
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Boas práticas e equívocos nas medidas de atividade catalítica de materiais em termocatálise Best practices and pitfalls in the measurements of the catalytic activity of materials in thermo-catalysis |
|
Leandro MartinsI; Laura L. SilvaI; Luiz G. PossatoII,*
I. Departamento de Engenharia, Física e Matemática, Instituto de Química, Universidade Estadual Paulista, 14800-060 Araraquara - SP, Brasil Recebido em 17/03/2023 *e-mail: gustavo.possato@unesp.br As catalysis grows as a multidisciplinary research area, many questions arise regarding the procedures used to determine the activity of materials in different chemical reactions. To correct inconsistencies, whether in definitions, concepts, or experimental methods, some catalysis science journals have been publishing editorials warning less-aware authors. In this work, we present some common concepts and procedures in thermo-catalysis, reviewing good practices and pointing out frequent failures that make it difficult to reliably compare the activity between materials of the same family and results from the scientific literature. INTRODUÇÃO As modificações da estrutura, textura e composição de um material cerâmico têm o objetivo de potencializar suas funcionalidades. Uma das grandes aplicações é em termocatálise. Após a síntese e caracterização, a próxima etapa que indicará o seu potencial como catalisador é o teste de atividade catalítica. É a partir dos resultados destes testes que parâmetros cinéticos importantes tais como conversão, seletividade e ordem de reação são determinados. Buscam-se também informações como desativação e regeneração, sinterização ou lixiviação da fase ativa, correlações de interação metal suporte e sensibilidade da reação à estrutura da fase ativa. O teste catalítico deve ser realizado de modo que se consiga obter a maior quantidade possível de informações confiáveis que permitirão avançar no seu desenvolvimento até que se alcance a aplicação em grande escala. Neste trabalho são apresentadas as informações que podem ser obtidas do estudo catalítico de materiais cerâmicos conduzidos em microrreatores, discutindo os procedimentos e as possíveis falhas de conceitos e procedimentos que podem levar a resultados que dificultam a correta comparação entre eles e também entre resultados da literatura científica. Algumas revistas da área de materiais e catálise vêm publicando editoriais alertando para alguns equívocos na condução de experimentos.1 Há a proposta de redação handbooks em catálise com coletâneas de boas práticas como requisito mínimo para a condução de experimentos padronizados, o que melhoraria a importância e facilitaria a utilização de dados publicados na obtenção dos modelos cinéticos.2 À medida que a catálise cresce como uma área multidisciplinar, esses tutoriais representam um recurso valioso e útil, especialmente para aqueles que estão iniciando no tema ou menos familiarizados com os procedimentos. Definições de "energia de ativação" e "energia de ativação aparente" A Figura 1 mostra que, na presença de um catalisador X, estranho à estequiometria de uma dada reação reversível de R formando P, surge uma nova rota reacional energeticamente mais favorável e o processo é mais rápido. A reação catalítica é um conjunto de etapas elementares. O catalisador intervém em algumas destas etapas e é recuperado ao final do ciclo para interagir com uma nova molécula de reagente R. Em decorrência da definição, pode-se supor que o ciclo catalítico seja interminável. No entanto, na prática, isto não é verificado, pois em maior ou menor grau ocorre a sua desativação. Neste caso, há a necessidade de regeneração ou substituição do catalisador.
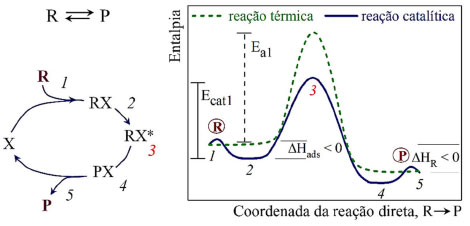 Figura 1. Ciclo catalítico, em que R se adsorve no catalisador X, formando o complexo ativado RX* e convertendo-se em P; e diagrama de entalpia versus coordenada de reação para uma reação térmica e catalítica (mostrado o sentido da reação direta R → P)
No diagrama de entalpia-coordenada de reação da Figura 1 é mostrado o percurso de uma reação química térmica (linha tracejada) em que a entalpia do reagente R está acima da entalpia do produto P, ou seja, neste exemplo trata-se de uma reação exotérmica (ΔHr < 0). Comparando-se com uma reação catalítica (linha contínua), observa-se que o estado inicial e o estado final são os mesmos, ou seja, a variação da energia livre de reação (ΔG) é igual nos dois processos e a posição de equilíbrio não é alterada pelo catalisador. Conhecidos a energia de ativação da reação direta Ea1 e o calor de reação ΔHr, pode-se calcular a energia de adsorção associada ao ponto 2 (Ea2), pois Ea2 = Ea1 - ΔHr. Com relação ao processo catalítico, há um poço de entalpia entre os pontos 1 e 2, que representa a adsorção de R em X, antes de passar pelo complexo ativado RX* no ponto 3. A adsorção química de R, por tratar-se de uma reação química, também exibe uma energia de ativação antes de atingir o poço de entalpia em 2 (ou em 4, quando tratar-se da reação inversa). A adsorção é um processo invariavelmente exotérmico, ou seja, apresenta o poço de entalpia. A explicação é que a energia livre de Gibbs diminui para qualquer evento espontâneo de acordo com a relação ΔGads = ΔHads - TΔSads. A adsorção deve sempre acompanhar uma diminuição da entropia (ΔSads < 0 e -TΔSads > 0), pois o número de graus de liberdade de uma molécula adsorvida é menor do que na fase fluida. Finalmente, na situação em que ΔGads < 0, ΔHads < 0. A "energia de ativação" (Ecat1) da reação catalítica direta é calculada como a diferença de energia entre os pontos 3 e 2. Na condução dos experimentos de catálise, são utilizadas as informações de taxa de consumo de reagentes e formação de produtos para construção do gráfico de Arrhenius, ln(r) versus 1/T. Türks et al. 3 mostraram de forma didática o cálculo da energia de ativação em catalisadores Ni/Al2O3 para a redução do CO2 a CH4. Roduner4 mostra a relação matemática entre Eap1 e Ecat1. Desta forma, o que se utiliza na prática é a diferença entre 3 e 1, o que se denomina de "energia de ativação aparente" (Eap1). O valor de Eap1 é menor do que Ecat1. Para obter o valor de Ecat1 deve-se obter a energia de adsorção ΔHads, o que não é determinado nos experimentos de catálise. A distinção entre "energia de ativação" (Ecat1) e "energia de ativação aparente" (Eap1) não se dá apenas devido a etapa de adsorção. Essa distinção também ocorre em casos que a sequência de etapas elementares da reação catalítica é mais complexa, como por exemplo, na síntese da NH3. O mecanismo experimentalmente conhecido para a síntese de amônia em Fe e Ru é um mecanismo de Langmuir-Hinshelwood no qual a primeira etapa é a etapa determinante da taxa de reação.5,6 N2 + 2* → 2N* em que * denota o sítio vazio e X* (X = N, NH, NH2 e NH3) as espécies ligadas aos sítios.
