
 |
 |
 |
 |
 |
Revisão
|
|
| Agentes antineoplásicos biorredutíveis: avanços e desafios após 20 anos Bioreductive antineoplasic agents: advances and challenges after 20 years. |
|
Pedro A. L. Santana; Gabriel V. L. Marques; Mariana F. P. Cortezzi; Ricardo J. Alves*; Renata B. de Oliveira*
Departamento de Produtos Farmacêuticos, Faculdade de Farmácia, Universidade Federal de Minas Gerais, 31270-901 Belo Horizonte - MG, Brasil Recebido em: 16/06/2023 *e-mail: renatabo.ufmg@gmail.com; dylancover@gmail.com Solid tumors are frequently affected by the presence of hypoxic cells, originating anomalous and disordered cell growth. This subpopulation presents resistance to conventional chemotherapy agents and radiotherapy, due to the difficulties of reaching the target region, the permanence in the G0 phase of cell cycle and the low local concentration of oxygen. The use of hypoxia activated prodrugs becomes, in this scenario, an effective and selective alternative in cancer therapy, though not yet available for clinical use in solid tumor treatment. Twenty years after the publication of the paper intitled "Agentes Antineoplásicos Biorredutíveis: Uma Nova Alternativa para o Tratamento de Tumores Sólidos" in the Química Nova journal (2002), the current essay aims to identify the advances in the pre-clinical and clinical studies regarding the development of new hypoxia-directed drugs and the improvement of the molecules previously studied. Moreover, to clarify the main challenges for the implementation and success of this drug class, in order to identify the hindrances for the preparation of a bioavailable, safe and selective drug. INTRODUÇÃO O câncer é uma doença resultante do acúmulo de mutações ocorridas durante o processo de divisão em células somáticas. Essas mutações levam à proliferação e diferenciação celular anormal que resulta na formação do tumor. O câncer está geralmente relacionado a propensões genéticas e a agentes ambientais, tais como indicadores demográficos e hábitos que configuram fatores de risco. O GLOBOCAN, portal on-line da IARC (International Agency for Research on Cancer), estima que em 2040 a incidência de câncer alcance o número de 27,5 milhões de casos, o que representa um crescimento de 61,7% em comparação com o relatado no ano de 2020.1 Tumores sólidos, como de intestino, mama, ovário, próstata, pulmão, entre outros, são os de maior incidência e mortalidade dos casos de câncer no Brasil, tendência que reflete o cenário mundial. Complicações relacionadas à ocorrência de tumores sólidos e sua malignidade são atribuídas ao crescimento celular acelerado e desorganizado. Essa proliferação celular cria regiões com diferentes níveis de vascularização (Figura 1). A periferia do tumor apresenta maior contato com a irrigação sanguínea e por esse motivo está em condições de normóxia. A região central do tumor, que se trata da mais deficiente em perfusão, encontra-se em estado de anóxia e em consequente necrose, devido à ausência total de oxigenação. A zona intermediária corresponde, por fim, à área caracterizada pelo abastecimento parcial de oxigênio, o que qualifica o processo de hipóxia.2
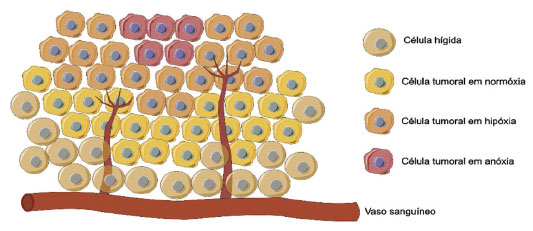 Figura 1. Representação da estrutura de um tumor sólido e da presença de células em normóxia, hipóxia e anóxia
A condição de hipóxia é um fator que dificulta o tratamento de neoplasias por diversos fatores. Além da baixa perfusão sanguínea, que dificulta a distribuição dos fármacos, essas subpopulações celulares se encontram principalmente na fase G0 do ciclo celular e, dessa forma, tornam-se menos sensíveis a algumas classes de quimioterápicos, principalmente àqueles que atuam em células em rápida divisão.3 Ademais, o emprego da radioterapia (RT) é similarmente dificultado, uma vez que a ação citotóxica da radiação é dependente da presença de oxigênio.3 Ao agir nas células da periferia e eliminá-las, os agentes antineoplásicos convencionais podem expor a região em hipóxia à vascularização, de forma a tornar as células ali presentes viáveis para a divisão celular, o que possibilita o tumor a estar em um processo contínuo de atividade e proliferação.4 Em 1972, foi proposta que a condição de hipóxia favorece a ocorrência de reações de redução quando comparada a regiões de células bem oxigenadas.5 Surgiu, portanto, a ideia de se planejar pró-fármacos cuja ativação ocorreria especificamente em ambientes em hipóxia, dando origem à classe dos fármacos biorredutíveis. Dessa forma, é possível alcançar uma maior seletividade do fármaco e consequentemente reduzir a ocorrência de efeitos adversos.6 Tendo em vista a necessidade de se desenvolver pró-fármacos que atendessem aos requisitos necessários para a ativação e a seletividade em células tumorais em hipóxia, foram escolhidas estruturas apresentando grupos biorredutíveis que, após sofrerem redução in vivo, originariam espécies reativas e citotóxicas. Classes químicas como as das mostardas nitroaromáticas, nitroimidazóis, N-óxidos e quinonas demonstraram potencialidade para serem utilizadas e trouxeram para a indústria protótipos interessantes. Apesar disso, compostos dessas classes tiveram, em determinado momento, seus estudos interrompidos, seja por ausência de eficácia ou segurança, mas que ainda servem como modelo para o desenvolvimento de análogos otimizados que possam ser utilizados na clínica.7 Com base no artigo publicado no ano de 2002 intitulado "Agentes Antineoplásicos Biorredutíveis: Uma Nova Alternativa para o Tratamento de Tumores Sólidos", de Oliveira e Alves,8 objetiva-se com esta revisão analisar e identificar novas alternativas e avanços no planejamento dos pró-fármacos biorredutíveis após 20 anos, com foco nas principais modificações em comparação às moléculas estudadas nas décadas anteriores.
