
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Determinação de etil-glicuronídeo e etil-sulfato em sangue por UPLC-MS/MS: aplicação clínica em vítimas de trauma Determination of ethyl-glucuronide and ethyl-sulfate in blood by UPLC-MS/MS: clinical application in trauma victims |
|
Isabela Ritter OttI; Mariane TegnerII; Fernando Engel GerbaseII,III; Octaviano Pereira Zago SagriloIII; Rafael LindenI,II; Marina Venzon AntunesI,II,*
I. Laboratório de Análises Toxicológicas, Universidade Feevale, 93525-075 Novo Hamburgo - RS, Brasil Recebido em: 21/02/2022 *e-mail: marinaantunes@feevale.br We developed a UPLC-MS/MS method for the quantification of the ethanol metabolites ethyl-glucuronide (EtG) and ethyl-sulfate (EtS) in blood samples. Sample preparation was a protein precipitation with acetonitrile 0.1% formic acid containing internal standards (IS) 0.5 µg mL-1 EtG-D5 and EtS-D5. Chromatographic separation was performed in a fluoro-phenyl column, mobile phases were 0.1% formic acid in water (A) and acetonitrile (B) in gradient elution from 5% B to 70% B, with 10 min run time. Ionization was performed with electrospray source at 550 °C in negative mode, quantification mass transitions of m/z EtG 221- 75, EtS 125- 80.1, EtS-D5 130- 79.9, and EtG-D5 226- 75.1. The method was linear from 0.1 to 18 µg mL-1 for EtG and 0.02 to 6 µg mL-1 for EtS, specific, with no interfering peaks during the blank samples analyses. The method was precise and accurate, the matrix effect was compensated with the IS (-12% a + 6.1%). EtG and EtS were analyzed in 28 blood samples from trauma patients with positive alcoholaemia. The concentrations of EtG and EtS ranged from 0.25 to 14.11 µg mL-1 and 0.13 to 5.53 µg mL-1, with high correlation with ethanol concentrations, r=0.739 and r=0.817 (p <0,001), respectively. INTRODUÇÃO O etanol é o mais comum dos álcoois, obtido através da fermentação de açúcares. Por ser uma substância psicoativa de consumo legal, sua ingesta é comum e pode causar dependência. De acordo com a Organização Mundial da Saúde, o álcool já causou mais de 3 milhões de mortes por ano em todo o planeta, sendo responsável por 10% das mortes entre pessoas de 15 a 49 anos de idade.1 A relação álcool e trauma se destaca pela grande capacidade da substância de causar acidentes. Já que possui propriedades de alterar o comportamento, perder o senso de autocrítica, inibir o córtex do cérebro causando o efeito depressor como cansaço e sono, percepção alterada principalmente sensorial, visão borrada, tempo de reação prejudicado, entre outras consequências.2 O consumo de etanol também causa a possibilidade de gerar problemas sociais, familiares, uma série de doenças hepáticas, cardiovasculares e neoplásicas, bem como provocar acidentes como por exemplo de trânsito e de trabalho, além de poder levar a brigas violentas, suicídios e lesões fatais.1 Assim é possível notar a importância de avaliar o uso nocivo de etanol com a ocorrência de acidentes. Para isso testam-se métodos variados para confirmar a presença de seus metabólitos de uso recente e até mesmo do próprio etanol na corrente sanguínea e outros fluídos corporais, para que assim se possa ter uso, tanto na área forense quanto na área clínica. A maior parte do álcool biotransformado pelo organismo é eliminado pela via oxidativa. Após o consumo, o metabolismo que ocorre em menor quantidade é o não oxidativo. Assim, sua metabolização se dá mais lentamente, trazendo então uma vantagem na análise dos metabólitos não oxidativos que, consequentemente, ficam