
 |
 |
 |
 |
 |
Artigo
| Innovative gas chromatographic determination of formaldehyde by miniaturized extraction and on-fiber derivatization, via SPME and SPME arrow |
|
Stefano DugheriI,*; Giovanni CappelliII; Jacopo CeccarelliII; Niccolò FanfaniIII; Lucia TrevisaniII; Donato SquillaciII; Elisabetta BucalettiII; Riccardo GoriIV; Nicola MucciII; Giulio ArcangeliII
I. Industrial Hygiene and Toxicology Laboratory, University Hospital Careggi, Florence 50139, Italy Recebido em: 09/05/2022 *e-mail: stefano.dugheri@unifi.it Formaldehyde (FA) is a carbonyl compound, ubiquitous in the environment and among the most widespread pollutants: it has exhibited toxic properties and is classified as a human carcinogen. FA is released from several sources, both temporary (e.g., combustion processes) and permanent (e.g., building products). This work proposes an innovative fully-automated application of headspace solid-phase microextraction (SPME) with on-fiber derivatization for the analysis of airborne FA emitted from liquid solutions or solid manufacts, in static mode, via gas chromatography-mass spectrometry. The method was tested in a wide range of airborne FA concentrations, using SPME and SPME Arrow fibers: the inter-day LOD and LOQ for SPME and SPME Arrow were evaluated, resulting in 0.072 and 0.215 ppm and 0.014 and 0.042 ppm, respectively. Moreover, other conventional detectors, such as Electron Capture Detector (ECD), Thermoionic Specific Detector (TSD), Photoionization Detector (PID), and Flame Ionization Detector (FID), were tested to set an analytical method to meet different requirements. The sensitivity and linearity of PID, FID and MS were comparable, while TSD and ECD were not suitable for the developed method, due to issues of response or linearity. MS results to be the most suitable and perfoming detector, however PID and FID result to be cheaper valid alternatives. INTRODUCTION Formaldehyde (FA) is a ubiquitous environmental chemical, one of the most relevant among the 187 hazardous air pollutants (HAPs), labeled by the US Environmental Protection Agency (EPA)1 and it represents over 50% of the HAPs-related cancer risks in the US.2 FA in indoor air is originated from several sources, which can be classified into temporary (e.g., cooking, combustion), intermitting (e.g., air cleaners, indoor chemistry), and permanent sources (e.g., building products, wood-based manufacts).3,4 Acute exposure to FA via inhalation causes local irritation in mucous membranes, including the eyes, the nose, and the upper respiratory tract. Sensory and olfactory irritations of the eyes and upper airways have been described as acute reversible effects of FA exposure in >50% of the exposed population, including laboratory staff, teachers, and students.5-9 Coughing, wheezing, chest pains, and bronchitis are other acute effects.10 Among the chronic effects related to FA exposure, cancer is the most significant disease.11 The quantitative estimated carcinogenic risk for inhalation exposure to FA ranges over orders of magnitude: about the low level of FA exposure (0.001 to 0.1 ppm-1.2 and 120 µg m-3), the Chemical Industry Institute of Toxicology (CIIT) predicted an additional risk of upper respiratory tract cancer for non-smoker equal to 2.3×10-10-2.7×10 8,12 while in high-level exposure scenarios, the inhalation unit risk (the upper-bound excess lifetime cancer risk estimated to result) is equal to 1.3×10-5 for 1 µg m-3 exposure. Moreover, FA may cause some adverse effects on the central nervous system,13 sensitization, contact dermatitis, irritant effects on the mucosal surface of the upper airways and eyes,14-16 and bronchial asthma;17 a prolonged inhalation of FA at low levels, e.g., <1 ppm, is unlikely to result in a chronic pulmonary injury.18 Several nations accepted international guidelines which individuate references intervals for the quantity of FA in indoor or outdoor (0.1 mg m-3) environment,19 emitted from manufacts (0.05 0.13 mg m-3),20 and contained in cosmetics,21 textile,22 toys,23 food and beverage.24 A huge number of analytical methods for determining airborne FA concentration have been developed.25 The current, validated methods for detecting gaseous FA are based on either active or passive sampling: the former using 2,4-dinitrophenylhydrazine (DNPH), as derivatizing agent on a filter or cartridge,26,27 subsequently analyzed by Liquid Chromatography (LC) or Gas Chromatography (GC),28,29 via chemical extraction; the latter using O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA), as a reagent on the solid sorbent,30 such as Solid-Phase Micro Extraction (SPME) fiber,31,32 and thermal desorption in the injection port of gas chromatograph. SPME, patented by Pawliszyn in 1989,33 is considered one of the major breakthroughs in 20th-century analytical chemistry.34-40 SPME, a solvent-free miniaturized technique, is not exhaustive and allows to combine sampling, isolation, and enrichment in one step in total automatized mode, esulting suitable for Green Analytical Chemistry (GAC).41,42 Another aspect of the greenness43 of the SPME technique is the possibility to perform an "on-fiber derivatization": in 1998 Martos and Pawliszyn35 proposed, for the sampling of FA, to functionalize the fiber by exposing it to the headspace (HS) of a 4-mL vial containing an aqueous doping solution of PFBHA, minimizing the amounts of chemicals employed for the analyses. The PFBHA kinetics showed that the derivatization reaction with FA follows a first-order rate,35 it is immediate and produces O-(pentafluorobenzyl)oxime (FA-oxime) (Figure 1).44
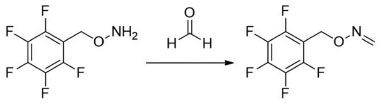 Figure 1. Chemical reaction of O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine (PFBHA) and formaldehyde
To overcome the lack of stability and robustness of SPME, in 2015 SPME Arrow has been developed: it contains greater phase volume than SPME fibers and consists of a stainless-steel cylinder with an internal rod and an outer sheath to safeguard the sorbent from mechanical damage and minimize the analyte loss.45 To enable the automatic identification, increase the stability, and the full automation of SPME managing,46 Fast Fit Assemblies (FFAs)-SPME fibers has been developed and proposed in 2009 by Chromline (Prato, Italy). Recently, FFA technology was extended to SPME Arrow. This work aims to develop an automated analytical method that allows the rapid, high throughput, and green analysis of FA by on-fiber derivatization with PFBHA. A comparison between SPME and SPME Arrow analysis is presented, highlighting the benefits and drawbacks of both approaches. In addition, three fundamental aspects as the choice of the derivatizing reagent, SPME fiber coating, and chromatographic column were considered. Moreover, different procedures to perform the on-fiber derivatization, as well as tests to reduce the blank signal, were evaluated. It is proposed a comparison of the analytical performances using GC instruments equipped with different detectors: Mass Spectrometer (MS), Flame Ionization Detector (FID), Photoionization Detector (PID), Electron Capture Detector (ECD), and Thermoionic Specific Detector (TSD).
