
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Espectroscopia de fluorescência: dos fundamentos à influência dos parâmetros instrumentais para análises de corantes orgânicos e nanopartículas inorgânicas Fluorescence spectroscopy: from fundamentals to the influence of instrumental parameters for analysis of organic dyes and inorganic nanoparticles |
|
Rafael S. MourãoI; Brener R. Carvalho ValeI,II; André F. Vale FonsecaI; Thaís A. S. CarvalhoI; Marco A. SchiavonI,*
I. Departamento de Ciências Naturais, Universidade Federal de São João del-Rei, 36301-160 São João del-Rei - MG, Brasil Recebido em 18/04/2023 *e-mail: schiavon@ufsj.edu.br Fluorescence spectroscopy is an extremely sensitive analytical technique that has been widely used to elucidate problems that require low detection limits. Although fluorescence spectroscopy is widespread in industry and academia, there is a lack of teaching materials demonstrating its basic principles, as well as the influence of instrumental parameters involved in the technique, which limit its correct use and the full potential of it. For this reason, this work aims to explore the principles of fluorescence spectroscopy and correlate them in function of different instrumental variables to carry out a fluorescence experiment. The spectrofluorometer parameters such as the excitation wavelength, excitation and emission slits, step, integration time, detection geometry as well as concentration of the samples were varied to illustrate how the best resulting spectra can be registered free of artifacts. To do so, two organic dyes with structured vibronic bands (pyrene) and another with unstructured bands (rhodamine 6G) were used. Another study carried out involved the use of 3D spectra, excitation/emission spectra, of cadmium telluride (CdTe) quantum dots. In this case, it was possible to show the excitation spectrum of nanomaterials, collecting emissions at different wavelengths, as well as analyzing the particle size distribution. INTRODUÇÃO Espectroscopia de fluorescência O uso de radiação eletromagnética em técnicas espectroscópicas tem promovido um avanço incrível no entendimento da estrutura da matéria. Essas técnicas analisam a radiação que é absorvida, emitida ou espalhada pelo analito. Estes dados são fornecidos por aparelhos chamados espectrômetros, que são bastante utilizados nos laboratórios de pesquisa, ensino, controle de qualidade em empresas e diversos equipamentos comerciais estão disponíveis para esses fins. Os dados espectroscópicos são geralmente representados por um espectro, que é a representação gráfica da resposta de interesse em função da energia (comprimento de onda, frequência, número de onda, etc. ). Por ser uma técnica extremamente sensível, mesmo amostras com baixas concentrações podem ser facilmente estudadas, o que torna a técnica muito usada na análise de numerosos compostos de interesse farmacêutico, ambiental e industrial.1 Além disso, a maioria das técnicas espectroscópicas não costumam ser destrutivas à amostra.2 A espectroscopia de fluorescência é uma técnica que se destaca nesse cenário por várias razões. Ela é uma técnica analítica extremamente sensível e tem sido amplamente utilizada na resolução de problemas que requerem baixos limites de detecção.3 A sensibilidade desta técnica é consideravelmente maior em comparação às outras, como por exemplo, a espectrofotometria UV/Visível, apresentando limites de detecção de até três ordens de grandeza menores. O limite de detecção de espectroscopia de fluorescência é da ordem de ng mL-1, devido a sua alta sensibilidade e alta relação sinal-ruído.4-6 Outro ponto a ser ressaltado é a simplicidade instrumental e o baixo custo de manutenção de análise quando comparados com outras técnicas analíticas.7 Além disso, o tempo característico envolvido no processo de fluorescência é da ordem de 10-9 s, o que faz com que ela também seja muito atraente para o estudo de fenômenos relacionados às propriedades biofísicas de moléculas biológicas.2 Luminescência De modo geral, os processos que ocorrem entre a absorção e a emissão de luz são ilustrados pelo diagrama de Jablonski.8 O diagrama de Jablonski é comumente utilizado para discutir a absorção e emissão de luz. Além disso, é usado em uma variedade de formas para ilustrar vários processos moleculares que podem ocorrer em estados excitados. Um diagrama típico de Jablonski4,9 é apresentado no material suplementar (Figura 1S). A energia térmica em temperatura ambiente não é suficiente para povoar significativamente os estados eletrônicos excitados, pois a diferença de energia entre os estados S0 e S1 (ver definição no material suplementar, Figura 1S) é muito grande para a população térmica de S1, S0 → S1 >> kb T, onde kb é a constante de Boltzman e T a temperatura termodinâmica. Dessa forma, usa-se radiação ao invés de calor para a indução da fluorescência, sendo que após a absorção de luz, vários processos podem ocorrer. Um fluoróforo é geralmente excitado para algum nível vibracional mais alto de S1 ou S2, ocorrendo interações entre esses dois estados eletrônicos. É chamado de conversão interna quando o elétron decai de S2 para S1 em um processo não radiativo. Esse processo é conhecido como conversão interna (S2 → S1) e geralmente ocorre dentro de 10-12 s ou menos. O elétron também pode relaxar dentro de um mesmo estado eletrônico por meio dos estados vibracionais até atingir o nível vibracional fundamental do estado eletrônico excitado. Esse processo é chamado de relaxamento vibracional e acontece de forma não radiativa e rápida (10-13 a 10-11 s). Dessa forma, a emissão de fluorescência resulta de um estado excitado termicamente equilibrado, ou seja, do estado vibracional de menor energia de