mais tempo no indivíduo que teve consumo de álcool.3 Esse metabolismo será o foco desse estudo. Por meio desta via não oxidativa, os metabólitos resultantes são: etilglicuronídeo (EtG), etilsulfato (EtS), fosfaditiletanol (PEth) e etil ésteres de ácidos graxos (EEAGs ou FAEE). Esses produtos podem ser detectados de horas até dias no organismo do indivíduo que ingeriu álcool, mas sua detecção irá depender da dose consumida.3 Os biomarcadores de meia vida curta, que ficam presentes na corrente sanguínea no uso recente de etanol, são o etilglicuronídeo (EtG) e o etilsulfato (EtS),4 com tempo de meia vida da eliminação de 3,6 e 3,3 horas, respectivamente.5 Ambos metabólitos têm sido detectados em urina como indicadores de abstinência ao álcool, com janela de 48 a até 130 horas, já o EtG em cabelo tem sido monitorado para avaliar o consumo crônico de álcool. No sangue são detectados por cerca de 8-12 horas e na urina por até 48 horas.4 A determinação dos metabolitos não oxidativos do álcool em sangue pode ser considerada como indicadores de consumo recente, com maior tempo de meia-vida e janela de detecção em relação a alcoolemia. Nesse contexto, é relevante a disponibilidade de metodologias analíticas para quantificação sanguínea do EtG e EtS. A análise dos metabolitos em vítimas de trauma no Brasil não foi explorada até o presente momento. Assim, esse estudo propõe o desenvolvimento e validação de uma metodologia simples e rápida para determinação sanguínea do EtG e EtS, relacionando com alcoolemia em pacientes vítimas de trauma.
PARTE EXPERIMENTAL Reagentes e soluções Os padrões de EtG, EtS, EtG-D5 e EtS-D5 foram obtidos da Toronto Research Chemicals (Toronto, Canadá). Metanol e ácido fórmico em grau HPLC foram adquiridos da Merck (Darmstadt, Alemanha). A água purificada adquiriu-se através de um sistema purificador Elga Purelab Ultra® da Veolia Labwater (High Wycombe, Reino Unido). A acetonitrila foi obtida da Honeywell - Riedel-de-Haën (Charlotte, EUA). Preparo das soluções e amostras controles As soluções calibradoras com a mistura dos dois analitos foram preparados através da dissolução em metanol da solução mãe nas seguintes concentrações de EtG 2; 5; 10; 20; 40; 80; 160; 240; 360 µg mL-1 e EtS 0,4; 1; 2; 5; 10; 20; 40; 80 e 120 µg mL-1. As soluções foram diluídas 1:20 (v/v) em sangue isento dos analitos, nas concentrações finais de 0,1; 0,25; 0,5; 1,0; 2,0; 4,0; 8,0; 12 e 18 µg mL-1 para EtG e 0,02; 0,05; 0,1; 0,25; 0,50; 1,0; 2,0; 4,0; 6,0 µg mL-1. Os controles de qualidade foram preparados em metanol da mesma forma, nas concentrações de EtG e EtS respectivamente: o controle de qualidade alto (CQA) 278 e 100 µg mL-1, o controle de qualidade médio (CQM) 100 e 36 µg mL-1e o controle de qualidade baixo (CQB) 3,3 e 1,2 µg mL-1. Os controles em sangue obtiveram as seguintes concentrações de EtS e EtG: CQA 5 e 13,9 µg mL-1, CQM 1,8 e 5 µg mL-1 e CQB 0,1 e 0,2 µg mL-1. Preparo das amostras Preparou-se as amostras através da precipitação de proteínas em sangue. Uma alíquota de 25 µL do sangue total foi adicionado a 100 µL do precipitante 0,1% ácido fórmico em acetonitrila, contendo 0,5 µg mL-1de EtG-D5 e EtS-D5. Após agitação em vórtex por 30 segundos, as amostras foram centrifugadas a 15.000 rpm por 10 minutos. O sobrenadante foi transferido para vial, diluído com água ultrapura (1:1, v/v) e 5 µL injetados em sistema de cromatografia liquida de ultra eficiência associado a espectrometria de massas em sequencial (UPLC-MS/MS). Equipamentos e condições cromatográficas As amostras foram