
 |
 |
 |
 |
 |
Artigo
| Dual promotion of Cu/ZnO catalysts modified by acetylacetone precursors for CO2 hydrogenation to methanol |
|
Junjie XianI,II,; Yuntian XuI,II,; Gaocheng QuI,II; Wei NaI,II; Hua WangI,II; Wengui GaoI,III,*
I. Faculty of Metallurgy and Energy Engineering, Kunming University of Science and Technology, 650093 Kunming, China Recebido em 27/02/2023 *e-mail: gao_wengui@126.com It is challenging to reveal the catalytic mechanism of Cu/ZnO catalysts with multivalent copper and improve the performance of CO2 hydrogenation to methanol. We synthesized Cu/ZnO catalysts with different Cu:Zn ratios using acetylacetone salts as the precursors, in which Cu, Cu2O, CuO coexist. The catalytic activity was significantly associated with the abundance of Cu+ active sites on the surface and the formation of more Cu+-ZnO interfaces. When the ratio of Cu:Zn = 5:4, the catalyst without reduction pretreatment showed good performance, with a CO2 conversion rate of 12.9%, methanol selectivity of 85.1%, and the space-time yield of 0.238 gMeOH gcat-1 h-1. It was suggested that Cu+ tends to closely bind with ZnO, and the preparation method leads to more Cu+-ZnO interfaces. The synergistic interaction between Cu+ and ZnO promoted the adsorption and dissociation of CO2 and H2, verifying its dual effect on CO2 and H2 and promoting CO2 hydrogenation to methanol. INTRODUCTION The conversion and utilization of carbon dioxide have been widely studied.1-5 In recent years, much progress has been made in directly converting carbon dioxide hydrogenation into high-value-added compounds.6-15 Many prestigious journals have been devoted to studying carbon dioxide utilization technologies, such as Greenhouse Gases: Science & Technology and Journal of CO2 Utilization.16-19 At the moment, the quantity of CO2 that can be recycled is quite restricted, necessitating more research and development activities, which necessitate the development of chemical conversion routes for large-scale CO2 utilization.20 However, CO2 hydrogenation is thermodynamically limited due to its high stability and low reactivity. The key to converting CO2 into chemicals is to activate stable CO2 molecules, which generally require catalysts to participate in chemical reactions. CO2 hydrogenation to methanol may have different reaction pathways, and the active site for Cu-based catalysts has been controversial. There are currently three views on the active centre of copper-based catalysts for the preparation of methanol: Cu0-Cu+ is the active centre;21,22 Cu0 is the active centre,23 and Cu+ is the active centre.24,25 Particularly, the strong interaction between Cu and ZnO promoted Cu dispersion and CO2 adsorption to enhance catalytic activity.26 Additionally, the fact that the migration of Zn species to Cu surface generates active sites, oxygen defects or Cu-Zn surface alloy may facilitate CO2 hydrogenation to methanol.27 Moreover, some other researchers think that copper metal alone is inactive for carbon monoxide hydrogenation and that the amount of Cu+ on the surface of alkali-doped unloaded copper catalysts affects the methanol synthesis activity.28,29 Cu2O is easily reduced to metallic Cu with H2,30 but the presence of H2O or CO2 can maintain copper in its Cu+ state,31,32 and ZnO will stabilize Cu+ species.33 Although these details have been reasonably well established, the active centre of the copper species has been the subject of debate.34,35 It has been reported that both Cu+ and Cu0 species are necessary for methanol production, and the ratio of Cu+:Cu0 determines the specific activity of methanol is also a new idea.36 However, Okamoto et al. 37 investigated the active species of the CuO-ZnO catalyst using characterization such as XPS and hypothesized that the CuO-Cu+ two-dimensional layer formed the active site for methanol synthesis at low temperature and high pressure. Acetylacetonate salt is a readily available and inexpensive compound widely used as homogeneous catalysts such as epoxidation, esterification, and benzylation of various alcohols.38,39 Saberi et al. 40 found that the CuII-acac complex anchored on magnetic nanoparticles is an efficient magnetically separable and stable catalyst. Moreover, Coskun et al. 41 prepared the catalysts of CuII and NiII acetylacetonates anchored onto Siral 80, which immobilized derivatives exhibited significant CO2 conversion efficiency. It is unfortunate, a sudden reduction in the conversion after the second usage. Toyir et al. 42 reports new gallium-promoted copper-based catalysts prepared by co-impregnation of methoxide-acetylacetonate (acac) precursors from methanolic solutions onto silica and zinc oxide supports. The maximum value for the activity was 378 g MeOH kgcat-1 h-1 at 543 K, with a selectivity of 88% towards methanol production. Chen et al. 43 reported that a highly efficient synthesis of cyclic carbonates from olefins and CO2 has been achieved by the use of a molybdenyl acetylacetonate (MoO2(acac)2) quaternary ammonium salt catalytic system with tert-butyl hydroperoxide as an oxidant through a one-pot multistep process. This study reveals the reason for enhanced catalytic performance and stability in the Cu/ZnO catalyst with acetylacetone-modified precursors. The Cu/ZnO catalyst was well mixed via the hydrothermal method with CuII and ZnII acetylacetonates (Scheme 1).