S1. Portanto, o surgimento da fluorescência é acompanhado do decaimento dos processos de conversão interna e relaxamento vibracional, e isso indica que, a fluorescência independe da energia de excitação, conhecido como regra de Kasha, a qual será discutida em mais detalhes posteriormente. O tempo de vida da fluorescência de moléculas é de aproximadamente 10-8 s.4 No estado S1, as moléculas também podem sofrer uma conversão de spin para o primeiro estado tripleto T1, processo conhecido como cruzamento intersistema. Por ser um estado de maior multiplicidade, T1 é localizado em menor energia em relação a S1. A conversão de S1 para T1 é proibida e acontece com menor probabilidade e com tempos de vida da ordem de 10-9 a 10-8 s. Geralmente este processo de cruzamento intersistema compete com as taxas de decaimento da fluorescência. Além disso, a taxa constante de uma transição radiativa do estado tripleto, processo chamado de fosforescência, são várias ordens de magnitude menores (10-3 s) do que aquelas para a fluorescência. Os átomos pesados facilitam o cruzamento intersistemas devido ao acoplamento spin-orbita, e, dessa forma, aumentam os rendimentos quânticos da fosforescência. Portanto, moléculas contendo átomos pesados como iodo e bromo são geralmente fosforescentes.4 No caso da fluorescência, o spin do elétron em S1 está emparelhado com o elétron no estado fundamental. Por isso, o retorno ao estado fundamental é permitido por spin e ocorre rapidamente pela emissão de um fóton. Já na fosforescência a emissão de luz ocorre a partir de estados excitados tripletos, nos quais o elétron em T1 e o elétron em S0 tem a mesma orientação de spin. Essas transições são proibidas, e por isso, as taxas de emissão são lentas entre 103 a 100 s-1.4 De forma geral, o fóton de emissão fluorescente possui menor energia do que o fóton de excitação, por causa das possíveis perdas de energia nas transições dos estados vibracionais do estado eletrônico excitado, permitindo que os espectros de absorção e emissão sejam distinguidos. Cada fluoróforo possui um comprimento de onda de excitação e um comprimento de onda de emissão característico, sendo a distância entre eles chamada de deslocamento Stokes, como pode ser observado na Figura 1. A fluorescência de moléculas com pequenos deslocamentos Stokes é de difícil detecção, enquanto que moléculas com grandes deslocamentos Stokes tem fluorescência mais facilmente detectável.2
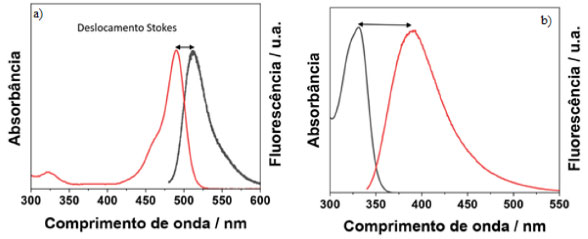 Figura 1. Espectros de emissão e absorção de um fluoróforo com (a) pequeno deslocamento Stokes e outro (b) com grande deslocamento Stokes
Perdas de energia entre excitação e emissão sempre são observadas para moléculas fluorescentes em solução. Uma das ocorrências comuns do deslocamento Stokes é o decaimento rápido para o nível vibracional mais baixo de S1. Os fluoróforos geralmente decaem para níveis vibracionais mais altos de S0, o que resulta em perda adicional de energia de excitação por meio da termalização do excesso de energia vibracional. Outros efeitos também podem fazer com que os fluoróforos exibam mais alterações do deslocamento Stokes, como efeitos do solvente, reações do estado excitado, formação de complexos e/ou transferência de energia.4 Espectrofluorímetro O esquema de um espectrofluorímetro convencional pode ser observado no material suplementar (Figura 2S). Considerando um instrumento ideal de um espectrofluorímetro, os espectros de emissão registrados representam a intensidade emitida ou a taxa de emissão de fótons em cada comprimento de onda, ao longo de um intervalo predeterminado pelas larguras das fendas e pelo monocromador de emissão. Ele é obtido experimentalmente excitando o analito em um determinado comprimento de onda e a fluorescência deste analito é coletada pelo monocromador de emissão ao longo de uma faixa selecionada. De modo equivalente, o espectro de excitação representa a dependência da intensidade de emissão do fluoróforo em um dado comprimento de onda de emissão em função do comprimento de onda de excitação.4,10 Embora a técnica seja relativamente simples, existem diversos parâmetros que precisam ser controlados a fim de se obter um espectro livre de artefatos experimentais, os quais são efeitos instrumentais não relacionados à amostra, e que se manifestam no espectro obtido. Por exemplo, a intensidade da lâmpada de xenônio depende do comprimento de onda e, por isso, o espectro de excitação precisa ser corrigido para que as diferentes intensidades não distorçam a forma do espectro. Controle da largura das fendas de excitação e emissão, correção da resposta da fotomultiplicadora em função do comprimento de onda, e concentrações adequadas do fluoróforo são outros parâmetros experimentais que ajudam a suprimir distorções do espectro medido. Muitas dessas informações experimentais não são bem difundidas na área, permitindo que usuários menos experientes possam fazer conclusões equivocadas sobre a análise de um determinado material. Desta forma, neste trabalho visou-se avaliar diversos parâmetros experimentais em um espectrofluorímetro como comprimento de onda de excitação, fendas de excitação e emissão, passo, tempo de integração, geometria de detecção, bem como a concentração do analito, e avaliar o efeito destes parâmetros no espectro obtido, utilizando moléculas orgânicas fluorescentes como compostos de referência. Além disso, foi analisado também uma amostra de nanopartículas inorgânicas, pontos quânticos de telureto de cádmio (CdTe), como um exemplo de amostra mais complexa. Neste caso, foi possível mostrar o espectro de excitação do nanomaterial estudado, coletando emissões em diferentes comprimentos de onda, bem como analisando a distribuição de tamanho de partícula.