AGENTES BIORREDUTÍVEIS Diferentes classes de agentes biorredutíveis podem ser utilizadas na clínica a partir de diferentes mecanismos de ação. A seguir, o desenvolvimento das principais classes de agentes biorredutíveis que se encontram em estudos clínicos atualmente será abordado. Agentes biorredutíveis nitrados O planejamento de um candidato a fármaco, para ativação em condições de hipóxia, apresentando um grupo nitro em sua estrutura é uma estratégia bastante promissora, em razão da possibilidade de biorredução deste grupo funcional. Este grupamento é conhecido por ser toxicofórico e, frequentemente, relacionado com hepatotoxicidade, ocorrência de efeitos adversos e mutagênese.9 Porém, ao pensar em agentes antineoplásicos, a classe desperta interesse por apresentar potencial radiossensibilizador, isto é, capaz de promover sensibilização celular mesmo na ausência de oxigênio, pois é capaz de gerar e mimetizar as ações produzidas pelas espécies reativas de oxigênio (ROS). Surge, portanto, a aplicabilidade de substâncias nitradas como agentes terapêuticos no tratamento de neoplasias.10 Reações de redução são favorecidas em ambientes com baixa concentração de oxigênio. Em condições de hipóxia, o produto de redução por um elétron, o ânion nitro radical, possui meia vida maior pois não é reoxidado pelo oxigênio molecular, o que possibilita sua redução completa a hidroxilamina ou amina (Figura 2).11 Na presença de oxigênio a reação origina um ciclo fútil, no qual o grupo nitro original é restaurado.12 Com isso, a proposta de se desenvolver um pró-fármaco capaz de ser ativo apenas após redução garante a seletividade de agentes neoplásicos para tumores sólidos.8
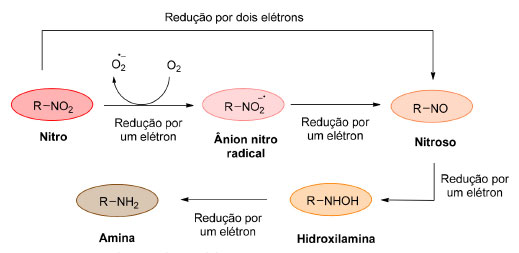 Figura 2. Esquema de redução do grupo nitro pelo metabolismo celular
Mostardas nitroaromáticas Mostardas nitrogenadas são compostos conhecidos desde o início do século XX devido ao seu planejamento a partir da estrutura do gás mostarda, substância altamente tóxica utilizada como arma química durante a Primeira e Segunda Guerras Mundiais. Como exemplo, a mecloretamina, a mostarda nitrogenada mais simples, difere do gás mostarda apenas pela substituição do átomo de enxofre deste pelo de nitrogênio ligado a um grupo metila (Figura 3). A ação tóxica do gás mostarda sobre tecidos e células sanguíneas também é discutida desde o início do século passado e alguns desses efeitos citotóxicos também são comuns às mostardas nitrogenadas.13-15
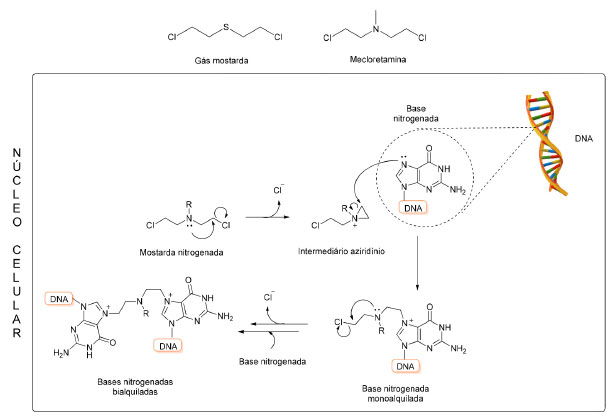 Figura 3. Estruturas químicas do gás mostarda e mecloretamina e representação do mecanismo de alquilação do material genético pelas mostardas nitrogenadas
O potencial antineoplásico dessa classe foi inicialmente descrito por Berenblun, Kendal e Orr (1936), ao observar a ação inibitória do crescimento de lesões relacionadas ao câncer em camundongos.16,17 A partir disso, diversos mecanismos de ação foram propostos para explicar a eficácia da classe no tratamento do câncer. Hoje, sabe-se que as mostardas nitrogenadas atuam como agentes alquilantes do DNA e formam ligações cruzadas com a fita dupla do material genético (Figura 3),18 o que inviabiliza o processo de divisão celular. O mecanismo citostático é ainda acrescido do potencial citotóxico, o que garante o sucesso da classe.19 O estudo do metabolismo da mostarda SN 23862 demonstrou a ocorrência de uma ciclização intramolecular, após redução do grupo nitro, com a formação de um metabólito com atividade significativamente menor (Figura 4).20
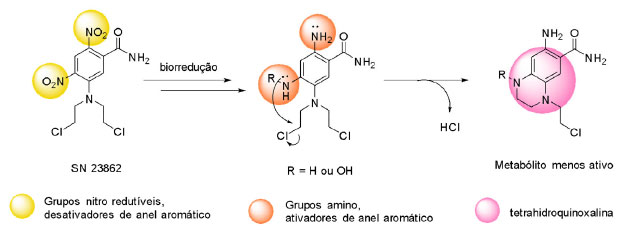 Figura 4. Representação do mecanismo de formação da tetra-hidroquinoxalina após biorredução da mostarda SN23862
Com o objetivo de superar essa limitação e obter novas mostardas nitroaromáticas mais potentes e seletivas e que não formassem o metabólito tetra-hidroquinoxalina após bioredução, Denny et al.21 e Atwell et al.22 realizaram um estudo com diversas mostardas nitroaromáticas ligadas a uma amida contendo um álcool de cadeia curta na cadeia lateral e seus correspondentes ésteres fosfatos. Diversas substâncias foram sintetizadas a partir das substituições propostas (Figura 5) e testadas para a determinação da seletividade frente a linhagens de células tumorais humanas de cólon, pulmão, ovário e mama em condições de normóxia e hipóxia.21,22
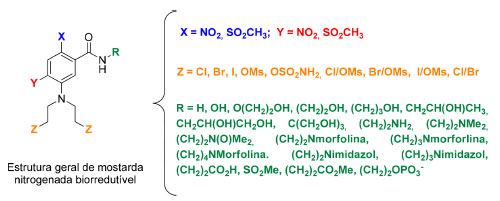 Figura 5. Estrutura geral de mostardas nitrogenadas biorredutíveis representando a variação dos substituintes
Os estudos conduzidos por Atwell et al.22 visaram identificar mostardas nitroaromáticas que pudessem ser utilizadas como pró-fármacos em gene-directed enzyme prodrug therapy (GDEPT). Nesse caso, uma nitroredutase NAD(P)H NAD(P)H de Escherichia coli, insensível ao oxigênio (E. coli nfsB nitroreductase - NTR) foi utilizada, favorecendo a redução do grupo nitro em posição para à mostarda nitroaromática, evitando, assim, a formação do metabólito tetra-hidroquinoxalina. A partir desses estudos a mostarda dinitroaromática bromomesilada, PR-104A (Figura 6), foi definida como a mais promissora com atividade antitumoral potente contra linhagem de células de carcinoma de cólon humano (WiDr) (IC50 = 0,78 ± 0,06 µmol L-1), alta seletividade para células que expressam NTR (NTR+ve) (razão NTR-ve/NTR+ve > 500) e com propriedades físico-químicas adequadas (solubilidade em meio de cultura = 1,7 mmol L-1 e logP = 0,46) para difusão e penetração nos compartimentos extravasculares. Posteriormente, a estrutura da mostarda PR-104A foi otimizada com a obtenção de seu éster fosfato PR-104, com o intuito de aumentar sua hidrossolubilidade.21,23