MICRORREATORES UTILIZADOS NOS TESTES CATALÍTICOS Para diminuir os custos com a construção de equipamentos, o consumo de materiais, de resíduos a serem descartados e os riscos (incêndio, explosões e emissões), os experimentos iniciais são realizados em reatores de escalas reduzidas. Pode-se utilizar desde um microrreator (volume de 2,5 a 25 mL), um reator em escala de bancada (60-700 mL) até um reator de planta piloto (5-40 L). No desenvolvimento do processo industrial, é natural que se avaliem os aumentos de escala, pois além da atividade catalítica, há a necessidade de avaliação do efeito das condições de reação (pressão, temperatura, agitação, forma de mistura, reuso, uso de solvente etc. ). Um exemplo é como as partículas são disponibilizadas em sua forma final (forma e estrutura do catalisador, porosidade e aditivos) e os efeitos fluidodinâmicos que surgem com o aumento de escala. Os reatores podem ser descontínuos ou batelada (Figuras 2(a), 1S, 2S e 3S) e contínuos (Figuras 2(b), 4S, 5S, 6S e 7S).7
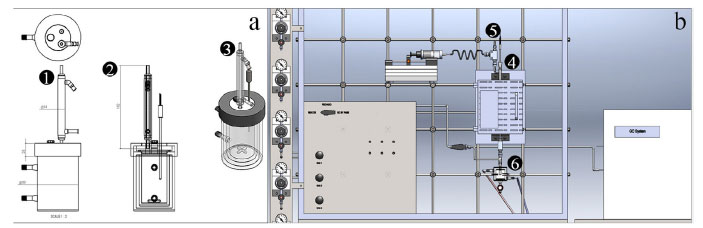 Figura 2. (a) Exemplo de reator batelada encamisado contendo: (1) entrada para alimentação/retirada de amostra, (2) entrada para termopar e (3) entrada para condensador. (b) Sistema de avaliação catalítica contínuo. Em (4) encontra-se o reator dentro do forno, (5) e (6) os sistemas de alimentação do reator e condensador de compostos pouco voláteis, respectivamente
Como mostrado na Figura 2(a), o reator batelada pode ser ou não encamisado, ter entrada para alimentação e retirada de amostras, conter dispositivos de controle de temperatura, como um termopar e condensador acoplado. Nos reatores industriais contínuos, não é comum utilizar catalisadores em pó ou finamente divididos, pois o empacotamento do leito causaria alta perda de carga ao escoamento do fluido, conforme previsto pela equação de Ergun.8 Desta forma, industrialmente, prefere-se utilizar catalisadores granulados. No entanto, o transporte de massa do fluido à superfície ativa do catalisador granulado e o transporte de calor influenciam na taxa de reação, seletividade e até mesmo no mecanismo da reação. Nos testes iniciais com os microrreatores contínuos, o objetivo é medir a cinética intrínseca da reação. Para isto, as limitações de transporte são minimizadas utilizando uma pequena quantidade de catalisador em pó, geralmente entre 50-200 mg, formando uma lâmina de alguns milímetros de altura. O microrreator contínuo, operado nesta condição, é denominado de "reator diferencial", em contraste ao reator tubular que contém catalisador ao longo de seu comprimento. No reator diferencial idealizado, excluem-se os fenômenos de transporte de massa e calor que modifiquem a taxa de reação, que é aproximadamente constante em todos os pontos do leito. Visto que as taxas dependem da concentração, esta suposição é geralmente razoável somente para pequenas conversões e para reatores de pequena profundidade, assim como para pequenos gradientes de temperatura. Por outro lado, quando uma pequena massa de catalisador é utilizada e o reator operado em altas conversões, não pode ser considerado um reator diferencial, pois poderá haver formação de gradientes de concentração ao longo do pequeno leito. Para reações altamente exotérmicas ou endotérmicas, o catalisador pode ser misturado com uma pequena quantidade de carbeto de silício ou quartzo, que são materiais quimicamente inertes e estáveis nas condições reacionais e ajudam a minimizar a formação de pontos quentes e gradientes de temperatura. Pode-se utilizar diluentes com granulometria diferente do catalisador para facilitar a separação por peneiramento ao final da reação. O carbeto de silício é preferido devido a sua maior condutividade térmica, 55, em comparação com o quartzo que é de apenas 5 W m-1 K-1.9 Mesmo com o uso de diluentes térmicos inertes, por conta das questões termoquímicas das reações, a temperatura do reator deve ser monitorada com o uso de um termopar colocado em contato com a amostra de catalisador, pois pode diferir da temperatura do forno. Os microrreatores contínuos são constituídos de um tubo de vidro, Figura 2(b) no ponto (4), para temperaturas de até 500 ºC, ou quartzo, contendo um vigreux no centro para apoiar a lã de vidro ou quartzo que servirá de suporte para a lâmina de catalisador em pó. Quando a reação a ser avaliada ocorre em altas pressões, os microrreatores contínuos são constituídos de aço inox. O reator pode ser alimentado com reagente na forma de gás ou líquido, sendo que na entrada geralmente é colocado um misturador líquido-gás com aquecimento, adequado para vaporizar substâncias voláteis, ponto (5) da Figura 2(b). Geralmente, um aquecimento através de um resistor do tipo fio ou fita é suficiente para vaporizar e evitar a condensação de substâncias voláteis, tais como o metanol e o etanol. Para substâncias menos voláteis, o correto é alimentar por um vaporizador com controle de temperatura e isolar termicamente o reator e as tubulações de entrada e saída. Nas reações, é adequado utilizar um gás de arraste para garantir a homogeneização dos reagentes pelo leito catalítico. A diluição também evita a condensação das substâncias voláteis. Os reagentes líquidos podem ser alimentados ao misturador líquido-gás ou vaporizador por meio de uma bomba seringa ou tipo HPLC, em vazões típicas de 0,1 a 2 mL h-1. Alternativamente, para vazões menores, pode-se utilizar um saturador e no cálculo da vazão volumétrica da corrente de gás saturada utiliza-se a equação de Antoine, ln(P) = A - B/(T + C),10 em que A, B e C são constantes da substância pura, P a pressão de vapor e T a temperatura. Por utilizar uma menor vazão volumétrica de reagente na corrente saturada, o saturador garante uma melhor estabilidade de composição ao longo do tempo. A retirada de alíquotas para análise dos efluentes de um microrreator contínuo pode ser feita de duas formas. Para reações que ocorrem em fase gás ou com o uso de reagentes voláteis, a retirada de alíquotas é feita por meio de válvulas automáticas, pneumáticas ou elétricas. Quando há a presença de compostos pouco voláteis coloca-se um separador líquido-gás encamisado com controle de temperatura na saída do reator, em uma temperatura típica de 1 ºC. Deve-se atentar que, mesmo para compostos pouco voláteis, uma parte pode ser transportada pela corrente gasosa para fora do separador líquido-gás, incorrendo em desvios no balanço de massas. O dispositivo de condensação está representado na Figura 2(b) no ponto (6). Se o microrreator for o descontínuo, há que se atentar para uma boa agitação, feita por meio de uma barra de agitação magnética, garantindo a turbulência necessária. É possível verificar a restrição ao transporte de massa na catálise conduzindo experimentos com diferentes velocidades de agitação até que não se observe mudanças na atividade catalítica, indicando que a conversão é governada apenas pelas propriedades químicas do catalisador. Na maioria das aplicações, o reator é utilizado para reações com reagentes e produtos em fase líquida. Se os líquidos forem voláteis, é adequado que o reator seja fechado para evitar a evaporação. O reator em vidro borossilicato geralmente tem um volume total interno de 50 mL e é encamisado para circulação de água com controle de temperatura. A temperatura no interior do reator pode ser verificada por meio de um termopar colocado em contato com a solução. Com relação a retirada de alíquotas, o ideal é retirar um volume pequeno, da ordem de 1 mL e com o cuidado de retirar líquido e catalisador de modo proporcional para evitar a concentração de catalisador no reator ao longo do tempo. Após a retirada da alíquota o catalisador deve ser separado imediatamente por centrifugação ou filtração para evitar o progresso da reação. Uma opção é utilizar um filtro para seringas. Se a fase líquida não puder ser analisada imediatamente, deve ser mantida refrigerada.
