
 |
 |
 |
 |
 |
Artigo
| Electron withdrawing group effect on biological activities of pyrimidine hybrids as potential anti-matrix metalloproteinase-7 |
|
Abel Kolawole OyebamijiI,*; Sunday Adewale AkinteluII; Kehinde Abraham OdeladeIII; Babatunde AdetuyiIII; Emmanuel Temitope AkintayoI,IV; Cecillia Olufunke AkintayoI,V; Banjo SemireVI; Jonathan Oyebamiji BabalolaVII
I. Department of Chemistry and Industrial Chemistry, Bowen University, 232101 Iwo, Osun State, Nigeria Recebido em 18/12/2022 *e-mail: abeloyebamiji@gmail.com We investigated the effect of both electron donating group and e//lectron withdrawing group on biological activity of pyrimidine-based compounds as metalloproteinase-7 inhibitors and predicting a library of drug-like compounds with potent cytotoxicity using in silico approach. The selected compounds were optimized and subjected to both docking as well as absorption, distribution, metabolism, excretion and toxicity (ADMET) analyzes. We observed that the addition of electron withdrawing group (-CF3) to the predicted pyrimidine-based compound induced a radical improvement in the hydrogen bond strength with Leu181 and Ala182 in matrix metalloproteinase-7. Also, communal orientation of 2-mercapto-4-(3-(trifluoromethyl)phenyl)-6-((4-(trifluoromethyl)phenyl)amino)pyrimidine-5-carbonitrile (PC10) and matrix metalloproteinase-7 showed improved binding tendency with calculated binding affinity value of -10.2 kcal mol-1 than other studied compounds. Our findings may open door for the design and development of library of efficient pyrimidine-based drug-like compounds as potential anti-cancer agents. INTRODUCTION The negative effect of colorectal cancer among humans has been reported to take third position amid cancer rating globally.1 It mostly occurs in the gastrointestinal tract and the rate at which it kills both men and women is alarming, which rendered it a deadly ailment that need urgent prevention.2-4 It was considered to be a recurrent ailment in human most especially women at the age of ≥ 65; although, this malignant neoplasa found among younger people have been attributed to several factors such as smoking, obesity, etc.5 Colorectal cancer can be categorized into three (3) such as molecular pathway involved, position and histological subtype. According to report by several scientists, series of commercially available drugs such as bevacizumab, capecitabine, 5-fluorouracil, etc. for treating patients with colorectal cancer has been recommended but due to high level of resistivity by this disease to the commercially available drugs, there is increase in the desire to find lasting solution to this menace via efficient therapeutic drug and this has drawn the attention of numerous researchers globally.6-8 Presently, several advanced measures have been implemented to curb this menace, yet, the number of cancer related death remain alarming. Matrix metalloproteinase-7 can also be referred to as Matrilysin. It was considered an enzyme which belongs to calcium and zinc-dependent endopeptidase and it can damage extracellular matrix proteins as well as ability to modify tissues linked with many human biological processes.9-11 Over forty years ago, the role of matrix metalloproteinase-7 in cancer metastasis was discovered and reports have shown that matrix metalloproteinase-7 possess a probable responsibility in tumor metastasis and inflammatory processes. It also cleaves cell surface molecules which therefore enhanced its tendency to influence a vast range of physiological and pathological processes of cancer and inflammatory diseases. As reported by Wielockx et al., matrix metalloproteinase-7 was linked with, and seems to support, the evolution from normal epithelium to adenomatous lesion.12 Also, matrix metalloproteinase-7 in conjunction with epithelial-mesenchymal transition stimulate establishment of fresh blood vessels sponsored by cancer cell.13 Thus, it enhances cancer development as well as its metastasis via capillary endothelium.14 Pyridine based compounds have been reported by several scientists to be potent drug-like compounds.15 These nitrogen containing compounds have the ability to form various non-covalent interactions (hydrogen bonds, pi-pi bond, van der Waals forces, pi-alky bond and dipole-dipole bonds) together with protein compounds.16 Series of the derivative of these compounds have anti-inflammatory, antitumor, and antioxidant activities which has drawn the attention of several researchers globally.17-19 Triazoles could also be found in molecular structure of several drugs such as anastrozole and letrozole which helps in treating postmenopausal breast cancer.20 Triazoles ring possess advantage of providing good water solubility and it has greater tendency to connect with several enzymes in cancer and this could be due to its capacity to form hydrogen bonds with amino acid residues present in the receptors.21-25 Therefore, this work was aimed at investigating the inhibiting activity of EDG or EWG attached to pyrimidine-based compounds as metalloproteinase-7 inhibitors and predicting a library of drug-like compounds with potent cytotoxicity.