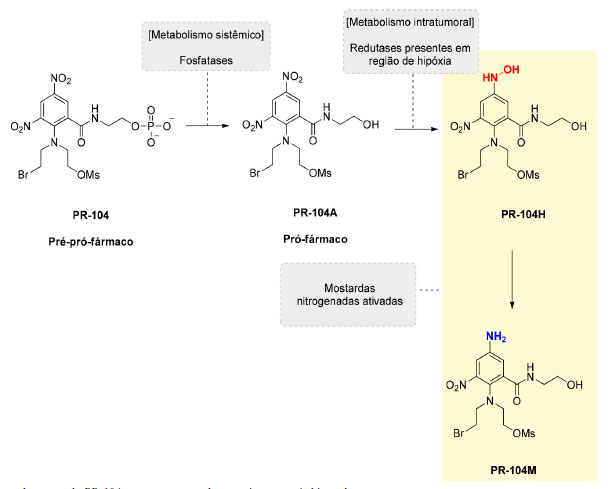 Figura 6. Estrutura da mostarda PR-104 e representação de sua ativação após biorredução
A mostarda PR-104 atua como um "pré-pró-fármaco", sendo convertida no pró-fármaco PR-104A após ação de fosfatases na circulação sistêmica e, em seguida, sendo convertida nas mostardas ativas PR104H e PR-104M após biorredução (Figura 6). O potencial terapêutico da PR-104 tem sido avaliado em diversos estudos clínicos de fase I, II e III, isolada ou em associação com radioterapia ou outros com outros quimioterápicos utilizados na clínica. Infelizmente, os resultados destes estudos têm demonstrado que a PR-104 pode provocar efeitos adversos dose-limitantes, como trombocitopenia, neutropenia e mielotoxicidade.24-26 Além disso, foi descoberto que, em certos tipos de tumores, a PR-104 pode ser ativada mesmo na presença de oxigênio, por enzimas da família aldo-ceto redutases (AKR), o que resultou em uma significativa implicação na continuidade no desenvolvimento clínico da PR-104.27 Na tentativa de se evitar estas limitações, a estrutura da PR-104 foi modificada resultando em uma nova geração de mostardas ativadas em hipóxia, sendo a mostarda CP-506 o exemplo mais promissor desta nova série. A sequência de bioativação da mostarda CP-506 está representada na Figura 7.28
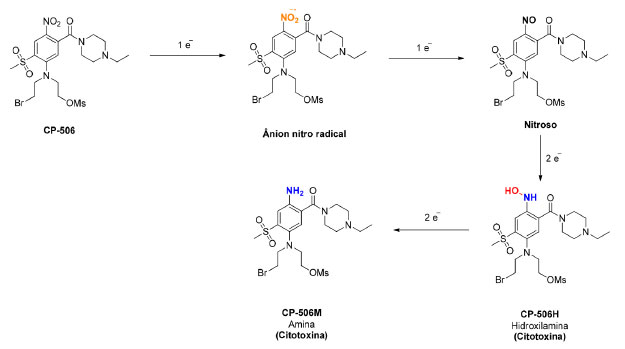 Figura 7. Estrutura da mostarda CP-506 e representação da sua sequência de bioativação
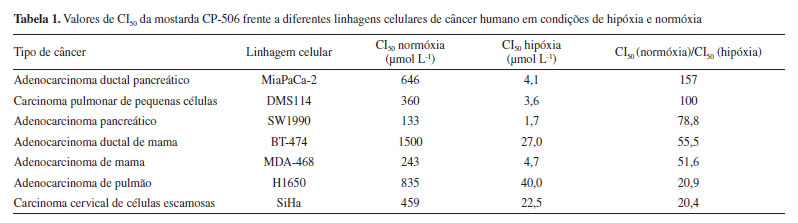
A CP-506 apresenta várias vantagens em relação à PR-104, sendo resistente ao metabolismo pela aldo-ceto redutase AKR1C3 e o seu sal mesilato é solúvel em água, evitando a necessidade da formação de um "pré-pró-fármaco" fosforilado, tendo potencial para ser absorvida por via oral. Além disso, a CP-506 é uma mostarda mononitrada e o grupo nitro está presente na posição para em relação à mostarda, evitando o metabolismo redutivo, seguido de auto-alquilação, do grupo nitro em orto, conforme mostrado na Figura 4.28 Van der Wiel et al.28 realizaram ensaios in vitro utilizando 15 linhagens de células tumorais diferentes para avaliação da citotoxicidade e determinação da seletividade da mostarda CP-506 em condições de hipóxia e normóxia. Para todas as linhagens testadas, os valores da concentração que inibe 50% do crescimento celular (CI50) foi significativamente maior em condições de normóxia do que em condições de hipóxia, sendo que a relação CI50 normóxia/CI50 hipóxia variou de 4 a 157. Os resultados obtidos com melhores índice de seletividade (> 20) estão mostrados na Tabela 1.
Adicionalmente, a atividade antitumoral in vivo da mostarda foi investigada em 15 modelos de tumor xenográfico em camundongos tratados com CP-506, por via intraperitoneal, nas doses de 600 ou 800 mg kg-1 por dia por um período de cinco dias consecutivos. Os modelos de tumor foram selecionados com base nos resultados de sensibilidade obtidos nos estudos in vitro (Tabela 1). CP-506 inibiu significativamente o crescimento do tumor em 13 dos 15 modelos testados, com uma taxa de inibição de 30,9 a 99,3%. Vale ressaltar que no caso do modelo xenográfico de adenocarcinoma de mama (MDA 468), o tratamento com a mostarda CP-506 resultou na regressão completa do tumor em 9 dos 10 animais tratados. Atualmente, voluntários estão sendo recrutados para participar de estudos clínicos de fase 1/2 para avaliação da eficácia da CP-506 em monoterapia ou associado com carboplatina ou com inibidores do checkpoint imunológico.29 Nitroimidazóis Os compostos nitroimidazólicos compõem um grupo de substâncias caracterizadas pela presença de um grupo nitro ligado a um anel imidazólico, tendo o metronidazol como exemplo mais conhecido, indicado para o tratamento de infecções parasitárias ou bacterianas. Desde a década de 1970 vem-se realizando estudos com os nitroimizadóis, e o próprio metronidazol teve sua eficácia comprovada contra células tumorais in vitro pela capacidade de produzir espécies citotóxicas após a redução do grupo nitro em ânion nitro radical. Entretanto, seu potencial como antineoplásico apresentou resultados não muito satisfatórios no que se refere à seletividade e ação in vivo.30 Ainda assim, a classe não foi completamente descartada para esta finalidade, sendo estudada atualmente como radiossensibilizadores. Os radiossensibilizadores são agentes químicos que podem aumentar à sensibilidade das células em hipóxia à radiação, intensificando o dano ao DNA e produzindo radicais livres indiretamente, mesmo na ausência de oxigênio31 (Figura 8).