CONVERSÃO, SELETIVIDADE, RENDIMENTO E DISTRIBUIÇÃO DE PRODUTOS Dispositivos de quantificação A determinação de reagentes e produtos na termocatálise é feita principalmente a partir de cromatografia em fase gasosa, pois os reagentes e os produtos estão muitas vezes na fase gasosa ou são líquidos que possuem baixo pontos de ebulição (100-200 ºC) e que podem ser levados à fase de vapor nos próprios cromatógrafos.11 Para compostos com ponto de ebulição maiores, utiliza-se a cromatografia líquida de alta eficiência (HPLC) para quantificação. A espectrometria de massas também é utilizada nas determinações, sobretudo em experimentos que requerem maior resolução temporal.12 Outras espectroscopias, tais como na região do infravermelho (FTIR), ultravioleta-visível e Raman são empregadas com menor frequência e têm caráter mais qualitativo do que quantitativo.13 As caracterizações por cromatografia constituem na separação dos produtos e reagentes em fase gás ou vapor por eluição em uma coluna com o auxílio de um fluido de arraste, geralmente o hélio. A composição do efluente do reator é determinante para a escolha do método de caracterização cromatográfico e do dispositivo de detecção. Podem ser usados detectores do tipo seletivos ou universais, destrutivos ou não destrutivos.14 Um dos principais detectores é o detector de ionização em chama (FID, flame ionization detector), pois apresenta alta sensibilidade e sua resposta geralmente é proporcional ao número de átomos de carbono da molécula. O FID é baseado na detecção de íons resultantes da combustão de compostos orgânicos em uma chama de hidrogênio e ar. Informações detalhadas sobre o FID são descritas por McWillian e Dewar15 e Holm.16 O FID não detecta compostos com ausência de ligação C-H, tais como CO, CO2 e H2O, sendo utilizado, neste caso, o detector de condutividade térmica (TCD, thermal conductivity detector). O TCD é um detector universal, que se baseia na condutividade térmica relativa de dois fluidos, geralmente um gás de referência puro, muitas vezes o hélio, e a amostra. Embora universal, o TCD é menos sensível do que o FID.17 O espectrômetro de massas (MS) pode ou não estar associado a um cromatógrafo. Quando associado, fornece o espectro de massas de cada composto, correspondente ao tempo de retenção. Quando não associado, a resposta será da decomposição da mistura dos compostos presentes. Com o espectrômetro, detecta-se os compostos através da abundância relativa m/z. Quando se utiliza para misturas, o ideal é o monitoramento em reações simples, com a participação de poucos compostos, pois pode ocorrer a sobreposição de fragmentos m/z resultantes da decomposição de compostos distintos. O detector pode atuar no modo varredura (corrente de íons monitorados - monitoring ion current (MIC)), em que ocorre varredura em uma faixa m/z, ou no modo seleção de determinado m/z (monitoramento do íon selecionado - select ion monitoring (SIM)). Esse último pode aumentar a sensibilidade de ppb do modo varredura para ppt.18 Outro detector é o de captura de elétrons (ECD, eléctron capture detector) não destrutivo e seletivo. Com uma recorrência menor nos experimentos de termocatálise, é aplicado a compostos com átomos com alta eletronegatividade, tais como halogênios, organometálicos, nitrilas ou nitrocompostos. A depender do analito, o ECD pode ser até um mil vezes mais sensível do que o FID e um milhão de vezes mais sensível que o TCD.19 Além destes, cerca de 60 tipos detectores já foram usados em cromatografia. Na catálise, os mais empregados são o FID, TCD e MS. As análises espectroscópicas como FTIR e Raman20 também podem ser utilizadas, geralmente quando há a necessidade de maior resolução temporal. Enquanto a cromatografia fornece o resultado de alíquotas de tempo em tempo (até alguns minutos), as análises espectroscópicas podem analisar em poucos segundos. Por isso, a detecção por espectroscopia é utilizada com frequência nos experimentos em condições "operando". 21 Infelizmente, apesar de fornecerem resultados valiosos para o estudo catalítico, essas técnicas podem depender de condições peculiares de análise, como reatores específicos, caminho óptico disponível e uso de lentes especiais para a detecção dos compostos. Métodos de quantificação Um ponto que pode ser fonte de equívoco na quantificação é em relação à curva analítica ou curva de calibração. As curvas constituem uma representação gráfica para a obtenção da correlação matemática entre a concentração dos analitos e a resposta gerada no equipamento, como área de pico na cromatografia ou absorbância em técnicas espectroscópicas. A resposta deve ter uma relação matemática com a concentração, sendo mais comumente uma reta ou, menos frequente, curvas não lineares. No reator batelada, a análise pode ser feita após a retirada de alíquotas do meio reacional. No reator contínuo, pode ocorrer de duas formas, com o uso de uma válvula automática com volume de injeção de alguns microlitros da mistura reacional ou por condensação em um separador líquido-gás encamisado e com circulação de fluido térmico proveniente de um banho termostático. A fração condensada deve seguir protocolo de análise igual ao que ocorre com o reator batelada. Pode-se fazer uma calibração externa, ou seja, quando há a resposta a um padrão de referência em diferentes concentrações. A curva deve ser construída em condições e concentrações semelhantes àquelas utilizadas no teste cinético. O ideal é que a curva seja obtida com o analito em diferentes concentrações no mesmo solvente utilizado nos experimentos.22 Uma vez que a resposta do detector depende do volume analisado, não deve ocorrer desvios significativos entre os volumes injetados. No entanto, como a quantidade de líquido a ser injetada é da ordem de microlitros, variações na posição no êmbolo da seringa ou pequenas bolhas levam a grandes imprecisões na análise. Estes desvios podem ser corrigidos com o uso de um padrão interno, que elimina a dependência da relação do analito com volume injetado. Para isto faz-se necessário a construção de uma curva analítica e o ajuste dos dados de ra/rp (resposta do analito por resposta do padrão) em função da razão das massas ma/mp. A resposta pode se referir à intensidade ou à área sob a curva do cromatograma, sendo mais correto o uso da área. Se a resposta for linear, obtém-se a relação ma/mp = a × ra/rp, em que a é o coeficiente angular. A massa de analito é dada pela relação ma = a × mp ×(ra × rp)/ ra × rp. O padrão interno deve ter estabilidade química e física, semelhança com analito, solubilidade no meio e ter tempo de eluição na cromatografia distinta dos demais compostos. Por exemplo, o n-butanol pode ser utilizado como padrão interno em reações que envolvam compostos hidroxilados, tais como metanol, etanol e glicerol, pois apresenta todos os requisitos anteriores. Procedimentos de quantificação Conversão A conversão do reagente (XR) é a razão em mol da quantidade de reagente consumida pela quantidade alimentada ao reator (Equação 1). A quantidade de reagente consumida é dada pela diferença entre nR0 (mol no início) e nR (mol ao fim do período avaliado). Para reações com dois reagentes ou mais, a conversão é sempre escrita com base no reagente limitante a partir da estequiometria da reação, pois ao considerar o reagente limitante a conversão estará compreendida entre 0 e 100%, à medida que for considerado o reagente em excesso, a sua conversão nunca atingirá 100% e não refletirá de forma adequada o avanço da reação. 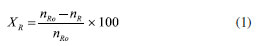 A Equação 2 mostra que a Equação 1 pode ser escrita em termos da vazão molar F (em mol por tempo) para reatores contínuos. 