analisadas em UPLC-MS/MS Acquity UPLC Class I TQD da Waters (Millford, EUA) com a coluna cromatográfica Acquity UPLC® Fluoro-Phenyl (100 x 2,1 mm; 1,7 µm) também da Waters, a 40 °C, com eluição em modo gradiente sendo a fase móvel (A) ácido fórmico 0,1% em água e (B) acetonitrila, com fluxo de 0,3 mL min-1. A composição da fase móvel inicial foi de 5% B até 0,5 minutos, com aumento linear de B até 80% em 8 minutos e voltando a 5% B em 8,5 minutos, com 1,5 minutos de equilíbrio. A ionização foi realizada em fonte eletrospray (ES) no modo negativo, com energia no capilar de 3,00 kV e temperatura de dessolvatação em 550 oC. A temperatura da coluna se manteve em 40 oC. O tempo de Dwell foi de 0,08 s. O gás de colisão em 54 L min-1. A energia de colisão foi de 2 V, o cone em 30 V para EtS e 35 V para EtG. As transições de massas monitoradas no modo MRM foram (m/z): EtS 125 → 80,1 quantificação e EtS 125 → 95,9 qualificação; EtG 221 → 75 quantificação e EtG 221 → 85 qualificação. Já para os PIs: EtS-D5 130 → 79,9 quantificação e 130 → 97,9 qualificação e para EtG-D5 226 → 75,1 quantificação e 226 → 84,85 qualificação. Validação do método O método foi validado segundo parâmetros essenciais para que pudesse obter confiabilidade e aceitabilidade da investigação dos dados prescritos nas resoluções da Agência Nacional de Vigilância Sanitária6,7 e Scientific working group for forensic toxicology (SWGTOX),8 tais como: seletividade, linearidade, sensibilidade, precisão e exatidão, estabilidade, efeito matriz, rendimento da extração e arraste. Seletividade Foram avaliadas amostras de 10 voluntários que não consumiram bebidas alcoólicas em 30 dias e foram processadas conforme descrito no item "preparação das amostras" para analisar a presença de picos cromatográficos e verificar se esses podem interferir na detecção dos analitos e dos padrões internos (PI). Linearidade Analisou-se amostras calibradoras no intervalo de 0,1 a 18 µg mL-1 para EtG e 0,02 a 6 µg mL-1 para EtS em sextuplicata. A linearidade foi obtida através da correlação entre as concentrações dos metabólitos e razões das suas áreas e seus padrões internos deuterados. Realizou-se teste F para avaliar a homocedasticidade da curva, comparando-se os valores do F-experimental (Fexp) com os valores de F-tabelado (Ftab) a 95% de intervalo de confiança e f1 = f2 (n-1) graus de liberdade, sendo n o número de réplicas (n=6). O Fexp foi calculado através da razão da variância dos calibradores no limite superior de quantificação e entre a variância do limite inferior de quantificação. Valores de Fexp > Ftab indicam heterocedasticidade significativa.9 A escolha dos modelos de ajuste ponderal da calibração foi realizada com base no coeficiente de determinação (r2) e no somatório dos resíduos. Critério para aceitação da linearidade foi r2 ≥0.99 e concentrações calculadas dos calibradores para cada nível entre ± 15% da concentração nominal.8,9 Sensibilidade Determinou-se através da obtenção da menor concentração dos metabólitos quantificado pelo equipamento com CV% < 20% e exatidão entre 80-120%. O menor ponto da curva de calibração foi analisado em triplicata em 3 dias. Ensaios de Precisão e Exatidão As amostras foram preparadas conforme descrito em "preparo das amostras". A precisão e exatidão foram definidas a partir de ensaios de 5 dias analisando os calibradores, os controles: baixo (0,2 µg mL-1 de EtG e 0,1 µg mL-1 de EtS), médio (5 µg mL-1 de EtG e 1,8 µg mL-1 de EtS) e alto (13,9 µg mL-1 EtG e 5 µg mL-1, de EtS). Calculou-se o coeficiente de