EXPERIMENTAL Chemicals and reagents Formaldehyde solution 4% (buffered, pH 6.9) (CAS 50-00-0), Methanol (CAS 67-56-1), hexane (CAS 110-54-3), and 2-propanol (CAS 67-63-0) were purchased by Sigma-Aldrich (Saint Louis, MO, USA). Formaldehyde O-(pentafluorobenzyl)oxime (FA-oxime) (CAS 86356-73-2) was purchased by GiottoBiotech (Sesto F.no, Italy). Polydimethylsiloxane/divinylbenzene (PDMS/DVB) (Cat. No. FFA57293-U) 65-μm FFA-SPME fibers and PDMS/DVB (Cat No. FFA27486) 120-μm FFA-SPME-Arrow 1.10 mm fibers were purchased by Chromline (Prato, Italy). Different batches of O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine hydrochloride (PFBHA·HCl) (CAS 57981-02-9) were supplied by Sigma-Aldrich (Saint Louis, MO, USA) and Alfa Aesar (Wardhill, MA, USA). MilliQ water 18 MΩ cm (mQ) was obtained from Millipore's simplicity system (Darmstadt, Germany), and further purified to eliminate aldehydes by PURE UV3 - 4-Stage UV Water Purification System purchased by Pure n Natural Systems, Inc. (Steamwood, IL, USA). The gases helium (99.999%), hydrogen (99.999%), nitrogen (99.999%), Pure Air (99.999%), and Ar-CH4 (5%, v/v, CH4) were obtained by Air Liquid (Paris, France). HeadSpace screw-top 20-mL glass Vials (HSV) (Part No: 5188-2753) and Hdsp cap 18 mm magnetic PTFE/Sil (Part No.: 5188-2759) were purchased by Agilent Technologies (Santa Clara, CA, USA). Instrumental In-line, two Varian CP3800 GC systems, each with two 1079 injectors (SCION Instruments NL BV, Amundsenweg, The Netherlands) - equipped with 0.75 mm internal diameter (i.d.) liner and 2 mm i.d. liner set backward with a customized nut - were used for SPME and SPME Arrow, respectively. The injection was carried out in split mode (1:20, split flow 20 mL min-1, injection temperature 250 °C). The chromatographic columns were Agilent J&W (Part No 122-38-32UI) DB 35-MS-UI GC Column (30 m × 0.25 mm, 0.25 μm). One of the GC instruments was coupled with a Varian Saturn 2200 Ion-Trap as the detector. The second instrument was coupled with the following detectors: Tandem OI 4450 PID/FID mod. OI 4450, Varian 63Ni ECD, and Varian TSD. The initial column temperature was set to 50 °C (1 min) and then increased by 10 °C min-1 to 260 °C. Helium, as the carrier gas, was used and set at 1.2 mL min-1 for MS, while for PID/FID, ECD, and TSD was set at 2.0 mL min-1. Gases for FID were nitrogen (30 mL min-1), pure air (300 mL min-1), and hydrogen (30 mL min-1). Gas make-up for ECD was Ar-CH4 set at 30 mL min-1. Full automation of these procedures was achieved using a CTC PAL3 System xyz Autosampler (CTC Analytics AG Industrie strasse 20 CH-4222, Zwingen, Switzerland) equipped with Multi Fiber eXchange (MFX) system (Chromline, Prato, Italy), to guarantee an automated routine from the exchange of FFA-SPME fibers (both SPME and SPME Arrow) to the injection. The autosampler was on-line to both GC systems. Calibration levels The working solutions of FA were freshly prepared in FA-free water by diluting the stock solution of FA (4%) up to a concentration of 500 mg mL-1. Two calibration curves (0-40 ppm for SPME and 40-400 ppm for SPME Arrow) were prepared by adding proper volumes of FA working solution to FA-free water, obtaining 1 mL of each calibration solution. Then, 3.2 µL of each calibration solution were dispensed in HSVs, to achieve a final concentration of airborne FA of 0, 5, 10, 20, 40, 80, 160, 400 ppm. Quantification of PFBHA and FA-oxime To determine the quantity of PFBHA, standard solutions were prepared by dissolving proper quantities of PFBHA in methanol. Direct injection (1 µL) of these solutions in the GC-MS system (containing 1, 5, 10, 20, 30, 40, and 80 µg of PFBHA) allowed getting a regression curve. This curve was used to quantify the amount of PFBHA loaded on both SPME and Arrow SPME fibers. In order to determine the quantity of FA-oxime present in the blank, standard solutions were prepared by dissolving proper quantities of FA-oxime in hexane. Direct injection (1 µL) of these solutions in the GC-MS system (containing 0.01, 0.1, 1, and 5 µg of FA-oxime) allowed to obtain the regression curve. Sample preparation with on-line SPME The sample was transferred into an HSV, and then the vial was placed into the xyz autosampler for routine processing. The procedure starts with the fiber derivatization: PDMS/DVB SPME and SPME Arrow fibers were doped exposing it under magnetic stirring (500 rpm) for 4 min in the HSV, containing 1 mL of a PFBHA FA-free water solution (50 mg mL-1, prepared by dissolving 50 mg of commercial PFBHA in 1 mL of FA-free water), after an agitation step of 5 min at 60 °C. Following the fiber derivatization, the HSV, containing calibration levels or unknown samples, was heated for 10 min at 60 °C, sampled for 20 s under agitation (500 rpm), and desorbed for 1 min in the injector. Then, the HSV was brought back, and a new routine procedure can start. Method performance evaluation To evaluate the precision and the accuracy of the method, by sampling with SPME, one control solution (1 mL) corresponding to 30 ppm was prepared and analyzed, following the procedure described above from the stock solution. Three different sets of calibration and standard solutions were prepared and analyzed on six different days to evaluate inter-day performances of the method, and average curves were constructed every day. To evaluate intraday performances, six different sets of calibration and standard solutions were prepared and analyzed sequentially. The calibration curve of FA was obtained by plotting the peak area versus the nominal concentration of each calibration solution. Least-squares linear regression analysis was applied to get the best fitting function between the calibration points. To attain reliable LOD and LOQ values, the standard deviation (SD) of the response and slope approach was employed. In fact, when the LOD values were checked as a signal-to-noise (s/n) evaluation approach, their value was strongly influenced by the stability and reproducibility of the background noise. Therefore, the estimated SDs of responses were calculated by the standard deviation of Y-intercepts (SDY-I) of regression curves. The precision was evaluated through the relative standard deviation (RSD%) of the quantitative data of the replicate analysis of the control solution. The accuracy was determined to calculate the yield between the determined and nominal amounts. Using SPME Arrow fiber, the evaluation of the method performances was conducted as described for SPME, while precision and accuracy were investigated by preparing one control solution (1 mL) corresponding to 320 ppm. Concentration ranges for the two calibration curves were the same as described in the Calibration levels section.