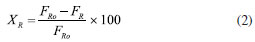 Há um equívoco frequente no cálculo da conversão. É possível encontrar estudos que calculam a conversão somente em função da quantidade de produtos formados e de reagente residual, conforme mostra a Equação 3. Este método é classificado como "conversão interna", pois na equação não aparece a quantidade em mol de reagente inicial (nR0), e o cálculo é feito apenas com as quantidades em mol de reagentes e produtos presentes no meio reacional. Na Equação 3, há a necessidade de correção por um termo ni, que pode ser o coeficiente estequiométrico da reação ou número de átomos de carbono das espécies envolvidas. O método fornece exatamente o mesmo valor das Equações 1 e 2, desde que todos os produtos possam ser quantificados. Quando há a formação e retenção de coque no catalisador ou de subprodutos desconhecidos, a conversão será subestimada. Outra questão é quando o sistema de quantificação é inadequado para a reação. Em reação com formação de compostos extremamente voláteis e gases, os compostos se perdem para a atmosfera após a abertura do reator e não são contabilizados no balanço dos componentes. 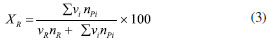 Seletividade A seletividade a um produto i (Si) é definida como a formação relativa de um determinado produto em relação ao reagente consumido, como definido na Equação 4. 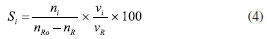 em que ni é o número de mol do produto i formado e nR0-nR a diferença entre a quantidade em mol de reagente alimentado e o que sai do reator, ou seja, a quantidade de reagente consumido. Os coeficientes ni são os números de átomos de carbono nas moléculas do produto e reagente. Um exemplo é na formação do éter dietílico (ni = 4) na reação de desidratação intermolecular com duas moléculas de etanol (ni = 2). Para formar uma molécula de éter dietílico é necessária a conversão de 2 moléculas de etanol, Si = (ni/(nR0 - nR)) × (ni/nR) × 100 = (1/2)) × (4/2) × 100 = 100%. Se não fossem considerados os termos ni a seletividade seria de 50%, mas em uma reação simples com um único produto a seletividade sempre será de 100%. Para reações múltiplas série-paralelo, a seletividade é dependente da conversão, e a forma correta de comparar a formação dos produtos é na condição de isoconversão. Por exemplo, considere uma reação consecutiva R → P → S, em que a primeira reação é muito mais rápida que a segunda. Se a taxa de R → P é rápida em relação à de P → S, o produto intermediário P se acumulará durante os estágios iniciais da reação e a seletividade para P aparecerá maior do que para S. Em tempos de reação mais longos, como a conversão de R se aproxima do fim, P irá produzir S, ou seja, a seletividade depende da conversão de S. Para que a condição de isoconversão seja atingida para um conjunto de catalisadores, a temperatura de reação e a massa de catalisador deverão ser variadas em uma série de experimentos. Uma forma controversa de calcular a seletividade é quando a quantidade de produto é dividida pela somatória de todos os outros produtos (Equação 5). A princípio, as Equações 4 e 5 fornecem os mesmos valores de seletividade, no entanto, quando houver a formação de coque a Equação 5 fornecerá seletividade maior do que a 4. O coque fica depositado no catalisador, não sai do reator, e não é quantificado. No entanto, o coque também é produto e a Equação 4 é mais adequada, mesmo que SSi não resulte em 100%. Na Equação 5, SSi sempre será igual a 100%, independente ou não da formação de coque.  As definições apresentadas são típicas da área da química básica e geralmente seguidas pela termocatálise, no entanto, há outras definições, como, por exemplo as que são apresentadas por Levenspiel.23 Nesta publicação, a seletividade é definida como a formação de um produto X em relação a um produto Y, seja instantânea ou global. A vantagem do método utilizado pela química básica é que a soma de todas as seletividades traz consigo informações sofre a formação de outros produtos (ou não identificados nas análises ou adsorvidos no catalisador como coque). Rendimento O rendimento (Yi, yield em inglês) fornece o quanto de um produto desejado é formado. É a multiplicação da seletividade pela conversão, mostrando a quantidade de produto produzida por quantidade de produto que poderia ser produzida na conversão total (Equação 6). O rendimento máximo de 100% será obtido quando a seletividade a i for 100% e a conversão de R for 100%.  Distribuição molar de produtos A Equação 7 mostra que a distribuição molar de produtos representa a quantidade molar relativa dos produtos da reação. Ou seja, para um produto i, é a quantidade formada de i, ni, dividida pela somatória de todos os produtos, incluindo i. A Equação 7 é similar à 5, com exceção dos coeficientes ni. O cálculo pode ser útil na determinação da distribuição de produtos em reações cujos produtos podem estar em fases diferentes, como os produtos na fase gás e fase líquida. Como consequência da definição, a somatória da distribuição molar de produtos sempre resulta em 100%. 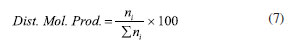 Se a distribuição molar de produtos for calculada em termos de massa de produtos, e os produtos devidamente agrupados, pode-se obter informação sobre o balanço de massas da reação, de forma coerente ao que ocorre com a seletividade. Há que se observar que algumas publicações se referem de forma equivocada a "distribuição molar de produtos" como "seletividade".
REUSO, ESTABILIDADE CATALÍTICA E BALANÇO DE CARBONO A termocatálise desempenhada por materiais cerâmicos frequentemente está relacionada a grandes processos de relevância industrial e, nestes casos, buscam-se materiais com longos períodos de atividade catalítica a fim de aumentar o lucro.24 A capacidade de produzir grandes volumes de produtos é crucial e isso se traduz na necessidade de catalisadores ativos. A estabilidade do catalisador é a sua capacidade de reter a função, que é avaliado por experimentos de reuso em reatores descontínuos ou experimentos de longa duração em reatores contínuos.25 Os estudos são essenciais para estabelecer o quão estável é sua atividade catalítica e por quanto tempo pode funcionar. Esses estudos também buscam elucidar os mecanismos que podem levar à perda da atividade catalítica, como a formação de coque, sinterização e instabilidade da estrutura do catalisador. O estudo de estabilidade em reatores descontínuos é conduzido reusando-se o catalisador em ciclos subsequentes. A retirada do catalisador do meio reacional pode ocorrer por filtração ou centrifugação e o ideal é que isto seja feito rapidamente para evitar o avanço da reação durante a manipulação. Há ainda que garantir que a massa de catalisador usada em cada ciclo se mantenha constante, o que nem sempre é possível, pois na separação do catalisador do meio reacional a cada ciclo pode-se perder massa. Uma estratégia é corrigir a massa de reagentes, de modo que a proporção massa de reagente por massa de catalisador permaneça constante a cada ciclo, ou seja, a cada ciclo a massa de meio reacional também diminui. O estudo de estabilidade em altas conversões ou em conversões de equilíbrio não é adequada, pois pode indicar uma certa estabilidade do catalisador ou desativação muito branda conforme resultados da Figura 3(a), que mostra a conversão de R em fase líquida após 6 h na reação. Na situação apresentada, nenhuma conclusão sobre o desempenho do catalisador é confiável. O problema é que a conversão é limitada pela disponibilidade de reagentes. Mais reagentes seriam convertidos se maior quantidade estivesse presente. A quantidade de catalisador é muito alta ou o tempo de reação demasiado longo. Portanto, a desativação só se tornará visível quando vários ciclos forem executados e a atividade catalítica diminuir significativamente. Geralmente, este comportamento é falsamente descrito como alta estabilidade e reusabilidade. Menores conversões são obtidas com diminuição da atividade catalítica, por meio de menor quantidade de catalisador ou menor temperatura de reação. Alternativamente, para informações mais precisas sobre a reusabilidade em reatores descontínuos, pode-se observar a atividade catalítica inicial, realizando experimentos em diferentes tempos de reação.26 A Figura 3(b) mostra que no início da reação a desativação é bastante considerável, estabilizando-se para os demais ciclos.