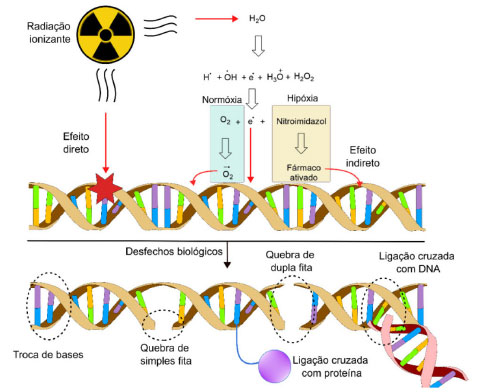 Figura 8. Representação esquemática do efeito direto e indireto da radiação ionizante
Alguns exemplos de fármacos nitroimidazólicos que já foram investigados em estudos clínicos como radiossensibilizadores e possíveis candidatos a pró-fármacos ativados em condições de hipóxia estão ilustrados na Figura 9.32
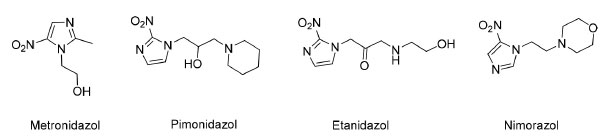 Figura 9. Exemplos de fármacos nitroimidazólicos
Mostarda 2-nitroimidazólica: desenvolvimento da evofosfamida Apesar da eficácia das mostardas nitrogenadas utilizadas na clínica, como a ciclofosfamida e a ifosfamida (Figura 10), estes fármacos apresentam baixa seletividade, resultando em efeitos adversos ao paciente.33 A ifosfamida, derivado da ciclofosfamida, é um pró-fármaco aprovado pela agência regulatória dos Estados Unidos da América, Food and Drug Administration (FDA) em 1998 e pela Agência Nacional de Vigilância Sanitária (Anvisa) em 2013. Sua biotransformação pelas enzimas da família do citocromo P450 (CYP 450) leva à liberação da mostarda ifosforamida (IPM) (Figura 10), comprovadamente eficaz, porém de baixa seletividade.34 Ao mesmo tempo, sabe-se que derivados 2-nitroimidazólicos apresentam alta afinidade por populações celulares em hipóxia. Com base nisso, Duan et al.11 visaram o planejamento de um composto híbrido contendo uma unidade 2-nitroimidazol e uma unidade derivada de uma mostarda nitrogenada, a bromo-ifosforamida (IPM-Br), capaz de liberar a toxina IPM-Br em condições de hipóxia, obtendo-se o composto TH-302, posteriormente chamado evofosfamida (Figura 11).
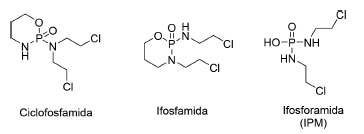 Figura 10. Estruturas químicas da ciclofosfamida, ifosfamida e ifosforamida (IPM)
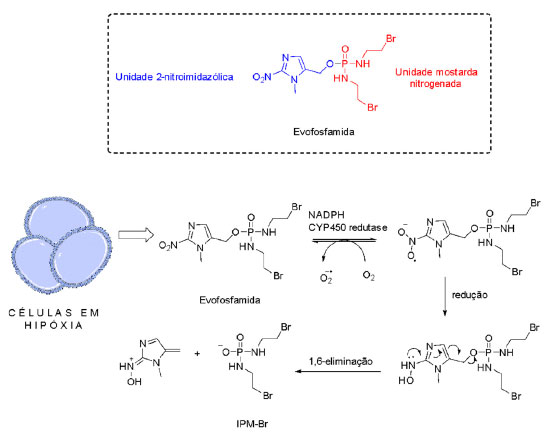 Figura 11. Estrutura da evofosfamida e mecanismo proposto para sua ativação
O mecanismo de ativação proposto para a evofosfamida é baseado na redução do grupo nitro da unidade nitromidazol por enzimas da família CYP450.35 Para a evofosfamida, a redução também indica a liberação da mostarda IPM-Br, agente alquilante que atuará na morte das células tumorais (Figura 11). Os resultados deste estudo demonstraram que a evofosfamida apresentou excelente ação antiproliferativa e seletividade em ensaios in vitro utilizando células humanas de câncer de pulmão H460 e de câncer de cólon HT29, em condições de hipóxia e normóxia (Tabela 2).11

Para avaliação da atividade in vivo da evofosfamida, estudos foram conduzidos utilizando camundongos com câncer pancreático MIA PaCa-2. A evofosfamida foi administrada na dose de 100 mg kg-1 dia-1 e o tratamento iniciou-se 3 dias após a indução do câncer nos animais. Adicionalmente, o desempenho terapêutico e o efeito sinérgico deste candidato a fármaco foram avaliados em associação com a gencitabina, fármaco cuja atividade antitumoral já é bem estabelecida. Os resultados obtidos com esta associação foram extremamente favoráveis, como a redução da progressão do tumor e aumento da proporção de sobreviventes.11 Em razão de sua excelente eficácia em animais, estudos clínicos com a evofosfamida estão em andamento. Estudos para a avaliação da associação da evofosfamida e gencitabina se desenvolveram até a fase II dos estudos clínicos. Borad et al.36 analisaram o potencial da combinação para o tratamento de câncer pancreático avançado ou metastático, o mais letal dos tumores sólidos. O desfecho primário analisado foi a incidência de sobreviventes sem progressão (SSP). O estudo se desenvolveu de modo open-label, multicêntrico e randomizado, entre junho 2010 e julho 2011 com 229 pacientes, para avaliar os benefícios da terapia de associação da evofosfamida e gencitabina em pacientes adultos com histórico prévio de câncer pancreático avançado confirmado e não tratado. Conforme desenho experimental ilustrado na Figura 11, os pacientes tratados com a associação de evofosfamida e gencitabina foram divididos em grupos que receberam diferentes doses de evofosfamida (240 ou 340 mg m-2) mantendo a dose de gencitabina (1000 mg m-2). A avaliação do tumor e determinação dos níveis séricos de evofosfamida e do metabóito ativo IPM foram realizadas a cada 8 semanas. Os resultados determinantes do desfecho demonstram a ação favorável da associação. Desfechos secundários como