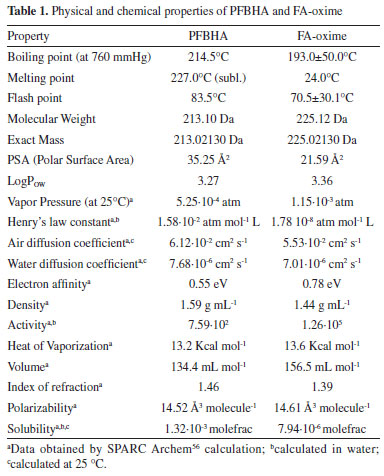
RESULTS AND DISCUSSION On-fiber derivatization reagent choice In recent years, the attention toward rapid, miniaturized, and solvent-free analytical techniques has significantly grown. In the field of the GAC,43 several analytical methods have been reported for the determination of FA and the derivatization step is necessary to improve the sensitivity. Simple GC methods and SPME with on-fiber derivatization have been applied to the determination of aldehydes in air, foodstuffs, biological samples, and water.31,37 Ho and Yu47 demonstrated the feasibility of the collection and analysis of airborne carbonyls by on-sorbent derivatization and thermal desorption. Similar attempts have shown successful analysis of carbonyls using PFBHA, 2,4,6-trichlorophenylhydrazine (TCPH), and pentafluorophenylhydrazine (PFPH) as derivatization agents.41 DNPH and 2-hydroxymethylpiperidine (HMP) were also considered candidates for on-fiber SPME derivatization agents but yielded a significant number of peaks from which the product of the reaction could not be identified.35 As reported in the literature, SPME fiber loaded with TCPH and the application of PID,48 ECD,49 and MS50 allowed the revelation of the formed hydrazones. Gioti et al.50 revealed serious background contamination of the FA peak present in the system. Al Azzam et al.51 evaluated the effect of temperature on reaction yield, showing that the peak area rises with an increase in temperature (from 50 to 70 °C) and then gradually decreases (71-90 °C) for the decomposition of the derivative. Moreover, the sampling method based on the ability of PFPH to react almost quantitatively with carbonyl compounds to form hydrazones was investigated by Bourdin et al.44 and Stashenko et al.52 Using PFPH, typical FA concentrations observed were approximately 65 µg L-1 (compared to PFBHA's 25 µg L-1), showing a higher level of impurity in the derivatization reagent,53 as well as persistent background contamination problems. These issues caused rather large fluctuations in FA peak areas52 and the presence, in high proportion, of two by-products: pentafluorobenzene and 2,3,4,5,6-pentafluoroaniline.44 PFBHA was finally chosen as the derivatization reagent as only one by-product was formed with this compound; considering the proportion of by-products formed no memory effect was observed at 250 °C. The physical and chemical properties of PFBHA and FA-oxime are presented in Table 1.54,55
SPME fiber selection The criterion for SPME fiber selection was to select a coating that would load the highest mass of PFBHA. Absorption processes of liquid-phase coatings (for example PDMS and polyacrylate) or adsorption processes of solid, porous phases (e.g., Carboxen (CAR) and DVB, in which the thick structure of the coating enables the analyte to be fixed in the pores of the solid phase only) occur on the SPME fiber surface. The amount of analyte extracted with the absorptive coating is less than with the adsorptive coating, hence porous phases were preferred.57 Effective use of the theory minimizes the number of experiments that need to be performed, but the assumption of ideal conditions required by the mathematical modeling needs to be verified. Therefore, the distribution constants estimated from physico-chemical tables or by the structural unit contribution method can predict trends in SPME analysis. To calculate n, that is the mass (ng) of PFBHA adsorbed in a sampling time t (s), using a porous coating, the theory of heat transfer can be applied:57,58  where Dg is the PFBHA diffusion coefficient in air, A is the surface of the needle opening (for SPME and SPME Arrow 0.00086 and 0.0063 cm2, respectively), Cg is the concentration of the analyte in the 20-mL vial (ng mL-1), and δ the thickness of the boundary layer surrounding the SPME fiber coating (cm), defined by Equation 2: 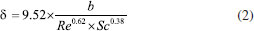 where the Reynolds number (Re) is expressed as 2ub v-1 (where u is the linear air speed (cm s-1), v the air viscosity, 0.014607 cm2 s-1, b the radius of the SPME fiber - 65 μm PDMS/DVB = 0.013 cm for SPME and 120 μm PDMS/DVB = 0.024 cm for Arrow SPME), and Schmidt's number (Sc) is defined by v Dg-1. Theoretically, linear air speeds higher than 10 cm s-1 yield a δ value close to 0 and invalidate Equation 2.58 Martos and Pawliszyn35 exposed the SPME fibers to an aqueous HS of a derivatization solution (17 mg mL-1) and revealed that PFBHA on the CAR/PDMS fiber coating was 2 times higher than the quantity observed for the PDMS/DVB fiber.59 They showed that there was no advantage in using CAR/PDMS, given all its affiliated issues, since the coating proved to be less rugged than PDMS/DVB coating for a significant peak tailing. Bourdin et al.44 indicated that CAR-based coatings were proven to induce the formation of by products during the thermal desorption step, thus rejecting the PDMS CAR DVB triple-phase SPME fibers. The porous phases DVB and CAR have similar surface areas: 750 and 720 mg m-2, respectively. The main difference between these two coatings is the much higher percentage of micropores in CAR than in DVB: the latter is primarily mesoporous, allowing to reach an adsorption equilibrium rapidly. In contrast, the rate of adsorption onto the CAR-containing SPME fibers is slower, probably due to the diffusion in micropores, which would act as a limiting step.44 The PDMS/DVB SPME fiber for GC was used for more than 150 loading and desorption steps without failure, while SPME Arrow was used for more than 300 cycles. For the aforementioned reasons, a PDMS-DVB SPME fiber in association with PFBHA was finally chosen. GC Column selection and optimizing separation The degradation product of PFBHA loaded on the SPME fiber, identified as 2,3,4,5,6-pentafluorobenzyl alcohol (PFBA),44 was directly adjoining that of FA-oxime when 5% phenyl-methylpolysiloxane capillary (30 m × 0.25 mm, 0.25 μm) column was used. Alternative GC columns were considered by several authors. The low/mid-polarity cyanopropyl-phenyl- methylpolysiloxane GC column, as 14% cyanopropyl-phenyl60 and 50% cyanopropyl-phenyl,31,61 were adopted with the limitation of not being allowed with some GC detectors and maximum temperature permitted of 240 °C. The 60-meters capillary (0.25 mm, 1 μm) column coated with 5% phenyl-methylpolysiloxane has been successfully used,44,62,63 but its length increases the analysis time. First, we propose a 30-meters 35% phenyl-methylpolysiloxane capillary (0.25 mm, 0.25 μm) column for its robustness, maximum operating temperature (360 °C), and compatibility with all the proposed detectors. Moreover, using this column, we observed separation between FA-oxime and PFBA, as reported in Figure 2. In these experimental conditions, FA-oxime showed a retention time of 6.5 min; the monitored qualifier ions were m/z of 195 and 225, and the quantifier ion was 181 as m/z.