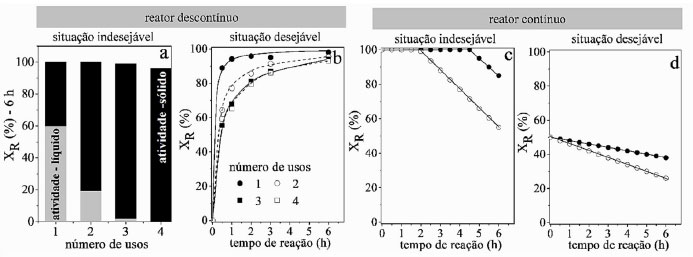 Figura 3. Resultados hipotéticos em reator descontínuo e contínuo: (a) conversões de reuso após 6 h de reação, mostrando que há lixiviação para a fase líquida nos primeiros usos; (b) conversão em diferentes tempos de reação; (c) reator contínuo com alta conversão de reagentes e com desativações aparentemente iguais e (d) com conversão menor, revelando que as desativações são diferentes
Uma pergunta importante e frequente aparece nos trabalhos de catálise heterogênea em fase líquida: "a catálise ocorre no meio homogêneo ou heterogêneo?". A pergunta surge porque há vários exemplos em que a fase ativa do catalisador sólido é lixiviada e solubilizada na fase líquida, atuando como catalisador. Esta questão foi apresentada no estudo de Sheldon et al. 27 e uma forma de respondê-la é retirar o sólido do meio reacional na metade da conversão de equilíbrio e acompanhar o avanço da reação.28 Se a reação for completamente interrompida, então a catálise é heterogênea. Se continuar é porque há espécies cataliticamente ativas solúveis e a catálise é homogênea. Há outra forma de verificar se há espécies ativas lixiviadas na fase líquida conforme mostra a Figura 3(a): retirar o sólido, adicionar novos reagentes na fase líquida e acompanhar a conversão ao longo do tempo para cada novo ciclo.29 No exemplo apresentado ocorre lixiviação parcial de fase ativa, que diminui consideravelmente ao longo do uso. Desta forma, a conversão observada no primeiro ciclo é devido à catálise homogênea e heterogênea. No último ciclo há somente contribuição da catálise heterogênea, indicando estabilidade do catalisador para diversos usos. Com relação aos experimentos de estabilidade em reatores contínuos, os testes também não devem ser realizados em condições de conversão total ou no equilíbrio (Figura 3(c)). A melhor prática é conduzir experimentos em baixa conversão e fora da conversão de equilíbrio, como avaliar o desempenho com conversões abaixo de 50% da conversão de equilíbrio (Figura 3(d)), a fim de discernir de forma confiável a atividade catalítica estável ou decrescente. Adicionalmente, nos experimentos em reatores contínuos, um equívoco frequente é utilizar uma mesma amostra de catalisador para obter resultados de conversão e seletividade em diferentes temperaturas, negligenciando que a amostra sofre modificação com o tempo. O aproveitamento de uma mesma amostra pode resultar em um falso resultado. Se a conversão estiver longe do equilíbrio, o aumento de temperatura do reator sempre resultará em um aumento da conversão, de acordo com o que foi estabelecido por Arrhenius, ocultando desta forma alguma desativação, caso ocorra. Se fosse utilizada uma nova amostra de catalisador a cada temperatura a conversão seria maior. Vários fatores podem alterar a atividade do catalisador como formação de coque, sinterização e, até mesmo mudanças na forma de partículas ativas. Este fato pode ser verificado por meio de experimentos de atividade catalítica com temperaturas crescentes e decrescentes com a mesma amostra.23 Se ocorrerem histereses, então há alguma modificação e deve-se realizar experimentos com uma nova amostra a cada temperatura. Os experimentos isotérmicos são conduzidos monitorando-se a conversão ao longo do tempo, permitindo-se obter a conversão no tempo zero. Desta forma, dados de conversão em função da temperatura podem ser obtidos com precisão, com a conversão se referindo à conversão no tempo zero. Os experimentos conduzidos desta forma permitem também obter informações sobre a desativação a cada temperatura. A confiabilidade dos dados de reatividade é um ponto que desperta atenção e uma das formas de monitorar se o experimento está sendo executado corretamente com relação à calibração dos sistemas de detecção e quantificação é por meio do balanço de elementos (principalmente o carbono). Comumente, o balanço de carbono (BC) em reatores contínuos é obtido a partir da somatória de número de mol de átomos de carbono alimentado ao reator (SCentrada) e que sai do reator (SCsaída), conforme a Equação 8. Na entrada do reator, geralmente alimenta-se apenas reagente puro, de modo que o termo SCentrada pode ser escrito como SCreagente entrada. Na saída do reator há a presença de reagentes não convertidos e produtos, e o termo SCsaída pode ser escrito como SCreagente saída + SCprodutos. 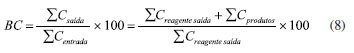 BC é um número compreendido entre 0 e 100%. Para reações com ausência na formação de coque, o valor de BC menor que 100% indica erro na calibração do detector, o que impactará também nos cálculos de conversão, seletividade e distribuição de produtos. Independentemente da formação ou não de coque, o valor acima de 100% indica erro de calibração. O BC é um parâmetro de controle da qualidade do experimento que está sendo executado. Com relação às reações contínuas com a formação de coque, os valores de BC ao longo do tempo indicam a quantidade de coque acumulada ao longo do tempo. A Figura 4 mostra que o catalisador 1 acumula menos coque do que o catalisador 2, pois a região hachurada de 1, que é a área entre 100% e a curva de BC, é menor para o catalisador 1. As curvas indicam também que a deposição de coque é maior no início da reação, aproximando-se de 100% para o catalisador 1 e indicando que a deposição de coque diminui com aumento do tempo de reação. Isto deve ocorrer pela menor taxa de reação como reflexo da desativação. Deste modo, o BC além de um parâmetro de controle é também um indicativo da quantidade de carbono acumulado no catalisador.
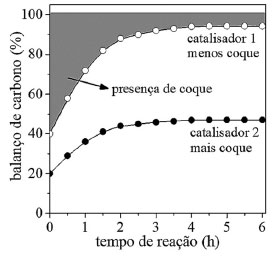 Figura 4. Comparação do balanço de carbono em função do tempo em um reator contínuo; a região hachurada indica que a quantidade acumulada de coque é menor para o catalisador 1
É importante notar que o balaço do elemento próximo a 100% pode ser enganoso em conversões baixas, pois o numerador e o denominador da Equação 8 se aproximam. Para reações com conversões abaixo de 5%, o ideal é utilizar o BC fornecido pela Equação 9. 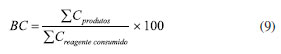
FREQUÊNCIA DE REAÇÃO A frequência de reação (turnover frequency - TOF) de um catalisador é definida pelo número de ciclos completos, sendo adsorção do reagente, transformação química e dessorção do produto, por sítio ativo e por unidade de tempo.30 Os valores de TOF representam de forma geral a média da atividade dos sítios catalíticos. Os valores podem ser baixos na ordem de 10-5 s-1 (um ciclo completo por dia) ou tão alto quanto 109 s-1 (comparável à taxa de colisão cinética de um gás a 10 bar). De forma bruta, os valores de TOF dos catalisadores heterogêneos costumam ser da ordem de um ciclo por sítio e por segundo.30 A vida útil do catalisador pode ser definida como o número de ciclos completos observados antes que o catalisador pare de operar a uma taxa aceitável, podendo ser curta, como no craqueamento catalítico de frações do petróleo, ou muito longa, correspondendo a até 109 rotações na síntese de amônia. Para estudos mais detalhados, muitas vezes é desejável relatar as atividades catalíticas como taxas por sítio de superfície (ou seja, TOFs), com o número de sítios de superfície medidos com a adsorção de moléculas sonda na superfície do sólido (por exemplo, adsorção de H2 ou CO para titular sítios de metal, adsorção de amônia ou piridina aos sítios ácidos). Às vezes, o TOF é confundido com a constante cinética k, sendo usado indistintamente com o mesmo significado, uma vez que a taxa de reação (r = TOF × [Cat]) depende da concentração de catalisador. Entretanto, esta comparação não é válida, pois o TOF pode variar com a concentração dos reagentes.31 O TOF deve ser uma medida de frequência, conforme a definição, e não simplesmente dos reagentes consumidos ou produtos formados. Por exemplo, se existir um catalisador que pode atingir a mesma frequência para duas reações diferentes, R → P e 2R → P, a segunda consumirá o dobro de reagente, embora o TOF deva ser o mesmo. Dessa forma, para o segundo caso, o TOF deve ser calculado como -1/(2[Cat]) × d[R]/dt). Como a variação da concentração