PARTE EXPERIMENTAL Reagentes Cloreto de cádmio monohidratado (99%, Vetec); hidróxido de sódio (97%, Synth); telureto de sódio (99%, Sigma-Aldrich); ácido 3-mercaptopropiônico (99%, Sigma-Aldrich); borohidreto de sódio (97%, Nuclear); L-glutationa reduzida (98%, Sigma-Aldrich); pireno (98%, Sigma-Aldrich); rodamina 6G (98%, Sigma-Aldrich); sulfato de quinina (98%, Sigma-Aldrich) e fluoresceína (98%, Sigma-Aldrich). Espectrofluorímetro e espectrofotômetro O espectrofluorímetro utilizado neste trabalho foi um Horiba Jobin Yvon, Fluorolog-3. A fonte de excitação consiste de uma lâmpada de xenônio de 450 W. Uma representação esquemática do exterior do equipamento e o diagrama esquemático do design do fluorímetro utilizado se encontram no material suplementar (Figura 3S). Os espectros de excitação de fotoluminescência foram corrigidos. Para esta correção uma porção do feixe de excitação é direcionado a um fotodetector, o qual registra a intensidade em função do comprimento de onda (IR(λ)). O sinal de fotoluminescência em função dos diferentes comprimentos de onda de excitação (IF(λ)) é então dividido, simultaneamente, por IR(λ). As análises de espectroscopia de absorção molecular UV-Visível foram realizadas em um espectrofotômetro (Shimadzu, UV-2450/2550) com feixe duplo. As análises de espectroscopia de fluorescência e absorção foram feitas em cubetas de quartzo (Hellma) com 10,00 mm de caminho óptico. Experimentos de variação de parâmetros instrumentais Os experimentos desse trabalho foram realizados no espectrofluorímetro Fluorolog-3, sendo que para os experimentos de variação de parâmetros utilizou-se uma amostra de pireno 0,5 µmol L-1 em tolueno e outra amostra de rodamina 6G 0,5 µmol L-1 em água. Para o experimento de variação da fenda de emissão, foram realizadas 5 variações no tamanho da fenda de emissão, sendo elas: 0,3; 0,5; 1,0; 3,0 e 5,0 nm. Em todos os casos a fenda de excitação foi fixada em 0,3 nm e o passo fixado em 0,4 nm. A fim de melhorar a relação sinal ruído dos espectros, variou-se o tempo de integração dessas análises, sendo que na utilização da fenda de emissão em 0,3 nm foi mantido o tempo de integração em 0,8 s; para a fenda de emissão em 0,5 nm o tempo de integração foi fixado em 0,4 s; já para as fendas de emissão em 1,0, 3,0 e 5,0 nm, o tempo de integração foi mantido em 0,1 s. Estudando-se a fenda de excitação, foram feitas 3 variações no tamanho da mesma, sendo elas: 1,0; 3,0 e 5,0 nm, sendo que para todos os casos, a fenda de emissão foi fixada em 0,3 nm e o passo foi fixado em 0,2 nm. O tempo de integração utilizado foi 0,2 s, em todos os casos. Para o estudo do passo, foram realizadas duas variações, sendo elas 0,2 e 1,0 nm. Mantendo-se a fenda de emissão em 1,0 nm, a fenda de excitação em 3,0 nm e o tempo de integração em 0,1 s. Todas as variações citadas acima foram realizadas tanto para o pireno, varrendo o espectro de emissão de 330 à 500 nm, quanto para a rodamina 6G, tendo o espectro de emissão varrido de 460 à 680 nm. Experimento da avaliação do efeito de concentração e extinção Para este experimento, utilizou-se uma amostra de sulfato de quinina 1,0 µmol L-1 e outra amostra de rodamina 6G 0,5 µmol L-1, ambos com o solvente sendo a água. A fim de se obter diversas concentrações das amostras, foram realizadas algumas diluições com alguns valores de absorbância desejados que variaram de 0,03 até 1,20 no topo da banda de absorção. Os espectros de absorção foram registrados com um espectrofotômetro sendo utilizadas diluições sucessivas para preparo das soluções para densidades ópticas de interesse. Para o experimento utilizando-se o espectrofluorímetro, foram obtidos os espectros de fluorescência sendo utilizados os seguintes parâmetros para as amostras de rodamina 6G: a excitação em 346 e 510 nm; fenda de excitação de 1 nm; o monocromador de emissão foi variado de 515 até 690 nm; fenda de emissão de 0,3 nm; passo de 0,5 nm e tempo de integração de 0,1 s. Para as amostras de sulfato de quinina, os parâmetros foram: excitação em 300 e 330 nm; fenda de excitação de 2 nm; a emissão foi varrida de 340 até 550 nm; fenda de emissão de 0,3 nm; passo de 0,5 nm com tempo de integração de 0,1 s. Sendo que para ambas as amostras, o experimento foi realizado utilizando a geometria de detecção face frontal (FF) e ângulo reto (RA). Experimento utilizando espectros 3D Foram utilizadas amostras dos corantes fluoresceína 0,5 µmol L-1, sulfato de quinina 1,0 µmol L-1 e pireno 0,5 µmol L-1, sendo que também foram sintetizados e utilizados pontos quânticos de CdTe para a execução desse experimento, com suspensões com densidade óptica de 0,3 em 400 nm. Os espectros 3D foram gerados no espectrofluorímetro Fluorolog-3, de forma automática e os parâmetros utilizados para as análises das amostras se encontram na Tabela 1S do material suplementar. Síntese dos pontos quânticos de CdTe/MPA e CdTe/GSH Os pontos quânticos (PQs) de telureto de cádmio (CdTe) foram sintetizados via rota coloidal. Para execução deste procedimento foi utilizado 0,4 mmol (0,0805 g) de CdCl2.H2O, o qual foi solubilizado em 80 mL de água deionizada obtida do equipamento Milli-Q Direct 8/16 System da Millipore, em um béquer. Posteriormente, foram adicionados 0,8 mmol (69,8 µL) do ligante ácido 3-mercaptopropiônico (MPA) para a síntese de CdTe/MPA e 0,163 g do ligante glutationa reduzida (GSH) para a síntese de CdTe/GSH. Em seguida, o pH da solução foi ajustado para 10 com hidróxido de sódio (1,0 mol L-1). Nesta mesma solução, foram acrescentados 0,4 mmol (0,00886 g) de Na2TeO3 e 1 mmol (0,0378 g) de NaBH4. Essa mistura foi transferida para balão de três bocas e aquecida à 100 ºC por um sistema controlado por refluxo, a fim de manter a concentração constante. Quando a temperatura atingiu os 100 ºC, foram cronometrados 30 min de tempo de síntese.11
RESULTADOS E DISCUSSÃO Variação de parâmetros utilizados no espectrofluorímetro Ao se trabalhar com um espectrofluorímetro é comum se deparar com alguns parâmetros que podem ser alterados para que haja um melhor resultado na análise a ser realizada. Dentre os parâmetros, variou-se e estudou-se diferentes larguras de fenda de excitação e de emissão. Também foi avaliado a resolução espectral do monocromador, que resumidamente é definida como o número de passos em uma determinada faixa de comprimento de onda analisada pelo equipamento. E por fim, outro parâmetro a ser citado, porém, não sendo analisado de forma individual foi o tempo de integração, sendo este, o tempo em que o equipamento acumula o sinal em um determinado comprimento de onda. Para mostrar a influência da fenda de emissão e excitação, alem da resolução espectral do monocromador, foram escolhidas duas moléculas, uma com bandas vibrônicas estruturadas e outra com banda não estruturada. Usou-se então, o pireno e a rodamina 6G, respectivamente. Na Figura 2(a) são apresentados os espectros de emissão do pireno, variando-se apenas a fenda de emissão, sendo que a fenda de excitação foi fixada em 0,3 nm e o passo fixado em 0,2 nm. As intensidades foram normalizadas, isto é, as intensidades foram divididas pelo valor máximo da emissão do espectro, para fins de comparação. Quanto menor a fenda de emissão, melhor a resolução das bandas. Essa melhor resolução permite identificar cada uma das bandas separadamente uma das outras. Como desvantagem, quanto menor a fenda, menor a relação sinal ruído. No entanto, essa relação sinal ruído pode ser melhorada aumentando-se o tempo de integração. Foi observado que a largura a meia altura do espectro não se alterou com as variações nos tamanhos das fendas.
