
 |
 |
 |
 |
 |
Artigo
|
|
| Fenilpropanoides com ação ANTI-Trypanosoma cruzi isolados de Baccharis ligustrina C. DC. (ASTERACEAE) Anti-Trypanosoma cruzi Phenylpropanoids Isolated from Baccharis ligustrina C. DC. (ASTERACEAE) |
|
Matheus L. SilvaI; Leila GimenesII; Paulete RomoffIII; Marisi G. SoaresIV; Fernanda F. CamiloV; Erica Valadares de C. LevattiVI; Andre G. TemponeVI ; João Henrique G. LagoI,*
I. Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-170 São Paulo - SP, Brasil Recebido em 22/06/2022 *e-mail: joao.lago@ufabc.edu.br In the present work, dried aerial parts of Baccharis ligustrina (Asteraceae) were subjected to microwave assisted extraction (MAE) using aqueous solution of 1-butyl-3-methylimidazolium bromide (BMImBr) and the obtained extract was successively partitioned using hexane and EtOAc. Using reduced amounts of extracts and efficient chromatographic steps, four acyl C6C3 derivatives (n-hexacosyl ferulate, n-hexacosyl, n-octacosyl, and n-triacontyl p-coumarates) were obtained from hexane phase whereas two C6C3 acids (ferulic and p-coumaric) were obtained from EtOAc phase. Isolated phenylpropanoids were evaluated against amastigote forms of parasite Trypanosoma cruzi. As result, it was observed that p-coumaric and ferulic acids were inactives whereas alkyl derivatives displayed EC50 values of 6.5 µmol L-1 (n-octacosyl p-coumarate), 9.3 µmol L-1 (n-triacontyl p-coumarate), 15.7 µmol L-1 (n-hexacosyl p-coumarate), and 32.2 µmol L-1 (n-hexacosyl ferulate). All tested compounds displayed reduced toxicity against NCTC cells (CC50 > 200 mmol L-1). INTRODUÇÃO O gênero Baccharis apresenta ampla distribuição em diferentes regiões do Brasil e conta com aproximadamente 500 espécies, das quais cerca de 20% são utilizadas na medicina popular para tratamento de inflamações, males do fígado, reumatismo e úlceras.1,2 Tais espécies são encontradas nas regiões sul e sudeste do país, principalmente em regiões de altitude (a cerca de 1800-2000 m do nível do mar), que são biomas com predominância de gramíneas e arbustos.3 Do ponto de vista químico, espécies de Bacharis são produtoras de diferentes metabólitos dos quais se destacam flavonoides, diterpenos de esqueletos caurano, labdano e clerodano e também de triterpenoides, principalmente de esqueletos oleanano e ursano.4-6 Além desses metabólitos, merece destaque a ocorrência de tricotecenos, que são derivados diterpênicos com importantes ações farmacológicas.7 Dentre as espécies estudas quimicamente encontra-se B. ligustrina, distribuída amplamente nos estados de Minas Gerais e São Paulo, e que se mostrou constituída essencialmente por dois triterpenos (ácidos oleanólico e ursólico) e dois flavonóides (naringenina e hispidulina).8 Como parte dos nossos estudos visando a prospecção de compostos bioativos em espécies de Baccharis,9-11 as partes aéreas de B. ligustrina foram submetidos à extração assistida por micro-ondas (MAE) usando solução aquosa de 1-butil-3-brometo de metilimidazólio (BMImBr) e o extrato assim obtido foi extraído sucessivamente com hexano e com AcOEt. Após diferentes processos de fracionamento cromatográfico em pequena escala foram obtidos, de ambas as fases de partição, seis derivados fenilpropanoídicos: ácido ferúlico (1), ácido p-cumárico (2), ferulato de n-hexacosila (3), p-cumarato de n-hexacosila (4), p-cumarato de n-octacosila (5) e p-cumarato de n-triacontila (6). Adicionalmente, os derivados C6C3 isolados (1-6) foram avaliados quanto a ação antiparasitária, em especial frente as formas amastigotas de Trypanosoma cruzi, protozoário causador da doença de Chagas. Essa doença, endêmica no Brasil, conta com um único fármaco disponível no país, o benznidazol, extremamente tóxico e com baixa eficácia, especialmente na fase crônica da doença.12 Frente a essa problemática, a busca de novos protótipos moleculares para o desenvolvimento de futuros fármacos para a doença de Chagas baseados em produtos naturais oriundos da biodiversidade brasileira consiste em uma abordagem importante, particularmente devido a ampla diversificação estrutural dos metabólitos especiais encontrados em espécies vegetais.13,14
RESULTADOS E DISCUSSÃO Partes aéreas de B. ligustrina foram submetidas à extração assistida por micro-ondas (MAE) com solução aquosa de brometo de 1-butil-3-metilimidazólio (BMImBr) e o material assim obtido foi extraído com hexano e com AcOEt. O fracionamento cromatográfico de ambas as fases permitiu a obtenção de seis fenilpropanoides (1-6), como mostrado na Figura 1.