de R, d[R]/dt, é negativa, no cálculo aparece o termo negativo. O TOF varia com as concentrações dos reagentes e às vezes também dos produtos. Portanto, expressar o TOF sem descrever as concentrações das espécies envolvidas é enganoso. Visto que o TOF é definido como "moléculas reagindo por sítio ativo por tempo", não há uma descrição clara de quanto tempo deve ser considerado. Se o tempo for calculado até, por exemplo, 80% de consumo do reagente, o resultado será muito diferente de considerar até 20% de consumo, pois o TOF depende da concentração do reagente. Desta forma, a rigor, o TOF deve ser uma medida instantânea e na mesma concentração de reagentes. Se for considerado uma reação R → P ocorrendo em um reator descontínuo, e o uso de dois catalisadores distintos com diferentes atividades catalíticas, a concentração do reagente R variará em função do tempo de forma distinta para ambos os catalisadores. Esta é uma característica do próprio reator descontínuo. Desta forma, o mais adequado é medir o TOF no tempo zero (TOF0), quando a concentração do reagente é igual para todos os catalisadores. A Equação 10 mostra o cálculo do TOF0 para um reator descontínuo, em que NR0 é o número de moléculas de R no início, NS é o número de sítios ativos expostos na superfície (acessíveis) e (dXR%/dt) a derivada da conversão no tempo zero.31 A equação de XR(%) = f(t) pode ser ajustada aos pontos experimentais em função do tempo utilizando a Equação 11, em que os termos a e b são obtidos após ajuste da curva aos pontos experimentais. Desta forma, (dXR%/dt)t=0 será igual a a/b. 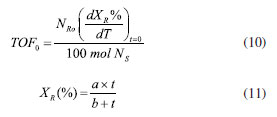 Para ser mais rigoroso, Kozuch e Martin32 propõem que os valores de TOF0, inspirados pelas funções termodinâmicas padrão clássicas, como ΔGº, devem ser estimados na condição de concentração de reagente de 1 mol L-1 (para reações em fase líquida) ou 105 Pa (para reações em fase gás) e 273,15 K. Entretanto, utilizar concentração definida de reagente não é fácil, pois nas condições apontadas a atividade do catalisador pode ser desprezível e a forma de superar a baixa atividade catalítica seria utilizar massas maiores de catalisador, o que na grande maioria dos experimentos seria impossível. Um outro ponto no cálculo do TOF, além da questão cinética, é a determinação do número de sítios Ns expostos na superfície do catalisador. Medidas de quimissorção,33 titulação e análise do empacotamento de partículas por espectroscopia XANES34 foram empregados para medir os valores Ns de catalisadores heterogêneos em reações termoquímicas. Entretanto, os métodos para determinar NS assumem que todos os sítios expostos são igualmente ativos, o que não é verdade para reações sensíveis à estrutura. Na realidade, em catálise heterogênea, a suposição de que todos os sítios têm atividade catalítica idêntica é uma proposição necessária ao cálculo. Adicionalmente, os sítios ativos podem mudar com as condições experimentais, tais como temperatura e pressão. Assim, qualquer valor de TOF é na melhor das hipóteses uma aproximação. Nos casos em que a apresentação de resultados na forma de TOF apresentar limitações, pode-se utilizar a taxa geral da reação catalítica normalizada pelo número total de sítios ativos (site time yield - STY).
IMPRECISÃO NA MEDIDA DE VAZÃO VOLUMÉTRICA DE GASES A vazão volumétrica é a quantidade de determinado fluido que passa por tempo por uma seção de uma tubulação. A vazão descreve a rapidez com a qual um volume ou massa escoa. Pode ser descrita como vazão volumétrica (L min-1, por exemplo) ou vazão mássica (kg min-1, por exemplo). O termo "fluxo" é equivocadamente utilizado, pois refere-se à quantidade de uma grandeza que atravessa uma superfície por unidade de tempo (kg m-2 min-1, por exemplo). No cálculo vetorial, o "fluxo" é uma quantidade escalar, definida como a integral da superfície do componente perpendicular de um campo vetorial sobre uma superfície. O uso equivocado de "fluxo" deve ter sua origem na tradução errada de flow rate, que significa vazão em inglês. No entanto, não há problemas em se referir a aumento ou diminuição de fluxo volumétrico, a questão é quantificar a vazão volumétrica (por exemplo, 100 mL min-1) e chamar esta grandeza de fluxo volumétrico. Com relação à vazão, se for conhecida a área Ast da seção transversal da tubulação em m2 (Ast = π × r2, em que r é o raio interno do reator tubular) e a velocidade de escoamento v em m s-1, a vazão volumétrica é obtida em m3 s-1. As unidades de vazão mássica e volumétrica são reciprocamente conversíveis, sabendo-se a densidade do fluido, sendo que a densidade de um líquido raramente depende das condições de operação dos reatores. Já para os gases, a densidade depende da pressão e da temperatura. Quando a medida da vazão volumétrica for feita a temperatura e pressão ambiente, a vazão deveria ser transformada nas condições CNTP, ou seja, 273,15 K e 101325 Pa. Na falta desta mudança, a pressão e temperatura deveriam ser indicadas. Os instrumentos que realizam as medidas de vazão volumétrica são os MFCs (do inglês mass flow controlers, controladores de vazão mássica) que automaticamente fornecem vazões em NL min-1 (normal litro por minuto). É preciso ficar atento, pois o MFC é calibrado para um determinado gás. Se outro gás for utilizado, a calibração precisa ser refeita com o risco de medir a vazão errada. Os rotâmetros e bolhômetros são equipamentos mais baratos e acessíveis, mas podem não fornecer as vazões em NL min-1.
CRITÉRIOS WEIZ-PRATER E MEARS PARA DIFUSÃO INTERNA E EXTERNA Os dados cinéticos coletados com várias amostras de catalisadores podem sofrer influência de efeitos difusivos internos (critério de Weiz-Prater, CWP) e externos (critério de Mears, CM). Para determinar se a difusão interna controla a reação catalítica, a Equação 12 é útil, pois utiliza os valores medidos da taxa de reação em mol (tempo)-1 (massa de catalisador)-1, -r'A (obs). O critério resulta na equação CWP, em que rc é a densidade do catalisador, R o raio da partícula, CAs a concentração do reagente A na superfície do catalisador, e De a difusividade efetiva.35 O termo De é o que inspira mais cuidado e é o mais difícil de ser obtido, pois depende do regime difusional nos poros de catalisadores macro, meso ou microporosos.36 Para sólidos macroporosos, as colisões intermoleculares prevalecem em comparação com os choques com as paredes do catalisador e podemos utilizar modelos que se aproximam da difusão molecular. Para os outros dois regimes, De depende principalmente dos tamanhos relativos das moléculas e dos poros. Para os materiais com poros menores, em que as colisões das moléculas com as paredes são mais frequentes pode-se estimar a difusão de Knudsen. Para sistemas com microporos, não existem equações para determinar De, sendo necessário na maioria das vezes realizar medidas experimentais. Na equação, se CWP for menor do que 1 não existem limitações difusivas, mas se CWP for maior do que 1 a difusão interna limita a reação química. 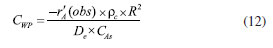 O critério de Mears (CM), da mesma forma que o critério de Weiz-Prater, utiliza a taxa de reação medida para verificar se a transferência de massa da fase fluida para a superfície do catalisador pode ser desprezada. O critério resulta na equação CM, em que rb é a densidade do leito de catalisador, R o raio da partícula, na ordem da reação, CAb a concentração do reagente A na fase fluida, e kc o coeficiente de transferência de massa.37 O coeficiente kc pode ser calculado, por exemplo, partir da correlação de Thoenes-Kramers, Equação 13.38 Na equação, se CM for menor do que 0,15 não existem limitações difusivas, mas se CM for maior do que 0,15 a difusão externa limita a reação química. 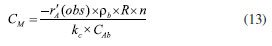 Deve-se observar que os critérios podem ser atingidos nas duas equações se o termo -r'A(obs) no numerador for baixo, ou seja, se a reação for conduzida em temperaturas baixas, na condição em que a conversão do reagente seja baixa. Este é outro motivo para evitar avaliar a atividade catalítica de materiais em altas conversões.
