
 |
 |
 |
 |
 |
Revisão
|
|
| Utilização de modelos in silico para avaliação da toxicidade de resíduos de agrotóxicos, fármacos e metabólitos em águas naturais Workflow of in silico models to evaluate the toxicity of pesticide residues, drugs and metabolites in natural waters |
|
Marcelo S. Alves; Lívia Streit*; Tânia Mara Pizzolato
Instituto de Química, Universidade Federal do Rio Grande do Sul, 91501-970 Porto Alegre - RS, Brasil Recebido em 18/01/2023 *e-mail: livia.streit@ufrgs.br Constantly we have to deal with an exponential increase in the amount of different chemical compounds that industries synthesize and manufacture, such as drugs, pesticides, persistent organic pollutants, preservatives, and personal hygiene products. Many of these chemical substances are biologically active and interact with biomolecules, such as proteins, through specific mechanisms that lead to different biological responses. Due to the inherent risk of many compounds to the environment and to humans, their toxicological activities must be evaluated. In order to evaluate the biological activity of molecules, three types of experiments can be used: in vivo (e.g., animal tests); in vitro (e.g., cell tissue culture); and in silico simulations. In vivo and in vitro experiments are time-consuming and expensive processes, in addition to causing ethical discussions and debates. An alternative to avoid these setbacks is the use of in silico models. The scientific community thus begins to use in silico models as a possible alternative, developing a large number of models and strategies capable of predicting the toxicological properties of several chemical compounds. This review discusses methods that employ in silico models to assess and predict the possible effects of contaminants found in natural waters. INTRODUÇÃO O estudo da toxicidade de um composto é determinado pela exposição de ecossistemas simples e complexos a substâncias químicas e os efeitos decorrentes desta exposição, que vão de efeitos leves a graves, incluindo a morte de organismos.1 Esses efeitos, reversíveis ou irreversíveis, são influenciados pelas propriedades de absorção, exposição, distribuição, metabolismo e excreção do composto em estudo ou seus metabólitos. Essas propriedades, por sua vez, determinam a interação dos compostos e seus metabólitos com macromoléculas celulares.2 Os principais objetivos na avaliação toxicológica são o diagnóstico, tratamento e prevenção de intoxicações por xenobióticos. Um dos principais pontos que cerca o estudo da toxicologia de um determinado composto reside no fato de as interações de natureza complexa poderem ser estudadas e mapeadas. Metodologias e princípios das áreas da biologia, química e informática podem ser empregados a fim de compreender o quanto os sistemas vivos são resilientes aos efeitos tóxicos de determinados compostos.3 As metodologias mais utilizadas para avaliar a toxicidade de uma molécula frente a um organismo vivo são os experimentos in vivo e in vitro. Os estudos toxicológicos in vivo são aqueles em que vários compostos são testados em organismos vivos, geralmente animais não-humanos e plantas, enquanto os estudos de toxicidade in vitro incluem a análise científica sobre o efeito desses compostos em células de cultura de mamíferos ou bactérias. Ao realizar um experimento in vivo, as interações ocorrem com um organismo intacto, ou seja, considera-se a fisiologia desse organismo. A avaliação do efeito adverso é feita através de um biomarcador, podendo ocorrer dentro do organismo ou em amostras retiradas do organismo. Quando o experimento é realizado in vitro, a interação tóxica aparece no contexto fisiológico do sistema in vitro. Devido às particularidades de cada avaliação, grandes diferenças podem ser observadas em comparação com os estudos toxicológicos in vivo.4 No âmbito da toxicologia, os modelos in vivo e in vitro são frequentemente referidos como modelos padrão para a avaliação de toxicidade de produtos químicos aos organismos vivos. Os dados gerados a partir desses modelos de estudo são geralmente extrapolados para os sistemas biológicos humanos, a fim de fornecer dados de exposição para uso e consumo humano. No entanto podem apresentar falhas e limitações, pois o comportamento, a fisiologia e o ambiente dos animais diferem para os seres humanos. Embora os modelos in vitro possam corrigir essa questão da extrapolação dos dados para os seres humanos, eles falham ao tentar representar toda a fisiologia do sistema biológico. O sistema celular é representativo de apenas um tipo de célula, sem nenhuma interação célula-célula, o que impede a mimetização de sistemas biológicos humanos.5 Uma abordagem que está em crescente utilização e tem como objetivo prever a toxicidade de compostos químicos nos ecossistemas, economizando tempo e dinheiro e contornando a questão ética, é o conjunto de metodologias in silico. O termo in silico é uma expressão que sugere "simulação computacional" com referência a problemas e/ou experimentos biológicos.6 A indisponibilidade de dados para a avaliação de risco e restrições quanto à utilização de animais estão aumentando cada vez mais a procura por abordagens computacionais. O princípio destes modelos é que compostos com estruturas similares devem se comportar de forma similar. Uma vez que o modelo é construído, pode ser utilizado como ferramenta preditiva no design de fármacos, avaliação de riscos ao meio ambiente e análise de riscos para todos os compostos cujas estruturas são similares às estruturas daquelas utilizadas no modelo. A modelagem in silico é um avanço no estudo da toxicologia no que tange a predição de avaliação de risco e está baseada em um conjunto de abordagens metodológicas. Dentre estas metodologias pode-se citar as seguintes: relação estrutura-atividade (SAR: structure-activity relationship), relação estrutura-atividade quantitativa (QSAR: quantitative structure-activity relationship), formação de categorias (category formation), métodos de extrapolação e interpolação (read-across, trend analysis), redes neurais (neural networking modelling) e árvores de decisão (decision tree). Computadores e softwares, cada vez mais rápidos e modernos, permitem a modelagem molecular de diferentes formas, como modelos básicos de valência, representações gráficas, modelos de nuvens eletrônicas, e estruturas em três dimensões. Os softwares utilizados para simulações físico-químicas baseiam-se em algoritmos que são capazes de calcular descritores moleculares, que são representações matemáticas de uma molécula provenientes de uma transformação da informação estrutural disponível da molécula em estudo. São tipicamente números que representam características da estrutura molecular (ex.: massa molecular), e podem ser, desde propriedades físico-químicas simples até impressões digitais moleculares complexas.7 Esses descritores auxiliam na determinação das interações entre moléculas e organismos vivos através de correlações entre a propriedade da estrutura química do composto e medidas de sua toxicidade para uma área específica, como por exemplo a carcinogenicidade. Os algoritmos procuram, de uma forma geral, correlações entre as propriedades da estrutura química de um composto e uma medida de sua atividade. Uma vez que a estrutura de um composto é quantificada em um conjunto de descritores moleculares, os algoritmos podem ser capazes de estabelecer uma relação matemática entre o composto e sua atividade. Essa resposta, que é produzida na modelagem computacional, é chamada de endpoint. Existem modelos in silico para um grande número de fenômenos de toxicidade, como: mutagenicidade, carcinogenicidade, biodegradabilidade, entre outros. Entre estes, a mutagenicidade e a carcinogenicidade, embora muito complexas, estão entre as mais amplamente estudadas.