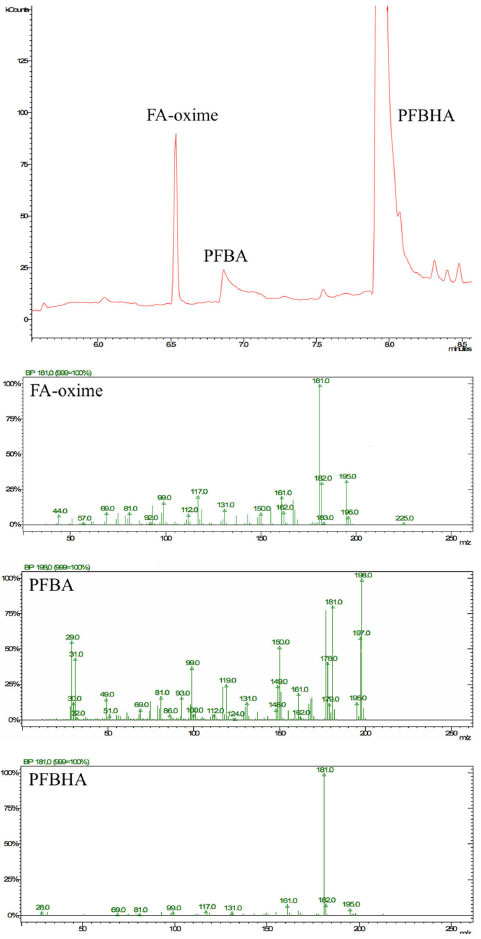 Figure 2. Chromatogram and mass spectra of formaldehyde O-(pentafluorobenzyl)oxime (FA-Oxime), 2,3,4,5,6-pentafluorobenzyl alcohol (PFBA), and O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine (PFBHA)
PFBHA loading and FA blank level reduction In order to maximize the loaded quantity of PFBHA on the SPME and SPME Arrow fibers, three approaches were compared: HS of a PFBHA water solution, HS of neat PFBHA, and dipping in an acid solution of PFBHA. The HS of 1 mL of FA-free water solution of PFBHA (17 mg mL 1) was firstly tested. Heating (60 °C) the HSV for 5 min, sampling for 1 min at 60 °C (both under stirring) the quantity of PFBHA detected was 19 µg. Tsai et al.31 used 1 mL of PFBHA water solution (17 mg mL 1) in a 4-mL vial, exposing the SPME fiber for various times in the HS of the vial at room temperature: the loaded PFBHA after 1 min was about 6 µg. Analogously, Deng et al.64 performed the doping of the fiber using 1 mL of PFBHA water solution (11 mg mL-1) in a 4-mL vial, stirring the solution at 1100 rpm at room temperature; by sampling for 1 min, the quantity of loaded derivatizing reagent was about 6 µg. Wang et al.53 demonstrated that working with the same concentration of PFBHA solution we used, heating at 50 °C in a 4-mL vial, the quantity of loaded derivatizing reagent was about 17 µg. The gap in these results is probably because of the different equilibrating temperatures: the efficiency of vaporization of PFBHA turns out to be enhanced at 50-60 °C. To quantify the SPME fiber loading using neat PFBHA, the solid derivatizing reagent (20 mg) was heated at 3 different temperatures (60 °C, 100 °C, and 200 °C) and the SPME fiber was exposed for 1 min in the headspace of the HSV. The loading of PFBHA at 60 and 100 °C was not comparable to the value obtained from the headspace of the PFBHA solution (ca. 3 µg vs 19 µg): that may be due to a poor sublimation efficiency at those temperatures. Moreover, at 200 °C the doping of the fiber was almost zero: at this temperature, the desorption of the PFBHA from the SPME fiber occurs. Lastly, we tested the dipping of the SPME fiber in a solution containing 100 mg of PFBHA in 10 mL of H2SO4 0.05M, as indicated by Egli et al.65 for SPME Arrow: the solution was heated 1 h at 90 °C, the fiber was dipped 1 min in the solution, and then exposed 30 min in the HS of a blank vial before the injection. However, also this test produced poor results in terms of loaded PFBHA (6 µg). These tests highlighted that the use of the HS-on-fiber derivatization with PFBHA in water solution is inevitable, hence we chose this loading method. In order to lower the FA blank level of PFBHA, attempts of pretreatments were performed: recrystallization from 2-propanol,66 liquid-liquid extraction (water/hexane),30 and sublimation of neat PFBHA. None of these tests reduced the blank level, which remained steady at 10 ± 3 ng. All the tests were carried out on every batch of derivatizing reagent purchased, and no significant differences were observed, highlighting that reducing the blank signal is not achievable via further purification of commercial PFBHA. Method development The focus was moved to adjusting the concentration of the PFBHA solution to maximize the loaded derivatizing agent. Two different concentrations were tested with SPME: 50 and 100 mg mL-1. At room temperature, the water solubility of PFBHA is 50 mg mL-1, while our tests showed total solubilization for the 100 mg mL-1 solution at 60 °C. The quantity of loaded PFBHA, equilibrating at 60 °C for 5 min and sampling for 1 min, was almost the same in both solutions and it was comparable to the 17 mg mL-1 solution (ca. 20 µg). In these loading conditions, the kinetic between FA and PFBHA follows the first-order rate, as shown in Equation 3, permitting a quantitative sampling of the airborne FA: 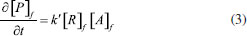 where [P]f, [R]f, and [A]f are the concentration of the product, derivatization reagent, and analyte in the fiber respectively, and k' is the reaction rate constant.53 In our analytical conditions, about 90 minutes pass between sequential fiber functionalization: the throughput using the 17 mg mL-1 solution did not provide a suitable number of processed samples. Using SPME, the 50 and 100 mg mL-1 solutions were employed for ca. 45-50 analyses, corresponding to ca. 3 days, without a significant decrease of the loaded PFBHA. After that time, the stability of the PFBHA solution worsens independently of the concentration. For these reasons, further tests were carried out using the 50 mg mL-1 solution. Moreover, between the 1st and the 30th injection a reduction by 50% of the blank FA-oxime was obeserved. In these conditions, it is possible to remove a major percentage of FA-oxime for each iteration from the PFBHA solution: this may be due to the difference in volatility between the two compounds. We also observed that a single vial of PFBHA can be sampled for about 8-10 injections with the SPME Arrow since the vial septum is quickly deteriorated by the tip of the fiber. The greater surface of the adsorbent phase allows to load a quantity of PFBHA double than SPME (see Figure 4), with a proportional increase in the level of FA-oxime. On the other hand, the high quantity of adsorbed PFBHA enables the detection of a higher load of airborne FA, without significant consumption of PFBHA, making this fiber more suitable for higher amounts of FA and/or in presence of interferences.