seletividade, resposta objetiva ao tumor e aumento da sobrevida geral também expuseram dados favoráveis à evofosfamida. É possível afirmar, portanto, que a combinação das substâncias é benéfica à terapêutica de pacientes com câncer pancreático (sobreviventes sem progressão de 3,6 meses para gencitabina, 5,6 meses para gencitabina + evofosfamida 240 mg m-2 e 6 meses para gencitabina + evofosfamida 340 mg m-2).36 A partir destes resultados, diversos estudos surgiram para determinar a aplicabilidade da evofosfamida como possível agente antineoplásico biorredutível. Em sua maioria, os ensaios decorrem da avaliação da ação do candidato a fármaco em associação com uma opção terapêutica já utilizada para o tratamento do câncer, com o intuito de obter o melhor desfecho possível. Nesse sentido, estudos foram conduzidos na tentativa de alcançar resultados que possibilitem a aprovação da evofosfamida como terapia adicional ao câncer.37 Para o tratamento de câncer de mama, principal câncer letal em mulheres em todo o mundo, o prognóstico é pessimista, além de comumente associado à ocorrência de metástase.38 O estudo realizado por Liapis et al.39 foi pioneiro na demonstração da eficácia da evofosfamida para este tipo de tumor e apresentou resultados promissores. Inicialmente, o estudo foi desenvolvido in vitro pela utilização de linhagens de células humanas de câncer de mama MCF-7, MDA-MB-453, MDA-MB-468, T47D e ZR-75, conduzido em condições de hipóxia e normóxia para avaliação da seletividade. Nas cinco linhagens estudadas foi possível observar o efeito tóxico mínimo que a evofosfamida apresenta sobre células em normóxia, enquanto exibe capacidade de inviabilizar o crescimento celular em células em hipóxia, o que indica uma alta seletividade.39 Estudos in vivo também foram conduzidos, utilizando-se modelos xenográficos de câncer de mama humano em camundongos. O estudo se desenvolveu com a comparação da atividade da evofosfamida e do paclitaxel isoladamente e da ação de ambos em associação.39 O paclitaxel é um fármaco aprovado pela FDA em 1992 e pela Anvisa em 2006 para tratamento de câncer de mama e atua inibindo o processo mitótico pela ação sobre os microtúbulos envolvidos na divisão celular.40 A análise se desenvolveu pela quantificação do surgimento e crescimento de câncer por bioiluminescência e registradas as diferenças entre os grupos de camundongos nos quais foram administrados veículo, evofosfamida, paclitaxel ou a associação de evofosfamida e paclitaxel. Animais que receberam tratamento com a evofosfamida ou com o paclitaxel apresentaram redução significativa no crescimento do tumor, sendo que os animais que receberam a combinação de ambos mostraram uma redução ainda maior quando comparado aos agentes administrados individualmente.39 Em outro estudo realizado por Spiegelberg et al.,41 foi avaliada a ação e segurança da utilização da evofosfamida como radiossensibilizador em radioterapia. Neste estudo foram utilizadas linhagens celulares de adenocarcinoma de esôfago (OE19) e carcinoma de células escamosas (OE21). O estudo se desenvolveu in vivo a partir da inserção de modelos xenográficos em camundongos imunocompetentes, posteriormente tratados com o candidato a fármaco em associação com a técnica convencional (radiação por raios-X a 20 Gy). Para controle, usou-se solução salina e sham, uma radiação de baixa energia que não possui ação biológica, somente usada como controle em ensaios. A evofosfamida foi administrada na dose de 50 mg kg-1, por via intraperitoneal. A associação da evofosfamida com a radioterapia resultou em uma redução significativa do crescimento do tumor em ambos os carcinomas quando comparado à radioterapia isoladamente. Essa combinação pode, portanto, representar uma abordagem promissora no aumento do índice terapêutico para pacientes com câncer de esôfago.41 A ação da evofosfamida também foi estudada na pediatria para o tratamento de tumores no cérebro. No estudo de Bailleu et al.,42 linhagens de glioma dos tipos DIPG007, DIPG0013, DIPG0014, KNS42, SF188 e UW479 foram cultivadas em meios sob uma concentração de 21% O2 (normóxia) e 1% O2 (hipóxia) e posteriormente submetidas a concentrações crescentes de evofosfamida, a fim de obter os valores de CI50 em ambas as condições (Tabela 3).