METHODS Computational details As shown in Figure 1, the steps involved in this work were clearly itemized pictorially.
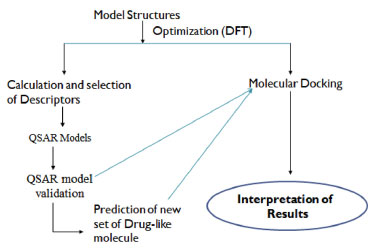 Figure 1. Pictorial overview of the work
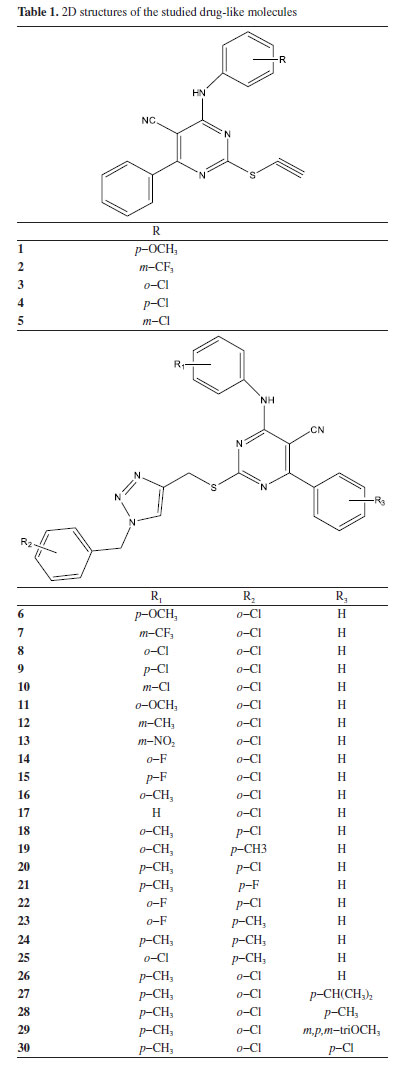
Pharmacokinetics of the studied compounds Series of pharmacokineticfactors for absorption, distribution, metabolism, excretion and toxicity (ADMET) were considered in this work using ADMETlab software.26 In this work, different categories of ADMET properties were considered such as physicochemical property; medicinal chemistry; absorption; distribution; metabolism; excretion; toxicity; environmental toxicity; tox21 pathway and toxicophore rule. Ligand preparation via density functional theory method Two dimensional (2D) structures of thirty (30) compounds were obtained from Wang et al.,27 using PaDEL 2.21 software28 and the three-dimensional structure of the studied compounds were achieved using Spartan 14.29 The 3D structures were optimized and during this process, every conformation of the studied drug-like compound (Table 1) and (Supplementary Material - Section 4) was considered. Also, the energy minimization was done for all the studied compounds using the appropriate force field.
Development and validation of QSAR model The quantitative structure activity relationship (QSAR) model developed was accomplished using multiple linear regression approach. In this section, one thousand four hundred and thirty-four (1434) two dimensional (2D) descriptors were obtained using PaDEL 2.21 software which were subjected to screening to identify the descriptors that best describe the anti-matrix metalloproteinase-7 activities of 1,2,3-triazole-pyrimidine based compounds. The columns of the descriptors with constant values were removed and the remaining descriptors were screened and tested. The observed inhibition concentration (IC50) of the studied 1,2,3-triazole-pyrimidine based compounds served as dependent variable while the calculated descriptors served as independent variables. The acceptable developed model was expected to possess squared correlation coefficient (R2) which must be in-line with 0.5 ≤ ϰ ≤ 1; however, the reliability of the developed model is not only a function of the squared correlation coefficient which therefore required validation of developed QSAR model (Equation 1). The factors considered for QSAR model validation were adjusted R2 ≤ 0.6, cross-validation (CVR2) ≤ 0.5, mean square error, P-Value. The developed model was used to predict the cytotoxicity of library of new set of compounds before subjecting them to further study. The IUPAC name of the studied compounds were 4-(3-ethoxyphenyl)-6-((4-ethoxyphenyl)amino)-2-mercaptopyrimidine-5-carbonitrile (PC1), N-(4-((6-(3-acetamidophenyl)-5-cyano-2-mercaptopyrimidin-4-yl)amino)phenyl)acetamide (PC2), N-(3-(5-cyano-2-mercapto-6-((4-propionamidophenyl)amino)pyrimidin-4-yl)phenyl)propionamide (PC3), N-(4-((6-(3-butyramidophenyl)-5-cyano-2-mercaptopyrimidin-4-yl)amino)phenyl)butyramide (PC4), 2-mercapto-4-(3-(2-methylprop-1-en-1-yl)phenyl)-6-((4-(2-methylprop-1-en-1-yl)phenyl)amino)pyrimidine-5-carbonitrile (PC5), 4-(3-acetylphenyl)-6-((4-acetylphenyl)amino)-2-mercaptopyrimidine-5-carbonitrile (PC6), 2-mercapto-4-(3-propionylphenyl)-6-((4-propionylphenyl)amino)pyrimidine-5-carbonitrile (PC7), 4-(3-butyrylphenyl)-6-((4-butyrylphenyl)amino)-2-mercaptopyrimidine-5-carbonitrile (PC8), 3-(5-cyano-2-mercapto-6-((4-sulfophenyl)amino)pyrimidin-4-yl)benzenesulfonic acid (PC9) and 2-mercapto-4-(3-(trifluoromethyl)phenyl)-6-((4-(trifluoromethyl)phenyl)amino)pyrimidine-5-carbonitrile (PC10).  Mean dependent variable = 31.37167, S.D. dependent variable = 4.462024, Sum squared resid = 22.39133, S.E. of regression = 1.312406, R-squared = 0.951102, Adjusted R-squared = 0.913489, F(10, 13) = 25.28613, P-value(F) = 6.74 × 10-7. Target identification, selection and preparation In this work, matrix metalloproteinase-7 (PDB ID: 2y6d)30 (Table 2, Figure 2) was selected and downloaded from protein data bank after a thorough literature search. Appropriate tools in PyMOL software31 were used to treat and prepare the downloaded receptor. Also, the crystallographic water as well as the small molecules embedded in the receptor were deleted and saved as clean receptor. The possible missing side chains were corrected using Swiss PDB Viewer 4.1.0 version32 and subjected to AutoDock Tool33 for further processing. The calculated value for the centre and size in X, Y and Z directions that show the binding site were -23.062, 8.911, and 10.599 (for centre) and 48, 40 and 40 (for size) respectively. The binding affinity for the studied complex was accomplished using AutoDock Vina software.34

 Figure 2. Three-dimensional (3D) structure of matrix metalloproteinase-7
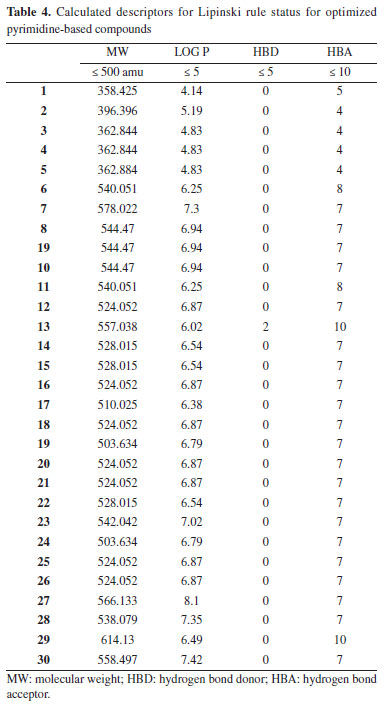
RESULTS AND DISCUSSION Pharmacokinetics of the drug-like molecules The 2D structures of thirty compounds were modelled and subjected to ADMETlab software in order to investigate their drug-likeness. Drug-likeness determination of any compounds at the initial stage of drug discovery remains a crucial step in drug design and discovery.35 As shown in Table 3, it was observed that compounds 1-5 completely obeyed Lipinski rule of five (LO5) (MW ≤ 500 amu; Log P ≤ 5; HBD ≤ 5; HBA ≤ 10) while the values for two descriptors (molecular weight and lipophilicity) for compounds 6-30 were higher than the standard value described by Lipinski rule of five. This showed that compounds 1-5 have tendency to exhibit good absorption/permeability compare to compounds 6-30; thus, it shows that compounds 6-30 can be administered intravenously.36 The value for compounds 1-5 which were within the acceptable range showed that these ligands can effortlessly overcome blood brain barrier; compounds 6-30 may experience difficulty in overcoming cell membrane.