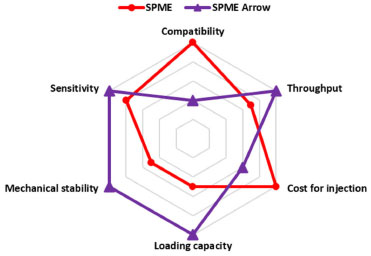 Figure 3. Radar chart fiber comparison
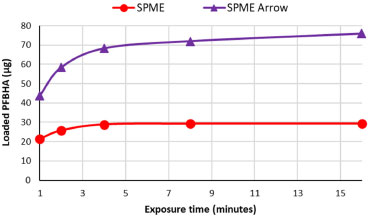 Figure 4. O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine (PFBHA) loading comparison between SPME and SPME Arrow fiber as a function of exposure time
For its design, SPME Arrow possesses higher mechanical stability that ensures longer life, higher sample throughput, and better sensitivity over SPME fibers.25 On the other hand, the higher cost of the SPME Arrow fiber and the necessity of specific injectors and liners, make the SPME still more versatile. The main practical differences between SPME and SPME Arrow fibers are compatibility and loading capacity. For these reasons, we consider the use of SPME preferable until it guarantees a suitable loading capacity; instead, to extend the range of concentration enhancing the applicability of the method in different contexts, SPME Arrow is mandatory. To our knowledge, this is the first work in which SPME Arrow was tested to increase the upper limit of quantitation (ULOQ). These features are reported in Figure 3: a numeric value, in a 1-10 range (1-3 for regular, 4-7 for discrete, and 8-10 for excellent), was assigned to each of the six parameters discussed above. The cost for a single injection is estimated considering 100 injections. The derivatization time was also investigated for both SPME and SPME Arrow fiber. Using the 50 mg mL-1 PFBHA solution, the SPME fibers were exposed in the headspace of the vial for 1, 2, 4, 8, and 16 min, and then analyzed. Plotting the loaded PFBHA with the exposure times, it can be observed that the loaded PFBHA grows until 4 min, then it remains ca. stable for SPME, while SPME Arrow shows a slight growth for superior times, as illustrated in Figure 3. Considering the tests described above, the construction of the calibration curve (using SPME fiber) was tested using different volumes of opportune solutions to reach 0, 5, 10, 20, and 40 ppm of airborne FA. A sample with free-FA water was used as 0 ppm level. Three different curves were prepared dispensing 1.6, 3.2, or 6.4 µL respectively in HSV. From the comparison of the response of the samples in terms of sensitivity (the slope of linear regression, m) and the R2 of each regression curve, the volume of 3.2 µL has been chosen as the preferable one: its calibration curve shows the higher slope (m = 52987) and the better R2 among the three proposed (Supplementary material, Table 1S). The slight variations observed in the results of the other volumes tested could be due to the potential higher incidence of the error dispensing 1.6 µL, while the use of 6.4 µL conducted to not complete vaporization of FA solution, as observed in some standard samples at the end of the analytical session. Consequently, 3.2 µL of calibration solutions corresponding to 40, 80, 160, and 400 ppm of airborne FA were prepared, sampled with SPME Arrow, and analyzed to obtain the regression curve for higher concentrations. The sampling time chosen was 20 s to guarantee a high throughput. The use of three observations for each calibration level of both SPME and SPME Arrow curves was retained acceptable, considering the reliable values of RSD% obtained (below 10%, ranging from 1.79-5.75%). An SPME-GC method with on-fiber derivatization for the determination of FA in manufacts was already present in the literature;32 in this study, the analyses were performed manually, and it demonstrated that this technique is well suited for the quantitative analysis of FA, suggesting the possibility of automatization. In fact, one of the focuses of our work was the development of a full automatized process, from the sample conditioning to the injection, using an xyz autosampler. This approach permitted to improve efficiency, reproducibility, robustness, and throughput of the samples analyzed. Performance result assessment The performances of the method were evaluated as described in Experimental for both SPME and SPME Arrow fibers via GC-MS. Daily LOD was calculated by multiplying 3.3 and the ratio between the standard deviation of water dispensed in HSV and the intercept of the average daily curve. LOQ is three times the LOD, RSD% for each day was calculated as the ratio between the standard deviation and the daily average of the control level. Daily accuracy was evaluated by the CV%, defined as the ratio between the daily average of the experimental values of the control level (i.e. the concentration obtained via GC-MS) and the theoretical value (i.e. the nominal concentration of the control level), multiplied by 100. In Table 2, the inter-day Accuracy is the average of the six daily CV% of the control level, each obtained from the 3 repetitions. The intra-day Accuracy is the CV% resulting from six repetitions in the same day. Inter-day and intraday performances evaluations are reported in Table 2.
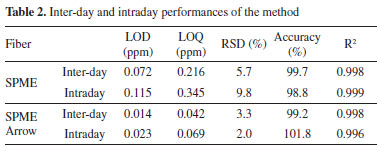
Both methods showed good linearity (R2); LOD (and consequently LOQ) is lower for SPME Arrow, confirming the major sensitivity of this fiber. Regarding precision and accuracy, no significant differences were highlighted comparing the fibers. LOD values were calculated for 20 s sampling time. The LOD of the method developed with SPME on GC-MS system was in line with the results from other studies: Bourdin et al.,44 Dugheri et al. and Martos and Pawliszyn35 showed methods with LOD in the ppb range. Martos and Pawliszyn35 demonstrated a linearity between the oxime mass formed and the exposure time, up to 90 s. To lower the LOD, a higher sampling time can be considered. Concerning the robustness of the method, it was evaluated using the F-test.67 Different sets of three calibration curves were prepared and analyzed with GC-MS, modifying the experimental conditions, and the variances obtained were compared. As pre-analytical variable, the change of the operator preparing the calibration levels was investigated. The other statistic populations were constructed changing the GC setting: replacing the column with a new one, renewing the injection port (septum and liner), and using split and splitless injection mode. The values obtained for F coefficients were in accordance with the theoretical values, at a confidence level of 95%. The data of F-test are available in Supplementary Material (Table 2S, Figures 5S and 6S). Comparison of detector performances In order to introduce an overview of sensitivity, robustness, specificity, cost, and portability, the method developed using MS as a detector was applied to PID, FID, ECD, and TSD, with SPME. Analytical parameters (LOD, LOQ, and R2) were calculated as described in the Performance result assessment. The chromatograms are showed in the Supplementary Material (Figures 7S and 8S). In the range of investigated concentration (0-40ppm), the calibration curves showed good linearity and sensitivity, as shown in Table 3. The highest sensitivity, defined by the slope of the equations, was showed by the MS detector. PID and FID showed comparable sensitivity and linearity , but lower than the GC-MS one.