METODOLOGIAS IN SILICO PARA AVALIAÇÃO DA TOXICIDADE Relações estrutura-atividade quantitativas (QSAR) Os modelos in silico, especificamente os de relações estrutura-atividade (SAR) e de relações estrutura-atividade quantitativas (QSAR), estão se tornando uma alternativa para auxiliar na investigação da toxicidade de compostos químicos. No início dos anos 1960, Hansch e Fujita, e Free e Wilson desenvolveram o conceito das relações estrutura-atividade quantitativas (QSAR), assumindo que as moléculas com estruturas semelhantes possuem atividades semelhantes.8,9 Uma contribuição importante do método de Free e Wilson é a quantificação da observação de que a mudança de um substituinte em uma posição de uma molécula geralmente é independente do efeito da mudança do substituinte em uma posição diferente.10 Cum-Brown e Fraser introduziram a ideia de correlacionar a estrutura química de um determinado composto com suas propriedades fisiológicas em sistemas biológicos.11 Estudos por QSAR fornecem também suporte nas áreas regulatórias como uma ferramenta de avaliação dos riscos apresentados pelos compostos ao meio ambiente e ao ser humano. As propriedades físico-químicas de compostos podem ser descritas quantitativamente se forem expressas por meio de parâmetros físico-químicos ou descritores estruturais adequados.12 A construção dos modelos QSAR exige a construção de um conjunto ou matriz de dados contendo a medida quantitativa da atividade biológica, os parâmetros físico-químicos, e estruturais capazes de descrever as propriedades dos compostos avaliados.13 Os métodos QSAR buscam uma relação matemática entre um grupo de descritores moleculares, usados para descrever cada molécula presente em um conjunto de substâncias, e seus valores de toxicidade, ou endpoints. A relação é derivada da origem do conjunto de compostos, que possuem valores de endpoints conhecidos, obtidos por inúmeras medidas experimentais. Este conjunto é conhecido como conjunto de treinamento (training set). Os três principais componentes dos modelos QSAR são: as propriedades a serem modeladas, a informação química e o algoritmo que relaciona as propriedades e as informações químicas. É importante destacar que um modelo QSAR não pode alcançar predições que sejam mais exatas que os dados originais. A seleção cuidadosa dos dados estruturais é uma etapa essencial para obter modelos com alta predição. Um modelo quantitativo QSAR é representado por meio de uma equação matemática que relaciona as propriedades dos compostos investigados com suas atividades biológicas e que possui significância estatística.14 Desenvolvimento de modelos Modelos in silico para predição de toxicidade são desenvolvidos a partir de uma série de etapas, como mostradas na Figura 1.
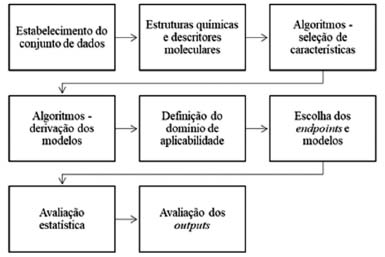 Figura 1. Workflow da avaliação in silico utilizando metodologia QSAR
Conjunto de dados Os dados utilizados na construção dos modelos devem possuir alta qualidade, pois possíveis erros, por exemplo na estrutura química, serão replicados para os modelos QSAR utilizados, e a predição será afetada diretamente. Compostos inorgânicos, isômeros, organometálicos, e mistura de substâncias devem ser removidos do conjunto de dados. Compostos de mesma estrutura, ou estruturas errôneas devem ser retirados do conjunto pois levam a erros no cálculo dos descritores moleculares. A verificação da homogeneidade dos dados é uma das primeiras etapas que deve ser seguida quando se aplica modelagem in silico, a fim de se obter dados confiáveis. Uma verificação manual dos dados deve ser realizada, pois auxilia na criação de um conjunto de dados de alta qualidade.15 Os conjuntos de dados podem ser divididos em: treinamento, validação e cego. O conjunto de treinamento é composto de dados que são utilizados na derivação do modelo. O conjunto de validação é independente do de treinamento e é utilizado para avaliar a previsibilidade do modelo. O conjunto cego, ou de avaliação externa, consiste de compostos com características conhecidas, e que não fazem parte nos conjuntos de treinamento e de validação. Estruturas químicas e descritores moleculares A utilização dos modelos QSARs passa obrigatoriamente pela utilização de descritores moleculares. De acordo com Todeschini e Consonni o descritor molecular é o resultado de um procedimento lógico e matemático que transforma a informação química codificada dentro de uma representação simbólica de uma molécula, em um número ou um resultado de algum experimento padronizado.16,17 É um valor numérico extraído, através de tratamentos matemáticos e que expressam características físico-químicas, estruturais e biofísicas de uma molécula. Para compostos químicos, informações como peso molecular, ou fragmentos característicos das moléculas, por exemplo, podem ser utilizados para descrever as substâncias. Os descritores moleculares podem ser classificados em: constitucionais (ex.: peso molecular e números de ligações duplas), que não fornecem informações sobre a geometria molecular; topológicos (ex.: ligações entre os átomos), que fornecem informações sobre a conectividade entre os átomos; geométricos (ex.: momento de inércia) que são relacionados ao arranjo espacial; os derivados de química quântica (ex.: momento de dipolo) que descrevem efeitos eletrônicos; e os de propriedades físico-químicas (ex.: LogP) que derivam de medidas experimentais. Antes de calcular os descritores químicos, a fórmula química pode ser representada através de determinados formatos, como SMILES (do inglês simplified molecular input line entry system) e InCHI (do inglês International Chemical Identifier). O formato SMILES é bastante utilizado e codifica informações sobre a arquitetura molecular. Essa representação é mais compacta do que os diferentes gráficos moleculares tradicionalmente usados em análises quantitativas de relações estrutura-atividade (QSAR). A escolha de como representar uma molécula está intimamente ligada com a finalidade do estudo.18 Algoritmos