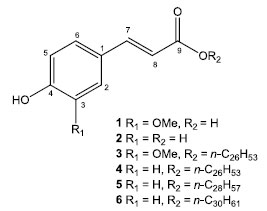 Figura 1. Estruturas dos compostos 1-6 identificados em B. ligustrina
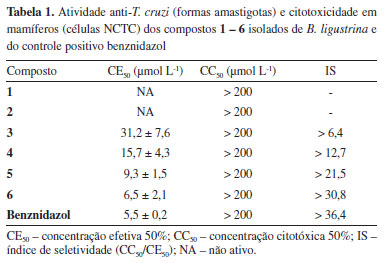
Os espectros de RMN de 1H dos compostos 1 e 2 mostraram-se muito semelhantes devido à presença de dois dupletos em δ 6,35/6,28 (J = 16,0 Hz, H-8) e em δ 7,49/7,48 (J = 16,0 Hz, H-7) atribuídos a hidrogênios de sistemas carbonílicos a,b-insaturados em configuração trans. No caso de 2 foram observados também dois dupletos em d 7,50 (J = 8,6 Hz, H-2/H-6) e 6,78 (J = 8,6 Hz, H-3/H-5), referentes a um sistema aromático 1,4-dissubstituido enquanto que, para 1, foram observados três sinais em δ 7,27 (d, J = 2,5 Hz, H-2), 7,08 (dd, J = 8,2 e 2,5 Hz, H-6) e 6,78 (d, J = 8,2 Hz, H-5), indicativos da ocorrência de um anel aromático 1,3,4-trissubstituído. No caso de 1, foi observado um sinal adicional em δ 3,81 (3H), relativo a um grupo metoxílico ligado ao anel aromático. Os espectros de RMN de 13C mostraram sinais referentes a carbonos sp2 entre δ 113-159 (C-1 a C-8), a carbonos carbonílicos (C-9) em δ 168,0 e 167,9 para 1 e 2 além de um sinal em δ 55,7, relativo ao grupo metoxílico de 1. A comparação dos dados espectrais obtidos com aqueles descritos na literatura15,16 permitiu caracterizar os compostos 1 e 2 como ácidos ferúlico e p-cumárico, respectivamente. Apesar das similaridades com os espectros de RMN de 1H dos compostos 1 e 2, aqueles registrados para 3 e 4 mostraram um sinal intenso em d 1,2 além de dois tripletos em d 4,18 (J = 6,7 Hz, H-1') e em d 0,89 (J = 6,5 Hz), sugerindo a ocorrência de ferulato/p-cumarato de cadeia longa. Os espectros de RMN de 13C de 3 e 4 mostraram sinais referentes aos carbonos carbonílicos (C-9) em cerca de d 167, sinais atribuídos aos carbonos carbinólicos (C-1') em aproximadamente d 65, enquanto aqueles referentes à cadeia metilênica longa e ao grupo metílico terminal foram observados em d 29 e d 14, respectivamente. Do mesmo modo que definido para 4, os espectros de RMN de 1H dos compostos 5 e 6 indicaram a ocorrência de ésteres alquílicos do ácido p-cumárico devido aos sinais em d 7,43 (d, J = 8,5 Hz, H-2/H-6), 6,85 (d, J = 8,5 Hz, H-3/H-5), 6,30 (J = 16,0 Hz, H-8), 7,63 (J = 16,0 Hz, H-7) e 4,19 (t, J = 6,6 Hz, H-1'). Além desses, foram observados sinais referentes a hidrogênios de cadeia carbônica saturada em d 1,2 (sl) e 0,89 (t, J = 6,5 Hz). Finalmente, as extensões das cadeias dos compostos 3 - 6 foram definidas por espectrometria de massas com ionização por electrospray, cujos espectros apresentaram picos referentes aos íons [M - H]- em m/z 557,4563 (3), 527,4446 (4), 555,4774 (5) e 583,5094 (6) consistentes com as fórmulas moleculares C36H62O4 (erro -1,26), C35H60O3 (erro -3,41), C37H64O3 (erro -0,54) e C39H68O3 (erro 0,69), respectivamente. Assim, após comparação com dados descritos na literatura,17,18 as estruturas foram definidas como ferulato de n-hexacosila (3), p-cumarato de n-hexacosila (4), p-cumarato de n-octacosila (5) e p-cumarato de n-triacontila (6). Os compostos 1 e 4 foram isolados, respectivamente, de Baccharis uncinella19 e de B. sphenophylla9 ao passo que a ocorrência