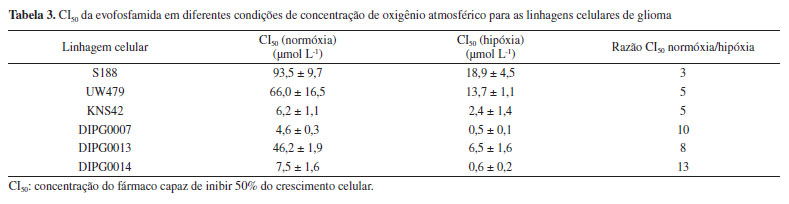
A partir dos resultados obtidos nos estudos in vitro, Bailleu et al.42 deram prosseguimento aos estudos para a avaliação do efeito sinérgico entre a evofosfamida e fármacos comumente indicados na pediatria oncológica (etoposido, doxorrubicina ou SN38, metabólito ativo do irinotecan), in vitro utilizando as linhagens de células de glioma SF188 e DIPG007. A avaliação da resposta conjunta foi configurada como interação do tipo sinergismo moderado, ainda que benéfico. Além dos estudos pré-clínicos descritos, a evofosfamida, também, vem sendo avaliada em diversos estudos clínicos de fase II e fase III contra uma variedade de tipos de câncer, como monoterapia ou em combinação com quimioterápicos convencionais. Alguns desses estudos clínicos não demonstraram uma melhora significativa na sobrevida dos pacientes tratados com evofosfamida como monoterapia ou em combinação, quando comparada ao tratamento convencional (por exemplo: estudos clínicos TH-CR-406/SARC021, NCT01746979).43 Derivado nitroimidazólico associado a um inibidor de tirosina quinase: desenvolvimento da tarloxotinibe Outro exemplo de derivado nitroimidazólico com potencial uso como agente biorredutível é o TH-4000 (Figura 12), anteriormente descrito como PR-610 e posteriormente nomeado como tarloxotinibe (nomeado com base na expressão "targeting low oxygen"). TH-4000 foi citado pela primeira vez por Patterson et al.44 em 2016 e trata-se de um pró-fármaco biorredutível de atuação em receptores de tirosina quinase (TK) de fator de crescimento epidermal (EGFR). Receptores de tirosina quinase são responsáveis pela comunicação intracelular e estão relacionadas com a interação com fatores de crescimento e promoção da proliferação celular. Esse processo, quando desequilibrado, pode levar à promoção do crescimento celular desordenado e à oncogênese.45 Desse modo, o desenvolvimento do tarloxotinibe partiu da inspiração da união entre um derivado nitroimidazólico com um inibidor de TK, com farmacocinética e ação em tumores sólidos otimizadas. A presença de uma carga positiva permanente na estrutura do tarloxotinibe dificulta sua penetração nos tecidos normais oxigenados, o que favorece a sua ação após redução no microambiente tumoral em hipóxia e aumenta a janela terapêutica. O tarloxotinibe-E (TH-4000E) consiste no fármaco ativo responsável pela promoção do efeito antineoplásico, originado pela redução do pró-fármaco tarloxotinibe (Figura 12).44
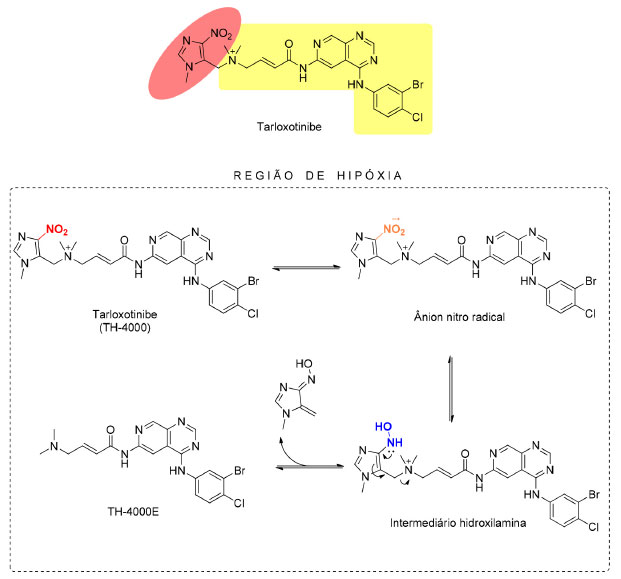 Figura 12. Estruturas químicas do afatinibe e tarloxotinibe e mecanismo de ativação do tarloxotinibe em região de hipóxia
Estudos realizados por Estrada-Bernal et al.46 visaram avaliar a eficácia do tarloxotinibe, in vitro e in vivo, como inibidor dos diferentes receptores de TK da família ErBb: HER1 (EGFR), HER2, HER3 (NRG1) e HER4. As mutações nesses receptores representam de 10 a 40% dos casos de adenocarcinoma pulmonar. Nos estudos in vitro foram feitas comparações dos valores de CI50 do tarloxotinibe (pró-fármaco) e do tarloxotinibe-E (ativo), assim como dos inibidores de TK gefitinibe, afatinibe e osimertinibe, em oito diferentes linhagens celulares, incluindo linhagens isoladas de pacientes com mutação de inserção no éxon 20 no gene EGFR. O tarloxotinibe-E apresentou valores de IC50 significativamente menores (IC50 = 0,3 ± 3 a 667 ± 25 nmol L-1) do que os dos três fármacos utilizados como referência em todas as linhagens testadas. Como esperado, o tarloxotinibe foi o menos ativo (IC50 = 307 ± 4,2 a > 10.000 nmol L-1) quando testado em condições de normóxia, confirmando a necessidade de ativação deste pró-fármaco para exercer sua ação.46 Nos estudos in vivo foram utilizados modelos xenográficos múltiplos em camundongos divididos em grupos que receberam veículo, afatinibe (6 mg kg-1 diariamente por via oral), ou tarloxotinibe (48 mg kg-1 por injeção intraperitoneal uma vez por semana, durante 4 semanas). Tarloxotinibe foi capaz de induzir a regressão ou inibir o crescimento do tumor em todos os modelos avaliados.46 No estudo também é investigado o perfil farmacocinético do tarloxotinibe e de seu metabólito ativo, de modo a demonstrar a seletividade e a capacidade de conversão in vivo do pró-fármaco no fármaco ativo em hipóxia. Neste estudo foi confirmado níveis significativamente mais elevados do tarloxotinibe-E no tecido tumoral do que no plasma ou na pele. Adicionalmente, um paciente com adenocarcinoma de pulmão com uma mutação HER2 exon 20 demonstrou uma resposta clínica expressiva ao tratamento com tarloxotinibe. Com base nos resultados desses estudos foi demonstrado o grande potencial do tarloxotinibe como um agente antineoplásico biorredutível.46 Estudos clínicos com o tarloxotinibe foram desenvolvidos e alcançaram a fase II, conforme reportado por McLean et al.47 Pacientes elegíveis para o ensaio deveriam obrigatoriamente portar câncer escamoso celular cutâneo (CSCC) ou carcinoma escamoso de cabeça e pescoço (HNSCC), recorrentes ou metastáticos, com laudo cito ou histológico e serem maiores de 18 anos de idade. O ensaio se desenvolveu de forma open-label e multicêntrica, entre 12 de agosto de 2015 a 1º de fevereiro de 2017, em centros de saúde nos EUA e na Austrália. Trinta voluntários foram divididos em três grupos de acordo com os atributos do tumor, como (CSCC ou HNSCC) e estágio (estável, progressivo ou não avaliável).46 Foram reportados eventos adversos de gravidade baixa, moderada e alta, sendo os mais comuns o prolongamento do intervalo QT (incidência = 11,37%) e rash cutâneo (incidência = 6,20%). A sobrevida livre de progressão e a sobrevida geral foram obtidas a partir dos dados coletados em um período de 12 meses.47 O tarloxotinibe representa o candidato a fármaco biorredutível mais recente dentre os participantes da fase de estudos clínicos. Atualmente, possui 19 patentes, foi avaliado em três ensaios clínicos e, atualmente, voluntários estão sendo recrutados para novo ensaio clínico para tratamento de tumor de pulmão de células não pequenas, de acordo com os resultados da busca na plataforma desenvolvida pelo Chemical Abstracts Service (CAS). O tarloxotinibe pode ser considerado um forte candidato a pró-fármaco para o tratamento de tumores sólidos, dado os resultados que expressam eficácia, seletividade e segurança. A atividade de fármacos inibidores de receptores de fatores de crescimento celular vai além do ambiente de hipóxia que acomete os tumores sólidos, mas envolve a interação com os alvos que podem apresentar diferentes graus de mutação, responsável pelo surgimento da neoplasia. Assim, é possível citar como desafio ao desenvolvimento deste candidato a fármaco como opção terapêutica, o tempo curto para o aprimoramento da substância e a alta complexidade envolvida para a excelência da ação a nível molecular.