de seleção de descritores A seleção de características que descrevem de forma otimizada as moléculas em estudo permite diminuir o número de descritores moleculares, visando diminuir a possibilidade de correlação aleatória e de derivação de modelos de alta complexidade, difíceis de serem interpretados. Esta seleção permite uma interpretação mais simples dos resultados obtidos pelos modelos QSAR aplicados. Algoritmos para derivação de modelos Ao longo das décadas, muitos algoritmos para derivação dos modelos foram desenvolvidos a fim de fornecerem respostas inequívocas. Esses algoritmos podem ser classificados como lineares e não-lineares. Os lineares correlacionam, através de uma função linear, a atividade e os descritores moleculares. São de fácil interpretação e de alta precisão para pequenos conjuntos de dados de compostos semelhantes. Já os algoritmos não-lineares são funções não-lineares das relações de estrutura-atividade com os descritores de entrada. Esses são mais precisos para conjuntos de dados maiores, mas são de difícil interpretação. Dentre os métodos lineares pode-se destacar: mínimos quadrados parciais, regressão linear múltipla, regressão logística, e análise discriminante linear. Dentre os métodos não-lineares pode-se destacar: máquinas de vetores de suporte, k-vizinhos mais próximos, redes neurais artificiais, árvores de decisão, e métodos de conjunto. Outra característica importante dos modelos é a natureza dos dados de resposta, podendo ser aqueles que trabalham com respostas classificadas (ex.: mutagênico e não-mutagênico), e aqueles que trabalham com dados contínuos. Definição do domínio de aplicabilidade Para a utilização dos modelos in silico, e por consequência os modelos QSAR, é necessário definir o domínio de aplicabilidade do estudo, para que sua abordagem seja válida. O domínio de aplicabilidade de um modelo QSAR é um espaço físico-químico, estrutural, biológico, de conhecimento ou de informação, sobre o qual o conjunto de treinamento do modelo foi desenvolvido, e para o qual é aplicável fazer previsões para novos compostos em estudo, que possuam características semelhantes a do conjunto de treinamento, com confiabilidade.19 Um determinado modelo QSAR só deve ser utilizado para fazer predições dentro desse domínio de aplicabilidade. Parâmetros de confiabilidade estatística Para ser considerado preditivo, significativo e adequado para aplicações dentro do domínio de aplicabilidade, um modelo QSAR deve passar por uma avaliação estatística, que compõe uma série de parâmetros. O modelo é testado em relação a um conjunto de teste externo para garantir que não ocorra um sobreajuste ao conjunto de treinamento. Algumas ferramentas estatísticas são utilizadas para indicar se determinados modelos podem ser aplicados fora do espaço do qual o modelo foi originado. Métodos de classificação, regressão, e aprendizado supervisionado podem ser utilizados na construção dos modelos. Os métodos de classificação atribuem novos objetos (ex.: compostos a serem avaliados) a duas ou mais classes. Pode-se citar os seguintes métodos de classificação: mínimos quadrados parciais, análise discriminante linear, árvores de decisão, particionamento recursivo e máquinas de vetor de suporte. Os métodos de regressão utilizam dados contínuos como uma variável de resposta. Pode-se citar os seguintes métodos de regressão: regressão linear múltipla, mínimos quadrados parciais; máquinas de vetor de suporte, e redes neurais artificiais. As metodologias QSAR também utilizam o procedimento de validação cruzada e calculam o coeficiente de determinação (q2). Valores de q2 acima de 0,5 indicam um modelo altamente preditivo. O desempenho estatístico de qualquer modelo está relacionado à incerteza e variabilidade dos dados originais usados para construir o modelo. Os modelos QSAR utilizam uma grande variedade de métodos estatísticos e de descritores moleculares. Podem ser modelos preditivos, focados na precisão da previsão, e modelos descritivos focados na interpretabilidade. Avaliação dos outputs Os outputs discutidos a seguir são baseados, como referência, nos modelos desenvolvidos pela plataforma VEGA. Índice de domínio de aplicabilidade O índice de domínio de aplicabilidade descreve o grau de pertencimento de um composto ao domínio de aplicabilidade do modelo, com valores que variam de zero a 1, sendo zero sem associação e 1, com associação completa ao modelo. Produtos químicos com valores menores que 0,7 são considerados como potencialmente não pertencentes ao domínio de aplicabilidade. Compostos que estão no intervalo de 0,7 a 0,9, estão em uma região crítica, pois podem estar fora do domínio. Valores maiores ou iguais a 0,9 indicam moléculas que devem ser consideradas como pertencentes ao domínio de aplicabilidade do modelo. Índice de similaridade O índice de similaridade leva em consideração o grau de similaridade entre o composto avaliado e os três compostos mais semelhantes. Valores próximos a 1 indicam que o quimiotipo do composto avaliado está bem representado pelo conjunto de treinamento do modelo. Índices mais baixos podem indicar que a previsão é uma extrapolação. Neste caso, a previsão não pode ser apoiada pela avaliação de produtos químicos semelhantes. Este fato não significa que a previsão esteja errada, mas que deve-se reunir mais elementos para apoiar os resultados do modelo. Índice de concordância O índice de concordância fornece informações sobre a concordância entre o valor previsto para o composto avaliado e os valores experimentais dos três produtos químicos mais semelhantes. Valores próximos de zero podem indicar uma previsão não confiável e a possível identificação de uma região no espaço químico cujo comportamento não é adequadamente descrito pelo modelo, sendo necessária uma avaliação cuidadosa dos compostos que dão origem a previsões conflitantes. Um ou mais análogos estruturais podem ser caracterizados por valores experimentais que estão em desacordo com a previsão para o