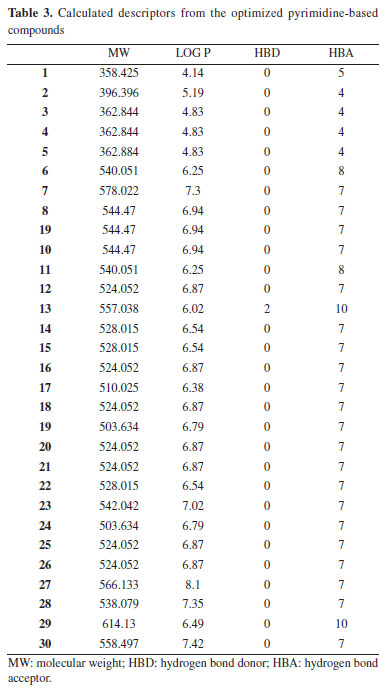
More so, the calculated Log P values that is within the acceptable range (ϰ ≤ 5) signifies the ability of the studied compounds to dissolve in aqueous media. Thus, in this work, compounds 1, 3-5 were with within the acceptable range while compound 2 was higher than 5 which is the limit for the standard with 0.19. Also, the calculated Log P value for compounds 6-10 were higher than the acceptable range for Log P. According to Peter et al., positive Log P value indicated that the ligand has the strength to permeate through biological membrane while negative Log P value for any ligand reveal that the compound has strength to dissolve in water. Thus, compounds 1, 3-5 have good strength to dissolve in lipid and permeate through biological membrane.37 The calculated hydrogen bond donor and hydrogen bond acceptor for the entire studied compounds were within acceptable range (Table 4). The calculated value for calculated human intestinal absorption (HIA) values ranges from 0.004-0.657 and this agreed well with calculated HIA value for the referenced drug (verapamil). Also, all the predicted compounds posses the ability to penetrate blood brain barrier (BBB) and the output values for all predicted compounds as well as the referenced drug (verapamil) revealed that they have the probability of being BBB+. More so, the potential interaction between the predicted compounds and CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 agreed well with the interaction between the studied referenced drug and CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 (Supplementary Material - Section 1). Also, other calculated ADMET properties were shown in Supplementary Material.
Quantitative structure activity relationship study In this work, the cytotoxicity of the studied compounds was explored and investigated using the developed QSAR model.38 The descriptors obtained from two-dimensional (2D) structure of the studied compounds using PaDEL software were used to developed the QSAR model as shown in Equation 1 and (Supplementary Material - Section 2). The studied drug-like molecules were divided into two sets (training (70%) and test (30%)) via Kennard stone algorithm approach using Dataset Division GUI 1.2 software.39 Compounds 2, 3, 5-14, 16-22, 24, 25, 27-29 were used as training set while compounds 1, 4, 15, 23, 26 and 30 served as test set to confirm the reliability of the developed model. The 2D descriptors from the training set were screened and the selected descriptors were used to develop the studied QSAR model as shown in Equation 1. As shown in Table 5, the predicted IC50 using the developed QSAR model via the descriptors from training set were close to the observed IC50. This showed that the predicting ability of the developed QSAR model was effective and this was confirmed via the calculated squared correlation coefficient (R2) (R2 = 0.951102) which fell within the range of accepted value for squared correlation coefficient (0.5 ≤ ϰ ≤ 1) (Figures 3 and 4). According to Oyebamiji et al.,40 calculated squared correlation coefficient (R2) alone cannot be used to ascertain the potency of any developed QSAR model, thus, QSAR validation is required. The factors considered for QSAR validation were adjusted R2, log-likelihood, mean dependent variable etc. which were observed to be within acceptable range;41 this supported the reliability of the developed model and predicting ability.