dos compostos 2, 3, 5 e 6 está sendo descrita pela primeira vez no gênero Baccharis. No entanto, tais compostos foram descritos anteriormente em outras espécies vegetais tais como Gnetum cleistostachyum (2),20 Bauhinia manca (3 - 5),17 Tapirira guianensis (4 e 5)21 e Dendrobium fuscescens (6).18 Finalmente, neste estudo, foi avaliada a efetividade contra formas clínicas não replicativas (amastigota intracelular) de T. cruzi dos compostos 1-6 isolados de B. ligustrina. Para tanto, foram determinados os valores de CE50 e de CC50 (Tabela 1), esses últimos frente a fibroblastos murinos (células NCTC), necessários para o cálculo dos índices de seletividade desses compostos. Como observado, os ácidos carboxílicos livres 1 e 2 se mostraram inativos (CE50 > 200 µmol L-1), enquanto os ésteres 3 e 4 apresentaram potencial com CE50 de 32,2 e 15,7 µmol L-1, respectivamente, todos sem demonstrar citotoxicidade em células de mamífero até a concentração máxima testada (CC50 > 200 µmol L-1). No caso de 4, o potencial frente a formas intracelulares de T. cruzi foi similar ao obtido anteriormente (CE50 = 16.9 µmol L-1).9 No caso dos ésteres 5 e 6, ambos com reduzida toxicidade (CC50 > 200 µmol L-1), os valores de EC50 determinados foram 9,3 e 6,5 mmol L-1, sendo, no caso de 6, similar ao do controle positivo benznidazol (EC50 = 5,5 µmol L-1). Tais resultados sugerem que a presença do grupo alquílico é crucial para a atividade frente aos amastigotas sendo potencial intensificado com o aumento da cadeia lateral. Tal observação estaria possivelmente associada ao aumento da lipofilicidade dos ésteres em relação ao ácido p-cumárico uma vez que os valores de log Po/w foram determinados como 1,29 (2), 10,59 (4), 11,28 (5) e 12,07 (6). Tal característica pode fazer com que tais compostos adquiram um caráter anfifílico permitindo, portanto, que atravessem a membrana celular e atinjam os parasitas no interior das células.22,23 Tais resultados permitem sugerir, no futuro, a realização de estudos de relação estrutura/atividade com ésteres do ácido p-cumárico contendo diferentes grupos alquílicos seguido da avaliação da atividade frente as formas amastigotas de T. cruzi. Apesar de estudos similares terem sido realizados contra formas extracelulares do parasita,24 nosso trabalho teve um enfoque na forma clínica e intracelular do parasita, os amastigotas, uma vez que são considerados alvos preferenciais em estudos de Drug Discovery.25
PARTE EXPERIMENTAL Procedimentos gerais Experimentos de extração assistida por micro-ondas (MAE) foram realizados em um forno de micro-ondas MAS-I (2450 MHz, Sineo Microwave Chemistry Technology Company, Xangai, China) com uma potência máxima fornecida de 1000 W sendo a temperatura interna monitorada por uma sonda infravermelha. Nas separações cromatográficas em coluna aberta foram utilizados gel de sílica (Merck, 230-400 mesh) e gel de Sephadex LH-20 (Amersham-Bioscience), enquanto nas análises com camada fina foi utilizado gel de sílica 60 PF254 (Merck) (0,25 mm) seguido de revelação com luz UV (254 e 366 nm). As separações por CLAE foram efetuadas em cromatógrafo Dionex