composto alvo. Por exemplo, uma inspeção visual pode identificar facilmente a presença de um alerta estrutural tóxico específico dentro da estrutura do(s) análogo(s) estrutural(is). Logo, dois compostos que são semelhantes do ponto de vista químico podem diferir pela presença/ausência de alertas estruturais, e este fato pode explicar diferenças nos valores de suas propriedades. Índice de precisão O índice de precisão informa sobre a confiabilidade no espaço estudado, levando em consideração a precisão da classificação dos três compostos mais semelhantes. Valores baixos alertam sobre a falta de precisão preditiva. Nesse caso, modelos adicionais devem ser estudados, para verificar se eles possuem melhor precisão. Índice de fragmentos centrados no átomo O índice de fragmentos centrados no átomo leva em consideração a presença de um ou mais fragmentos que não são encontrados no conjunto de treinamento ou que são fragmentos raros. Um valor de índice igual a 1 implica que todos os fragmentos centrados no átomo do composto alvo foram encontrados no conjunto de treinamento. Por outro lado, um valor inferior a 0,7 implica que um número proeminente de fragmentos centrados no átomo do composto alvo não foi encontrado nos compostos do conjunto de treinamento ou são fragmentos raros do conjunto de treinamento. Nesta situação é recomendável executar modelos adicionais para auxiliar na interpretação dos dados. Índice de intervalo de descritores de modelo O índice de intervalo de descritores de modelo verifica se os descritores calculados para o composto avaliado estão dentro da faixa de descritores do conjunto de treinamento e teste. O índice tem valor 1 se todos os descritores estiverem dentro do intervalo, zero se pelo menos um descritor estiver fora do intervalo. Este índice é calculado apenas para o modelo CAESAR. A plataforma VEGA - software para predição de endpoints Dentre os softwares disponíveis para avaliação da toxicidade utilizando descritores químicos, um dos mais amplamente utilizados são os que compõem a plataforma VEGA (https://www.vegahub.eu/), que permite aplicar diversos modelos QSAR aos compostos em avaliação, informando como saída, relatórios contendo as predições e todas as informações sobre a aplicabilidade do modelo. Atualmente, existem 59 modelos implementados na plataforma VEGA. Dentre os modelos pode-se citar os modelos IRFMN/Antares, CAESAR, ISS e IRFMN/ISSCAN-CGX, para avaliação da carcinogenicidade; os modelos CONSENSUS, CAESAR, SarPy e KNN para avaliação da mutagenicidade; o modelo CAESAR para estudo da toxicidade reprodutiva; e o modelo IRFMN para avaliação da biodegradabilidade imediata. O software VEGA utiliza como dados de entrada os dados em formato SMILES - simplified molecular input line entry specification (especificação de entrada de linha de entrada molecular simplificada), ou o formato SDF - structure data format (formato de dados de estrutura). Dentre os endpoints para avaliação da toxicidade, pode-se destacar a mutagenicidade, carcinogenicidade, toxicidade reprodutiva e a biodegradabilidade imediata. Carcinogenicidade O efeito tóxico referente à carcinogenicidade envolve o processo de transição de células normais a cancerosas que podem envolver alterações genéticas ou não-genéticas. Os carcinógenos podem ser classificados em genotóxicos, que causam danos ao DNA, e epigenéticos, que não causam dano direto ao DNA. Estes carcinógenos podem induzir o câncer por quaisquer vias de exposição (ex.: ingestão), e dependem das condições de exposição, tais como via, duração e nível de exposição. Miller propôs que os carcinógenos genotóxicos são eletrófilos, enquanto os não genotóxicos podem atuar através de uma variedade de mecanismos. Ao longo dos anos, através de testes experimentais com animais pôde-se identificar vários grupos funcionais e alertas estruturais carcinogênicos.20 A avaliação da carcinogenicidade através de modelos QSAR e a identificação de alertas estruturais para carcinógenos não genotóxicos diferem de acordo com o mecanismo de ação específico. Estes modelos fornecem previsões baseadas no conhecimento adquirido do conjunto de treinamento que foi usado para desenvolver o modelo. Dentre os modelos para estudo da carcinogenicidade, pode-se citar: IRFMN/Antares, CAESAR, ISS, IRFMN/ISSCAN-CGX. O modelo CAESAR utiliza um conjunto de dados verificados duplamente, utilizando uma grande série de descritores químicos. Mutagenicidade Muitos compostos químicos e misturas dos mesmos podem induzir à alteração no genoma dos organismos vivos, resultando em mutações que podem ser pontuais ou numéricas. A avaliação dos dados sobre a mutagenicidade é uma importante informação para o estudo das características toxicológicas de um composto. Na década de 1970, Bruce Ames idealizou o Teste de Ames, que se baseia na capacidade de as espécies de Salmonella typhimurium e Escherichia coli recuperarem a habilidade de sintetizar um aminoácido essencial frente ao efeito mutagênico de produtos químicos aos quais são expostos. Este teste é utilizado ainda hoje devido a sua reprodutibilidade interlaboratorial, por ser aplicável a diferentes compostos, e pelo seu custo benefício. Existem três principais categorias de ferramentas in silico para a previsão do potencial mutagênico: modelos QSAR, que são baseados em descritores numéricos (ex.: coeficientes de partição, descritores topológicos, contagens de grupos funcionais), sistemas baseados em regras que são construídas através de alertas estruturais (ex.: fragmentos moleculares que estão associados à ocorrência de resultados adversos) e modelos híbridos combinando essas duas abordagens. Modelos baseados em todas essas abordagens estão implementados na plataforma VEGA. Dentre os modelos para estudo da mutagenicidade, pode-se citar: CAESAR, CONSENSUS, ISS, KNN/Read-Across e SarPy/IRFMN. O modelo CAESAR é constituído de um banco de dados de 4225 substâncias, das quais 3380 foram utilizadas para construir o modelo, e 845 para testar o