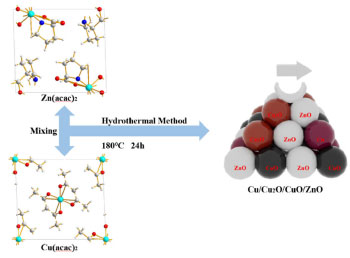 Scheme 1. Schematic diagram of the preparation of a Cu/Zn catalyst using the acetylacetonate salt precursor
EXPERIMENTAL Materials Cu(NO3)2·3H2O (analytical purity, Sinopharm Group, China), Zn(NO3)2·6H2O (analytical purity, Sinopharm Group, China), NaOH (analytical purity, Sinopharm Group, China), acetylacetone C5H8O2 (Tianjin Xingfu Fine Chemical Research Institute, China), self-prepared acetylacetone copper Cu(C5H8O2)2 and zinc acetylacetonate Zn(C5H8O2)2. Preparation of Cu-ZnO catalysts with different copper-zinc ratios First, CuII acetylacetonate and ZnII acetylacetonate were prepared by co-precipitating Cu(OH)2 and Zn(OH)2 with acetylacetonate, respectively. Then, as an example, take Cu: Zn = 6:3 (ratio of the metal ions) and twice as much NaOH as metal ions. We weighed 0.06 mol of Cu(C5H8O2)2 and 0.03 mol of Zn(C5H8O2)2 and then weighed 0.18 mol of NaOH and prepared into a 300 mL aqueous solution while placing them in a hydrothermal reactor. The hydrothermal reactor was placed in an oven and reacted at a temperature of 180 ºC for 24 h. After the reaction is over, the reactor was cooled to room temperature. The solution in the reactor was then filtered and washed five times with deionized water, the filter cake was dried in an oven at 70 ºC for 24 h, and residual organic were roasted in a nitrogen atmosphere at 300 ºC to give the catalyst, recorded as CZ-6:3 (Scheme 1). The catalyst obtained by only changing the metal ions ratio of copper and zinc by the same method is 5:4 and 4:5, named CZ-5:4, CZ-4:5. Then the catalyst with Cu:Zn of 5:4 prepared by conventional hydrothermal method with nitrates was used, and the pure copper catalyst prepared by the same method using only copper acetylacetonate, named as normal, Cu 100%. Catalyst characterization Catalyst characterization means XRD (X-ray diffractometer) method was used to characterize the surface and structure of the catalyst. Measurements were performed on a desktop XRD (Rigaku D/max-Rc, Japan) equipped with a CuKα radiation source, at 40 kV and 100 mA, in the scanning angle (2θ) range 10-90o at a scanning speed of 2o min-1. The specimen was prepared by packing around 0.2 g of sample powder in a glass holder. Different valence composition of the surface metal of the sample was determined using XPS (X-ray photoelectron spectroscopy, Thermo Kalpha, USA) using Al-Kα radiation (1486.6 eV, 6 mA × 12 KV) and a correction method based on surface contamination C1s (284.8 eV). H2-TPR (temperature program reduction), O2-TPO (temperature program oxidize), CO2-TPD (temperature programmed desorption), and H2-TPD were carried out on a Quanta chrome automated chemisorption analyzer (ChemBET pulsar TPR/TPD, USA). For H2-TPR, 0.03 g of pellets (20-40 mesh) of catalyst was loaded in a quartz U-tube, heated from room temperature to 200 oC at the heating rate of 10 ºC min-1, and maintained for 30 min in He flows. Then, the samples were cooled to room temperature, then heated to 900 oC in a 10% H2/Ar flow at a 30 mL min-1. The H2 consumption in the H2-TPR process was continuously monitored as a function of increasing temperature using a thermal conductivity detector (TCD). The O2-TPO program, in contrast to the H2-TPR program, only changes the action gas (4% O2/He); the rest remains unchanged. For CO2 temperature-programmed desorption (CO2-TPD for short), 0.1 g of catalyst of pellets (20-40 mesh) were loaded into a quartz U-tube, the gas flow rate was 30 mL min-1, and the heating rate was 10 ºC min-1. Moreover, they are reduced at 300 ºC and adsorbed at 40 ºC. The CO2-TPD characterization step of the catalyst without reduction will only bypass the first step, the H2 reduction step and the rest remains unchanged. The H2-TPD program, in contrast to the CO2-TPD program, only changes the action gas (H2); the rest remains unchanged. The SEM (scanning electron microscope) and TEM (transmission electron microscope) measurements were performed on a Tecnai G2 TF30 S-Twin high-resolution transmission electron microscope (FEI Company, Netherlands). In-situ DRIFTS analyze species adsorbed on catalyst surfaces (diffuse reflectance infrared Fourier transform spectroscopy, VERTEX 70v, Bruker, Germany). In CO2 adsorption, the catalyst was first reduced in a 10% H2/Ar atmosphere at 300 oC for 30 min and then purged with high-purity Ar gas at 300 oC for 30 min at a flow rate of 30 mL min-1. After cooling to the desired temperature, the CO2 gas is switched to the reaction cell, and the FTIR spectrometer continuously records the functional groups on the sample surface. The catalysts that have not undergone H2 pre-reduction will reduce the first step of H2 reduction and remain unchanged. For CO2 methanation, the catalyst is reduced in the same atmosphere for 30 min, followed by argon purging for 30 min. After that, the CO2/H2 (1:3) gas is switched to the reaction cell, and the FTIR spectrometer continuously records the functional groups on the sample surface. The catalysts that have not undergone H2 pre-reduction will reduce the first step of H2 reduction and remain unchanged. Catalytic performance evaluation Catalytic experiments were performed in a fixed-bed continuous flow microreactor (self-built). The catalyst activity evaluation process is divided into two stages with 0.5 g catalyst: reduction and reaction. Reduction stage: at atmospheric pressure, the preheater was set at 200 ºC, the reactor is set at 300 ºC, the reactor is injected with industrial hydrogen, the flow rate was set at 100 mL min-1, and 300 ºC for 120 min. Reaction stage: At when the end of the reduction process, hydrogen gas was switched to a CO2/H2 standard gas with a molar ratio of 1:3. The gas flow rate was 100 mL min-1, the reactor pressure was adjusted to 3 MPa, the reactor temperature was set to 280 ºC and GHSV = 6000 h-1. After stabilizing the reaction system, the products were detected and analyzed online using a gas chromatograph (Agilent Technologies 6890, USA). Then, all catalysts were evaluated by pre-reduction(r-) and non-reduction(ir-), respectively. If the evaluation of the activity of the catalyst with H2 pre-reduction, the catalysts are named r-CZ63, r-CZ54, and r-CZ45. The evaluation of the activity of the catalysts without H2 pre-reduction is unchanged except for the reaction stage without going through the reduction stage and directly entering the reaction stage, and they are named ir-CZ63, ir-CZ54, ir-CZ45, and all catalysts react to thermodynamic equilibrium (reaction for 8-9 h). Under the experimental conditions presented herein, the products are mainly methanol and some CO, while the amount of methane is tiny and negligible. The following are the possible reaction formulas 1 to 3 involved in CO2 hydrogenation to methanol. 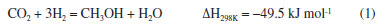 Reverse water gas reaction (RWGS): 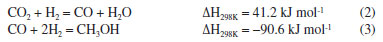 The calculation method of CO2 conversion rate X (CO2), methanol selective S (MeOH), and methanol yield Y (MeOH) is shown in the following formulas 4 to 6, which the nin and nout represent the amount of incoming and outgoing material, and m and t represent mass and time, respectively. CO2 conversion rate: 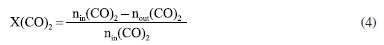 Methanol selectivity: 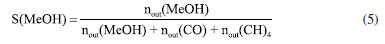 The STY of methanol was defined as the mass of methanol generated per gram catalyst per hour and calculated as follows: STY of methanol: 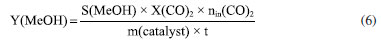
RESULTS AND DISCUSSIONS Catalyst activity evaluation Figure 1 exhibits the catalytic performance of Cu-ZnO catalysts' CO2 conversion rate and methanol selectivity with different copper-zinc ratios. Figure 2 is the CO2 methanol space-time yield picture of Cu-ZnO catalysts with different copper-zinc ratios. As shown in the Figure 1, the CO2 conversion rate of all catalysts shows an upward trend as the copper-zinc ratio increases, reaching a maximum of about 12.9% at ir-CZ63; when it is not reduced, it is at ir-CZ54 to the maximum reach is 12.5%. Except for the CZ-6:3 catalyst, the CO2 conversion rate of the other catalysts is greater than that of the reduced catalyst when it is not reduced. All catalysts without H2 pre-reduction have higher methanol selectivity than H2 pre-reduced. The copper-zinc ratio is raised and subsequently lowered with each catalyst. When the ir-CZ54, the non-reduced methanol selectivity achieves a maximum of approximately 85%, and the reduced methanol selectivity reaches 77%. And it was used Aspen to simulate the whole process, and calculate the thermodynamic limit of the reaction. It is found that the performance of the catalyst has basically reached the thermodynamic limit.