CONCLUSÕES Os agentes antineoplásicos biorredutíveis estão em constante avanço. Em mais de duas décadas, as substâncias originalmente estudadas e evidenciadas sofreram modificações estruturais com o objetivo de otimização da estrutura, visando aumento de potência, seletividade e segurança, de forma a possibilitar a aprovação de fármacos eficazes e seguros para o uso na clínica. De forma geral, os agentes antineoplásicos biorredutíveis sofreram modificações consideráveis nos últimos vinte anos. Oliveira e Alves8 citaram, na época, a possibilidade do emprego de outras classes para o tratamento de tumores sólidos, como quinonas e N-óxidos. Para as quinonas, encontra-se como entrave para o uso terapêutico a dificuldade de se garantir a seletividade e, dessa forma, a segurança adequada. A classe é empregada como agente antibacteriano, como os derivados de benzoquinona e naftoquinona. O efeito sobre bactérias torna a ampliação da indicação terapêutica de quinonas um risco para a resistência bacteriana e para a promoção de pressão seletiva de microrganismos resistentes, alvos de preocupação para a saúde pública atual. Já para os N-óxidos, Oliveira e Alves8 citam como exemplo a tirapazamina, cujos ensaios clínicos não são atualizados desde década de 2010. A razão pode estar relacionada ao alto índice de efeitos adversos relatados.48 É possível identificar os avanços no desenvolvimento de pró-fármacos biorredutíveis nos últimos 20 anos. O atraso no andamento e na concretização da classe não deve ser motivo de desistência, pois os resultados demonstram a possibilidade de se atingir uma nova expectativa de vida aos pacientes com diversos tipos de câncer. Historicamente, a química medicinal avança em diferentes velocidades entre as diversas classes terapêufticas. A obtenção de novos fármacos é um processo longo e dispendioso, que envolve equipes multiprofissionais e estruturas complexas para adequação e sucesso. A continuidade e o propósito dos estudos não devem ser perdidos ou desestimulados, pois o aumento na qualidade e na sobrevida de pacientes oncológicos é responsabilidade dos profissionais da saúde com os pacientes e a população.
AGRADECIMENTOS Os autores agradecem a Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) pelo auxílio financeiro (PPM 00417-17), à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pela bolsa de produtividade de R. B. O.
REFERÊNCIAS 1. Internacional Agency for Research on Cancer, https://gco.iarc.fr/today, acessado em Setembro de 2023. 2. Rakotomalala, A.; Escande, A.; Furlan, A.; Meignan, S.; Lartigau, E.; Front. Endocrinol. 2021, 12, 742215. [Crossref] 3. Allen, C.; Her, S.; Jaffray, D. A.; Adv. Drug Delivery Rev. 2017, 109, 1. [Crossref] 4. Taylor, E.; Zhou, J.; Lindsay, P.; Foltz, W.; Cheung, M.; Siddiqui, I.; Hosni, A.; Amir, A. E.; Kim, J.; Hill, R. P.; Jaffray, D. A.; Hedley, D. W.; Sci. Rep. 2020, 10, 1683. [Crossref] 5. Lin, A. J.; Cosby, L. A.; Shansky, C. W.; Sartorelli, A. C.; J. Med. Chem. 1972, 15, 1247. [Crossref] 6. Li, Y.; Zhao, L.; Li, X. F.; Front. Oncol. 2021, 11, 700407. [Crossref] 7. Luo, X.; Li, A.; Chi, X.; Lin, Y.; Liu, X.; Zhang, L.; Su, X.; Yin, Z.; Lin, H.; Gao J.; Bioconjugate Chem. 2021, 32, 983. [Crossref] 8. Oliveira, R. B.; Alves, R. J.; Quim. Nova 2002, 25, 976. [Crossref] 9. Ang, C. W.; Jarrad, A. M.; Cooper, M. A.; Blaskovich, M. A. T.; J. Med. Chem. 2017, 60, 7636. [Crossref] 10. Bonnet, M.; Hong, C. R.; Gu, Y.; Anderson, R. F.; Wilson, W. R.; Pruijn, F. B.; Wang, J.; Hicks, K. O.; Hay, M. P.; Bioorg. Med. Chem. 2014, 22, 2123. [Crossref] 11. Duan, J. X.; Jiao, H.; Kaizerman, J.; Stanton, T.; Evans, J. W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.; Ma, H.; Li, X.; Yang, Z.; Hoffman, R. M.; Ammons, W. S.; Hart, C. P.; Matteucci, M.; J. Med. Chem. 2008, 51, 2412. [Crossref] 12. Wardman, P.; Curr. Med. Chem. 2001, 8, 739. [Crossref] 13. Krumbhaar, E. B.; Krumbhaar, H. D.; J. Med. Res. 1919, 40, 497. [Crossref] 14. Gilman, A.; Philips, F. S.; Science 1946, 103, 409. [Crossref] 15. Wintrobe, M. M.; Am. J. Med. 1948, 4, 313. [Crossref] 16. Berenblum, I.; Kendal, L. P.; Orr, J. W.; Biochem. J. 1936, 30, 709. [Crossref] 17. Garai, O.; Postgrad. Med. J. 1948, 24, 307. [Crossref] 18. Polavarapu, A.; Stillabower, J. A.; Stubblefield, S. G. W.; Taylor, W. M.; Baik, M. H.; J. Org. Chem. 2012, 77, 5914. [Crossref] 19. Fu, D.; Calvo, J. A.; Samson, L. D.; Nat. Rev. Cancer 2012, 12, 104. [Crossref] 20. Palmer, B. D.; van Zijl, P.; Denny, W. A.; Wilson, W. R.; J. Med. Chem. 1995, 38, 1229. [Crossref] 21. Denny, W. A.; Atwell, G. J.; Yang, S.; Wilson, W. R.; Patterson, A. V.; Helsby, N. A.; WO 2005/042471 A1 2005. 