mesmo. Toxicidade reprodutiva A avaliação sobre a toxicidade reprodutiva representa uma análise relevante para o entendimento toxicológico de substâncias e seus efeitos durante o período gestacional, e na fertilidade masculina e feminina. Estes efeitos podem ser embriotóxicos/fetotóxicos, como defeitos estruturais, desenvolvimento mental ou físico prejudicado, aborto, etc. Agências reguladoras consideram importante que sejam feitas avaliações quanto ao efeito toxicológico à saúde humana, quanto a este endpoint. Em relação a sua avaliação através dos modelos QSAR, é o de maior complexidade, duração, e custos, pois envolve várias questões e inúmeros métodos experimentais. O modelo CAESAR é um modelo QSAR disponível no software VEGA para a avaliação da toxicidade reprodutiva. Biodegradabilidade imediata A biodegradabilidade imediata pode ser definida com um teste de triagem em que uma alta concentração do composto em estudo é utilizado e a biodegradação é avaliada através de parâmetros não-específicos em condições aeróbicas. Considera-se que, quando em torno de 60% de uma substância se degrada em 28 dias, ela é considerada facilmente biodegradável.21 A biodegradabilidade de um composto químico é um importante parâmetro na avaliação de efeitos toxicológicos ao longo do tempo ao meio ambiente e ao ser humano. Depende de vários fatores, como condições ambientais, a estrutura química do composto avaliado e a biodisponibilidade do mesmo.22 Os modelos QSAR foram desenvolvidos para várias aplicações, como propriedades ambientais (ex.: degradação); propriedades físico-químicas (ex.: coeficiente de partição); e propriedades ecotoxicológicas (ex.: toxicidade a abelhas). Dentre os modelos QSAR disponíveis, alguns dos mais largamente empregados em toxicologia computacional são os modelos CAESAR, IRFMN, ANTARES, ISS, ISSCAN-CGX, CONSENSUS, SarPy e KNN, disponíveis no software VEGA. Nesta seção, serão descritos alguns modelos QSAR disponíveis na plataforma VEGA a partir da sua aplicação preditiva para diferente endpoints de interesse. Modelos para predição da mutagenicidade CONSENSUS Fornece uma predição qualitativa da mutagenicidade baseado no Teste de Ames, aplicando uma abordagem de consenso baseado nos modelos CAESAR, SarPy, ISS e KNN. O algoritmo utiliza o domínio de aplicabilidade de cada modelo, sendo que a avaliação final será influenciada pelos modelos que forneçam os resultados mais confiáveis. Como predição, o modelo classifica a molécula em avaliação como mutagênica e não-mutagênica. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. CAESAR O modelo fornece uma predição qualitativa da mutagenicidade baseado no Teste de Ames a partir do modelo de classificação SVM (do inglês, support vector machine), retornando predições de mutagênico ou não-mutagênico. Ainda, o SVM é associado a um modelo que reconhece falsos negativos a partir de 12 alertas estruturais de mutagenicidade. Dois grupos de alertas estruturais são utilizados: se nenhum alerta estrutural para mutagenicidade for reconhecido no composto de interesse, um segundo conjunto com 4 alertas estruturais será verificado e, se algumas correspondências forem encontradas com o segundo conjunto, a previsão é "mutagênico suspeito". O relatório do modelo CAESAR fornece as seguintes informações: resumo da predição, domínios de aplicabilidade (compostos similares e escores de domínio), e fragmentos moleculares relevantes. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 1 são apresentados os dados estatísticos do modelo.

ISS Fornece uma predição qualitativa da mutagenicidade baseado no Teste de Ames. O modelo foi construído como um conjunto de regras para mutagenicidade retirados do trabalho de Benigni e Bossa (ISS), com 69 alertas estruturais de mutagenicidade e utiliza um algoritmo de Decision Tree. Se pelo menos uma estrutura indicadora de mutagenicidade for encontrada no composto de interesse, a predição "mutagênico" é dada; caso contrário, uma previsão de "não-mutagênico" é fornecida. O modelo é compatível com os formatos de moléculas SDF e SMILES como dados de entrada. Na Tabela 2 são apresentados os dados estatísticos do modelo.

KNN/Read-Across O modelo executa uma comparação em um conjunto de 5770 compostos, fornecendo uma predição qualitativa baseada no Teste de Ames. O conjunto de dados foi desenvolvido pelo Istituto di ricerche farmacologiche Mario Negri, e pelo Ministério da Saúde do Japão. O modelo de comparação foi construído com a aplicação istKNN e é baseado no índice de similaridade desenvolvido na plataforma VEGA. O índice leva em conta vários aspectos estruturais dos compostos, como o número de átomos, de ciclos, de heteroátomos, de átomos de halogênio e de fragmentos específicos (como grupos nitro). O valor do índice varia de 1 (semelhança máxima) a zero. Com base neste índice de similaridade estrutural, os quatro compostos do conjunto de dados resultantes mais semelhantes ao composto a ser previsto são levados em consideração; compostos com um valor de similaridade inferior que 0,7 são descartados. Se nenhum composto se enquadrar nessas condições, nenhuma previsão é fornecida. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 3 são apresentados os dados estatísticos do modelo.

SarPy/IRFMN O modelo fornece uma predição qualitativa da mutagenicidade baseado no Teste de Ames. Foi construído com o software SarPy através dos dados de mutagenicidade do modelo de mutagenicidade CAESAR.23 O modelo compreende dois conjuntos de regras para mutagenicidade (112 regras) e não-mutagenicidade (93 regras). Se pelo menos uma regra de mutagenicidade for encontrada no composto de interesse, uma predição de "mutagênico" é dada; se apenas uma ou mais regras de não-mutagenicidade forem correspondentes, uma predição de "não-mutagênico" é dada; se nenhuma regra corresponder ao composto dado, uma predição "possível não-mutagênico" é dada. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 4 são apresentados os dados estatísticos do modelo.