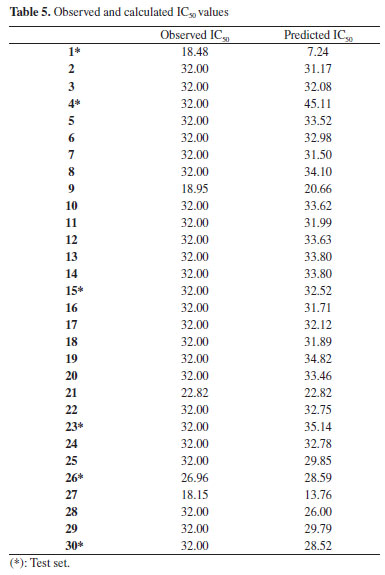
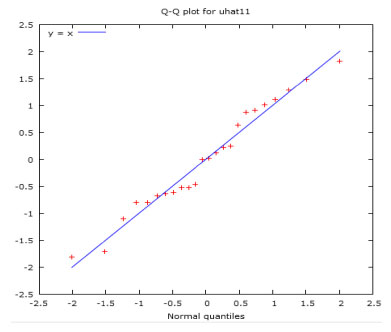 Figure 3. Correlation between the observed IC50 and predicted IC50
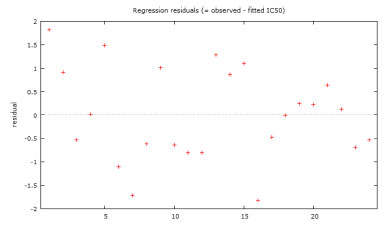 Figure 4. Plot of residual (observed IC50-predicted IC50) values versus observed IC50
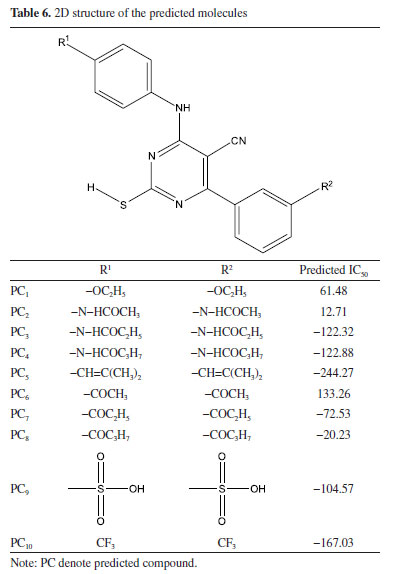
More so, ten (10) molecular derivatives were attached to the parent compound used in this work to form new set of compounds (PC1-PC10). Electron donating group compounds (EDG) were attached to the parent compound to form compounds PC1-PC5, while electron withdrawing group compounds were attached to the parent compound to form compounds PC6-PC10 (Table 6). These compounds were attached to the parent compound in order to alter the reactivity of the studied parent compound. As shown in Table 6, the reliability of the developed model was also confirmed via the predicted IC50 value for compound PC1-PC10. According to Oyewole et al., compound with lower IC50 value indicated compound with better cytotoxicity; therefore, all the predicted compounds proved to have better cytotoxicity than the studied thirty (30) compounds except PC1 and PC6.42 This showed the effect of the electron donating and electron withdrawing groups attached to the parent compound; however, -OC2H5 (PC1) and -COCH3 (PC6) prove not to donate/withdraw enough electron into/from the molecule thereby resulted to a compound with less efficient cytotoxicity.
Molecular docking study The molecular docking study was executed on pyrimidine-based compounds as inhibitor of matrix metalloproteinase-7. The studied compounds (pyrimidine-based compounds) were observed to be efficient matrix metalloproteinase-7 inhibitors with binding affinity value of -8.2 kcal mol-1 to -10.2 kcal mol-1 and these proved to be more potent than the inhibitory activity of verapamil against matrix metalloproteinase-7 (-7.3 kcal mol-1). As reported by Erazua et al., lower binding affinity value for any ligand indicated better efficiency to inhibit the target; therefore, compound PC10 with -10.2 kcal mol-1 was expected to inhibit matrix metalloproteinase-7 than other studied compounds.43 This showed the effectiveness of the attached electron withdrawing groups on the parent compound in the interaction against matrix metalloproteinase-7. The binding mode of 2-mercapto-4-(3-(trifluoromethyl)phenyl)-6-((4-(trifluoromethyl)phenyl)amino)pyrimidine-5-carbonitrile (PC10) in the binding site of the studied receptor revealed the formation of three (3) hydrogen bond contact through the interaction of the Halogen (Fluorine) with amino acid residues Leu181 and Ala182 as shown in Figures 5 and 6.