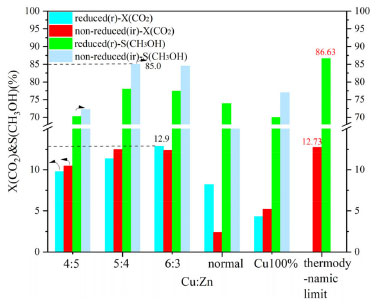 Figure 1. XCO2 and SCH3OH patterns of Cu-ZnO catalysts with different copper-zinc ratios
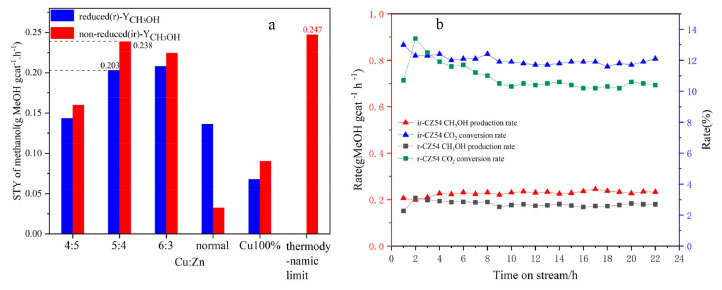 Figure 2. (a) STYCH3OH patterns of Cu-ZnO catalysts with different copper-zinc ratios and (b) during the 24 h stability test on CZ-5:4 catalyst
As shown in Figure 2(a), the methanol yield was produced by combining the catalyst CO2 conversion rate and methanol selectivity. Although the reduced and unreduced CO2 conversion rates were similar to or exceeded in the high Cu:Zn ratio catalysts, the methanol selectivity was higher for both unreduced catalysts. The methanol yields of all catalysts were higher than that of the reduction catalysts. Furthermore, the methanol yield of the catalysts increased and then decreased as the Cu:Zn ratio increased. The methanol yield reached a maximum of 0.238 gMeOH gcat-1 h-1 for the non-reduction and 0.203 gMeOH gcat-1 h-1 for the reduction catalysts at Cu:Zn ratio is 5:4. And it far exceeds the catalysts prepared by conventional hydrothermal methods (0.136 gMeOH gcat-1 h-1, 73.9% and 8.2%). However, the catalysts with different Cu-Zn ratios prepared using acetylacetonate salt precursors have higher methanol yields than normal Cu-ZnO catalysts and Cu 100% catalyst, possibly due to the synergistic effect of bimetallic species in the catalysts. In the CO2 hydrogenation to methanol reaction, the catalytic performance of all non-reduced catalysts is superior to that of the reduced catalysts, with Cu+ in the non-reduced catalysts primarily contributing to the improvement in methanol selectivity and yield. Therefore, these catalysts can be used for direct CO2 hydrogenation to produce methanol without reduction pretreatment, reducing hydrogen energy consumption, simplifying operation, and lowering production costs, with the CZ-5:4 catalyst showing the best catalytic performance. As shown in Figure 2(b), there may be a problem of catalyst deactivation in the reaction under high temperature and pressure. During the stability test, the CZ-5:4 catalyst basically stabilizes after 4 h and gradually reaches thermodynamic equilibrium. The Cu+-ZnO interface generation and copper valence distribution Figure 3 shows the XRD patterns of fresh Cu-ZnO catalysts with different Cu-Zn ratios, in which the main diffraction peaks of the catalyst at 2θ of 29.5o, 36.3o and 42.3o correspond to the Cu2O phase (110), (111), and (200) surfaces (PDF05-0667); and the main diffraction peaks of 43.3o, 50.5o at 2θ belong to the cubic Cu surfaces of phase (111) and (200) (PDF85-1326). The characteristic diffraction peaks at 2θ at 35.8o, 38.9o, and 48.7o correspond to the (11-1), (200), and (20-2) crystallographic planes of CuO (PDF65-2369). Moreover, the primary diffraction peaks at 31.5o, 34.5o, and 36.3o at 2θ correspond to the (100), (002), and (101) crystallographic planes of ZnO (PDF41-1634), respectively. In order to investigate the role of low-valent copper and to exclude the effect of ZnO in it, the Cu 100% catalyst prepared by the same method only with copper acetylacetonate was set as a control. As can be seen from the figure, the main component of the Cu 100% catalyst is Cu2O. All Cu-ZnO catalysts displayed characteristic diffraction peaks of Cu, Cu2O, CuO, and ZnO, showing that the catalysts included Cu, Cu2O, CuO, and ZnO. The diffraction peak signal intensity of Cu and CuO is relatively weak, indicating that the surface content of Cu and CuO on the surface of the catalyst may be minimal or suitably distributed, and the pattern mainly suggests that it is present in the form of monovalent copper, that is, Cu2O. The Cu2O diffraction peak on the CZ-5:4 catalyst migrates to a high angle. According to the Bragg equation, the Cu2O angle increases, the crystal plane spacing d decreases, and the crystal cell parameter decreases. Furthermore, compared with the Cu 100% catalyst, the Cu2O peak of the CZ54 catalyst still shifted at a high angle, and this may be due to the change of the Cu2O lattice spacing after more ZnO doping. At the same time, compared with CZ-4:5, its ZnO peak also shifts to a high angle, which proves that inter-metal doping leads to the reduction of cell parameters.
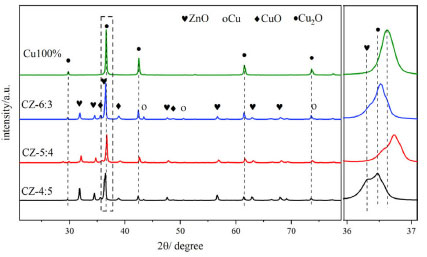 Figure 3. XRD patterns of Cu-ZnO catalysts with different Cu:Zn ratios and partial enlargement
The catalysts all display a few micron aggregates formed of many particles, accompanied by the emergence of many particles of holes and caves, and no fixed morphology is produced, as shown in the TEM and SEM diagram of CZ-5:4 (Figure 4(a), 4(b), 4(e)). The holes created in hydrothermal processes by the breakdown of catalyst precursors facilitate the movement of reactants and products during the reaction. Figure 4(c) shows TEM lattice sizes of 0.260 and 0.245 nm correspond to ZnO (002) and Cu2O (111), respectively. Figure 4(d) shows TEM lattice sizes of 0.281 and 0.173 nm correspond to ZnO (200) and Cu2O (211), respectively. In Figure 4(c) and 4(d) we can find the Cu2O-ZnO interface, which may be related to strong metal-support interaction. Then, the acetylacetonate precursor preparation catalyst can form dense nanostructures and abundant overlapping Cu2O-ZnO interfaces. The SEM of Figure 4(e) shows that the catalysts are mainly a lamellar structure, and the EDX surface sweep at the sheet-to-sheet growth interface shows that the Cu and Zn oxides are evenly distributed at the interface, establishing a mutually expanding Cu2O-ZnO interface. The creation of the Cu2O-ZnO interface may be attributed to intimate contact between small-sized metal Cu+ species and ZnO nanoparticles, consistent with the XRD and TEM results shown above (Figures 3, 4(c) and 4(d)). Therefore, Cu-ZnO catalysts prepared with acetylacetone as a precursor can promote the Cu-ZnO interface, while Cu+ is more easily doped with ZnO intensively, thus forming an interaction between metal Cu+ and ZnO to promote the catalytic reaction.