variação (CV%) através de análise de Variância, o valor máximo aceito de CV até 15% e exatidão entre 85 e 115%. Estabilidade Foram preparadas amostras CQB e CQA, analisadas em triplicata no dia do preparo conforme o item "preparação das amostras" e após 3 ciclos congelamento e descongelamento - 20 oC. Variações de até ±15% sobre as concentrações encontradas em relação às obtidas antes do congelamento foram consideradas aceitáveis. Efeito matriz e rendimento da extração Foram preparadas 10 amostras8 brancas com concentrações conhecidas dos controles baixo e alto e separadas em conjunto A (com o analito no sangue), conjunto B (com o analito no precipitante) e conjunto C (somente o precipitante e analitos), de modo que pudesse ser avaliado o efeito matriz na análise, através do percentual obtido da divisão do valor mais alto pelo mais baixo da variação das amostras. O efeito matriz (EM) foi definido através da comparação de amostra de sangue com os calibradores de EtG e EtS (conjunto B) e somente precipitante com analitos (conjunto C), de forma a utilizar a fórmula: EM = [100%-B/C%]. O rendimento da extração foi calculado a partir razão da área do analito obtida nas amostras controle (conjunto A) e pelo protocolo da extração do branco adicionado dos padrões no precipitante (conjunto B) usando a fórmula RE=A/B%. Arraste (Carry-over) Cinco amostras brancas, isentas dos analitos foram preparadas e injetadas de forma alternada com amostras CQA, verificando a ocorrência de arraste. Como critério aceitável foi a presença de picos nas amostras brancas com as áreas não superiores a 20% das encontradas nas amostras de controle de qualidade limite inferior de quantificação. Aplicação do método As amostras utilizadas na validação clínica da metodologia foram provenientes de vítimas de trauma, maiores de 18 anos, atendidas no Hospital Geral de Novo Hamburgo no ano de 2019 com alcoolemia positiva. O estudo foi aprovado pelo Comitê de Ética em Pesquisa da Universidade Feevale (CEP). As coletas de sangue foram feitas no atendimento do paciente no departamento de emergência e armazenadas em 2 tubos com capacidade de 4 mL, um deles contendo anticoagulante EDTA e outro contendo fluoreto. As amostras permaneceram armazenadas a -80 ºC até a análise laboratorial. Do total de 238 pacientes que concordaram em participar do estudo, 28 tiveram resultado positivo para alcoolemia e foram selecionadas para a determinação dos níveis sanguíneos de EtG e EtS. A alcoolemia foi determinada em sangue fluoretado por cromatografia a gás com detector de ionização em chama (GC-FID) em técnica de headspace. Análise estatística Foi realizada análise descritiva das variáveis, as concentrações de etanol, EtG e EtS em sangue foram correlacionadas em teste de correlação de Pearson, com P<0,05 considerado estatisticamente significativo. Foi utilizado o software IBM SPSS Statistics versão 26.0.
RESULTADOS E DISCUSSÃO Cromatografia e preparação da amostra Com a coluna cromatográfica de recheio fluoro-phenyl foi possível a separação adequada dos analitos, sem picos interferentes (Figura 1). A coluna de fase reversa com grupamento flúor-fenil, menos apolar do que fase tradicional octadecilsilica, foi escolhida pois permitiu adequada retenção do EtG,10 composto mais polar, e do EtS, empregando modo gradiente de fase móvel. Na condição empregada, o UPLC-MS/MS se mostrou eficiente para a detecção de EtG e EtS em sangue, permitindo análise em 10 minutos, com tempo de retenção do EtG e EtG-D5 de 2,0 minutos e do EtS e EtS-D5 5,3 minutos.