Modelos para predição da carcinogenicidade CAESAR O modelo fornece uma predição qualitativa do potencial carcinogênico de acordo com exigências específicas na regulamentação de produtos químicos. O modelo foi construído como uma "Rede Neural Artificial de Contrapropagação". O resultado da rede neural consiste de dois valores: positivo e não-positivo, ambos no intervalo [0,1] e com soma igual a 1. Esses valores representam quanto o neurônio (na rede neural) no qual o composto previsto cai pertence à classe de compostos carcinogênicos ou não-carcinogênicos. O maior entre esses dois valores leva à previsão de carcinogênico ou não-carcinogênico. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 5 encontra-se os dados estatísticos do modelo.

IRFMN/Antares O modelo fornece uma predição qualitativa do potencial carcinogênico (presença de efeitos cancerígenos em ratos machos ou fêmeas). Os dados são provenientes de informações de efeito carcinogênico em ratos, obtidos a partir de um procedimento de validação cruzada, finalizando com a extração de um conjunto de 127 alertas estruturais relacionadas à atividade carcinogênica representando fragmentos moleculares. Se pelo menos uma regra for compatível com o composto fornecido, uma previsão de "carcinogênico" é fornecida. Caso contrário, é fornecida uma previsão de "possível não-carcinogênico". O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 6 encontra-se os dados estatísticos do modelo.

IRFMN/ISSCAN-CGX O modelo fornece uma predição qualitativa do potencial carcinogênico (presença de efeitos cancerígenos em ratos machos ou fêmeas), e diferencia-se do modelo IRFMN/Antares quanto ao conjunto de regras de atividade carcinogênica empregado. O modelo foi construído como um conjunto de regras, extraído com o software SarPy de um conjunto de dados obtidos do Istituto Superiore della Sanità, (Carcinogenicidade ISS), e da Carcinogenicidade Genotoxicidade Experiência (CGX). O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 7 encontra-se os dados estatísticos do modelo.

ISS O modelo fornece uma predição qualitativa do potencial carcinogênico de acordo com específicos requisitos regulatórios. O modelo foi construído como um conjunto de regras extraído do trabalho de Benigni e Bossa (ISS), conforme implementado no software ToxTree, com 56 alertas estruturais de carcinogenicidade e utiliza um algoritmo de Decision Tree.24 Se pelo menos uma estrutura indicadora de carcinogenicidade for encontrada no composto de interesse, a predição "carcinogênico" é dada; caso contrário, uma previsão de "não-carcinogênico" é fornecida. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 8 encontra-se os dados estatísticos do modelo.

Modelos para predição da toxicidade reprodutiva CAESAR O modelo fornece uma predição qualitativa da toxicidade reprodutiva, baseado na classificação binária do FDA, em que as categorias A e B são consideradas "não-tóxico", as categorias C, D e X são consideradas "tóxico". O modelo é baseado no método de classificação Random Forest, e é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 9 encontra-se os dados estatísticos do modelo.

Modelos para predição da biodegradabilidade imediata IRFMN O modelo é baseado nos dados de teste do OECD TG 301C e fornece uma avaliação qualitativa das propriedades de biodegradabilidade imediata. Foi desenvolvido utilizando o software SarPy, do Istituto di ricerche farmacologiche Mario Negri e Politecnico di Milano. O modelo foi construído como um conjunto de regras, extraídas do conjunto de treinamento com o software SarPy. Foram obtidos sete conjuntos de regras diferentes de fragmentos relacionados à atividade de biodegradabilidade imediata a partir de análise estatística e por especialistas na área. O modelo em geral é conservador, e no caso da presença de fragmentos conflitantes a previsão é para não biodegradabilidade imediata. O esquema lógico do modelo é de que uma substância é sempre considerada não-biodegradável se pelo menos um fragmento relacionado com não-biodegradabilidade é encontrado, mesmo que fragmentos facilmente biodegradáveis sejam encontrados. Isso significa que uma parte do composto é de qualquer maneira persistente. O modelo é compatível com os formatos de moléculas SDF e SMILES, como dados de entrada. Na Tabela 10 encontra-se os dados estatísticos do modelo.

Utilização de modelos QSAR para análise de risco Modelos in silico são cada vez mais citados dentro de estruturas regulatórias como forma de cumprir os pré-requisitos de análise de risco sem a necessidade de utilizar métodos in vitro e in vivo. No contexto da toxicologia, atualmente, há uma pressão da sociedade contra a utilização de animais na avaliação de riscos, ligada à filosofia 3R - Replacement, Refinement and Reduction (substituição, refinamento e redução). No Brasil e no mundo há uma crescente mobilização e resposta de organizações aos testes, buscando uma revisão de protocolos e regulamentações, com o objetivo de estimular o desenvolvimento, validação e aplicação de métodos alternativos. Em 2008 foi aprovada a Lei Arouca no Brasil, que estabeleceu regras para os ensaios que utilizassem animais. Em 2014 ocorreu a criação do Conselho Nacional de Controle de Experimentação Animal (CONCEA), que estabeleceu a Resolução Normativa nº 17/2014, apresentando 17 métodos que devem ser implantados nos laboratórios do país, em um prazo de cinco anos. A CONCEA passou a ser responsável por credenciar instituições para criação e utilização de animais destinados a fins científicos e estabelecer normas para o uso e cuidado dos animais. Na União Europeia, o Comitê Científico de Segurança do Consumidor (SCCS) discute o papel e a aplicação dos QSARs e orienta que estas metodologias devem ser aplicadas sempre que possível, para obter estimativas de toxicidade, antes de qualquer teste experimental.25 Muitos modelos QSAR estão disponíveis para uma variedade de endpoints, e foram desenvolvidos para uso regulatório, de acordo com os critérios de qualidade e validação especificados no documento de orientação da OCDE (Organização para Cooperação e Desenvolvimento Econômico). De acordo com este documento, um modelo QSAR deve fornecer: um endpoint definido, um algoritmo inequívoco, um domínio de aplicabilidade definido, robustez, previsibilidade e uma interpretação mecanística. A Agência Europeia de Produtos Químicos publicou recentemente um documento que descreve como utilizar e informar os resultados de modelos QSAR. Inclui exemplos práticos em algumas plataformas in silico, como a OCDE QSAR, EPISuite e VEGA. Com a crescente aplicação dos modelos QSAR a uma grande quantidade de endpoints, a preocupação passou a residir ao fato da utilização de vários modelos juntos, a fim de obter resultados mais