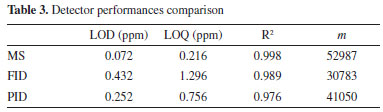
In particular, the sensitivity of the PID was greater compared to the FID, because of the specificity of this technique towards aromatic compounds.68 In fact, to avoid the saturation of the detector, it was necessary to lower the sensitivity of PID by two orders of magnitude, thus explaining the slightly lower LOD compared to FID (Table 3). The LOD observed for FID and PID are acceptable for our experimental conditions. However, other studies showed lower LODs, especially for FID detector, due to higher sampling times.69 Thus, a possible sensitivity improvement in our method could be represented by increasing the exposure time of the derivatized fiber. As for TSD, the response of the calibration levels was not proper for our operative conditions, hence no deeper studies were performed. ECD showed very high sensitivity, but the range of linearity is 0-3 ppm, reducing the field of applicability of the detector. Despite the chance of working at a low concentration of FA (the LOD is 0.063 ppm), due to the absence of linearity in our investigated range, this detector was not suitable for our method. The robustness was verified using the F-test, evaluating sets of curves, analyzed in triple, for each detector, varying the same experimental conditions used for the developed MS method robustness. The F coefficients observed were in line with the theoretical values, at a confidence level of 95% (Supplementary material, Table 3S and 4S and Figures 9S e 10S). In our opinion, MS results to be the most suitable detector since, in presence of a complex matrix, other substances potentially present can be identified in the same chromatographic run. Nevertheless, the use of other detectors is favorite due to the high overall cost of the MS instrument and the requirement of high trained operators. In fact, in this context PID and FID result to be cheaper alternatives. As far as portability is concerned, all the detectors tested can be equipped with a portable on-field GC instrument. Miniaturization is a technological trend that has been implemented in all kinds of analytical instrumentation, enabling the conversion of large bench-top instruments into portable instruments. This technology has been increasingly applied to GC, LC, and mass spectrometry for the last 10 years, resulting in savings in both time and costs throughout the monitoring process. With recent developments, portable MSs are becoming more prevalent and will eventually find their way from research laboratories to control laboratories to consent on site or at-line measurements.70-77 Person-portable chromatographs are generally smaller and lighter than conventional transportable chromatographs, and use less energy. They are usually powered by batteries and equipped with small gas tanks. Nowadays, different alternatives, compatibles with SPME injection modules, are present on the market: Griffin G510 (Teledyne FLIR LLC, Wilsonville, OR, USA), HAPSITE ER (INFICON AG, Bad Ragaz, Switzerland), and Torion T-9 (PerkinElmer Torion Technologies, Waltham, MA, USA). Among them, Torion Technologies developed an SPME syringe, called the Custodion, that allows high-throughput full automated analytical sessions, launching the chromatographic run. Conversely, these new technologies are still very expensive. For this reason, PID represents a good compromise in terms of performance, costs, and security, since it requires only the carrier gas and the electricity, with no need for pumping stages (MS), hydrogen (FID), nor a radioactive source (63Ni, ECD), even if a portable GC-PID system with the SPME interface, to our knowledge, is still not commercialized. These considerations are resumed in Figure 5, in which a numeric value, in a 1-10 range (1-3 for regular, 4-7 for discrete, and 8-10 for excellent), was assigned to each of the five parameters discussed above.
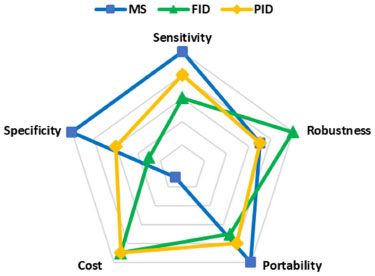 Figure 5. Radar chart detector comparison
CONCLUSIONS Solid-phase microextraction techniques such as SPME and SPME Arrow, using PFBHA for on-fiber derivatization, are valuable approaches for the determination of FA. Different parameters affecting the extraction-derivatization were carefully studied and optimized, and the method showed satisfactory results in terms of linearity, precision, accuracy, and sensitivity. The SPME and SPME Arrow method were developed and tested on standard solutions, and their applicability was successfully evaluated. Lastly, the use of automated and dedicated xyz robotic systems allowed high throughput screening in GC analyses, compatible with different detection methods (MS, FID, and PID), covering different analytical and practical requirements. SUPPLEMENTARY MATERIAL In supplementary material, available on http://quimicanova.sbq.org.br as a PDF file, with free access, are presented the Figure 1S to 10S and the Table 1S to 4S. The Total-Ion Current (TIC) chromatogram in EI mode is reported in Figure 1S. The mass spectra of FA-oxime, PFBA, and PFBHA, showing a retention time of 6.50, 6.85, and 7.95, respectively, are reported in Figures 2S to 4S. The target compounds were identified by comparison with literature data. In Table 1S are reported the R2 and the slope values of the calibration curves obtained with 1.6, 3.2, 6.4 μL of FA solution. In Table 2S, 3S and 4S are showed the data of robustness evaluation with F test. The Figures from 5S, 6S, 9S, 10S are the graphical representations of the Ftests, while Figures 7S and 8S show the chromatograms obtained with FID and PID detectors.
ACKNOWLEGMENTS The study group thanks the Laboratorio per l'Innovazione e per l'applicazione della Robotica nel Monitoraggio degli Ambienti Naturali, di vita e di lavoro (LIROMAN) of PIN - Polo Universitario città di Prato (Prato, Italy). The LIROMAN lab has provided the structures and the instruments to optimize the automation of the analytical method.