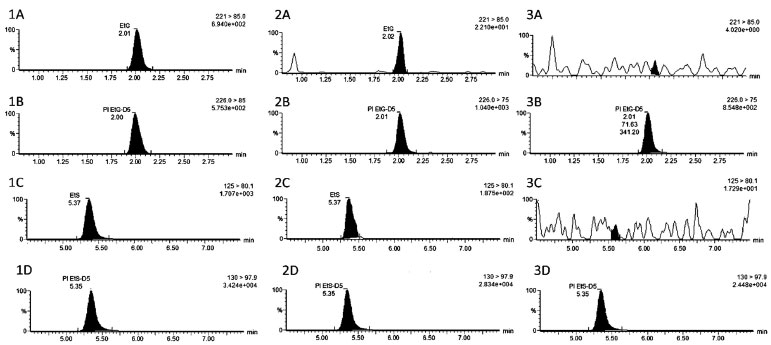 Figura 1. Cromatogramas obtidos a partir das análises de sangue total precipitado. Etilsulfato. Padrão interno EtS D5. Etilglicuronídeo. Padrão interno EtG D5. Sendo paciente 4,15 µg mL-1 de EtG e 1,4 µg mL-1 de EtS (1), sendo 1A EtG, 1B EtG-D5, 1C EtS e 1D EtS-D5; CQB 5 µg mL-1 de EtG e 1,8 µg mL-1 de EtS (2), sendo 2A EtG, 2B EtG-D5, 2C EtS e 2D EtS-D5; sangue branco (3), sendo 3A EtG, 3B EtG-D5, 3C EtS e 3D EtS-D5
O tempo de corrida cromatográfica é um aspecto relevante no ensaio, sendo determinante para a produtividade e relação custo-benefício do ensaio. O tempo de corrida do presente estudo foi semelhante ao estudo de Nguyen et al.11 de 8 minutos e superior ao proposto por Sidqey et al.12 de 4,5 minutos (Tabela 1). Nguyen et al.11 utilizaram coluna HILIC BEH Amida (150 mm x 2,1 mm, 1,7-mm) Waters, com tempos de retenção para EtG e EtS de, respectivamente 6 e 1,9 minutos. A coluna utilizada no estudo apresenta interação hidrofílica, semelhante à fase normal, porém com uso de água e acetonitrila como fase móvel, de forma a reter mais o EtG. Semelhantemente, Sidqey et al.12, utilizaram coluna HSS T3 C18 (2,1 mm x 100 mm, 1,8 μm), com total de corrida foi de 4,5 minutos e tempos de retenção para EtG e EtS foram de respectivamente, 1,53 e 1 minuto. Entretanto, no estudo o preparo das amostras foi precipitação de proteínas seguido de filtragem e concentração de extrato, sendo mais dispendioso. Já o estudo de Zhang et al.13 teve corrida de 25 minutos, com coluna de fase reversa tradicional C18, com eluição do EtG no tempo morto estando mais sujeito a efeito matriz, neste estudo os autores realizaram ainda a detecção de outros metabolitos do etanol, o fosfatidiletanol e etil-ésteres de ácidos graxos.
O emprego de ácido fórmico na fase móvel se deu buscando a melhoria na simetria dos picos cromatográficos e sensibilidade da análise. Foi testada fase móvel sem a adição do ácido, porém com baixa resolução cromatográfica. Essa estratégia tem sido adotada por outros autores na análise de EtG e EtS em diferentes matrizes biológicas, incluindo cabelo10 e urina,14 além do sangue.11-13 A técnica utilizada para preparo da amostra através da precipitação de proteínas do sangue foi rápida, simples e de fácil preparo, com sensibilidade adequada mesmo com pequeno volume de amostra de 25 µL. O preparo da amostra foi semelhante a outros previamente propostos, porém nos trabalhos de Nguyen et al.11 e Sidqey et al.12 houve etapa de concentração de extrato, o que não ocorre na presente metodologia, mostrando uma vantagem com relação aos estudos mencionados. Zhang et al.13 também empregaram a precipitação de proteínas sem concentração, com mistura de acetona e acetonitrila (1:4, v/v), com recuperação satisfatória para EtS, porém inferior a 30% para o EtG (Tabela 1). Validação do método O método foi linear nos intervalos de 0,1 µg mL-1 a 18 µg mL-1 para os metabólitos EtG e de 0,02 a 6 µg mL-1 para EtS. Os dados da curva de calibração possuem heterocedasticidade significativa, foi utilizado modelo ponderal 1/x, com somatório dos resíduos em 1,3x10-5 e 1,1x10-7 e r2=0,994 e r2=0,994 para EtG e EtS, respectivamente (Tabela 2).