 Figure 4. (a, b) TEM diagram; (c, d) HRTEM plot; (e) SEM diagram and its EDX element are mapped of CZ-5:4
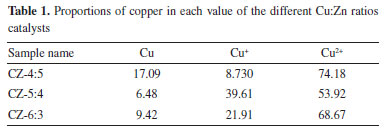
XPS characterization was performed to analyze the chemical states of species on the catalyst's surface. Figures 5(a) and 5(b) shows the Cu 2p and Zn 2p XPS spectra of the catalysts with Cu:Zn = 6:3, 5:4, 4:5, which show Cu 2p spectra exhibit typical core peaks located Cu 2p3/2 at ~ 932 eV and Cu 2p1/2 at ~ 952 eV and two satellite peaks.44 Since the spectrum of copper contains two satellite peaks, it is proved that the catalyst surface has CuO. Figure 5(b) depicts Zn 2p peaks at ~ 1021 and ~1044 eV, which correspond to Zn 2p3/2 and Zn 2p1/2 peaks of Zn2+ species in bulk ZnO, respectively.45 Figures 5(c) and 5(d) shows the Cu 2p3/2 XPS and Cu LMM spectra with peaks at ~ 932 eV attributed to Cu+ and Cu0, ~ 934 eV attributed to Cu2+; and ~ 568 eV attributed to Cu0; ~ 570 eV attributed to Cu+.44 The major component on the catalyst surface remains Cu2+, and there are Cu2O and Cu0, according to the calculation of the splitting peaks in Figure 5(c), which is consistent with the XRD results. Then, the calculated copper occupancy ratio of each valence state of the catalysts was obtained, as shown in Table 1. It can be seen that the CZ-5:4 catalyst has more low-valent copper, and its low-value copper binding energy is lower than that of CZ-4:5, indicating an increase in the electron density of Cu species on the surface of the CZ-5:4 catalyst. This can be explained by the doping of Cu2O to ZnO to form defects,45 which also makes Cu2O more easily to participate in the reaction, which is consistent with the XRD results shown. And it is clear that the CZ-5:4 catalyst has the most Cu+ content, as well as the best catalytic effect, implying that Cu+ plays an important role in the catalytic process.
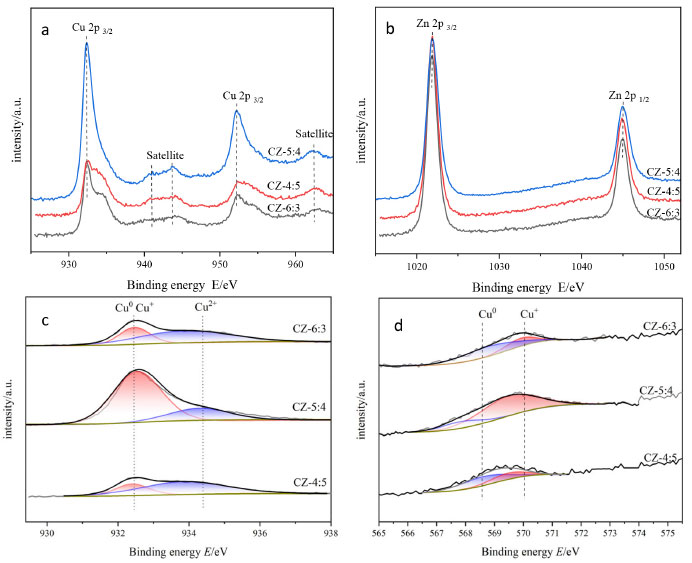 Figure 5. (a) the Cu 2p; (b) the Zn 2p; (c) Cu 2p3/2; (d) the Cu auger electron spectra of the catalysts with different Cu:Zn ratios
Moreover, surface oxygen species of the catalysts with Cu:Zn = 6:3, 5:4, 4:5 were identified by O1s spectra (Figure 6). The peaks at ~ 531, ~ 532, and ~ 533 eV on the catalysts in the energy spectrum correspond to the lattice oxygen species (OL), other weakly bound oxygen species (Oad), and surface hydroxyl groups (OOH), respectively.18,46 Notably, the binding energy of lattice oxygen in the CZ-5:4 catalyst is slightly lower than that of the other catalysts, which makes it easier for electrons to be excited on the oxygen defects. Further, more oxygen vacancies are generated by promoting electron transfer at the Cu-ZnO interface. Similarly, the oxygen species (O1s) on the surface of the CZ-5:4 catalyst have a more active chemical state. The chemical composition of the peak around 532 eV is associated with surface-adsorbed oxygen species (Oad). Typically, the ratio of Oad/Ototal may be used to calculate relative oxygen vacancy concentrations.47 The relative oxygen vacancy concentration of the catalyst is calculated as shown in Table 2. The proportionate order of oxygen vacancies is 19.76 (CZ-4:5) < 31.48 (CZ-6:3) < 40.43 (CZ-5:4). Since the concentration of oxygen vacancies on the surface of the CZ-5:4 catalyst is much larger than the other ratios, the catalytic performance and activity can be improved. The Cu/ZnO catalysts prepared as acetylacetonate salt precursors all have high surface relative oxygen vacancy concentrations. The decrease in O1s binding energy also indicates the formation of a strong interaction between Cu and ZnO, resulting in oxygen defects on the Cu surface partially covered by ZnOx. Also, Cu and ZnO are fully doped to form a Cu+-ZnO interface, which is corroborated by SEM analysis (Figures 4(e) and 4(f)) and published literature.
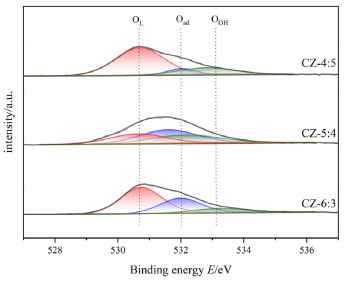 Figure 6. O1S spectra of the catalysts with different Cu:Zn ratios
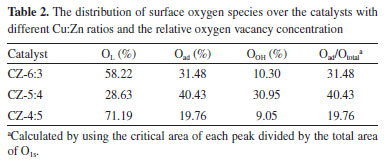
The copper valence ratio before the reaction is known, and we need to know the catalyst state after the reaction. Figure 7 shows Cu LMM spectra of the CZ-5:4 catalysts with reduction or not after the reaction. We can find the CZ54 catalysts after the reaction with reduction that although the Cu+ and Cu0 total content increased for both catalysts, the Cu+ content in the unreduced catalyst remained high compared to the reduced catalyst. Moreover, the Cu+ had high binding energy after the reaction of the unreduced catalyst. Presumably, the binding energy of Cu+ increased with the tight binding of ZnO. In combination with the results of the activity assessment (Figures 1 and 2), the higher Cu+ presence of the unreduced catalyst improves the methanol selectivity while having more active material after the reaction, indicating that the catalyst has good stability.