Ultimate 3000, em coluna Luna Phenomenex C18 (5 µm, 250 mm × 10 mm, 120Å). Os espectros de massas de alta resolução foram registrados em espectrômetro Bruker Daltonics q-ToF Maxis, operando por ionização por electrospray, em modo negativo. Os espectros de ressonância magnética nuclear foram registrados em espectrômetro Varian Inova (1H: 500 MHz e 13C: 125 MHz). CDCl3 e DMSO-d6 foram utilizados como solventes e como referência interna o sinal residual do solvente. Material vegetal As partes aéreas de B. ligustrina DC. (Asteraceae) foram coletadas em Campos do Jordão/SP, Brasil em junho de 2017 recebendo o código de coleta SISGEN A4123E4. Uma exsicata foi preparada e comparada com aquela de número SPSF37589, catalogada no Herbário D. Bento Pickel, do Instituto Florestal de São Paulo (PMSP). Extração e isolamento dos constituintes químicos As partes aéreas de B. ligustrina, secadas e moídas (25 g), foram submetidas à extração assistida por micro-ondas (MAE) com 50 mL de mistura contendo H2O:BMImBr 1:1 (v/v) durante 15 min a 60 °C, sendo o líquido iônico brometo de 1-butil-3-metilimidazólio (BMImBr) preparado conforme descrito na literatura.26 Após esse procedimento, a solução foi filtrada e extraída sucessivamente com hexano (3 × 25 mL) e com AcOEt (3 × 25 mL). Após secagem com Na2SO4 e evaporação dos solventes sob pressão reduzida, foram obtidos 121 mg e 412 mg das fases em hexano e em AcOEt, respectivamente. Parte da fase em n-hexano (100 mg) foi submetida a fracionamento cromatográfico em gel de sílica utilizando-se misturas de n-hexano/AcOEt (9:1, 8:2, 7:3, 1:1, 6:4 e 3:7) fornecendo seis frações (H1 - H6). A fração H2 (12 mg) foi purificada via CLAE (C18, MeOH:H2O 3:7 com 0,1% de HCOOH), fornecendo 3 (5,1 mg), 4 (2,9 mg), 5 (0,7 mg) e 6 (0,5 mg). Parte da fase em AcOEt (150,0 mg) foi submetida à permeação em gel de Sephadex LH-20 eluída com MeOH fornecendo cinco frações (A1 - A5). A fração A2 (64,1 mg) mostrou-se composta por uma mistura de 1 + 2 enquanto a fração A3 (2,2 mg) mostrou-se constituída majoritariamente por 2. Parte de fração A2 (30,0 mg) foi purificada via CLAE (C18, MeOH:H2O 1:1 com 0,1% de HCOOH), fornecendo 1 (14,8 mg) e 2 (10,2 mg). Parasitas Formas tripomastigotas de Trypanosoma cruzi foram cultivadas em células LLC-MK2 (Rhesus Monkey Kidney Cells - ATCC CCL 7) em meio RPMI-1640, suplementado com 2% de soro fetal bovino (SFB) e mantidas a temperatura de 37 °C em estufa com 5% CO2.27,28 As formas amastigotas foram obtidas por meio da infecção de macrófagos aplicados a 1×105/poço com formas tripomastigotas na proporção de 5:1 parasitas/macrófagos e incubadas por quatro horas. Os parasitas foram contados em hemocitômetro e depois aplicados a 1×106/poço em placas de 96 poços. Células de mamíferos Fibroblastos de camundongo, NCTC clone 929 (American Type Culture Collection - ATCC CCL-1), foram fornecidas pela Seção de Culturas Celulares do Instituto Adolfo Lutz, São Paulo, e armazenadas em nitrogênio líquido a temperatura -70 °C. Posteriormente foram mantidas em meio M-199 suplementado com soro fetal bovino (SFB) 10%, sob a temperatura de 37 °C em estufa com 5% de