Os intervalos das curvas de calibração foram definidos buscando quantificar amostras em caráter clínico e forense, compreendendo concentrações entre 0,1 e 18 µg mL-1 para EtG e entre 0,02 e 6 µg mL-1 para EtS. Os intervalos de concentrações adotadas nas curvas de calibração são similares entre os autores, com exceção dos valores de Nguyen et al.11 em soro, que foram inferiores. No presente estudo, os resultados de sensibilidade, precisão e exatidão apresentaram valores adequados, dentro dos limites estabelecidos pelas agencias regulatórias6,7 e guias de validação de métodos analíticos8 (Tabela 2). O CQLIQ apresentou CV < 20% e exatidão entre 80-120%, bem como CQB, CQM e CQA apresentaram CV < 15% e exatidão entre 85-115%. A ionização com fonte electrospray em modo negativo para análise dos metabolitos do etanol tem sido sistematicamente utilizada na análise de EtG e EtS em diferentes matrizes biológicas, como cabelo, urina e sangue, com sensibilidade satisfatória.10-14 O método também foi específico, sem picos interferentes em 10 amostras brancas de sangue (Figura 1). O efeito matriz foi testado para avaliar a supressão ou aumento de sinal pela presença de componentes da matriz do sangue, estando dentro dos limites preconizados pela SWTOX8 de até ±25%. Foram observados resultados de -12% a + 6,1%, indicando compensação satisfatória com uso dos padrões internos deuterados, mesmo com preparo simples de amostra, baseado em precipitação de proteínas. Além disso, as recuperações dos analitos foram próximas a 90% para ambos analitos, semelhantemente a estudo de Nguyen et al.11 para amostras de soro em concentrações mais baixas. Na Tabela 1 confere-se uma comparação entre artigos sobre a análise desses metabólitos. Não apareceram picos nos tempos de retenção quando injetada amostras brancas entre níveis altos. Podendo ser observado que não houve arraste na injeção de uma amostra mais concentrada para a injeção seguinte. Com relação a estabilidade, foram avaliadas as concentrações dos analitos após três ciclos de congela e descongela (Tabela 3), com variação para o de EtG -10 a + 13% em relação à concentração inicial e para o EtS de -6 a 9%, indicando a viabilidade para a reanálise dos metabólitos nestas condições de armazenamento. Sidqey et al.12 demonstrou estabilidade de ambos analitos em sangue armazenados a 10 e -20 oC, por pelo menos 7 dias.

Aplicação do método O método foi aplicado em amostras clínicas de 28 pacientes vítimas de trauma com alcoolemia positiva previamente detectada em amostras colhidas no atendimento de emergência, representando 11,7% dos 238 pacientes que aceitaram participar do estudo. O tipo de trauma mais frequente entre os pacientes com alcoolemia detectável foi colisão de trânsito (53,6%) seguido por queda (28,6%) e violência interpessoal (17,9%). A maior parte dos traumas foi severo (39,3%) ou moderado (35,7%). Os metabólitos não oxidativos do etanol foram detectados em 27 das 28 amostras (Tabela 4), com exceção de 1 paciente com alcoolemia de 1,03 g L-1. As concentrações sanguíneas de EtS estiveram entre 0,12 e 3,1 µg mL-1 e de EtG entre 0,25 e 14,11 µg mL-1, enquanto a alcoolemia variou de 0,17 a 5,29 g L-1 Os metabolitos não oxidativos do etanol com janela curta de detecção no sangue tiveram correlação significativa com a alcoolemia, com r: 0,771 para EtG e r: 0,814 para EtS (p <0,001).