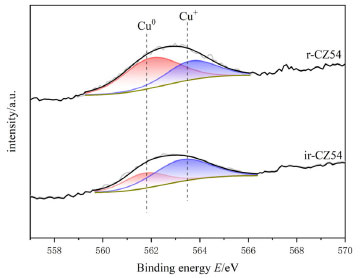 Figure 7. Cu LMM spectra of the CZ-5:4 catalysts after the reaction with reduction or not
Presence of the dual promotion of CO2 hydrogenation Promotion of CO2 adsorption reduction Figure 8 depicts the CO2-TPD spectra of Cu-ZnO catalysts with varying copper-zinc ratios. Because XRD reveals that Cu-ZnO catalysts generated by hydrothermal loading contain not only Cu2O but also Cu and CuO, a control CO2-TPD characterization can be performed by reduction and non-reduction pretreatment of the catalysts. There are three types of CO2 desorption peaks in the catalysts, with the low-temperature desorption peak α between 250~300 oC attributable to the functional group OH- on the catalyst's surface. The peak of medium temperature desorption β at 300~400 oC caused by the interaction of metal ions and oxygen atoms inside the catalyst (Zn-O, Cu-O) relates to bridging adsorption and is in the medium alkaline position. Because of the poor coordination of oxygen anions (O2-), horizontal adsorption has a strong alkaline position at high temperatures ranging from 450~650 oC.48 All non-reduced catalysts have a much larger desorption peak area than the reduced catalyst, and after reduction, only one desorption β peak appears. The desorption peaks of the non-reducing catalyst tend to lower first and then rise when the copper-zinc ratio decreases and the catalyst's copper-zinc ratio is 5:4 than the lowest temperature of the desorption peak at all peaks, which is more conducive to CO2 desorption.49 The figure also shows that among all the non-reducing catalysts, the catalyst CZ-5:4 has the biggest desorption peak β near the reaction temperature of CO2 hydrogenation to synthesize methanol, and the largest peak area, indicating that its CO2 adsorption performance is the best of these catalysts during the catalytic reaction. It is more advantageous for CO2 hydrogenation for methanol synthesis, consistent with the activity evaluation results.
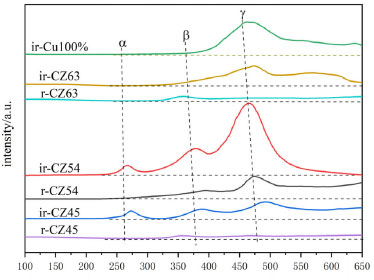 Figure 8. CO2-TPD patterns of the Cu-ZnO catalysts with different Cu:Zn ratios and pure copper cat
In addition, a control group of pure copper catalysts was created without the addition of zinc acetylacetonate, and it can be shown that the unreduced pure copper catalysts only had high-temperature desorption peaks with large peak areas. Lack of contact between internal metal ions and oxygen atoms results in less adsorption at low and medium temperatures, but its presence primarily as Cu+ makes it strong for CO2 adsorption. Furthermore, the reduced catalysts are primarily present as Cu0, whereas the unreduced catalysts are primarily present as Cu+. All catalysts performed better without reduction than after reduction, and the catalyst CZ-5:4 had the lowest desorption peak temperature and the biggest peak area of any catalyst. In conclusion, XPS shows that the catalyst CZ-5:4 has the largest Cu+ percentage. It is hypothesized that the increased contact between Cu+ and ZnO during the hydrothermal process of acetylacetone precursors increases the hydrogenation of CO2 to methanol. The catalyst was subjected to an in situ-DRIFS spectrogram experiment of CO2 adsorption in order to investigate further the surface morphology of CO2 adsorption on the catalyst. It is reported that CO2 desorbed in moderately and strongly basic sites promote the formation of bidentate and mono-dentate carbonates, respectively.50,51 And Figure 9(a) shows the Cu-ZnO catalyst with different copper-zinc ratios in the in-situ DRIFT spectra of CO2 adsorption of the catalyst. According to research, carbonates may be hydrogenated to generate formate and methoxylated, which are the significant intermediates in the hydrogenation of CO2 to methanol. As a result, the quantity of carbonate on the catalyst's surface influences its activity. In a CO2 environment, only carbonate adsorption peaks are observed on the sample surface.48 To begin with, the adsorption peak in the FTIR spectrum of the reduced catalyst is very weak in all of the samples, indicating that the adsorption of CO2 by the reduced catalyst is significantly weakened; the absorption peak is more evident when the catalysts without reduction, indicating that the catalyst without reduced for the CO2 adsorption is intense. Among all non-reducing catalysts, the catalysts CZ-6:3 and CZ-5:4 have a much bigger adsorption peak area than other catalysts, which is more favorable for CO2 absorption and has the same desorption result as for CO2-TPD. In all catalysts, the adsorption peaks around 1100 cm-1 correspond to the C-O bonding vibrations of methoxy, the adsorption peaks around 1220, 1280, and 1320 cm-1 belong to the C=O bonding vibrations of monodentate carbonate and bicarbonate species, respectively. Peaks at 1520 cm-1 are due to C=O bonding vibrations of bidentate carbonate, while peaks at 2865 cm-1 are due to O-H bonding vibrations of formate,48,52 and the not reduced catalyst has a prominent peak, which forms more formate, which is beneficial to the catalytic process of CO2 hydrogenation. The appearance of carbonate peaks is caused by unsaturated oxygen anions (O2-) (CO2 + O2- → CO32-). The strength of these carbonate peaks corresponds to the adsorption and activation capacity of CO2, and the catalysts ir-CZ63 and ir-CZ54 are significantly greater than that of other catalysts in adsorption and activation capacity of CO2.
