini, V.; Lynn, A. M.; Tiwari, M.; Eur. J. Med. Chem. 2017, 125, 736. 70. Sang, Z.; Wang, K.; Shi, J.; Cheng, X.; Zhu, G.; Wei, R.; Ma, Q.; Yu, L.; Zhao, Y.; Tan, Z.; Liu, W.; Eur. J. Med. Chem. 2020, 187, 111958. 71. Wu, J.; Pistolozzi, M.; Liu, S.; Tan, W.; Bioorg. Med. Chem. 2020, 28, 115324. 72. Bosak, A.; Gazić, I.; Vinković, V.; Kovarik, Z.; Chem. Biol. Interact. 2008, 175, 192. 73. Sang, Z.; Wang, K.; Shi, J.; Liu, W.; Tan, Z.; Eur. J. Med. Chem. 2019, 178, 726. 74. Krátký, M.; Stěpánková, S.; Vorčáková, K.; Vinsová, J.; Bioorg. Chem. 2018, 80, 668. 75. Roy, K. K.; Tota, S.; Tripathi, T.; Chander, S.; Nath, C.; Saxena, A. K.; Bioorg. Med. Chem. 2012, 20, 6313. 76. Benson, F. R.; Savell, W. L.; Chem. Rev. 1950, 46, 1. 77. Rastegari, A.; Nadri, H.; Mahdavi, M.; Moradi, A.; Mirfazli, S. S.; Edraki, N.; Moghadam, F. H.; Larijani, B.; Akbarzadeh, T.; Saeedi, M.; Bioorg. Chem. 2019, 83, 391. 78. Iraji, A.; Firuzi, O.; Khoshneviszadeh, M.; Tavakkoli, M.; Eur. J. Med. Chem. 2017, 141, 690. 79. Yazdani, M.; Edraki, N.; Badri, R.; Khoshneviszadeh, M.; Iraji, A.; Firuzi, O.; Bioorg. Chem. 2019, 84, 363. 80. Kaur, A.; Mann, S.; Kaur, A.; Priyadarshi, N.; Goyal, B.; Singhal, N. K.; Goyal, D.; Bioorg. Chem. 2019, 87, 572. 81. Marques, C. S.; López, Ó.; Bagetta, D.; Carreiro, E. P.; Petralla, S.; Bartolini, M.; Hoffmann, M.; Alcaro, S.; Monti, B.; Bolognesi, M. L.; Decker, M.; Fernández-Bolaños, J. G.; Burke, A. J.; Bioorg. Chem. 2020, 98, 103753. 82. Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A. T.; J. Med. Chem. 2012, 55, 1817. 83. Onishi, T.; Iwashita, H.; Uno, Y.; Kunitomo, J.; Saitoh, M.; Kimura, E.; Fujita, H.; Uchiyama, N.; Kori, M.; Takizawa, M.; J. Neurochem. 2011, 119, 1330. 84. Mishra, P.; Sharma, P.; Tripathi, P. N.; Gupta, S. K.; Srivastava, P.; Seth, A.; Tripathi, A.; Krishnamurthy, S.; Shrivastava, S. K.; Bioorg. Chem. 2019, 89, 103025. 85. Tripathi, A.; Choubey, P. K.; Sharma, P.; Seth, A.; Tripathi, P. N.; Tripathi, M. K.; Prajapati, S. K.; Krishnamurthy, S.; Shrivastava, S. K.; Eur. J. Med. Chem. 2019, 183, 111707. 86. Tripathi, P. N.; Srivastava, P.; Sharma, P.; Seth, A.; Shrivastava, S. K.; Bioorg. Med. Chem. 2019, 27, 1327. 87. Tripathi, A.; Choubey, P. K.; Sharma, P.; Seth, A.; Saraf, P.; Shrivastava, S. K.; Bioorg. Chem. 2020, 95, 103506. 88. Asif, M.; Mediterr. J. Chem. 2016, 5, 568. 89. Makhaeva, G. F.; Grigoriev, V. V; Proshin, A. N.; Kovaleva, N. V; Rudakova, E. V; Boltneva, N. P.; Serkov, I. V; Bachurin, S. O.; Dokl. Biochem. Biophys. 2017, 477, 405. 90. Gurjar, A. S.; Andrisano, V.; Simone, A. D.; Velingkar, V. S.; Bioorg. Chem. 2014, 57, 90. 91. Ujan, R.; Saeed, A.; Channar, P. A.; Larik, F. A.; Abbas, Q.; Alajmi, M. F.; El-Seedi, H. R.; Rind, M. A.; Hassan, M.; Raza, H.; Seo, S. Y.; Molecules 2019, 24, 1. 92. Skrzypek, A.; Matysiak, J.; Niewiadomy, A.; Bajda, M.; Szymański, P.; Eur. J. Med. Chem. 2013, 62, 311. 93. Gluszynska, A.; Eur. J. Med. Chem. 2015, 94, 405. 94. Ghobadian, R.; Nadri, H.; Moradi, A.; Bukhari, S. N. A.; Mahdavi, M.; Asadi, M.; Akbarzadeh, T.; Khaleghzadeh-Ahangar, H.; Sharifzadeh, M.; Amini, M.; Bioorg. Med. Chem. 2018, 26, 4952. 95. Mishra, C. B.; Gusain, S.; Shalini, S.; Kumari, S.; Prakash, A.; Kumari, N.; Yadav, A. K.; Kumari, J.; Kumar, K.; Tiwari, M.; Bioorg. Chem. 2020, 95, 103524. 96. Fang, L.; Chen, M.; Fang, X.; Liu, Z.; Gou, S.; Chen, L.; Bioorg. Med. Chem. 2016, 24, 886. 97. Choubdar, N.; Golshani, M.; Jalili-Baleh, L.; Nadri, H.; Küçükkilinç, T. T.; Ayazgök, B.; Moradi, A.; Moghadam, F. H.; Abdolahi, Z.; Ameri, A.; Salehian, F.; Foroumadi, A.; Khoobi, M.; Bioorg. Chem. 2019, 91. 98. Patel, D. V.; Patel, N. R.; Kanhed, A. M.; Teli, D. M.; Patel, K. B.; Joshi, P. D.; Patel, S. P.; Gandhi, P. M.; Chaudhary, B. N.; Prajapati, N. K.; Patel, K. V.; Yadav, M. R.; Bioorg. Chem. 2020, 101, 103977. 99. Huang, L.-K.; Chao, S.-P.; Hu, C.-J.; J. Biomed. Sci. 2020, 27, 18. 100. Gupta, G. L.; Samant, N. P.; Contemp. Clin. Trials 2021, 106549. 101. Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K.; Alzheimer's Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. 102. Bolognesi, M. L.; Cavalli, A.; Cavalli, A.; ChemMedChem 2016, 11, 1190. 103. Pirolla, N. F. F.; Batista, V. S.; Viegas, F. P. D.; Gontijo, V. S.; McCarthy, C. R.; Viegas-Jr, C.; Nascimento-Júnior, N. M.; Curr. Drug Targets. 2021, 22, 505. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access