Ainda sobre as concentrações de EtG e EtS, em Kwon et al.,15 avaliaram a cinética do EtG e EtS. Nesse estudo, após consumo de 1 g de etanol foram encontradas concentrações sanguíneas de 1,04 ± 0,30 µg mL-1 e 0,49 ± 0,17 µg mL-1, respectivamente. Em Halter et al.,16 voluntários que bebem socialmente e que consumiram de 0,5 a 0,78 g de etanol, tiveram níveis séricos de EtG de 0,266 a 1,08 µg mL-1 e EtS de 0,126 a 0,807 µg mL-1. Em Høiseth et al.17 foi avaliado o consumo de etanol em 13 pacientes recém admitidos em clínica de reabilitação para consumo de álcool, as concentrações de etanol variaram de não detectado a 3,7 g L-1, com concentrações de EtG e EtS em soro de 0,13 a 5,53 e 0,10 a 1,82 µg mL-1. No estudo de Dengiz et al.,18 também em pacientes traumatizados, porém exclusivamente vítimas de acidentes de transito, foram encontrados níveis médios de EtG, EtS e álcool similares aos encontrados em nosso estudo, de 2,72 µg mL-1, 0,66 µg mL-1 e 1,18 g L-1. Os dados do estudo demonstram uma associação entre níveis dos metabolitos não oxidativos EtG e EtS e a alcoolemia em vítimas de trauma. Esses achados suportam o uso dos biomarcadores na identificação do consumo recente de álcool em contexto clínico, em emergência. Em virtude de suas maiores janelas de detecção em relação à alcoolemia, podem ser relevantes para não identificar a intoxicação aguda, como identificar indivíduos sob efeito de ressaca. Uma limitação do estudo foi a análise retrospectiva das amostras armazenadas a -80 °C por um período de até 24 meses. Não há relatos sob a estabilidade a longo prazo do EtG e EtS em sangue armazenado nesta temperatura. Contudo, Høiseth et al.19 indicou boa estabilidade do EtG em amostras de sangue armazenadas -20 °C, tendo realizado estudo retrospectivo com amostras armazenadas nesta condição em até 5 anos. Outro aspecto a ser considerado é a necessidade do sistema de UPLC-MS/MS para a detecção e quantificação dos metabólitos do etanol em amostras de sangue com sensibilidade satisfatória. Diferentemente de países desenvolvidos, a disponibilidade desses equipamentos em hospitais no Brasil é bastante limitada. Apesar do custo envolvendo reagentes e solventes ser relativamente baixo, o investimento financeiro para a aquisição do equipamento, manutenção e necessidade de recursos humanos especializados são fatores que limitam a implementação de rotinas de análises toxicológicas em serviços de emergência, incluindo a metodologia proposta neste estudo. Contudo, a recente demanda pela realização de exames toxicológicos em matriz capilar aumentou o número de laboratórios privados estruturados com sistemas LC-MS/MS no Brasil. Além deles, laboratórios de pesquisa, Institutos de Perícia e Universidades que contam com infraestrutura poderiam fazer uso da metodologia em suas rotinas e em parceria com centros de saúde. Cabe ressaltar que além da aplicação em emergência, a quantificação dos metabolitos EtG e EtS em sangue é relevante no contexto forense e de toxicologia social.
CONCLUSÕES Foi desenvolvido e validado um método rápido, sensível e específico para análise dos metabólitos do etanol da via não oxidativa e de consumo recente, o EtG e EtS em UPLC-MS/MS. A preparação das amostras foi simples e eficiente para a detecção satisfatória dos analitos. O desempenho analítico do método mostrou-se adequado para às aplicações clínicas de avaliação de consumo de etanol em ambiente de emergência em vítimas de trauma.