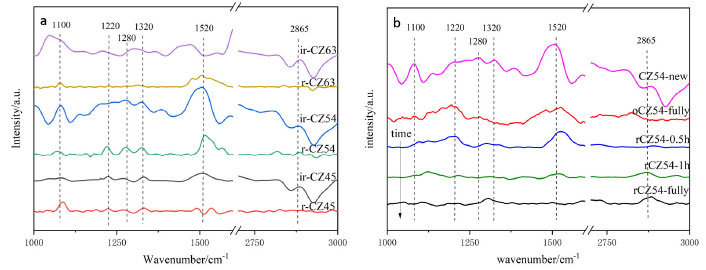 Figure 9. In-situ DRIFT spectra of the CO2 adsorption on (a) Cu-ZnO catalysts with different Cu:Zn ratios; (b) a new, fully oxidized and reduced difference time the CZ-5:4 catalyst
Figure 9(b) shows the in-situ DRIFTS of CO2 adsorbed by the CZ-54 catalyst at different reduction times and after oxidation. The telescopic vibrational peak of the bidentate carbonate species at 1520 cm-1 is significantly more intense when the catalyst is not reduced than the adsorption peak, and the catalyst is stronger for half an hour of reduction than for one hour and weakened with increasing reduction time, and the adsorption peak on the oxidized catalyst is reduced. The cuprous oxide in the catalyst was reduced to copper monomers, resulting in weakened adsorption and activation of CO2. On all catalysts, the adsorption peaks of the non-reduced catalysts were much larger than those of the reduced catalysts. The C=O bond vibrational peaks for the bicarbonate species at 2890 cm-1 were slightly higher for the fully oxidized than the fully reduced peaks, but both were much smaller than the original catalysts, presumably due to the reduced Cu+ in the catalysts, which weakened the ability to adsorb and activate CO2. In summary, all catalysts at the CO2 adsorption performance, the non-reduced catalysts are better than the pre-reduction catalysts, with the catalysts ir-CZ63 and ir-CZ54 having relatively strong CO2 adsorption performance, as well as the CO2-TPD results being consistent with the activity evaluation. CO2 is primarily adsorbed on the catalyst in the form of carbonate species, bicarbonate species, and formate species. Therefore, the formation of more Cu+-ZnO interfaces will have higher CO2 adsorption capacity and more easily generate carbonates. Promotion of H2 dissociation and oxidation Figure 10 illustrates the H2-TPR spectra of Cu-ZnO catalysts with varying copper-zinc ratios, and Table 3 lists the associated reduction peak area and temperatures. Because ZnO cannot be reduced at these temperatures, the Cu-ZnO catalyst reduction peaks correspond to copper species reduction. All the reduced peaks may be classified as four Gaussian peaks, denoted as peaks a, b, c, and d. Literature suggests that copper species in the body phase are more difficult to reduce than those in the fractional state. Copper reduction is made in steps, with Cu2+ first reduced to Cu+, then Cu+ reverting to Cu0.49-52 So that the low-temperature reduction peaks can be attributed to the reduction of surface-dispersed Cu2+ to Cu+, and the second-low temperature reduction peak b peak can be attributed to the body phase of Cu2+ is reduced to Cu+. The second-high reduced peak c peak belongs to the surface dispersed Cu+ reduced to Cu0, and the high-temperature reduction peak d peak is related to the reduction of bulk phase Cu+.
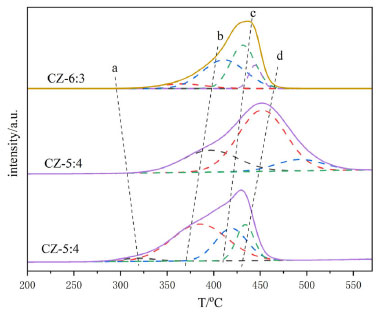 Figure 10. H2-TPR spectra of Cu-ZnO catalysts with different Cu:Zn ratio
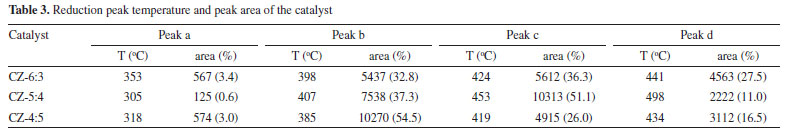
In order to better analyze the H2-TPR results, four Gaussian peaks were synthesized by fitting the reduction peaks, respectively attributed to the reduction peaks of dispersed CuO species and CuO species.53,54 As shown in Table 3, as copper content increases, the total area of the reduction peak increases and then decreases; the increase is because the high-valent copper content increases, so the hydrogen consumption increases, and the decrease may be because there is more Cu0 in the catalyst, the high-valent copper content is relatively reduced, and the hydrogen consumption decreases. Furthermore, because Cu0 has a high dissociation and adsorption capacity for H2, the catalyst is simpler to reduce, and its reduction temperature will be lowered. As shown in Figure 10, the acetylacetone precursor Cu/ZnO catalyst, in which the CuO reduction peak a and b moves above a higher temperature (305 oC, 407 oC), much higher than that of pure CuO and Cu/ZnO alone (200 oC, 225 oC; 193 oC, 203 oC),55 indicating CuO and ZnO have a strong metal support interaction (SMSI).56 Similarly, the temperature of Cu2O reduction peaks c and d also increases; Cu+ interacting with ZnO has a small crystal size but is difficult to reduce, possibly due to dispersed Cu+ species embedded in the ZnO matrix or Cu+ species in close contact with ZnO at the Cu/ZnO interface.57 The close contact and doping of Cu+ species with ZnO can be demonstrated by the elevated XRD angle and H2-TPR temperature. Compared with the Yttria-doped Cu/ZnO catalyst (10.8%, 81.8%), the Cu/ZnO catalyst by acetylacetone salt (12.9%, 85.1%) has higher conversion rate and methanol selectivity.55 In addition, among them, the CZ-5:4 catalyst has the largest proportion of peak c and better catalytic performance. Combining the peak strength of the reduction peak with activity evaluation, we can find that the more excellent peak c (the peak of surface dispersed Cu+ conversion to Cu0), the better the catalytic action. It shows that the Cu+ in the catalyst has a significant dissociation and activation ability for H2. The H2-TPD spectra of different copper-zinc ratio catalysts and pure copper catalysts are shown in Figure 11. The H2-TPD spectra of different Cu:Zn ratios and pure Cu catalysts are shown in Figure 11, which shows that there are mainly three temperature desorption peaks, named α, β, and γ peaks. When the peak strength and size of the three are compared, it is not difficult to see that the desorption peak of the CZ-5:4 catalyst is the largest, which can indicate that the adsorption capacity of H2 is the strongest, which is consistent with the activity characterization results, and also reveals that Cu+ has a better adsorption dissociation ability for H2. When compared to other copper-zinc ratio catalysts, the CZ-5:4 catalyst has the most apparent peak, which is known as the low-temperature adsorption point, which has the lowest temperature and is similar to the reaction, and it can also be said that the CZ-5:4 catalyst can adsorb more H2 and let more H2 at the catalytic reaction temperature, that is, the low adsorption point. Participate in the reaction, which further enhances the catalytic reaction. In addition, the Cu 100% catalyst of copper acetylacetonate precursor had good adsorption capacity for H2. The adsorption peaks were smaller and concentrated on the high-temperature adsorption sites compared to the ir-CZ54 catalyst. It indicates that the adsorption dissociation of H2 is mainly in the Cu+-ZnO sites, and the formation of more intermetallic interfaces is favorable for H2 dissociation and oxidation.