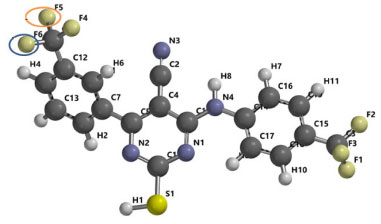 Figure 5. 3D structure of 2-mercapto-4-(3-(trifluoromethyl)phenyl)-6-((4-(trifluoromethyl)phenyl)amino)pyrimidine-5-carbonitrile (PC10)
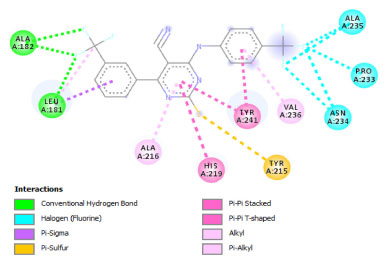 Figure 6. 2D structure of PC10 in active site of matrix metalloproteinase-7
As reported by Barhaghi et al.,44 the acceptable hydrogen bond was formed between two molecules when the distance between the two points was less than 3Å as well as when the angle D-H-A remain an obtuse angle. Therefore, the hydrogen bond formed by PC10-matrix metalloproteinase-7 complex has been observed to enhance the ability of PC10 to inhibit matrix metalloproteinase-7 than other studied compounds as well as verapamil (referenced drug) (Tables 7 and 8). Other observed types of the interaction between PC10-matrix metalloproteinase-7 complexes were Halogen (Fluorine), pi-sigma, pi-sulfur, pi-pi stacked, pi-pi T-shaped, alky, pi-alkyl. The 2D structures of the 1,2,3-triazole-pyrimidine based compounds-matrix metalloproteinase-7 complexes were shown in Supplementary Material - Section 3.
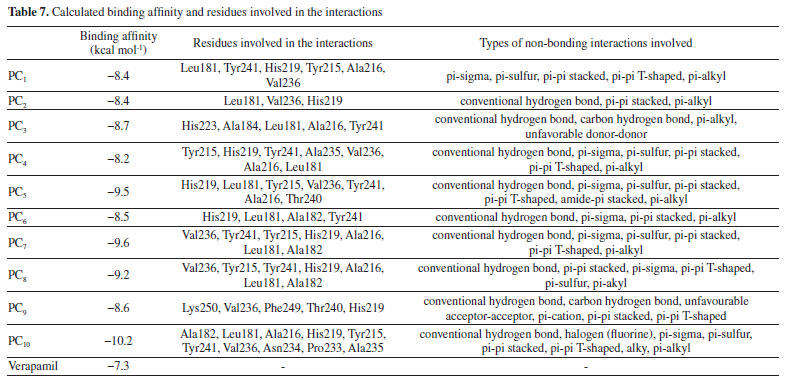

CONCLUSION The studied pyrimidine-based compounds against matrix metalloproteinase-7 were executed using in-silico approach. The two types of obtained descriptors (3D and 2D) were investigated and we confirmed anti-matrix metalloproteinase-7 activities of pyrimidine-based compounds. Also, addition of electron withdrawing group (-CF3) to pyrimidine-based compound enhanced a thorough advancement in the hydrogen bond strength with Leu181 and Ala182 located in binding site of matrix metalloproteinase-7. Also, the developed QSAR model was proved to be valid and reliable as the predicted % inhibition concentration (IC50) were closer to the observed IC50. Furthermore, the docking of 2-mercapto-4-(3-(trifluoromethyl)phenyl)-6-((4-(trifluoromethyl)phenyl)amino)pyrimidine-5-carbonitrile (PC10) and matrix metalloproteinase-7 resulted to existence of good binding tendency with calculated binding affinity value of -10.2 kcal mol-1 and this proved that it has higher tendency to inhibit matrix metalloproteinase-7 than other studied compounds. Also, the non-bonding properties observed for compound PC10 were Leu 181(HN):F; Ala182(HN):F and Ala 182(HN):F. More so, this work may give insight to designing and developing library of efficient 1,2,3-triazole-pyrimidine based drug-like compounds as potential anti-cancer agents.
AUTHOR CONTRIBUTION Conceptualization: O. A. K.; A. S. A.; B. J. O. Methodology: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Software: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Validation: O. A. K.; A. S. A; O. K. A; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Formal analysis: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Investigation: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Resources: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O. Data curation: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Writing - original draft preparation: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O. Writing - review & editing: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B.; B. J. O.; Visualization: O. A. K.; A. E. T.; A. C. O.; S. B.; B. J. O. Supervision: B. J. O. Project administration: O. A. K. Funding acquisition: O. A. K.; A. S. A.; O. K. A.; A. B.; A. E. T.; A. C. O.; S. B; B. J. O.
SUPPLEMENTARY MATERIAL The supplementary material is available at http://quimicanova.sbq.org.br, in pdf format, with free access.