AGRADECIMENTOS Os autores agradecem à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Universal processo 432940/2018-9) pelo apoio financeiro.
REFERÊNCIAS 1. World Health Organization; Alcohol, disponível em https://www.who.int/health-topics/alcohol#tab=tab_1, acessada em julho 2022. 2. Hoffmann, M. H.; Carbonell, E., Montoro, L.; Psicologia: Ciência e Profissão 1996, 16, 28. [Crossref] 3. Maenhout, T. M.; De Buyzere, M. L.; Delanghe, J. R.; Clin. Chim. Acta 2013, 415, 322. [Crossref] 4. Heier, C.; Xie, H.; Zimmermann, R.; IUBMB Life 2016, 68, 916. [Crossref] 5. Høiseth, G.; Morini, L.; Polettini, A.; Christophersen, A.; Mørland, J.; Forensic Sci. Int. 2009, 188, 52. [Crossref] 6. Resolução RDC no 166, de 24 de julho de 2017, disponível em https://www.in.gov.br/materia/-/asset_publisher/Kujrw0TZC2Mb/content/id/19194581/do1-2017-07-25-resolucao-rdc-n-166-de-24-de-julho-de-2017-19194412, acessada em julho 2022. 7. ANVISA; Resolução RDC no 27, de 17 de maio de 2012, disponível em http://bvsms.saude.gov.br/bvs/saudelegis/anvisa/2012/rdc0027_17_05_2012.html, acessada em julho 2022. 8. Scientific Working Group for Forensic Toxicology (SWGTOX); J. Anal. Toxic. 2013, 37, 452. [Crossref] 9. Almeida, A. M.; Castel-Branco, M. M. Falcão, A. C.; J Chromatogr. B 2002, 774, 215. [Crossref] 10. Bastiani, M. F.; Lizot, L. L. F.; da Silva, A. C. C.; Hahn, R. Z.; Dries, S. S.; Perassolo, M. S.; Antunes, M. V.; Linden, R. Forensic Sci. Int. 2020, 306, 110071. [Crossref] 11. Nguyen, V. L.; Paull, P.; Haber, P. S.; Chitty, K.; Seth, D.; Alcohol 2018, 67, 7. [Crossref] 12. Sidqey, D.; Liane, V. H.; Kristoffersen, L.; J Anal Toxic. 2021, 45, 378. [Crossref] 13. Zhang, X.; Zheng, F.; Lin, Z.; Johansen, S. S.; Yu, T.; Liu, Y.; Huang, Z.; Li, J.; Yan, J.; Rao, Y.; Anal. Chim. Acta 2017, 963, 68. [Crossref] 14. Sim, Y. E.; Kim, J. W.; Ko, B. J.; Kim, J. Y.J.; Anal. Sci. Technol. 2022, 13, 1. [Crossref]. 15. Kwon, M.; Choi, H. J.; Jo, Y. H.; Son, M. H.; Min, J. S.; Kim, N. Y.; Jung, J. E.; Forensic Sci. Int. 2019, 302, 109857. [Crossref] 16. Halter, C. C.; Dresen, S.; Auwaerter, V.; Wurst, F. M.; Weinmann, W.; Int. J. Legal Med. 2008, 122, 123. [Crossref]. 17. Høiseth, G.; Morini, L.; Polettini, A.; Christophersen, A. S.; Johnsen, L.; Karinen, R.; Mørland, J.; J. Anal. Toxicol. 2009, 33, 208. [Crossref] 18. Dengiz, H.; Daglioglu, N.; Goren, I. E.; Traf. Inj. Prevention 2020, 21, 371. [Crossref] 19. Høiseth, G.; Bernard, J. P.; Karinen, R.; Johnsen, L.; Helander, A.; Christophersen, A. S.; Mørland, J.; Forensic Sci. Int. 2007, 172, 119. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access