CO2.29 Determinação do potencial frente as formas amastigotas de T. cruzi Para determinar a atividade contra amastigotas intracelulares, macrófagos peritoneais de camundongos BALB/c foram infectados com formas tripomastigotas de T. cruzi. Os macrófagos obtidos da cavidade peritoneal de camundongos BALB/c foram plaqueados em uma lâmina de câmara de 16 poços (NUNC, 1 × 105/poço) e incubados por 24 h a 37 °C em uma incubadora umidificada com 5% de CO2. Tripomastigotas foram lavados em meio RPMI-1640, contados e utilizados para infectar os macrófagos (parasita:macrófago, razão = 10:1). Após 2 h de incubação a 37 °C em uma incubadora umidificada com 5% de CO2, os parasitas não internalizados foram removidos por lavagem com meio. Os compostos a serem testados (1-6) foram incubados com macrófagos infectados (48 h a 37 °C, 5% CO2) em concentração seriada usando benznidazol como droga padrão. Ao final do ensaio, as lâminas foram fixadas com MeOH e coradas com Giemsa antes da contagem em microscópio de luz (EVOS M5000,THERMO). Os valores de EC50 foram calculados conforme descrito previamente.27,28 Determinação da toxicidade frente a células de mamíferos A citotoxicidade dos compostos 1-6 foi avaliada utilizando-se células NCTC (clone 929), na concentração de 6×104 células/poço. As células foram incubadas com os compostos isolados, em diluição seriada, em meio M-199 e 10% de soro fetal bovino em placas de 96 poços. Em seguida, as células foram mantidas em estufa a 37 °C com 5% de CO2 por 48 h. Após esse período, foram adicionados 20 µL de resazurina a 10%, seguido de incubação por 20 h, com a leitura realizada da mesma forma descrita acima.29 Análise estatística Os dados obtidos representam a média e o desvio padrão de amostras duplicadas de dois ensaios independentes. Os valores de CE50 foram calculados usando curvas sigmoide dose-resposta no software GraphPad Prism 5.0.
MATERIAL SUPLEMENTAR Os espectros de RMN 1H, RMN 13C e de massas referentes aos compostos estudados no presente trabalho estão disponíveis em http://quimicanova.sbq.org.br em formato pdf, com acesso livre.
AGRADECIMENTOS Os autores agradecem à FAPESP (2021/02789-7), CNPq e CAPES pelo apoio financeiro para o desenvolvimento deste trabalho. F. F. C., A. G. T. e J. H. G. L. agradecem ao CNPq pelas bolsas de produtividade em pesquisa.
REFERÊNCIAS 1. Moreira, F. P. M.; Coutinho, V.; Montanher, A. B. P.; Caro, M. S. B.; Brighente, I. M. C.; Pizzolatti, M. G.; Monache, F. D.; Quim. Nova 2003, 26, 309. [Crossref] 2. Hischmann, G.; De Arias, A. R.; J. Ethnopharmacol. 1990, 29, 159. [Crossref] 3. Safford, H. D.; J. Biogeography 1999, 26, 693. [Crossref] 4. Emerenciano, V. P.; Ferreira, Z. S.; Kaplan, M. A. C.; Gottlieb, O. R.; Phytochemistry 1987, 26, 3103. [Crossref] 5. Jakupovic, J.; Schuster, A.; Ganzer, U.; Bohlmann, F.; Boldt, P. E.; Phytochemistry 1990, 29, 2217. [Crossref] 6. Boldt, P. E.; Baccharis (Asteraceae) a review of its taxonomy, phytochemistry, ecology, economic status. College Station, Texas, The Texas A & M University System, 1989. 