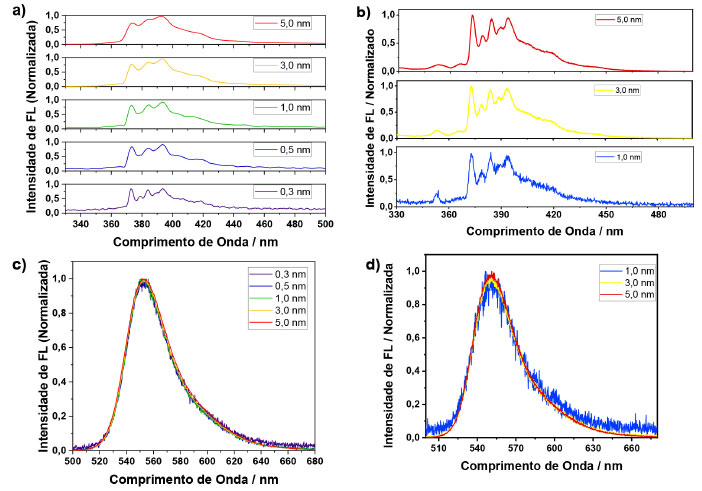 Figura 2. Espectros de emissão de uma amostra de pireno com (a) diferentes fendas de emissão e (b) variando-se a fenda de excitação. Espectros de emissão de uma amostra de rodamina 6G, (c) variando-se a fenda de emissão e (d) variando a fenda de excitação
Na Figura 2(b) são apresentados os espectros de emissão do pireno, fixando-se a fenda de emissão em 0,3 nm e o passo fixado em 0,2 nm, variando-se somente a fenda de excitação. Observou-se que a fenda de excitação apenas influenciou na relação sinal ruído do espectro de fluorescência, não afetando a resolução das bandas vibrônicas da molécula. Como conclusão dos experimentos variando-se as fendas de excitação e emissão, notou-se que a fenda de excitação é um parâmetro muito mais robusto do que a fenda de emissão. Muitas moléculas orgânicas não possuem bandas vibrônicas tão bem estruturadas quanto o pireno, e, portanto, poderiam ser menos suscetíveis a variação espectral dependendo da fenda utilizada. Pensando nisso, foi empregada a rodamina 6G, a qual não apresenta bandas vibrônicas definidas, utilizando-se a mesma metodologia. A Figura 2(c) apresenta os espectros de emissão da rodamina 6G, variando-se apenas a fenda de emissão, sendo que a fenda de excitação foi fixada em 0,3 nm e o passo fixado em 0,2 nm. Observou-se que, por ser um composto que justamente não possui bandas estruturadas, não houve mudança na forma do espectro, nem na largura à meia altura, porém houve uma diminuição na relação sinal ruído conforme diminuiu-se o tamanho da fenda de emissão, o que poderia ter sido reparado aumentando-se ainda mais o tempo de integração para fendas menores. Na Figura 2(d) são apresentados os espectros de emissão da rodamina 6G, sendo fixada a fenda de emissão em 0,3 nm e fixando o passo em 0,2 nm, variando-se somente a fenda de excitação. O resultado observado foi o mesmo obtido para o pireno, uma vez que, com o aumento da fenda de excitação, aumentou apenas a relação sinal ruído. Demonstrando-se menor influência sobre a forma espectral da molécula. Experimentos variando o parâmetro passo também foram realizados tanto para o pireno quanto para a rodamina 6G, não obtendo resultados tão significativos quanto os outros parâmetros estudados e os espectros podem ser observados no material suplementar (Figura 5S e 6S). Deste modo, quando é desejado aumentar a relação sinal ruído em um espectro de emissão, é importante que apenas a fenda de excitação seja aumentada, mantendo-se a fenda de emissão fixa, a fim de se evitar distorções espectrais que são meros artefatos de medida. Vale ressaltar que a largura espectral de um material carrega informações físico-químicas. Por exemplo, nanopartículas (NPs) apresentam espectros de emissão dependentes do tamanho, devido ao efeito de confinamento quântico.12 Considerando um ensemble com uma distribuição de partículas, é razoável considerar que a largura espectral esteja relacionada com a distribuição de tamanhos das nanopartículas. Portanto, evitar alargamento espectral devido a artefato experimental é de extrema importância para a caracterização do sistema. Com relação ao espectro do pireno, foi observado que um espectro registrado com uma fenda larga (> 0,5 nm) resulta em um espectro com baixa resolução, no qual as bandas vibrônicas da molécula não são resolvidas apropriadamente. Interações intermoleculares podem afetar largura do espectro e a presença ou ausência de uma banda vibrônica específica. Portanto, a fim de se extrair informações físico-químicas relevantes de uma dada molécula, o espectro deve ser medido de forma adequada. Estudo do efeito da concentração sobre a intensidade de fotoluminescência e processos de extinção Nesta etapa foi avaliado o efeito da concentração sobre a intensidade de fotoluminescência. Ao se realizar análises de fluorescência é esperado a ocorrência de processos de extinção (quenching) da fluorescência. Estes processos são diversos tipos de problemas que causam a diminuição da intensidade de fluorescência de uma substância. Uma indicação desse fenômeno é a falta de linearidade da intensidade de fluorescência com a concentração do fluoróforo, enquanto a absorbância é linearmente proporcional à concentração. Existem diversos tipos de processos de extinção, dentre eles, o efeito de filtro interno. Neste efeito, a extinção ocorre por conta da atenuação do feixe incidente ao longo do caminho óptico, o qual tem relação com a absorbância da amostra no comprimento de onda de excitação. Em soluções muito diluídas, tem-se absorbâncias muito baixas, e a intensidade de fluorescência é proporcional à concentração do fluoróforo. Nestas condições, a atenuação da luz incidente ao longo do caminho óptico pode ser desprezível. Já em concentrações altas, uma parte significativa da luz incidente é absorvida, e progressivamente, atenuada ao longo do caminho óptico. Deste modo a luz incidente pode ser extinguida antes de chegar ao ponto da cubeta onde a fluorescência é detetada. Por estas razões, percebe-se que este processo é apenas um artefato experimental. Quando há um aumento da concentração da amostra, tem-se não só o efeito de filtro interno, mas também interações intermoleculares. Estas interações são processos de transferência de energia radiativos (reabsorção) e não-radiativos (interação de dipolo),13 os quais têm relação com a sobreposição dos espectros de absorção e emissão da molécula.14 A diferença entre os dois está na distância entre o doador e o aceptor de energia, quando essa