REFERENCES 1. Lui, K. H.; Ho, S. S.H.; Louie, P. K.K.; Chan, C. S.; Lee, S. C.; Hu, D.; Chan, P. W.; Lee, J. C.W.; Ho, P. K.; Atmos. Environ. 2017, 152, 51. [Crossref] 2. Strum, M.; Scheffe, R.; J. Air Waste Manage. Assoc. 2016, 2, 120. [Crossref] 3. Salthammer, T.; Build. Environ. 2019, 150, 219. [Crossref] 4. Li, B.;Cheng, Z.; Yao, R.; Wang, H.; Yu, W.; Bu, Z.; Xiong, J.; Zhang, T.; Essah, E.; Luo, Z.; Shahrestani, M.; Kipen, H.; Build. Environ. 2019, 147, 540. [Crossref] 5. Garzillo, E. M.; Miraglia, N.; Pedata, P.; Feola, D.; Lamberti, M.; Int. J. Occup. Med. Environ. Health 2016, 29, 355. [Crossref] 6. Werner, S., Nies, E.; J. Occup. Med. Toxicol. 2018, 13, 28. [Crossref] 7. Yon, D. K.; Hwang, S.; Lee, S. W.; Jee, H. M.; Sheen, Y. H.; Kim, J. H.; Lim, D. H.; Han, M. Y.; Am. J. Respir. Crit. Care Med. 2019, 200, 388. [Crossref] 8. Alves, C. A.; Aciole, S. D. G.; Quim. Nova 2012, 35, 2025. 9. Pegas, P. N.; Evtyugina, M. G.; Alves, C. A.; Nunes, T.; Cerqueira, M.; Franchi, M.; Pio, C.; Quim. Nova 2010, 33, 1145. 10. Formaldehyde, available at https://www.epa.gov/sites/production/files/2016-09/documents/formaldehyde.pdf, accessed in August 2022. 11. Formaldehyde, available at https://monographs.iarc.who.int/wp-content/uploads/2018/06/mono100F-29.pdf, accessed in August 2022. 12. 1999 Priority Substances List Assessment Report - Formaldehyde, available at https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/ewh-semt/alt_formats/hecs-sesc/pdf/pubs/contaminants/psl2-lsp2/formaldehyde/formaldehyde-eng.pdf, accessed in August 2022. 13. Zendehdel, R.; Fazli, Z.; Mazinani, M.; Environ. Monit. Assess. 2016, 188, 1. [Crossref] 14. Nielsen, G. D.; Larsen, S. T.; Wolkoff, I.; Arch. Toxicol. 2017, 91, 35. [Crossref] 15. Lestari, K. S.; Humairo, M. V.; Agustina, U.; J. Environ. Public Health 2018, 2018. [Crossref] 16. Benharroch, D.; Benharroch, Y. B.; Respir. Med. Case Rep. 2020, 31, 101166. [Crossref] 17. Medical surveillance - Formaldehyde, available at https://www.osha.gov/laws-regs/regulations/standardnumber/1910/1910.1048AppC, accessed in August 2022. 18. Medical Management Guidelines for Formaldehyde, available at https://www.atsdr.cdc.gov/MHMI/mmg111.pdf, accessed in August 2022. 19. World Health Organization, Regional Office for Europe, Air quality guidelines for Europe, 2nd ed., 2000. 20. Airborne Toxic Control Measure to Reduce Formaldehyde Emissions from Composite Wood Products, available at https://ww2.arb.ca.gov/sites/default/files/barcu/regact/2007/compwood07/fro-atcmfin.pdf, accessed in August 2022. 21. European Parliament and of the Council. Regulation EC 1223/2009, 2009. 22. European Parliament and of the Council. Regulation EU 1513/2018, 2018. 23. European Parliament and of the Council. Regulation EU 1929/2019, 2019. 24. Bianchi, F.; Careri, M.; Musci, M.; Mangia, A.; Food Chem. 2007, 100, 1049. [Crossref] 25. Dugheri, S.; Mucci, N.; Cappelli, G.; Trevisani, L.; Bonari, A.; Bucaletti, E.; Squillaci, D.; Arcangeli, G.; J. Anal. Methods Chem. 2022, 1. [Crossref] 26. Jin, X. C.; Ballentine, R. M.; Gardner, W. P.; Melvin, M. S.; Pithawalla, Y. B.; Wagner, K. A.; Avery, K. C.; Sharifi, M.; Separations 2021, 8, 151. [Crossref] 27. Liu, X.; Mason, M. A.; Guo, Z.; Krebs, K. A.; Roache, N. F.; Atmos. Environ. 2015, 122, 561. [Crossref] 28. Szulejko, J. E.; Kim, K. H.; Trends Anal. Chem. 2015, 64, 29. [Crossref] 29. Yuniati, W.; Amelia, T.; Ibrahim, S.; Damayanti, S. ACS Omega 2021, 6, 28403. [Crossref] 30. Cancilla, D. A.; Chou, C. C.; Barthel, R.; Que Hee, S. S.; J. AOAC Int. 1992, 75, 842. [Crossref] 31. Tsai, S. W.; Chang, C. M.; J. Chromatogr. A 2003, 1015, 143. [Crossref] 32. Himmele, S.; Mai, C.; Schumman, A.; Hasener, J.; Steckel, V.; Lenth, C.; Int. J. Ion Mobil. Spec. 2014, 17, 55. [Crossref] 33. Arthur, C. L.; Pawlsizyn, J.; Anal. Chem. 1990, 62, 2145. [Crossref] 34. Nowak, P.M.; Wietecha-Posłuszny, R.; Pawliszyn, J. TrAC - Trends Anal. Chem. 2021, 138, 116223. [Crossref] 35. Martos, P. A.; Pawliszyn, J.; Anal. Chem. 1998, 70, 2311. [Crossref] 36. Filipiak, W.; Bojko, B.; TrAC - Trend. Anal. Chem. 2019, 115, 203. [Crossref] 37. Koziel, J. A.; Noah, J.; Pawliszyn, J.; Environ. Sci. Technol. 2001, 35, 1481. [Crossref] 38. Marini, F.; Bellugi, I.; Gambi, D.; Pacenti, M.; Dugheri, S.; Focardi, L.; Tulli, G.; Acta Anaesthesiol. Scand. 2007, 51, 625. [Crossref] 39. Dugheri, S.; Bonari, A.; Pompilio, I.; Mucci, N.; Montalti, M.; Arcangeli, G.; Rasayan J. Chem. 2016, 9, 657. 