7. Verdi, L. G.; Brighente, I. M. C.; Pizzolati, M. G.; Quim. Nova 2005, 28, 85. [Crossref] 8. Moreira, F. P. M.; Branco, A.; Pizzolatti, M. G.; Morais, A. A.; Monache, F. D.; Biochem. Syst. Ecol. 2002, 31, 319. [Crossref] 9. Silva, M. L.; Costa-Silva, T. A.; Tempone, A. G.; Lago, J. H. G.; Chem. Biodivers. 2021, 18, e2100466. [Crossref] 10. Costa-Silva, T. A.; Silva, M. L.; Antar, G. M.; Tempone, A. G.; Lago, J. H. G.; Phytomedicine 2021, 93, 153748. [Crossref] 11. Sessa, D. P.; Mengarda, A.; Simplicio, P.; Antar, G. M.; Lago, J. H. G.; Moraes, J.; J. Nat. Prod. 2020, 83, 3744. [Crossref] 12. Morillo, C. A.; Marin-Neto, J. A., Avezum, A.; N. Engl. J. Med. 2016, 374, 189. [Crossref] 13. Newman, D. J.; Cragg, G. M.; J. Nat. Prod. 2020, 83, 770. [Crossref] 14. Pirintsos, S.; Panagiotopoulos, A.; Bariotakis, M.; Daskalakis, V.; Lionis, C.; Sourvinos, G.; Karakasiliotis, I.; Kampa, M.; Castanas, E. Molecules 2022, 27, 4060. [Crossref] 15. Swisłocka, R.; Kowczyk-Sadowy, M.; Kalinowska, M.; Lewandowski, W.; Spectroscopy 2012, 27, 35. [Crossref] 16. Prachayasittikul, S.; Suphapong, S.; Worachartcheewan, A.; Lawung, R.; Ruchirawat, S.; Prachayasittikul, V. Molecules 2009, 14, 850. [Crossref] 17. Achenbach, H.; Stöcker, M.; Constenla, M. A.; Z. Naturforsch. 1986, 41, 164. [Crossref] 18. Talapatra, B.; Das, A. K.; Talapatra, S. K.; Phytochemistry 1989, 28, 290. [Crossref] 19. Bocco, B. M.; Fernandes, G. W.; Lorena, F. B.; Cysneiros, R. M.; Christoffolete, M. A.; Grecco, S. S.; Lancellotti, C. L.; Romoff, P.; Lago, J. H. G.; Bianco, A. C.; Ribeiro, M. O. Braz. J. Med. Biol. Res. 2016, 49, e5003. [Crossref] 20. Yao, C. S.; Lin, M.; Liu, X.; Wang, Y. H. J. Asian Nat. Prod. Res. 2005, 7, 131. [Crossref] 21. Correia, S. J.; David, J. P.; David, J. M.; Quim. Nova 2003, 26, 36. [Crossref] 22. Daina, A.; Michielin, O.; Zoete, V.; Sci. Rep. 2017, 7, 42717. [Crossref] 23. Hannesschlaeger, C.; Horner, A.; Pohl, P.; Chem. Rev. 2019, 119, 5922. [Crossref] 24. Lopes, S. P.; Castillo, Y. P.; Monteiro, M. L.; Menezes, R. R. P. P. B.; Almeida, R. N.; Martins, A. M. C., Sousa, D. P.; Int. J. Mol. Sci. 2019, 20, 5916. [Crossref] 25. Katsuno, K.; Burrows, J. N.; Duncan, K.; Hooft van Huijsduijnen, R.; Kaneko, T.; Kita, K.; Mowbray, C. E.; Schmatz, D.; Warner, P.; Slingsby, B. T.; Nat. Rev. Drug Discov. 2015, 14, 751. [Crossref] 26. Huddleston, J. G.; Visser, A. E.; Reichert, W. M.; Willauer, H. D.; Broker, G. A.; Rogers, R. D.; Green Chem. 2001, 3, 156. [Crossref] 27. Kesper Jr., N.; Almeida K. A.; Stolf, A. M. S.; Umezawa, E. S.; J. Parasitol. 2000, 86, 862. [Crossref] 28. Martins, L. F.; Mesquita, J. T.; Pinto, E. G.; Costa-Silva, T. A.; Borborema, S. E.; Galisteo Junior, A. J.; Neves, B. J.; Andrade, C. H.; Shuhaib, Z. A.; Bennett, E. L.; Black, G. P.; Harper, P. M.; Evans, D. M.; Fituri, H. S.; Leyland, J. P.; Martin, C.; Roberts, T. D.; Thornhill, A. J.; Vale, S. A.; Howard-Jones, A.; Thomas, D. A.; Williams, H. L.; Overman, L. E.; Berlinck, R. G.; Murphy, P. J.; Tempone, A. G.; J. Nat. Prod. 2016, 79, 2202. [Crossref] 29. Tada, H.; Shiho, O.; Kuroshima, K.; Koyama, M.; Tsukamoto, K.; J. Immunol. Methods 1986, 93, 157. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access