distância é curta, de até 20 nm, o processo ocorre de forma não radiativa, não existindo a presença de um fóton envolvido no sistema. Por outro lado, quando a distância é da mesma ordem que o fóton emitido pelo doador, o processo acontece de forma radiativa.15 Este último processo é bem simples, a emissão gerada pelo doador causa a excitação do aceptor, levando-o para o estado excitado.10 Como processos de reabsorção envolvem um fóton, este efeito distorce o espectro de emissão do doador na região de sobreposição com o espectro do aceptor. No caso da transferência de energia não radiativa, acontece uma interação do tipo dipolo-dipolo: o doador no estado excitado vai decair para o estado fundamental por indução do dipolo que existe quando o aceptor se encontra no estado fundamental, levando este último para o estado excitado sem a presença de um fóton. Devido à natureza do mecanismo, o espectro de emissão não é afetado. Este mecanismo foi teorizado por Förster, e normalmente, é chamado de transferência de energia de Förster.16 Na situação de altas concentrações do fluoróforo, a espécie doadora e aceptora são duas moléculas de um mesmo fluoróforo, uma no estado excitado outra no estado fundamental, que interagem entre si, produzindo uma homo transferência de energia. Outros dois tipos de processos de extinção intermolecular que podem ocorrer são aqueles de natureza dinâmica e estática. A extinção dinâmica (dynamicquenching, em inglês) é quando uma molécula se encontra no estado excitado, e durante o tempo que ela está no estado excitado até ela decair para o estado fundamental, uma outra molécula pode difundir no meio, colidindo e desativando o estado excitado da primeira. A extinção estática ou quenching estático é a associação de duas moléculas no estado fundamental, e que não produzem fluorescência quando são fotoexcitadas.4 A próxima etapa então foi a de avaliar o efeito da concentração sobre a intensidade de fotoluminescência. Para isso, usou-se duas amostras, uma com curto e outra com longo deslocamento Stokes: rodamina 6G e sulfato de quinina, respectivamente. Além disso, variou-se o comprimento de onda de excitação e a geometria de detecção do sistema. Foram avaliados os comprimentos de onda de excitação e a geometria de detecção para as amostras de sulfato de quinina, utilizando as geometrias face frontal (FF) e ângulo reto (RA), com comprimentos de onda de excitação em 300 e 330 nm, sendo que os espectros de fluorescência e absorção estão presentes no material suplementar (Figuras 7S e 8S). Em seguida, foi verificado a linearidade da absorbância com a emissão das amostras. Para a realização dos ajustes matemáticos, ajustou-se uma equação linear para os três primeiros pontos, e extrapolou-se essa reta a fim de se observar o desvio da linearidade dos dados experimentais com o aumento da concentração. A fim de garantir a efetividade desta estratégia, este procedimento foi também empregado nos espectros de absorção, os quais seguem perfeitamente um comportamento linear com a concentração, Figuras 8S e 11S (material suplementar). Portanto, desvios da linearidade apresentados nas Figuras 3 e 4 são, de fato, devido à extinção da fluorescência. A área dos espectros foi integrada e plotou-se a área integrada em função da absorbância no comprimento de onda de excitação, variando-se a geometria de detecção e o comprimento de onda de excitação. As Figuras 3(a) e (b) representam os dados com geometria face frontal com comprimentos de onda de excitação em 300 e 330 nm, respectivamente. As Figuras 3(e) e (d) representam os dados com geometria ângulo reto com comprimentos de onda de excitação em 300 e 330 nm, respectivamente.
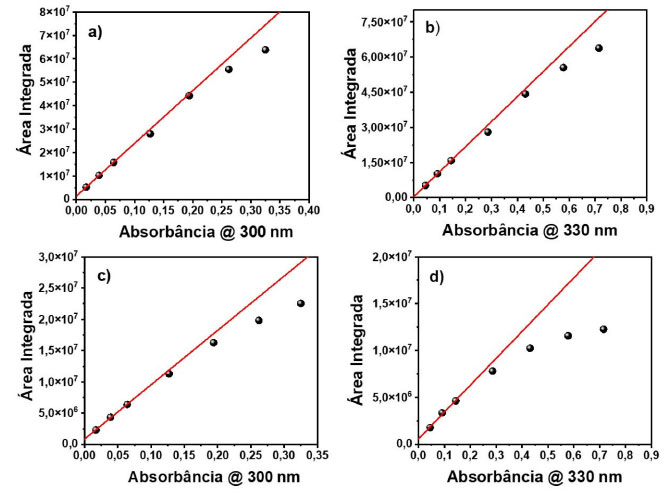 Figura 3. Gráficos da área integrada em função da absorbância com comprimento de onda de excitação em 300 nm e geometria FF e RA (a, c), comprimento de onda de excitação em 330 nm e geometria FF e RA (b, d) de amostras de sulfato de quinina
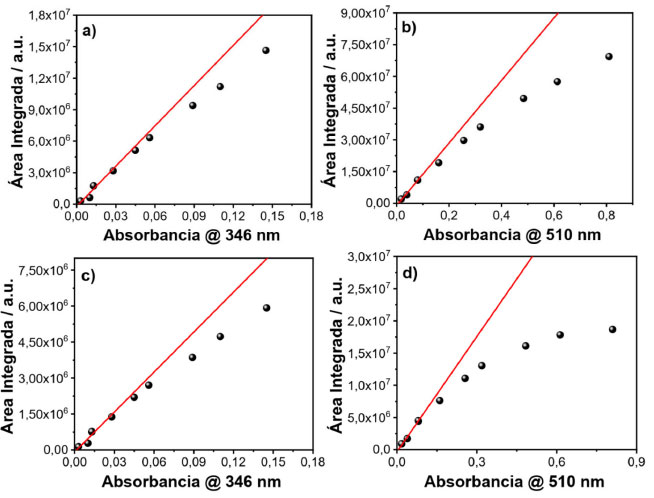 Figura 4. Gráficos da área integrada em função da absorbância com comprimento de onda de excitação em 346 nm e geometria FF e RA (a, c), comprimento de onda de excitação em 510 nm e geometria FF e RA (b, d) para amostras de rodamina 6G
Ao se comparar as áreas integradas de fotoluminescência em função da absorbância das soluções com geometria RA e FF, é possível perceber que para a geometria RA acontece uma pronunciada perda da linearidade em relação à geometria FF. Isso pode ser explicado pelo fato de que com a geometria RA pode estar havendo dois processos de quenching, filtro interno e interação intermolecular. Na geometria FF não acontece efeito de filtro interno, pois nessa geometria anula-se este efeito pelo fato de a detecção ocorrer na face da cubeta, podendo estar ocorrendo apenas o efeito de interação intermolecular: transferência de energia (radiativa ou não radiativa), quenching dinâmico ou estático. Como a transferência de energia, radiativa ou não-radiativa, envolve sobreposição espectral, não é esperado que ela seja dominante neste sistema, uma vez que a sobreposição entre absorção e emissão do sulfato de quinina é muito pequena. A fim de confirmar esta previsão foi realizado um experimento e os espectros de emissão em diferentes concentrações foram normalizados e plotados na Figura 9S (material suplementar).17 O mesmo procedimento variando a geometria de detecção e comprimento de onda de excitação foi empregado para rodamina 6G, corante com menor deslocamento Stokes e os espectros de emissão e