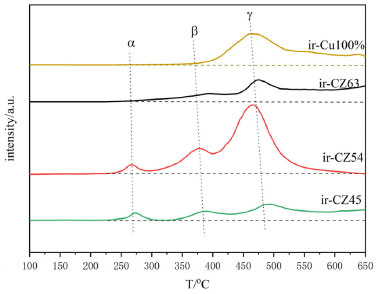 Figure 11. H2-TPD spectra of different Cu:Zn ratio catalysts and pure copper catalysts
Effect on CO2 hydrogenation process Figure 12 depicts the in-situ DRIFT spectra of CO2 hydrogenation reactions on Cu-ZnO catalysts with varying copper-zinc ratios. Moreover, the in-situ DRIFT spectral characterization for CO2 hydrogenation reactions can further measure the catalytic reaction pathways and the catalytic performance of the catalysts. A band of methanol C-H bonding vibrations was observed at around 2940 cm-1 for all catalysts, whether reduced or not, and the band of methanol C-O stretching vibration was also detected at around 1100 cm-1.48 Among them, the ir-CZ54 and r-CZ54 catalysts have higher methanol peaks. In addition, peaks corresponding to gaseous CO appeared in all catalysts at 2100 and 2200 cm-1, matching the standard spectral comparison of CO and indicating the generation of CO during the catalytic process.48 Intermediate product peaks appeared between 1200 and 1800 cm-1, and peaks at 1280 cm-1 belonged to bicarbonate C-O species. Peaks around 1520 and 1540 cm-1 are related to C-O bonding vibrations of bidentate carbonate species, whereas peaks at 1750 and 2890 cm-1 are attributed to the stretching vibration peaks of the formate species. It can be found that as the copper ratio increases, the carbonate and formate peaks increase and then decrease. There are also some weak peaks between 1200 and 1800 cm-1, which can be ascribed to water vapor formed from methanol synthesis processes.
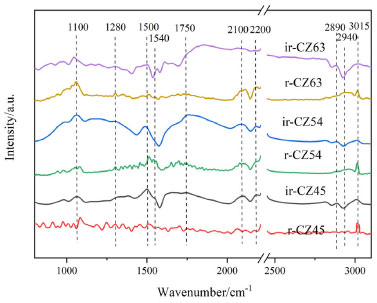 Figure 12. In situ DRIFT spectra of the CO2 hydrogenation reaction on Cu-ZnO catalysts with different Cu:Zn ratios
Whether or not the catalysts were reduced, and peaks of C-H bonding vibrations owing to methane appeared at 3015 cm-1 is not apparent in comparison. The methanol peak intensity of catalyst ir-CZ54 is relatively high among all catalysts, and the methane peak is small, showing that it produces the most methanol with high selectivity, consistent with the activity evaluation results. The results show that in the reaction of CO2 hydrogenation to methanol, the formate pathway of direct CO2 hydrogenation and the carbonate pathway of hydrogenation through CO intermediates can occur concurrently; the catalyst CZ-5:4 without reduction has the best catalytic performance of all catalysts. Combined with the previous CO2 in situ DRIFT adsorption analysis, we found the ir-CZ54 catalyst has the largest proportion of Cu+, forms more Cu+-ZnO interface, and has a strong CO2 adsorption capacity and the best H2 dissociation ability. It shows that Cu+-ZnO plays a dual function in the CO2 hydrogenation process. Moreover, Cu+ is in the intermediate valence state, and combining it with ZnO can promote CO2 reduction and H2 oxidation.
CONCLUSIONS A series of Cu/ZnO catalysts with different Cu:Zn ratios in the complex valence state were prepared by hydrothermal method using acetylacetonate salts as precursors. Then, the dual promotion effect of the acetylacetone precursor preparation method on CO2 hydrogenation to methanol in the Cu-ZnO system was investigated. Furthermore, it was found that the acetylacetone precursor preparation method significantly improved the reactivity and reaction sites of Cu/ZnO methanol synthesis catalysts. Notably, the catalytic efficiency of all proportions of the catalysts by H2 pre-reduction was lower than without reduction. The results indicate that copper in the catalyst exists in the form of CuO, Cu2O, and Cu, with Cu2O as the active substance. According to TEM observations, Cu+ is a copper species that is more easily doped into ZnO. The higher the proportion of Cu+, the better the adsorption performance of copper zinc catalysts for CO2. Although Cu 100% catalysts with higher Cu+ content cannot form a Cu+-ZnO interface, their CO2 adsorption ability is weak. In addition, the adsorption and dissociation ability of H2 is also related to the formation of more Cu+-ZnO interfaces. It forms more Cu+-ZnO active sites by the hydrothermal method of acetylacetone precursor. It has good adsorption and dissociation ability for both H2 and CO2 during the catalytic process, which has a dual promotion effect on CO2 reduction and H2 oxidation during CO2 hydrogenation to methanol due to the redox nature of its intermediate valence state. In conclusion, due to the limited reduction capacity of acetylacetone, the Cu+ percentage increases and then decreases as the Cu percentage increases. When the non-reduced CZ-5:4 catalyst with high copper-zinc dispersion has good performance and high stability for CO2 hydrogenation to methanol. The acetylacetone precursors' preparation method has industrial production prospects, and the Cu/ZnO catalyst can also be attempted to expand production to support it, but now it mainly focuses on the basic theoretical research of the reaction.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
ACKNOWLEDGMENTS The authors would like to thank Major R&D Special Project of Yunnan Province (No. 202302AG050005-2) and the Analysis and Testing Foundation of Kunming University of Science and Technology (2021M20202102053).
AUTHOR CONTRIBUTIONS J. X. , Y. X. , G. Q. , W. N. and H. W. were responsible for the resources; J. X. for conducting data curation and analysis; J. X. , Y. X. and W. G. for writing the original draft; W. G. for the conceptualization and writing review.