DETERMINAÇÃO DE PARÂMETROS TEXTURAIS DE CATALISADORES A fisissorção de nitrogênio a 77 K é o método padrão para análise da textura de catalisadores sólidos. A análise fornece uma isoterma de fisissorção, à qual podem ser aplicados diversos modelos matemáticos para obter informações relevantes à interpretação da avaliação catalítica, principalmente a área específica BET (SBET) e a distribuição de tamanho de poros. A obtenção de área específica para sólidos microporosos com poros menores que 2 nm a partir da equação de BET linearizada (Equação 14) inspira cuidados. Na equação, os parâmetros c e VM são ajustados a partir de dados experimentais de volume de gás adsorvido V e de pressão relativa P/P0. O parâmetro VM tem uma importância especial, pois representa o volume de adsorbato adsorvido por grama de sólido formando uma monocamada completa. Com o valor de VM, calcula-se o número de moléculas NM na monocamada. Se a área ocupada por uma molécula de diâmetro dm for s (o N2 ocupa uma área de 0,162 nm2), a área ocupada por NM moléculas será SBET = NM s, que representa a área específica do catalisador. Para os sólidos com poros de diâmetros D ≳ 10 × dm, o preenchimento dos poros envolve primeiro a formação de um filme adsorvido. Por outro lado, para os sólidos com poros pequenos D ~ dm, o preenchimento de poros ocorre através de um aumento abrupto do volume por causa da alta energia capilar. Não há espaço nos poros para a formação de várias camadas, que é a hipótese do modelo que permitiu a obtenção da equação de BET.39 Adicionalmente, o parâmetro c é uma relação exponencial da diferença das energias de adsorção da primeira camada (ΔHads,1) e de liquefação do adsorbato (ΔHlatente), de acordo com a Equação 15. Para a temperatura de condensação do nitrogênio a -196 ºC, pode-se calcular ΔHads,1, sabendo que ΔHlatente vale 5,59 kJ mol-1. O expoente da equação pode ser negativo ou positivo, mas o parâmetro c sempre será positivo. Valores de c da ordem de 0,01 mostram que a interação adsorvente-adsorbato é fraca, da ordem de 1 é considerada média e, da ordem de 10 é considerada forte. O parâmetro c negativo ocorre em sólidos microporosos, contrariando a definição da Equação 14.39 Infelizmente, vários estudos utilizam a equação de BET para sólidos microporosos, sem se atentar às limitações do método. Alternativamente, para um sólido microporoso pode-se determinar o volume de microporos e a sua área externa, ambos pelo método t-plot. 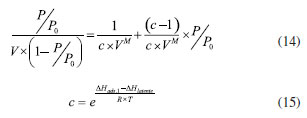 Finalmente, as distribuições de tamanho de poros normalmente são calculadas a partir da curva de dessorção da isoterma usando o método BJH.40 O método funciona bem, mas o menisco de nitrogênio líquido torna-se instável a uma pressão relativa de 0,42, levando ao fechamento forçado da histerese, que por sua vez leva a um pico artificial na distribuição de tamanho de poro em um tamanho de aproximadamente 4 nm. Isto não ocorre com a curva de adsorção.
OUTRAS CONSIDERAÇÕES A termocatálise desempenhada com os catalisadores sólidos é uma ciência muito diversa e outros fatores também influenciam o desempenho dos materiais e a reprodutibilidade dos ensaios catalíticos. 1. A síntese e a ativação de um catalisador devem ser documentadas com precisão, de forma que seja possível sua a reprodução fiel. A atividade catalítica é fortemente dependente da forma de preparo do catalisador. 2. O experimento catalítico deve ser comparado com uma amostra de referência. Este é um equívoco frequente. Por exemplo, um estudo que utilize materiais com desenho e concepção de poros definidos deve utilizar um material de igual composição e não poroso para destacar a influência dos poros na catálise. Pode-se utilizar também um catalisador comercial (benchmark) para comparação. 3. Os dados cinéticos devem ser vinculados às informações experimentais de forma detalhada, incluindo temperatura, pressão (para reações em fase gás), e variações nas composições iniciais. 4. É desejável que os estudos de catálise, quando possível, relatem resumidamente as condições experimentais mal sucedidas, o que é muito comum de ocorrer, de forma a auxiliar futuros experimentos. 5. O volume do microrreator descontínuo deve ser grande o suficiente para que possam ser retiradas alíquotas usando o mesmo experimento até o limite de restar ao final ao menos 50% do volume inicial evitando prejudicar a reprodutibilidade. 6. A ausência de limitações aparentes de transporte deve ser examinada. As limitações de transporte são minimizadas operando o reator em condições de baixa conversão e com regimes de escoamento turbulento (agitação alta em reatores descontínuos ou alta vazão volumétrica de gás de arraste em reatores contínuos). 7. Os experimentos devem ser realizados por tempo suficientemente longo para verificar o estado estacionário ou fenômenos de desativação/ativação do catalisador. 8. É aconselhável que sejam realizadas caracterizações de propriedades físicas e químicas (volume de poros, superfície específica, composição, etc. ) do material antes e após o uso em catálise. 9. É aconselhável que as propriedades físicas e químicas dos catalisadores sejam medidas sob ou em condições mais próximas à sua operação, pois as informações destes experimentos serão diretamente correlacionadas com os dados de atividade catalítica. Por este motivo os experimentos in situ e "operando" com coleta simultânea das características do catalisador e dados catalíticos no mesmo experimento são bastante valorizados. 10. Embora este trabalho tenha sido estruturado para reações catalíticas que sejam favorecidas por uma fonte de calor, a termocatálise, muitos fundamentos discutidos aqui também são aplicados às reações catalíticas que acontecem mediante a uma fonte de excitação elétrica e por fótons, a eletrocatálise e a fotocatálise, respectivamente. Devido à sua alta eficiência e adequação para produção em larga escala, a termocatálise, é frequentemente usada em processos industriais padrão (como a reação de deslocamento água-gás e síntese de amônia). A importância da eletrocatálise em numerosas aplicações industriais e processos de conversão de energia é bem conhecida. Vários questionamentos podem ser levantados para que a eletrocatálise seja conduzida de maneira adequada como a voltagem aplicada, tipo de reator e eletrodos. A fotocatálise é reconhecida como um método viável e promissor para produzir produtos químicos e energia que visa acelerar a conversão solar em química com a ajuda de catalisadores. Na fotocatálise, os desafios são em relação à fonte de excitação, como a faixa de radiação utilizada, desenvolvimento dos reatores e condições adequadas de análise dos produtos, visto que, em muitos casos, o rendimento da reação pode ser muito menor em comparação à termocatálise, por exemplo. Zhang et al. 41 discutem em uma revisão alguns catalisadores cujos sítios ativos são átomos isolados aplicados em termocatálise, eletrocatálise e fotocatálise e comparam essas três formas de favorecimento da reação catalítica.
AGRADECIMENTOS Os autores agradecem à FAPESP pelo financiamento dos projetos em catálise 17/21965-5, 18/01258-5 e 21/05246-4.