40. Cappelaro, E. A.; Yariwake, J. H.; Quim. Nova 2015, 38, 427. [Crossref] 41. Dugheri, S.; Massi, D.; Mucci, N.; Marrubini, G.; Cappelli, G.; Speltini, A.; Bonferoni, M. C.; Arcangeli, G.; Trends Environ. Anal. Chem. 2021, 29, e00116. [Crossref] 42. Dugheri, S.; Mucci, N.; Bonari, A.; Marrubini, G.; Cappelli, G.; Ubiali, D.; Campagna, M.; Montalti, M.; Arcangeli, G.; Acta Chromatogr. 2020, 32, 69. [Crossref] 43. Plotka-Wasylka, J.; Namiesnik, J.; Green Analytical Chemistry Past, Present and Perspectives; Springer Nature Singapre Pte Ltd: Singapore, 2019. 44. Bourdin, D.; Desauziers, V.; Anal. Bioanal. Chem. 2014, 406, 317. [Crossref] 45. Herrington, J. S.; Gòmez-Rìos, G. A.; Mayers, C.; Stidsen, G.; Bell, D. S.; Separations 2020, 7, 12. [Crossref] 46. Pacenti, M.; Dugheri, S.; Traldi, P.; Degli Esposti, F.; Perchiazzi, N.; Franchi, E.; Calamante, M.; Kikic, I.; Alessi, P.; Bonacchi, A.; Salvadori, E.; Arcangeli, G.; Cupelli, V.; J. Autom. Methods Manage. Chem. 2010, 1. [Crossref] 47. Ho, S. S. H.; Yu, J. Z.; Anal. Chem. 2002, 74, 1232. [Crossref] 48. Curylo, J.; Wardencki, W.; Chem. Anal. (Warsaw, Pol.) 2005, 50, 735. 49. Konidari, C. N.; Giannopoulos, T. S.; Nanos, C. G.; Staliska, C. D.; Anal. Biochem. 2005, 338, 62. [Crossref] 50. Gioti, E. M.; Fiamegos, Y. C.; Skalkos, D. C.; Stalikas, C. D.; J. Chromatogr. A 2007, 1152, 150. [Crossref] 51. Al Azzam, K. M.; Makahleh, A.; Saad, B.; Asian J. Chem. 2021, 33, 930. [Crossref] 52. Stashenko, E. E.; Puertas, M. A.; Salgar, W.; Delgado, W.; Martinez, J. R.; J. Chromatogr. A 2000, 886, 175. [Crossref] 53. Wang, Q.; O'Reilly, J.; Pawliszyn, J.; J. Chormatogr. A 2005, 1071, 147. [Crossref] 54. ChemSrc, available at https://www.chemsrc.com/en/cas/86356-73-2_830658.html, accessed in August 2022. 55. ChemSrc, available at https://www.chemsrc.com/en/cas/57981-02-9_955552.html, accessed in August 2022. 56. SPARC Archem, available at http://www.archemcalc.com/sparc.html, accessed in August 2022. 57. Pacenti, M.; Dugheri, S.; Gagliano-Candela, R.; Strisciullo, G.; Franchi, E.; Degli Esposti, F.; Perchiazzi, N.; Boccalon, P.; Arcangeli, G.; Cupelli, V.; Acta Chromatogr. 2009, 21, 379. [Crossref] 58. Koziel, J.; Jia, M.; Pawliszyn, J.; Anal. Chem. 2000, 72, 5178. [Crossref] 59. Pieraccini, G.; Bartolucci, G.; Pacenti, M.; Dugheri, S.; Boccalon, P.; Focardi, L.; J. Chromatogr. A 2002, 955, 117. [Crossref] 60. Iglesias, J.; Gallardo, J. M.; Medina, I.; Food Chem. 2010, 123, 771. [Crossref] 61. Liou, S. W.; Chen, C. Y.; Yang, T. T.; Lin, J. M.; Bull. Environ. Contam. Toxicol. 2008, 80, 324. [Crossref] 62. Saison, D.; De Schutter, D. P.; Delvaux, F.; Delvaux, F. R.; J. Chromatogr. A 2009, 1216, 506. [Crossref] 63. Dugheri, S.; Massi, D.; Mucci, N.; Berti, N.; Cappelli, G.; Arcangeli, G.; Arh. Hig. Rada Toksikol. 2020, 71, 178. [Crossref] 64. Deng, C.; Li, N.; Zhu, W.; Qian, J.; Yang, X.; Zhang, X.; J. Sep. Sci. 2005, 28, 172. [Crossref] 65. Egli, P.; Schilling, B.; Boehm, G.; Ingenious News 2015, 2, 1. 66. Spaulding, R.; Charles, M. J.; Tuazon, E. C.; Lashley, M.; J. Am. Soc. Mass Spectrom. 2002, 13, 530. [Crossref] 67. Dejaegher, B.; Heyden, Y. V.; J. Chromatogr. A 2007, 1158, 138. [Crossref] 68. Cavalcante, R. M.; de Andrade, M. V. F.; Marins, R. V.; Oliveira, L. D. M.; Microchem. J. 2010, 96, 337. [Crossref] 69. Koziel, J.A.; Pawliszyn, J., J. Air Waste Manag Assoc. 2001, 51, 173. [Crossref] 70. Lawton, Z. E.; Traub, A.; Fatigante, W. L.; Mancias, J.; O'Leary, A. E.; Hall, S. E.; Wieland, J. R.; Oberacher, H.; Gizzi, M. C.; Mulligan, C. C.; J. Am. Soc. Mass Spectrom. 2017, 28, 1048. [Crossref] 71. Zhang, X.; Zhang, H.; Yu, K.; Li, N.; Liu, Y.; Liu, X.; Zhang, H.; Yang, B.; Wu, W.; Gao, J.; Jiang, J.; Anal. Chem. 2020, 92, 4656. [Crossref] 72. Fiorentin, T. A.; Logan, B. K.; Martin, D. M.; Browne, T.; Rieders, E. F.; Forensic Sci. Int. 2020, 313, 110342. [Crossref] 73. Wang, W.; Wang, S.; Xu, C.; Li, H.; Y., X.; Hou, K.; Li, H.; Anal. Chem. 2019, 91, 10212. [Crossref] 74. Zhou, C.; Wu, H.; Zhang, X.; Zhang, Y.; Xie, W.; Xu, W.; Anal. Chem. 2019, 91, 8808. [Crossref] 75. Fedick, P. W.; Pu, F.; Morato, N. M.; Cooks, R. G.; J. Am. Soc. Mass Spectrom. 2020, 31, 735. [Crossref] 76. Pulliam, C. J.; Bain, R. M.; Osswald, H. L.; Snyder, D. T.; Fedick, P. W.; Ayrton, S. T.; Flick, T. G.; Cooks, R. G.; Anal. Chem. 2017, 89, 6969. [Crossref] 77. Wang, S.; Wang, W.; Li, H.; Xing, Y.; Hou, K.; Li, H.; Anal. Chem. 2019, 91, 3845. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access