absorção podem ser observados no material suplementar (Figura 10S). Em seguida, as áreas dos espectros (igualmente ao que foi feito para o sulfato de quinina) foram integradas e plotou-se a área integrada em função da absorbância, variando-se a geometria de detecção e o comprimento de onda de excitação. As Figuras 4(a) e (b) representam os dados com geometria FF com comprimentos de onda de excitação em 346 e 510 nm, respectivamente. As Figuras 4(c) e (d) representam os dados com geometria RA com comprimentos de onda de excitação em 346 e 510 nm, respectivamente. Comparando-se os espectros com geometria RA e FF, percebe-se que para a geometria RA, há uma maior perda da linearidade em relação à geometria FF, devido à geometria RA favorecer o efeito de filtro interno. Na geometria FF o efeito de filtro interno é mínimo e apenas efeitos de interação intermolecular estão envolvidos. A perda da linearidade ocorre de forma mais pronunciada com esta amostra em relação ao sulfato de quinina, pois como a rodamina 6G possui um curto deslocamento de Stokes, efeitos de reabsorção e interação de dipolos são mais pronunciados. Além de todos os quenchings citados, os efeitos de quenching dinâmico e estático também podem estar envolvidos nos resultados observados, no entanto, em uma menor extensão. A fim de demonstrar se o efeito de interação intermolecular está realmente acontecendo, utilizou-se os dados da rodamina 6G com geometrias FF e RA com comprimento de onda de excitação em 346 nm, e fez-se as normalizações das intensidades em função do comprimento de onda, os quais podem ser vistos no material suplementar (Figura 12S(a) e (b)). Com o aumento da concentração de rodamina 6G, foi observado uma supressão dos fótons de maior energia no espectro de emissão do corante, região de sobreposição com o espectro de absorção. Este efeito indica que reabsorção pode ser a origem do processo de quenching. Neste exemplo, foram utilizadas as duas geometrias de detecção, e como pode ser observado, a geometria RA favorece não somente maior o efeito de filtro interno, como indicado nas curvas da Figura 4, como também, efeitos de reabsorção, o qual é proporcional ao caminho óptico. Portanto, os diversos experimentos realizados sugerem que para se reduzir o efeito de filtro interno em uma medida de espectroscopia de fluorescência, existem duas possibilidades: mudança do comprimento de onda de excitação ou na geometria de detecção do sistema, como mostrado nas Figuras 3 e 4. No entanto, dependendo do deslocamento Stokes da amostra, a excitação em outro comprimento de onda pode não ser tão efetiva quanto a geometria FF para remoção do efeito de filtro interno e redução do efeito de reabsorção. Espectro de excitação 3D em corantes e PQs de CdTe A criação de espectros 3D também é uma funcionalidade que pode ser obtida em um espectrofluorímetro. Neste tipo de experimento ambos comprimentos de onda de excitação e emissão são variados. Com isso, é possível obter um espectro de excitação ou um espectro de emissão de uma determinada amostra. O objetivo de se fazer um espectro 3D é observar a dependência do espectro de emissão com as diferentes energias de excitação. De acordo com a regra de Kasha, o espectro de emissão de um fluoróforo não depende do comprimento de onda de excitação, uma vez que a fluorescência irá ocorrer a partir do nível vibracional mais baixo do estado excitado. Portanto, a dependência do espectro de emissão com o comprimento de onda de excitação indica a presença de uma mistura de fluoróforos no ensemble. Considerando a situação que existe somente um fluoróforo, o espectro de excitação corrigido será igual ao espectro de absorção. Entretanto, diferentemente do espectro de absorção que mede todo o ensemble e registra a média da absorbância em termos do coeficiente de absortividade molar (ε) e da concentração (c) de cada espécie i (Equação 1), o espectro de excitação permite registrar a variação da intensidade de emissão de um específico fluoróforo com o comprimento de onda de excitação, dando muito mais seletividade para a técnica. 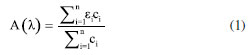 Em termos de otimização do espectro, caso a intenção seja utilizar o espectro 3D para obter um espectro de excitação com alta resolução, é aconselhado diminuir a fenda de excitação e aumentar a fenda de emissão. Caso o objetivo seja um espectro de emissão com alta resolução, basta diminuir a fenda de emissão e aumentar a fenda de excitação. Ou seja, é necessário melhorar a resolução espectral daquilo que se deseja obter. No material suplementar (Figura 13S) pode-se ver exemplos de espectros 3D coletados direto de um espectrofluorímetro, porém esses dados depois de tratados podem ser transformados em um espectro 2D comum. Neste estudo, trabalhou-se apenas com espectros de excitação. O objetivo foi exemplificar o poder da técnica diante de uma amostra complexa composta por diversos fluoróforos, como uma dispersão coloidal de PQs de CdTe. Inicialmente, foram estudados alguns corantes mais simples como a fluoresceína, sulfato de quinina e pireno para se demonstrar o funcionamento da técnica. Utilizando a amostra de fluoresceína e observando-se o espectro de emissão desse corante, Figura 5(a), foi registrado o espectro de excitação, coletando a emissão em diferentes pontos. Os comprimentos de onda de emissão coletados foram 490, 510, 530, 600 e 630 nm, e o resultado pode ser visto na Figura 5(b). Seguindo o mesmo procedimento, foi caracterizada uma solução de sulfato de quinina. O espectro de emissão e absorção dessa amostra pode ser visto na Figura 5(c). As emissões foram coletadas nos comprimentos de onda em 350, 370, 390, 450, 480, 500 e 520 nm e o espectro de excitação gerado pode ser visto na Figura 5(d). Inicialmente ocorre a mesma sobreposição observada para a fluoresceína, porém acima de 360 nm começa a surgir uma calda no espectro, a qual pode ser atribuída a dímeros de sulfato de quinina.18