REFERÊNCIAS 1. Editorial; Nat. Catal. 2020, 3, 471 [Crossref]; Debecker, D. P.; Hii, K. K.; Moores, A.; Rossi, L. M.; Sels, B.; Allen, D. T.; Subramaniam, B.; ACS Sustainable Chem. Eng. 2021, 9, 4936 [Crossref]; Schüth, F.; Ward, M. D.; Buriak, J. M.; Chem. Mater. 2018, 30, 3599 [Crossref]; Bligaard, T.; Bullock, R. M.; Campbell, C. T.; Chen, J. G.; Gates, B. C.; Gorte, R. J.; Jones, C. W.; Jones, W. D.; Kitchin, J. R.; Scott, S. L.; ACS Catal. 2016, 6, 2590 [Crossref]; Armor, J. N.; Appl. Catal. A. , 2005, 282, 1 [Crossref]; Kramm, U. I.; Marschall, R.; Rose, M.; ChemCatChem 2019, 11, 2563. [Crossref] 2. Trunschke, A.; Bellini, G.; Boniface, M.; Carey, S. J.; Dong, J.; Erdem, E.; Foppa, L.; Frandsen, W.; Geske, M.; Ghiringhelli, L. M.; Girgsdies, F.; Hanna, R.; Hashagen, M.; Hävecker, M.; Huff, G.; Knop-Gericke, A.; Koch, G.; Kraus, P.; Kröhnert, J.; Kube, P.; Lohr, S.; Lunkenbein, T.; Masliuk, L.; d'Alnoncourt, R. N.; Omojola, T.; Pratsch, C.; Richter, S.; Rohner, C.; Rosowski, F.; Rüther, F.; Scheffler, M.; Schlögl, R.; Tarasov, A.; Teschner, D.; Timpe, O.; Trunschke, P.; Wang, Y.; Wrabetz, S.; Top. Catal. 2020, 63, 1683. [Crossref] 3. Türks, D.; Mena, H.; Armbruster, U.; Martin, A.; Catalysts 2017, 7, 152. [Crossref] 4. Roduner, E.; Chem. Soc. Rev. 2014, 43, 8226. [Crossref] 5. Ertl, G. Em Fundamental and Applied Catalysis; Jennings, J. R. , ed.; Springer: Boston, 1991. [Crossref] 6. Zeinalipour-Yazdi, C. D.; Hargreaves, J. S. J.; Laassiri, S.; Catlow, C. R. A. C.; R. Soc. Open Sci. 2021, 8, 11. [Crossref] 7. Sie, S. T.; Blauwhoff, P. M. M.; Catal. Today 1991, 11, 103 [Crossref]; Sie, S. T.; ACS Symp. Ser. 1996, 634, 6 [Crossref]; Hickman, D. A.; Degenstein, J. C.; Ribeiro, F. H.; Curr. Opin. Chem. Eng. 2016, 13, 1. [Crossref] 8. Ergun, S.; Chem. Eng. Prog. 1952, 48, 89. 9. Munro, R. G.; J. Phys. Chem. Ref. Data 1997, 26, 1195 [Crossref]; Yoon, Y. G.; Car, R.; Srolovitz, D. J.; Phys. Rev. B 2004, 70, 012302. [Crossref] 10. Thomson, G. W. M.; Chem. Rev. 1946, 38, 1. [Crossref] 11. Collins, C. H.; Braga, G. L.; Bonato, P. S.; Fundamentos de Cromatografia, 1ª ed.; Editora da Unicamp: Campinas, 2006. 12. Possato, L. G.; Gonçalves, R. G.; Santos, R. M.; Chaves, T. F.; Briois, V.; Pulcinelli, S. H.; Martins, L.; Santilli, C. V.; Appl. Surf. Sci. 2021, 542, 148744. [Crossref] 13. Maksimov, P.; Laari, A.; Ruuskanen, V.; Koiranen, T.; Ahola, J.; RSC Adv. 2020, 10, 2369. [Crossref] 14. Pedroso, M. P.; Scientia Chromatographica 2011, 3, 145. [Crossref] 15. McWillian, I. G.; Dewar, R. A.; Nature 1958, 181, 760. [Crossref] 16. Holm, T.; J. Chromatogr. A 1999, 842, 221. [Crossref] 17. Budiman, H.; Nuryatini; Zuas, O.; Procedia Chem. 2015, 16, 465. [Crossref] 18. Stashenko, E. E.; Ferreira, M. C.; Sequeda, L. G.; Martínez, J. R.; Wong, J. W.; J. Chromatogr. A. 1997, 779, 360. [Crossref] 19. Zacheis, G. A.; Gray, K. A.; Kamat, P. V.; J. Phys. Chem. B 1999, 103, 2142. [Crossref] 20. Dreimann, J. M.; Kohls, E.; Warmeling, H. F. W.; Stein, M.; Guo, L. F.; Garland, M.; Dinh, T. N.; Vorholt, A. J.; ACS Catal. 2019, 9, 4308. [Crossref] 21. Cassinelli, W. H.; Martins, L.; Magnani, M.; Pulcinelli, S. H.; Briois, V.; Santilli, C. V.; RSC Adv. 2016, 6, 20453 [Crossref]; Passos, A. R.; Fontaine, C. L.; Martins, L.; Pulcinelli, S. H.; Santilli, C. V.; Briois, V.; Catal. Sci. Technol. 2018, 8, 6297. [Crossref] 22. Bahia Filho, O.; Prada, P. R.; Meneghesso, C.; Lanças, F. M.; Scientia Chromatographica 2011, 3, 251. [Crossref] 23. Levenspiel, O.; Engenharia das Reações Químicas, 1ª ed.; Blucher: São Paulo, 2000. 24. Schmal, M.; Catálise Heterogênea, 1ª ed.; Synergia: Rio de Janeiro, 2018. 25. Schlögl, R.; Angew. Chem. , Int. Ed. 2015, 54, 3465. [Crossref] 26. Jones, C. W.; Top. Catal. 2010, 53, 942. [Crossref] 27. Sheldon, R. A.; Wallau, M.; Arends, I. W. C. E.; Schuchardt, U.; Acc. Chem. Res. 1998, 31, 485. [Crossref] 28. Martins, L.; Cardoso, D.; Microporous Mesoporous Mater. 2007, 106, 8. [Crossref] 29. Martins, L.; Bonagamba, T. J.; Azevedo, E. R.; Bargiela, P.; Cardoso, D.; Appl. Catal. , A 2006, 312, 77. [Crossref] 30. Dumesic, J.; Huber, G. W.; Boudart, M. Em Handbook of Heterogeneous Catalysis: Online; Ertl, G.; Knözinger, H.; Schüth, F.; Weitkamp, J. , eds.; Wiley‐VCH Verlag: Weinheim, 2008. [Crossref] 31. Catuzo, G. L.; Santilli, C. V.; Martins, L.; Catal. Today 2021, 381, 215. [Crossref] 32. Kozuch, S.; Martin, J. M. L.; ACS Catal. 2012, 2, 2787. [Crossref] 33. Burcham, L. J.; Briand, L. E.; Wachs, I. E.; Langmuir 2001, 17, 6175. [Crossref] 34. Possato, L. G.; Pulcinelli, S. H.; Martins, L.; Briois, V.; Santilli, C. V.; ChemCatChem 2022, 14, e202200684. [Crossref] 35. Weisz, P. B.; Prater, C. D.; Adv. Catal. 1954, 6, 143. [Crossref] 36. Sievers, C.; Noda, Y.; Qi, L.; Albuquerque, E. M.; Rioux, R. M.; Scott, S. L.; ACS Catal. 2016, 6, 8286. [Crossref] 37. Mears, D. E.; Ind. Eng. Chem. Process Des. Dev. 1971, 10, 541. [Crossref] 38. Thoenes Junior, D.; Kramers, H.; Chem. Eng. Sci. 1958, 8, 271. [Crossref] 39. Zelenka, T.; Horikawa, T.; Do, D. D.; Adv. Colloid Interface Sci. 2023, 311, 102831 [Crossref]; Silva, L. L.; Cardoso, D.; Sievers, C.; Martins, L.; J. Phys. Chem. C 2020, 124, 2439. [Crossref] 40. Barrett, E. P.; Joyner, L. G.; Halenda, P. P.; J. Am. Chem. Soc. 1951, 73, 373. [Crossref] 41. Zhang, F.; Zhu, Y.; Lin, Q.; Zhang, L.; Zhang, X.; Wang, H.; Energy Environ. Sci. 2021, 14, 2954. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access