confiáveis.26 Algumas agências reguladoras, como a Agência de Proteção Ambiental dos Estados Unidos (US EPA), o Centro Europeu para a Validação de Métodos Alternativos (ECVAM) da União Europeia, a Agência para o Registro de Substâncias Tóxicas e Doenças (ATSDR) e o Comitê Científico de Toxicidade, Ecotoxicidade e Meio Ambiente da Comissão da União Europeia (CSTEE), recomendam a utilização de modelos QSAR para a avaliação de riscos.27 Obtenção de dados experimentais confiáveis A indisponibilidade de um banco de dados de propriedades/efeitos químicos de alta qualidade, algoritmos de mineração de dados poderosos, poder computacional e sistemas versáteis e confiáveis, são desafios para a aplicação dos modelos para avaliação da toxicidade química. Ao longo dos anos as metodologias in silico diversificaram-se e tornaram-se mais rigorosas para fornecer resultados relevantes, confiáveis e consistentes. No entanto os resultados obtidos ainda enfrentam o desafio de sua aceitação mais ampla, para reunir evidências para avaliação de riscos. A integração dos resultados de diferentes modelos QSAR e outras ferramentas in silico podem melhorar a confiança na predição. Ao comparar os resultados é fundamental considerar critérios-chave sobre evidência, concordância e incerteza que afetarão a relevância, confiabilidade e consistência dos resultados. As metodologias in silico compartilham fundamentos comuns, como a necessidade de dados de entrada e de treinamento de alta qualidade; a importância da documentação e métodos utilizados; e uma compreensão do domínio de aplicabilidade do modelo. Muitos dos problemas mais citados com o uso de abordagens in silico estão relacionados à necessidade de uma melhor documentação e justificativa de porque o método usado foi apropriado. Estudo de caso: resíduos de agrotóxicos e fármacos no meio ambiente Globalmente as perdas anuais na agricultura referente à ação de pragas chegam a 1 bilhão de toneladas, tendo nos pesticidas um aliado importante no crescimento da agricultura moderna, mas também um risco ao meio ambiente.28 No Brasil o artigo 41 do Decreto nº 4.074, de 04/01/2022 determina que as empresas que comercializam produtos agrotóxicos, componentes e afins, que possuam registro no Brasil, apresentem anualmente aos órgãos federais as quantidades produzidas, importadas, exportadas e comercializadas destes produtos. Este controle permite o acompanhamento por ingrediente ativo e classe de uso. Estas informações têm como objetivo melhorar o conhecimento sobre a utilização destes produtos, além de permitir a realização de estudos e tomada de decisões. Na área de controle ambiental permitem definir prioridades na escolha das substâncias para avaliação de impacto ambiental, contaminação das águas e do solo e de efeitos adversos à fauna. No campo da saúde, as informações permitem definir prioridades em pesquisas, monitoramento e fiscalização.29 Nos EUA o registro, distribuição, venda e uso de agrotóxicos é regulamentado com base na Lei Federal Inseticida, Fungicida e Rodenticida (FIFRA), que traz uma série de testes para avaliar o risco de pesticidas. Na União Europeia (UE), o Conselho do Parlamento Europeu publicou regulamentos e leis para instruir o trabalho de gestão de agrotóxicos (EC 1107/2009), e o Regulamento EU 283/2013 declarou os requisitos de dados e experimentos relevantes para avaliação de agrotóxicos (Reg. EU 283/2013). Os dados físico-químicos e tóxicos necessários para avaliação e registro dos agrotóxicos vieram de testes tradicionais in vivo e in vitro. Embora os experimentos tradicionais com animais são padrões para a avaliação de pesticidas, eles são caros a ponto de não atender à demanda de avaliação de toxicidade de forma rápida e eficiente. Logo há um movimento junto aos órgãos reguladores para utilização de métodos alternativos para avaliar o risco ambiental dos agrotóxicos. Na União Europeia, o regulamento Reg. EC 1907/2006 propõe a utilização de modelos in silico e in vitro para reduzir e substituir os experimentos in vivo na avaliação de produtos químicos. A Organização para Cooperação e Desenvolvimento Econômico (OCDE) e a EPA publicaram documentos de orientação relevantes para direcionar o desenvolvimento, validação e aplicação do modelo QSAR para avaliação de toxicidade de agrotóxicos no meio ambiente.30 No caso dos fármacos, a presença dos resíduos, no ambiente (em especial nas águas de rios, lagos e poços artesianos) está diretamente relacionada ao consumo humano e veterinário, efluentes urbanos não tratados e descarte inadequados destes medicamentos.31,32 Órgãos regulatórios como a EPA dos EUA, o Comitê Científico de Toxicidade, Ecotoxicidade e Meio Ambiente da Comissão da União Europeia (CSTEE) e regulamentos como Registro, Avaliação, Autorização e Restrição de Produtos Químicos (REACH) sob a União Europeia endossaram abordagens in silico para toxicidade e previsão de destino dos fármacos. No Brasil, a Portaria nº 2.914/2011, Anexo VII, publicada pelo Ministério da Saúde estabelece concentrações permitidas para substâncias químicas, como agrotóxicos, que podem afetar a potabilidade da água.33 Para resíduos de fármacos, não há limites nesta portaria e nem na Resolução CONAMA nº 357/05, devido à ausência de informações toxicológicas.34
CONCLUSÃO A modelagem in silico é um grande avanço no campo da toxicologia, pois através da utilização de algoritmos, possibilita a predição da toxicidade de compostos químicos nos ecossistemas, contornando as questões éticas que cercam as metodologias in vivo e in vitro, além de economizar tempo e dinheiro. Os algoritmos procuram correlações entre as propriedades da estrutura química e uma medida de sua atividade, e estabelecem uma relação matemática entre o composto e um grande número de fenômenos de toxicidade, como a mutagenicidade. Os modelos in silico são cada vez mais citados e utilizados em estruturas regulatórias como forma de cumprir os pré-requisitos de análise de risco sem a necessidade de utilizar métodos in vitro e in vivo, mostrando uma alta capacidade preditiva e confiabilidade nos dados produzidos. Os grandes desafios da aplicação da modelagem in silico são a disponibilidade de um banco de dados de propriedades/efeitos químicos de alta qualidade, algoritmos de mineração de dados poderosos, poder computacional e sistemas confiáveis. Ainda, o uso rigoroso e especializado de modelos QSAR é essencial para a previsão de endpoints toxicológicos, pois muitas vezes as estruturas químicas dos compostos de interesse estão fora do domínio de aplicabilidade dos modelos atualmente disponíveis. Neste caso, aconselha-se o uso concomitante de diferentes modelos aliado a uma cuidadosa interpretação dos resultados, considerando os parâmetros estatísticos de cada modelo.