22. Atwell, G. J.; Yang, S.; Pruijn, F. B.; Pullen, S. M.; Hogg, A.; Patterson, A. V.; Wilson, W. R.; Denny, W. A.; J. Med. Chem. 2007, 50, 1197. [Crossref] 23. Patterson, A. V.; Ferry, D. M.; Edmunds, S. J.; Gu, Y.; Singleton, R. S.; Patel, K.; Pullen, S. M.; Hicks, K. O.; Syddall, S. P.; Atwell, G. J.; Yang, S.; Denny, W. A.; Wilson, W. R.; Clin. Cancer Res. 2007, 13, 3922. [Crossref] 24. Jameson, M. B.; Rischin, D.; Pegram, M.; Gutheil, J.; Patterson, A. V.; Denny, W. A.; Wilson, W. R.; Cancer Chemother. Pharmacol. 2010, 65, 791. [Crossref] 25. Konopleva, M.; Thall, P. F.; Yi, C. A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; Jabbour, E.; Faderl, S.; Thomas, D.; Cortes, J.; Kadia, T.; Kornblau, S.; Daver, N.; Pemmaraju, N.; Nguyen, H. Q.; Feliu, J.; Lu, H.; Wei, C.; Wilson, W. R.; Melink, T. J.; Gutheil, J. C.; Andreeff, M.; Estey, E. H.; Kantarjian, H.; Haematologica 2015, 100, 927. [Crossref] 26. McKeage, M. J.; Gu, Y.; Wilson, W. R.; Hill, A.; Amies, K.; Melink, T. J.; Jameson, M. B.; BMC Cancer 2011, 11, 432. [Crossref] 27. Guise, C. P.; Abbattista, M. R.; Singleton, R. S.; Holford, S. D.; Connolly, J.; Dachs, G. U.; Fox, S. B.; Pollock, R.; Harvey, J.; Guilford, P.; Doñate, F.; Wilson, W. R.; Patterson, A. V.; Cancer Res. 2010, 70, 1573. [Crossref] 28. van der Wiel, A. M.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A. M.; Lieuwes, N. G.; Biemans, R.; Lin, X.; Fu, Z.; Kumara, S.; Jochems, A.; Ashoorzadeh, A.; Anderson, R. F.; Hicks, K. O.; Bull, M. R.; Abbattista, M. R.; Guise, C. P.; Deschoemaeker S.; Thiolloy, S.; Heyerick, A.; Solivio, J. M.; Balbo, S.; Smaill, J. B.; Theys, J.; Dubois, J. L.; Patterson A. V.; Lambin, P.; Mol. Cancer Ther. 2021, 20, 2372. [Crossref] 29. ClinicaTrials.gov, https://beta.clinicaltrials.gov/study/NCT04954599, acessado em Setembro de 2023. 30. Bryant, C.; Deluca, M.; J. Biol. Chem. 1991, 266, 4119. [Crossref] 31. Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S.; Int. J. Nanomed. 2021, 16, 1083. [Crossref] 32. Wardman, P.; Br. J. Radiol. 2019, 92, 20170915. [Crossref] 33. Borch, R. F.; Liu, J.; Schmidt, J. P.; Marakovits, J. T.; Joswig, C.; Gipp, J. J.; Mulcahy, R. T.; J. Med. Chem. 2000, 43, 2258. [Crossref] 34. Han, H. Y.; Choi, M. S.; Yoon, S.; Ko, J. W.; Kim, S. K.; Kim, T. W.; Int. J. Mol. Sci. 2021, 22, 12201. [Crossref] 35. Adams, G. E.; Cancer 1981, 48, 696. [Link] acessado em Setembro 2023 36. Borad, M. J.; Reddy, S. G.; Bahary, N.; Uronis, H. E.; Sigal, D.; Cohn, A. L.; Schelman, W. R.; Stephenson Junior, J.; Chiorean, E. G.; Rosen, P. J.; Ulrich, B.; Dragovich, T.; Del Prete, S. A.; Rarick, M.; Eng, C.; Kroll, S.; Ryan, D. P.; J. Clin. Oncol. 2015, 33, 1475. [Crossref] 37. Larue, R. T. H. M.; Van De Voorde, L.; Berbée, M.; van Elmpt, W. J. C.; Dubois, L. J.; Panth, K. M.; Peeters, S. G. J. A.; Claessens, A.; Schreurs, W. M. J.; Nap, M.; Warmerdam, F. A. R. M.; Erdkamp, F. L. G.; Sosef, M. N.; Lambin, P.; BMC Cancer 2016, 16, 644. [Crossref] 38. Instituto Nacional de Câncer José Alencar Gomes da Silva (INCA); A Situação do Câncer de Mama no Brasil: Síntese de Dados dos Sistemas de Informação, Rio de Janeiro, Brasil, 2019. [Link] acessado em Setembro 2023 39. Liapis, V.; Zinonos, I.; Labrinidis, A.; Hay, S.; Ponomarev, V.; Panagopoulos, V.; Zysk, A.; DeNichilo, M.; Ingman, W.; Atkins, G. J.; Findlay, D. M.; Zannettino, A. C. W.; Evdokiou, A.; Cancer Med. 2016, 5, 534. [Crossref] 40. Weaver, B. A.; Mol. Biol. Cell. 2014, 25, 2677. [Crossref] 41. Spiegelberg, L.; van Hoof, S. J.; Biemans, R.; Lieuwes, N. G.; Marcus, D.; Niemans, R.; Theys, J.; Yaromina, A.; Lambin, P.; Verhaegen, F.; Dubois, L. J.; Radiother. Oncol. 2019, 141, 247. [Crossref] 42. Bailleul, Q.; Navarin, P.; Arcicasa, M.; Bal-Mahieu, C.; Carcaboso, A. M.; Le Bourhis, X.; Furlan, A.; Meignan, S.; Leblond, P.; Cancers 2021, 13, 1804. [Crossref] 43. ClinicaTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01746979, acessado em Setembro de 2023. 44. Patterson, A. V.; Smaill, J. B.; Silva, S.; Guise, C. P.; Bull, M. R.; Jackson, V.; Pearce, T.; Davar, N.; WO 2016/090174 A1 2016. 45. Du, Z.; Lovly, C. M.; Mol. Cancer 2018, 17, 58. [Crossref] 46. Estrada-Bernal, A.; Le, A. T.; Doak, A. E.; Tirunagaru, V. G.; Silva, S.; Bull, M. R.; Smaill, J. B.; Patterson, A. V.; Kim, C.; Liu, S. V.; Doebele, R. C.; Clin. Cancer Res. 2021, 27, 1463. [Crossref] 47. McLean, L. S.; Morris, T. A.; Gramza, A.; Liu, S.; Khan, S. A.; Colevas, A. D.; Pearce, T.; Rischin, D.; Invest. New Drugs 2022, 40, 782. [Crossref] 48. Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y.; Mol. Cancer 2019, 18, 157. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access