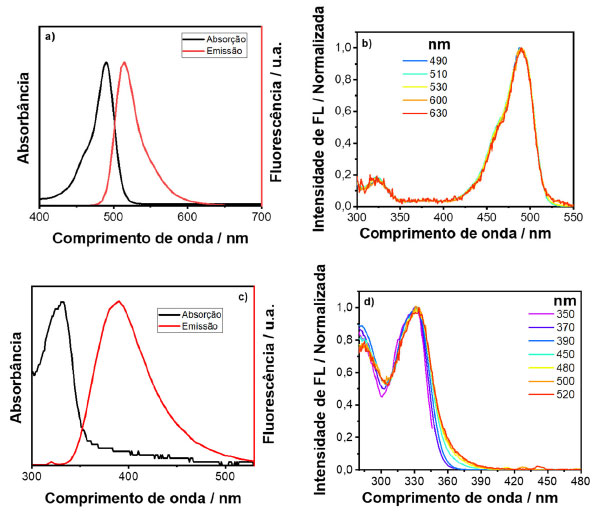 Figura 5. (a, c) Espectros de absorção e emissão para uma solução aquosa de fluoresceína e sulfato de quinina, respectivamente; (b, d) espectros de excitação gerados pelo espectro 3D para uma solução aquosa de fluoresceína e sulfato de quinina, respectivamente (nota: os comprimentos de onda de emissão provados estão indicados no inset das figuras)
Analisando os resultados obtidos, observou-se que todos os espectros de excitação, independente da seleção do comprimento de onda de emissão, se sobrepõem, indicando que todo o espectro de emissão se origina de uma mesma molécula. Na Figura 6(a) é apresentado o espectro de emissão e absorção do pireno. As emissões foram coletadas em 370, 390, 410, 430, 450 e 470 nm e o espectro de excitação gerado pode ser observado na Figura 6(b). Para este corante, basicamente houve uma sobreposição inicial dos espectros para comprimentos de onda de emissão provados abaixo de 430 nm. Vale ressaltar que o espectro de excitação revelou bandas de absorção que não estão presentes no espectro de absorção, por exemplo, a banda 1. A ausência dessa banda no espectro de absorção é devido a contribuição de várias transições que acontecem em energias próximas que se sobrepõem e não são resolvidas no espectro. Além disso, espalhamento Rayleigh dificulta a identificação de bandas localizadas na região do UV em um experimento de absorção no modo de transmissão, uma vez que a intensidade de espalhamento é proporcional ao inverso do comprimento de onda elevado à quarta potência (Iespalhamento α 1/λ4).
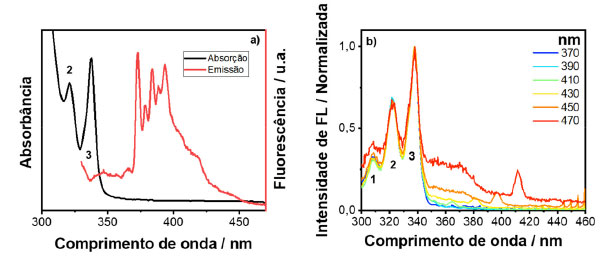 Figura 6. (a) Espectros de absorção e emissão; (b) espectros de excitação gerados pelo espectro 3D para uma solução aquosa de pireno (nota: os comprimentos de onda de emissão provados estão indicados no inset das figuras)
Ao analisar o espectro de excitação para energias maiores que 350 nm, observa-se o aparecimento de uma banda larga característica da formação de exciplex, o qual constitui da associação de uma molécula de pireno no estado excitado e outra no estado fundamental.4,19 A banda de absorção do exciplex só é possível de se observar no espectro de excitação, pois sua formação envolve uma espécie que se encontra no estado excitado. As bandas observadas em 400 e 420 nm são resultados de espalhamento Raman do solvente. Portanto, este exemplo demonstra o poder superior de um espectro de excitação de fotoluminescência em relação a um espectro de absorção. A nanopartícula escolhida para o estudo foi um ponto quântico de telureto de cádmio (CdTe) coloidal, em que as nanopartículas inorgânicas estão recobertas por moléculas de ácido 3-mercaptopropiônico (MPA), como ligantes de superfície. Os espectros de emissão e absorção do CdTe/MPA coloidal em meio aquoso podem ser vistos na Figura 7(a). Assim como feito para os corantes, as emissões foram provadas em alguns pontos, sendo eles 500, 520, 540, 560, 580, 600, 650 e 700 nm. Os espectros de excitação gerados podem ser observados na Figura 7(b).
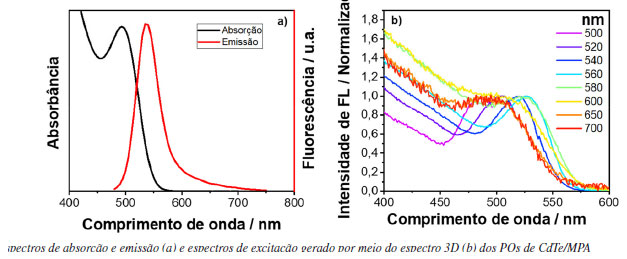 Figura 7. Espectros de absorção e emissão (a) e espectros de excitação gerado por meio do espectro 3D (b) dos PQs de CdTe/MPA
No caso dos PQs de CdTe, quando se prova a excitação em diferentes comprimentos de onda de emissão é possível fazer a distribuição de tamanhos de partículas do material, sendo que quanto mais deslocado para menor comprimento de onda, menor o tamanho do ponto quântico.20 Este tipo de resultado é semelhante ao que é obtido por meio de fluorescência de partícula única.21 Todavia, o que foi observado é que quando se monitora a emissão na região que possui bandas de armadilha (região acima de 600 nm), a qual vêm de uma distribuição de estados que se localiza abaixo do gap do material,22 essa região tem maior contribuição de partículas menores, pois partículas menores são mais suscetíveis a defeitos de superfície devido à alta relação de superfície por volume. Por isso, pode-se observar que o espectro de excitação provado na região de armadilha tem características de PQs de tamanhos menores, e por isso, o espectro se desloca para maiores energias
CONCLUSÕES Ao final da realização do presente trabalho foi possível avaliar, com sucesso, as diferentes variáveis que influenciam um espectro de fluorescência e suas funções, bem como o experimento de fluorescência. Ao que diz respeito ao estudo da variação dos parâmetros do espectrofluorímetro, foi observado que para a fenda de emissão, quanto maior o tamanho da fenda, pior é a resolução espectral obtida, ou seja, para se obter melhores resultados, deve-se usar fendas de emissão menores. A consequência do aumento da resolução do espectro é a perda da relação sinal-ruído, porém essa desvantagem é facilmente corrigida aumentando-se o tempo de integração ou o aumento na fenda de excitação, desde que não necessite de precisão no comprimento de onda de excitação. O parâmetro do passo não mostrou ser tão sensível para as amostras de corantes à temperatura ambiente. Na etapa de avaliação do efeito da concentração sobre a intensidade de