AGRADECIMENTOS Programa CAPES/PrInt - 041/2017 pelo suporte financeiro e Programa de Pós-Graduação do Instituto de Química da Universidade Federal do Rio Grande do Sul.
REFERÊNCIAS 1. Klaassen, C. D.; Mary O. A.; Casarett and Doull's Toxicology: The Basic Science of Poisons, 6th ed.; McGraw-Hill: New York, 2001. 2. Hodgson, E.; A Textbook of Modern Toxicology, 3rd ed.; John Wiley & Sons: New Jersey, 2004. 3. Yahya, F.; Hashim, N.; Ali, D.; Ling, T.; Cheema, M.; J. King Saud Univ., Sci. 2021, 33, 101254. [Crossref] 4. Blaauboer, J.; Toxicol. In Vitro 2017, 45, iii. [Crossref] 5. Hartung, T.; George D.; Toxicol. Sci. 2009, 111, 233. [Crossref] 6. Kar, S.; Sanderson, H.; Roy, K.; Green Chem. 2020, 22, 1458. [Crossref] 7. Benfenati, E.; Diaza, R.; Gini, G.; Cardamone, L.; Gocieva, M.; Mancusi, M.; Padovani, R.; Tamellini, L. In Theory, Guidance and Applications on QSAR and REACH; Benfenati, E., ed.; Orchestra, Italy, 2012, Part A. 8. Hansch, C.; Fujita, T.; J. Am. Chem. Soc. 1964, 86, 1616. [Crossref] 9. Free, S. M.; Wilson, J. W.; J. Med. Chem. 1964, 7, 395. [Crossref] 10. Ferreira, M. M. C.; J. Braz. Chem. Soc. 2002, 13, 742. [Crossref] 11. Brown, A. C.; Fraser, T. R.; J. Anat. 1868, 2, 224. [Link] acessado em maio 2023 12. Tavares, L. C.; Quim. Nova 2004, 27, 631. [Crossref] 13. Ferreira, M. M. C.; Montanari, C. A.; Gaudio, A. C.; Quim. Nova 2002, 25, 439. [Crossref] 14. Martins, J. P. A.; Ferreira, M. M. C.; Quim. Nova 2013, 36, 554. [Crossref] 15. Gramatica, P.; Cassani, S.; Sangion, A.; Green Chem. 2016, 18, 4393. [Crossref] 16. Todeschini, R.; Consonni, V.; Handbook of Chemoinformatics: From Data to Knowledge in 4 Volumes, WILEY‐VCH Verlag GmbH & Co. KgaA: Erlangen, 2008, p. 1004-1033. 17. Todeschini, R.; Consonni, V.; Handbook of Molecular Descriptors, 2nd ed.; John Wiley & Sons: New Jersey, 2008. 18. Toropov, A.; Benfenati, E.; Comput. Biol. Chem. 2007, 31, 57. [Crossref] 19. Jaworska, J.; Comber, M.; Van Leeuwen, C.; Auer, C.; Environ. Health Perspect. 2003, 10, 1358. [Crossref] 20. Miller, E. C.; Miller, J. A.; Cancer 1981, 47, 1055. [Crossref] 21. Lombardo, A.; Pizzo, F.; Benfenati, E.; Manganaro, A.; Ferrari, T.; Gini, G.; Chemosphere 2014, 108, 10. [Crossref] 22. Pizzo, F.; Lombardo, A.; Manganaro, A.; Benfenati, E.; Sci. Total Environ. 2013, 463-464, 161. [Crossref] 23. Ferrari, T.; Cattaneo, D.; Gini, G.; Bakhtyari, G. N.; Manganaro, A.; Benfenati, E.; SAR QSAR Environ. Res. 2013, 24, 365. [Crossref] 24. Benigni, R.; Bossa, C.; Mutat. Res. 2008, 659, 248. [Crossref] 25. Gellatly, N.; Sewell, F.; Comput. Toxicol. 2019, 11, 82. [Crossref] 26. Benfenati, E.; Chaudhry, Q.; Gini, G.; Dorne, J. L.; Environ. Int. 2019, 131, 105060. [Crossref] 27. Khan, K.; Benfenati, E.; Roy, K.; Ecotoxicol. Environ. Saf. 2019, 168, 287. [Crossref] 28. Prestes, O. D.; Friggi, C. A.; Adaime, M. B.; Zanella, R.; Quim. Nova 2009, 32, 1620. [Crossref] 29. https://www.ibama.gov.br/agrotoxicos/relatorios-de-comercializacao-de-agrotoxicos, acessada em maio 2023. 30. Yang, L.; Sang, C.; Wang, Y.; Liu, W.; Hao, W.; Chang, J.; Li, J.; Chemosphere 2021, 285, 131456. [Crossref] 31. Touraud, E.; Roig, B.; Sumpter, J. P.; Coetsier, C.; Int. J. Hyg. Environ. Health 2011, 214, 437. [Crossref] 32. Tlili, I.; Caria, G.; Ouddane, B.; Ghorbel-Abid, I.; Ternane, R.; Trabelsi-Ayadi, M.; Net, S.; Sci. Total Environ. 2016, 563-564, 424. [Crossref] 33. https://bvsms.saude.gov.br/bvs/saudelegis/gm/2011/prt2914_12_12_2011.html, acessada em maio 2023. 34. CONAMA; Resolução nº 357, de 17 de março de 2005. [Link] acessado em maio 2023 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access