REFERENCES 1. Granados-Romero, J. J.; Valderrama-Treviño, A. I.; Contreras-Flores, E. H.; Barrera-Mera, B.; Enríquez, M. H.; Uriarte-Ruíz, K.; Ceballos-Villalva, J. C.; Estrada-Mata, A. G.; Rodríguez, C. A.; Arauz-Peña, G.; International Journal of Research in Medical Sciences 2017, 5, 4667. [Crossref] 2. Dobre, M.; Dinu, D. E.; Panaitescu, E.; Bîrlă, R. D.; Iosif, C. L.; Boeriu, M.; Constantinoiu, S.; Ivan, R. N.; Ardeleanu, C. M.; Costache, M.; Rom. J. Morphol. Embryol. 2015, 56, 671. [Link] accessed in May 2023 3. Siegel, R. L.; Miller, K.; Jemal, A.; Ca-Cancer J. Clin. 2015, 65, 5. [Crossref] 4. Hayashi, T.; Konishi, I.; J. Clin. Med. Res. 2023, 15, 68. [Crossref] 5. Abdallah, A.; Abdelwahab, K.; Awny, S.; Zuhdy, M.; Hamdy, O.; Atallah, K.; Elfeky, A.; Hegazy, M. A. F.; Metwally, I. H.; Indian Journal of Surgical Oncology 2023, 14, 93. [Crossref] 6. Marmol, I.; de Diego, C. S.; Dieste, A. P.; Cerrada, E.; Yoldi, M. J. R.; Int. J. Mol. Sci. 2017, 18, 197. [Crossref] 7. Goldstein, D. A.; Zeichner, S. B.; Bartnik, C. M.; Neustadter, E.; Flowers, C. R.; Clin. Colorectal Cancer 2016, 15, 1. [Crossref] 8. Deng, Z.; Qin, Y.; Wang, J.; Wang, G.; Lang, X.; Jiang, J.; Xie, K.; Zhang, W.; Xu, H.; Shu, Y.; Zhang, Y.; Clin. Genet. 2020, 97, 25. [Crossref] 9. Verma, R. P.; Hansch, C.; Bioorg. Med. Chem. 2007, 15, 2223. [Crossref] 10. Löffek, S.; Schilling, O.; Franzke, C. W.; Eur. Respir. J. 2011, 38, 191. [Crossref] 11. Fanjul-Fernandez, M.; Folgueras, A. R.; Cabrera, S.; Biochim. Biophys. Acta, Mol. Cell Res. 2010, 1803, 3. [Crossref] 12. Wielockx, B.; Libert, C.; Wilson, C.; Cytokine Growth Factor Rev. 2004, 15, 111. [Crossref] 13. Seyfried, T. N.; Huysentruyt, L. C.; Crit. Rev. Oncog. 2013, 18, 43. [Crossref] 14. Nagase, H.; Visse, R.; Murphy, G.; Cardiovasc. Res. 2006, 69, 562. [Crossref] 15. Lazrak, F.; Essassi, E. M.; Rodi, Y. K.; Misbahi, K.; Pierrot, M.; Phosphorus, Sulfur, and Silicon 2004, 179, 1799. [Crossref] 16. Ezzat, K.; ChemistrySelect 2021, 6, 3041. [Crossref] 17. Song, M. X.; Deng, X. Q.; J. Enzyme Inhib. Med. Chem. 2018, 33, 453. [Crossref] 18. Basu, N. K.; Rose, F. L.; J. Chem. Soc. 1963, 5660. [Crossref] 19. Wu, G.; Gao, Y.; Kang, D.; Huang, B.; Huo, Z.; Liu, H.; Poongavanam, V.; Zhan, P.; Liu, X.; MedChemCommun 2018, 9, 149. [Crossref] 20. Zengin, M.; Tan, O. U.; Arafa, R. K.; Balkan A.; Bioorg. Chem. 2022, 121, 105696. [Crossref] 21. Turky, A.; Sherbiny, F. F.; Bayoumi, A. H.; Ahmed, H. E. A.; Abulkhair, H. S.; ArchPharm 2020, 12, e2000170. [Crossref] 22. Othman E. M.; Fayed, E. A.; Husseiny, E. M.; Abulkhair, H. S.; New J. Chem. 2022, 46, 12206. [Crossref] 23. Ashour, H. F.; Abou-zeid, L. A.; El-Sayed, M. A. A.; Selim K. B.; Eur. J. Med. Chem. 2020, 189, 112062. [Crossref] 24. Othman E. M.; Fayed, E. A.; Husseiny, E. M.; Abulkhair, H. S.; Bioorg. Chem. 2022, 123, 105762. [Crossref] 25. Othman, E. M.; Fayed, E. A.; Husseiny, E. M.; Abulkhair, H. S.; Bioorg. Chem. 