fotoluminescência, observou-se que para um material com curto deslocamento Stokes (rodamina 6G) houve um pronunciado efeito de interação intermolecular comparado com um material que tem um grande deslocamento Stokes (sulfato de quinina). Esse resultado já era esperado, pois a sobreposição espectral gerada pelos espectros de absorção e emissão de uma amostra aumenta, consideravelmente, a interação intermolecular tanto com transferência de energia radiativa (reabsorção), quanto com transferência não-radiativa (interação de dipolo). Utilizando a geometria de detecção face frontal, foi possível corrigir o efeito de filtro interno, diminuindo perdas na intensidade de fluorescência emitida do material, mostrando ser mais eficaz na correção desse quenching do que a geometria de detecção em ângulo reto. Quando foi variado o comprimento de onda de excitação, observou-se maiores efeitos de filtro interno quando excitado em uma região de maior absorção, resultando em maiores perdas de linearidade da concentração com a absorbância. Sendo que o mesmo foi observado tanto para a amostra com grande deslocamento Stokes, quanto para a amostra com curto deslocamento Stokes. Esta estratégia pode ser utilizada para amostras concentradas, no entanto, quando o deslocamento Stokes é pequeno, efeitos de reabsorção são ainda visíveis na geometria RA. A utilização de espectros 3D para gerar espectros de excitação também teve resultados importantes, pois foi possível mostrar o espectro de excitação de materiais, coletando emissões em diferentes comprimentos de onda, podendo mostrar a natureza das emissões observadas. No caso das moléculas orgânicas fluorescentes foi possível demonstrar a contribuição de exciplex na solução de pireno e resolver outras transições que não foram evidentes por meio do espectro de absorção. Além de que, no estudo utilizando-se PQs de CdTe, foi possível fazer a distribuição de partículas do material, podendo observar até a influência das bandas de armadilhas em partículas menores, devido a maior susceptibilidade a defeitos de superfície que essas partículas menores possuem. Em suma, a técnica de fluorescência é uma poderosa ferramenta para se investigar interações intermoleculares em sistemas complexos, desde a formação de dímeros de moléculas orgânicas até mesmo para processos de armadilhamento de portadores de carga em nanocristais.
MATERIAL SUPLEMENTAR Este trabalho contém material suplementar. Nele estão presentes informações que vão desde partes da introdução de forma a complementar o texto principal, até diversos resultados contendo imagens, gráficos e espectros que não foram possíveis de serem adicionados no texto principal, mas que também são de grande importância para o trabalho. Podendo ser acessado em:
AGRADECIMENTOS Os autores agradecem à CAPES, ao CNPq, à FINEP e à FAPEMIG pelo apoio recebido.
REFERÊNCIAS 1. Albani, J. R.; Principles and Applications of Fluorescence Spectroscopy, 1st ed.; Wiley Blackwell: New Jersey, 2007. 2. Pavoni, J. F.; Neves-Junior, W. F. P.; Spiropulos, M. A.; de Araújo, D. B.; Rev. Bras. Ensino Fis. 2014, 36, 1. [Crossref] 3. Wahab, M. F.; J. Chem. Educ. 2007, 84, 1308. [Crossref] 4. Lakowicz, J. R.; Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, 2006. 5. Crouch, S. R.; Holler, F. J.; Skoog, D. A.; Principles of Instrumental Analysis, 7th ed.; Cengage Learning: São Paulo, 2016. 6. Vaz, R.; Bettini, J.; Júnior, J. G. F.; Lima, E. D. S.; Botero, W. G.; Santos, J. C. C.; Schiavon, M. A.; J. Photochem. Photobiol. , A 2017, 346, 502. [Crossref] 7. Sotomayor, M. D. P. T.; Dias, I. L.; Lanza, M. R. V.; Moreira, A. B.; Kubota, L.; Quim. Nova 2008, 31, 1755. [Crossref] 8. Hinshelwood, C. N.; Physical Chemistry, 2nd ed.; Brooks Cole: California, USA, 1945. 9. Faleiros, M. M. : Fotoluminescência Excitada no Ultravioleta em Polímeros Conjugados; Dissertação de Mestrado, Universidade de São Paulo, São Paulo, Brasil, 2007. [Link] acessado em Agosto 2023 10. Valeur, B.; Molecular Fluorescence: Principles and Applications, 1st ed.; Wiley-VCH: Weinheim, 2002. 11. Vale, B. R. C. : Exciton and Charge Carrier Dynamics in Colloidal Semiconductor Nanocrystals; Tese de Doutorado, Universidade Federal de São João del-Rei, São João del-Rei, Brasil, 2017. 12. Alivisatos, A. P.; Science 1996, 271, 933. [Crossref] 13. Wan Ismail, W. Z.; Goldys, E. M.; Dawes, J. M.; Appl. Phys. B: Lasers Opt. 2016, 122, 1. [Crossref] 14. Förster, T.; Discuss. Faraday Soc. 1959, 27, 7. [Crossref] 15. Tartuci, L. G.; Domingos, L. F. T.; Bettini, J.; Vieira, K. O.; Raphael, E.; Vale, B. R. C.; Ferrari, J. L.; Schiavon, M. A.; J. Nanopart. Res. 2017, 19, 1. [Crossref] 16. Förster, T.; Delocalized Excitation and Excitation Transfer, Florida State University: Tallahassee, 1965. [Link] acessado em agosto 2023 17. Gutow, J. H.; J. Chem. Educ. 2005, 82, 302. [Crossref] 18. Kyker, G. C.; Cornatzer, W. E.; McEwen, M. M.; J. Biol. Chem. 1946, 162, 353. [Crossref] 19. Pensack, R. D.; Ashmore, R. J.; Paoletta, A. L.; Scholes, G. D.; J. Phys. Chem. C 2018, 122, 21004. [Crossref] 20. Janke, E. M.; Williams, N. E.; She, C.; Zherebetskyy, D.; Hudson, M. H.; Wang, L.; Gosztola, D. J.; Schaller, R. D.; Lee, B.; Sun, C.; Engel, G. S.; Talapin, D. V.; J. Am. Chem. Soc. 2018, 140, 15791. [Crossref] 21. Mondal, P.; Chakraborty, S.; Grandhi, G. K.; Viswanatha, R.; J. Phys. Chem. Lett. 2020, 11, 5367. [Crossref] 22. Vale, B. R. C.; Vieira, K. O.; Sousa, J. C. L.; Ferrari, J. L.; Schiavon, M. A.; Quim. Nova 2015, 38, 22. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access