REFERENCES 1. Liu, C.; Guo, Q.; Ye, J.; Sun, K.; Ge, Q.; CIESC Journal 2016, 67, 6. [Link] accessed in August 2023 2. Sun, H. Z.; Zhu, W. Q.; Song, M. X.; Wang, Q.; Liu, X. M.; Nasier, A.; Chem. Asian. J. 2013, 25, 8. [Crossref] 3. Gao, W.; Wang, H.; Han, C.; Liu, W.; Wang, Y.; Jia, M.; Chem. Ind. Eng. Prog. 2014, 33, 2963. [Crossref] 4. Centi, G.; Perathoner, S.; Catal. Today 2009, 148, 191. [Crossref] 5. Peng, H.; Zhihong, W. U.; Zhang, J.; Jing, L. U.; Chenxiao, W. U.; Peiqiang, L. I.; Yin, H.; Chem. Ind. Eng. Prog. 2014, 353, 1003. [Crossref] 6. Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J. G.; Chem. Rev. 2020, 120, 7984. [Crossref] 7. Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhang, T.; Chem. Soc. Rev. 2020, 49, 1385. [Crossref] 8. Zelong, L.; Yuanzhi, Q.; Jijie, W.; Hailong, L.; Mingrun, L.; Shu, M.; Li, C.; Joule 2019, 3, 570. [Crossref] 9. Asencios, Y. J. O.; Marcos, F. C. F.; Assaf, J. M.; Assaf, E. M.; Braz. J. Chem. Eng. 2016, 33, 627. [Crossref] 10. Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Min, Z.; Guo, X.; Song, C.; RSC Adv. 2018, 8, 7651. [Crossref] 11. Artz, J.; Müller, T. E.; Thenert, K.; Kleinekorte, J.; Leitner, W.; Chem. Rev. 2018, 118, 434. [Crossref] 12. Bulushev, D. A.; Ross, J. R. H.; Catal. Rev. 2018, 60, 566. [Crossref] 13. Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y.; Catal. Sci. Technol. 2017, 7, 4580. [Crossref] 14. Wang, W. H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; Fujita, E.; Chem. Rev. 2015, 115, 12936. [Crossref] 15. Xiong, Z.; Zhao, Y.; Zhang, J.; Zheng, C.; Chem. Ind. Eng. Prog. 2013, 32, 1043. [Crossref] 16. Plasseraud, L.; ChemSusChem 2010, 3, 631. [Crossref] 17. Goeppert, A.; Czaun, M.; Jones, J. P.; Prakash, G.; Olah, G. A.; ChemInform 2014, 43, 7995. [Crossref] 18. Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S. C.; Angew. Chem. , Int. Ed. 2011, 123, 2210. [Crossref] 19. Ye, J.; Liu, C.; Mei, D.; Ge, Q.; ACS Catal. 2013, 3, 1296. [Crossref] 20. Samrand, S.; Nor, A. S. A.; Mohammad, R. R.; J. CO2 Util. 2014, 5, 66. [Crossref] 21. Jinyao, L.; Jiangliu, S.; Dehua, H.; Qijian, Z.; Xiaohui, W.; Yu, L.; Qiming, Z.; Appl. Catal. , A 2001, 218, 113. [Crossref] 22. Okamoto, Y.; Fukino, K.; Imanaka, T.; Teranishi, S.; J. Phys. Chem. 1983, 87, 3740. [Crossref] 23. Shi, C.; Qiming, Z.; Dianqiu, L.; Guanghua, Y.; Chinese Journal of Petroleum (Petroleum Processing) 1987, 10, 25. (CA 67-56-1) 24. Herman, R. G.; Klier, K.; Simmons, G. W.; Finn, B. P.; Bulko, J. B.; Kobylinski, T. P.; J. Catal. 1979, 56, 407. [Crossref] 25. Hongbo, C.; Junxiu, C.; Hongbin, Z.; Qiduan, C.; J. Xiamen Univ. , Nat. Sci. 1990, 29, 411. (CA 67-56-1) 26. Din, I. U.; Shaharun, M. S.; Alotaibi, M. A.; Alharthi, A. I.; Naeem, A.; J. CO2 Util. 2019, 34, 20. [Crossref] 27. Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C. F.; Helveg, S.; Chorkendorff, I.; Sehested, J.; Science 2016, 352, 969. [Crossref] 28. Sheffer, G. R.; King, T. S.; J. Catal. 1989, 116, 488. [Crossref] 29. Sheffer, G. R.; King, T. S.; ChemInform 1989, 115, 376. [Crossref] 30. Yang, F.; Choi, Y.; Liu, P.; Hrbek, J.; Rodriguez, J. A.; J. Phys. Chem. C 2010, 114, 17042. [Crossref] 31. Herman, R. G.; Klier, K.; Simmons, G. W.; Finn, B. P.; Kobylinski, T. P.; ChemInform 1979, 10, 456. [Crossref] 32. Klier, K.; Chatikavanij, V.; Herman, R. G.; Simmons, G. W.; J. Catal. 1982, 74, 343. [Crossref] 33. Klier, K.; Eley, D. D.; Herman, P.; Paul, B. W.; Adv. Catal. 1982, 31, 243. [Crossref] 34. Shen, W. J.; Ichihashi, Y.; Matsumura, Y.; Catal. Lett. 2002, 83, 33. [Crossref] 35. Fujitani, T.; Nakamura, J.; Appl. Catal. , A 2000, 191, 111. [Crossref] 36. Liu, J. T.; Liu, S. Z.; Li, J.; Gao, Z. H.; Zuo, Z. J.; Wei, H.; J. CO2 Util. 2017, 20, 59. [Crossref] 37. Okamoto, Y.; Fukino, K.; Imanaka, T.; Teranishi, S.; J. Phys. Chem. B 1983, 87, 3740. [Crossref] 38. Ligtenbarg, A. G. J.; Hage, R.; Feringa, B. L.; Coord. Chem. Rev. 2003, 237, 89. [Crossref] 39. Bolm, C.; Coord. Chem. Rev. 2003, 237, 245. [Crossref] 40. Saberi, D.; Mahdudi, S.; Cheraghi, S.; Heydari, A.; J. Organomet. Chem. 2014, 772, 222. [Crossref] 41. Coşkun, S.; Taşçi, Z.; Ulusoy, M.; Yurdakoç, M.; Turk. J. Chem. 2014, 38, 600. [Crossref] 42. Toyir, J.; Ramírez, D. L. P. P.; Fierro, J. L. G.; Homs, N. S.; Appl. Catal. , B 2001, 34, 255. [Crossref] 43. Chen, F.; Dong, T.; Xu, T.; Li, X.; Hu, C.; Green Chem. 2011, 13, 2518. [Crossref] 44. Yang, B.; Deng, W.; Guo, L.; Ishihara, T.; Chin. J. Catal. 2020, 41, 1348. [Crossref] 45. Hu, X.; Qin, W.; Guan, Q.; Li, W.; ChemCatChem 2018, 10, 4438. [Crossref] 46. Xu, X.; Chen, D.; Yi, Z.; Jiang, M.; Wang, L.; Langmuir 2013, 29, 5573. [Crossref] 47. Ye, H.; Na, W.; Gao, W.; Wang, H.; Energy Technol. 2020, 8, 2000194. [Crossref] 48. Wang, Y.; Kattel, S.; Gao, W.; Li, K.; Liu, P.; Chen, J. G.; Wang, H.; Nat. Commun. 2019, 10, 1. [Crossref] 49. Chen, L. F.; Guo, P. J.; Qiao, M. H.; Yan, S. R.; Li, H. X.; Shen, W.; J. Catal. 2008, 257, 172. [Crossref] 50. Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S.; Catal. Commun. 2014, 45, 74. [Crossref] 51. Pokrovski, K.; Jung, K. T.; Bell, A. T.; Langmuir 2001, 17, 4297. [Crossref] 52. Kock, E. M.; Kogler, M.; Bielz, T.; KloTzer, B.; Penner, S.; J. Phys. Chem. C 2013, 117, 17666. [Crossref] 53. Gao, P.; Yang, H.; Zhang, L.; Zhang, C.; Zhong, L.; Wang, H.; Wei, W.; Sun, Y.; J. CO2 Util. 2016, 16, 32. [Crossref] 54. Lei, H.; Nie, R.; Wu, G.; Hou, Z.; Fuel 2015, 154, 161. [Crossref] 55. Qi, T.; Li, W.; Li, H.; Ji, K.; Chen, S.; Zhang, Y.; Mol. Catal. 2021, 509, 111641. [Crossref] 56. Qi, S. C.; Liu, X. Y.; Zhu, R. R.; Xue, D. M.; Liu, X. Q.; Sun, L. B.; Chem. Eng. J. 2022, 430, 132784. [Crossref] 57. Zhu, Y.; Kong, X.; Zhu, S.; Dong, F.; Zheng, H.; Zhu, Y.; Li, Y. W.; Appl. Catal. , B 2015, 166, 551. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access