2022, 127, 105968. [Crossref] 26. Oyebamiji, A. K.; Tolufashe, G. F.; Oyawoye, O. M.; Oyedepo, T. A.; Semire, B.; J. Chem. 2020, 2020, 1. [Crossref] 27. Wang, B.; Zhao, B.; Chen, Z. S.; Pang, L. P.; Zhao, Y. D.; Guo, Q.; Zhang, X. H.; Liu, Y.; Liu, G. Y.; Zhang, H.; Zhang, X. Y.; Ma, L. Y.; Liu, H. M.; Eur. J. Med. Chem. 2018, 143, 1535. [Crossref] 28. Salih, R. H. H.; Hasan, A. H.; Hussen, N. H.; Farouq, E. H.; Hadda, T. B.; Jamalis, J.; Almalki, F. A.; Adeyinka, A. S.; Coetzee, L. C. C.; Oyebamiji, A. K.; J. Mol. Struct. 2023, 1282, 135191. [Crossref] 29. Morakinyo, A. E.; Omoniyi, F. E.; Nzekwe, S. C.; Oyebamiji, A. K.; Adelowo, J. M.; Lawal, S. A.; Olumade, A. A.; Olopade, E. O.; Oyedepo, T. A.; Trop. J. Nat. Prod. Res. 2022, 6, 87. [Link] accessed in May 2023 30. Edman, K.; Furber, M.; Hemsley, P.; Johansson, C.; Pairaudeau, G.; Petersen, J.; Stocks, M.; Tervo, A.; Ward, A.; Wells, E.; Wissler, L.; ChemMedChem 2011, 6, 769. [Crossref] 31. Oke, A. M.; Adelakun, A. O.; Akintelu, S. A.; Soetan, E. A.; Oyebamiji, A. K.; Ewemoje, T. A.; Pharmacological Research - Modern Chinese Medicine 2022, 4, 100142. [Crossref] 32. Ibrahim, A. O.; Semire, B.; Adepoju, A. J.; Latona, D. F.; Oyebamiji, A. K.; Owonikoko, A. D.; Oladuji, T. E.; Odunola, O. A.; Biointerface Res. Appl. Chem. 2023, 13, 233. [Crossref] 33. Latona, D. F.; Oyebamiji, A. K.; Mutiu, O. A.; Olarinoye, E. F.; Trop. J. Nat. Prod. Res. 2022, 6, 416. [Crossref] 34. Waziri, I.; Kelani, M. T.; Oyedeji-Amusa, M. O.; Oyebamiji, A. K.; Coetzee, L. C. C.; Adeyinka, A. S.; Muller, A. J.; J. Mol. Struct. 2023, 1276, 134756. [Crossref] 35. Lipinski, C. A.; J. Pharmacol. Toxicol. Methods 2000, 44, 235. [Crossref] 36. Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J.; Adv. Drug Delivery Rev. 1997, 23, 3. [Crossref] 37. Peter, E.; Rohde, B.; Selzer, P.; J. Med.Chem. 2000, 43, 3714. [Crossref] 38. Elrayess, R.; Elgawish, M. S.; Elewa, M.; Nafie, M. S.; Elhady, S. S.; Yassen, A. S. A.; Pharmaceuticals 2020, 13, 370. [Crossref] 39. Khaldan, A.; El khatabi, K.; El-Mernissi, R.; Ghaleb, A.; Hmamouchi, R.; Sbai, A.; Bouachrine, M.; Lakhlifi, T.; J. Mater. Environ. Sci. 2020, 11, 429. [Link] accessed in May 2023 40. Oyebamiji, A. K.; Semire, B.; Curr. Pharm. Biotechnol. 2020, 21, 70. [Crossref] 41. Seketoulie, K.; Swapnil, B. P.; Seung, C. J.; Lett. Drug Des. Discovery 2020, 17, 618. [Crossref] 42. Oyewole, R. O.; Oyebamiji, A. K.; Semire, B.; Heliyon 2020, 6, e03926. [Crossref] 43. Erazua, E. A.; Akintelu, S. A.; Adelowo, J. M.; Odoemene, S. N.; Josiah, O. M.; Raheem, S. F.; Latona, D. F.; Adeoye, M. D.; Esan, A. O.; Oyebamiji, A. K.; Trop. J. Nat. Prod. Res. 2021, 5, 2022. [Crossref] 44. Barhaghi, M. S.; Crawford, B.; Schwing, G.; Hardy, D. J.; Stone, J. E.; Schwiebert, L.; Potoff, J.; Tajkhorshid, E.; J. Chem. Theory Comput. 2022, 18, 4599. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access