
 |
 |
 |
 |
 |
Assuntos Gerais
|
|
| O uso das ferramentas da análise da superfície de hirshfeld no estudo das ligações não-covalentes em complexos correlatos The use of hirshfeld surface analysis tools in the study of non-covalent bonds in related complexes |
|
Eduardo M. Iwaya; Avany J. F. Miorim; Dayane M. Reis*
Departamento Acadêmico de Química e Biologia, Universidade Tecnológica Federal do Paraná, 81280-340 Curitiba - PR, Brasil Recebido em: 01/08/2023 *e-mail: dayane@utfpr.edu.br The analysis of non-covalent interactions in crystalline structures through the Hirshfeld surface method using the CrystalExplorer® software has been widely used by the global scientific community over the past few decades. Although widely recognized, as far as we know, there has been no publication on this subject in the Química Nova journal. Therefore, in this work, we intended to demonstrate how these tools could be employed to study and compare the non-covalent bonds in 21 mononuclear complexes of the type [MY2(o-XPy)2], [MY2(m-XPy)2], and [MY2(p-XPy)2], where M = Zn2+ or Co2+, Y = Cl−, Br-, or I−; Py = pyridinic ligand ortho-, meta-, or para-monosubstituted by X, where X = Cl−, Br−, or I−. With a main emphasis on halogen bonds and π···π stacking interactions, the study demonstrates how Hirshfeld analysis can be used for a qualitative and quantitative comparison of non-covalent bonds, proving to be an important tool for a comprehensive understanding of the forces that stabilize these complexes in the solid-state. The study demonstrates the potential of Hirshfeld surface analysis as a valuable resource for synthetic chemists to select chemical species for rational synthesis and achieve desired properties or supramolecular arrays. INTRODUÇÃO Na química supramolecular, as ligações não-covalentes são as forças que permitem que moléculas individuais se organizem em estruturas mais complexas, como redes, agregados e supramoléculas.1 As propriedades químicas e físicas diferenciadas destas estruturas em relação às moléculas individuais torna esta ciência importante nas áreas da química de materiais,2,3 catálise,4,5 sensores,6,7 química biológica8,9 e em diversos outros ramos de aplicação.10,11 Na busca por estas características, muitas vezes é necessário um ajuste cuidadoso nas ligações não-covalentes, pois é crucial olhar para a estrutura cristalina em sua totalidade ao invés de focar em algumas interações não-covalentes pré-selecionadas.12 A observação da estrutura cristalina através das ferramentas da análise da superfície de Hirshfeld permite que seja possível observar todas as ligações não-covalentes responsáveis por manter a estrutura supramolecular. O método foi desenvolvido por R. W. G. Hirshfeld em 1977 e é uma forma de visualizar e analisar a distribuição da densidade eletrônica entre moléculas adjacentes.13 O cálculo utiliza os dados coletados na difração de raios X por monocristal e armazenados no arquivo CIF (Crystallographic Information File) em conjunto com os argumentos da teoria de partição dos cristais, que se baseia no fracionamento do cristal em diferentes regiões com base em suas densidades eletrônicas, para gerar uma superfície que define o volume ocupado pela densidade eletrônica da pró-molécula no cristal. Através de funções matemáticas das distâncias atômicas, a superfície fornece informações sobre as interações não-covalentes envolvendo a espécie química de interesse, que são representadas através de cores que indicam as regiões aceitadoras e doadoras destas interações.14 Uma das ferramentas desta análise permite que sejam geradas projeções bidimensionais da superfície de Hirshfeld em função de de e di, as distâncias em angstrons entre a superfície e o átomo mais próximo dentro (di) e fora (de) dela. Essas projeções, também chamadas de gráficos de impressão digital (tradução livre de fingerprint plots), permitem separar as diferentes ligações não-covalentes em gráficos bidimensionais distintos e obter a porcentagem de contribuição de cada uma delas para a superfície, permitindo uma análise minuciosa das contribuições não-covalentes que mantêm a estrutura supramolecular. Como o nome sugere, os gráficos de impressão digital são únicos para cada espécie química e, devido aos fundamentos matemáticos empregados, são independentes do número de átomos na estrutura, o que permite a comparação entre estruturas correlatas.14 Dentre as diversas ligações não-covalentes responsáveis pela manutenção da estrutura supramolecular estão a ligação de halogênio (LX), a ligação de hidrogênio e as interações do tipo empilhamento π···π,15 por exemplo, sendo todas estas forças fortes responsáveis pela organização supramolecular. Estas forças de atração podem dirigir a síntese, a catálise e os processos de reconhecimento molecular em sistemas biológicos ou artificiais.16 Em especial sobre a LX, esta é uma interação altamente direcional que ocorre entre um átomo de halogênio eletrofílico (X-, doador de ligação de halogênio) e um átomo nucleofílico (aceitador de ligação de halogênio).17 A direcionalidade, a hidrofobicidade, o ajuste da força e as dimensões do átomo doador são características únicas da LX que vem sendo usadas no design e na síntese de sistemas automontados. Neste contexto, as ferramentas da análise da superfície de Hirshfeld são aliadas no estudo comparativo de compostos de coordenação correlatos,18,19,20 buscando padrões estruturais capazes de orientar o químico sintético na escolha de pré-ligantes e outras espécies químicas que possam vir a participar do retículo cristalino com o intuito de atingir a relação entre a estrutura e a propriedade desejada. Apesar da reconhecida importância da análise de Hirshfeld pela comunidade científica global, a pesquisa na base de dados Web of Science mostra que ainda não há nenhuma publicação sobre este assunto no periódico Química Nova. A sua relevância é evidenciada quando não se restringe o periódico, aonde a pesquisa retorna 5728 resultados para a palavra-chave "Hirshfeld surface", e 1296 resultados para a combinação "Hirshfeld surface e supramolecular".21 Neste estudo, estruturas de complexos correlatos foram empregadas para demostrar como as ferramentas de Hirshfeld podem ser interessantes no suporte ao estudo dos aspectos supramoleculares de compostos de coordenação, com foco principal nas LX e no empilhamento π···π. O resultado mostra que a análise de Hirshfeld pode dar suporte ao estudo das ligações não-covalentes, à comparação entre diferentes estruturas e na determinação da presença ou ausência do empilhamento π···π em um retículo cristalino. Além disso, o estudo indica como a combinação de dados cristalográficos e de métodos computacionais de fácil acesso podem ser usados para guiar futuros trabalhos de síntese e contribui para a divulgação destas ferramentas de análise junto à comunidade científica brasileira.
METODOLOGIA COMPUTACIONAL O estudo foi delineado por uma pesquisa minuciosa na base de dados Cambridge Structural Database (CSD) por complexos mononucleares do tipo [MY2(o-XPy)2] (Figura 1S), [MY2(m-XPy)2] (Figura 12S) e [MY2(p-XPy)2] (Figura 18S), onde M = Zn2+ ou Co2+, Y = Cl−, Br− ou I− e Py = ligante piridínico monosubstituído em orto-, meta- ou para- por X, sendo X = Cl−, Br− ou I−. Foram encontradas 21 estruturas, que foram numeradas e separadas em grupos (A, B ou C) em função do átomo de halogênio Y (Tabela 1).
O software CrystalExplorer 21.5®14 foi empregado para o cálculo das superfícies de Hirshfeld (função dnorm e shape index) e dos gráficos bidimensionais de impressão digital. As superfícies de Hirshfeld foram mapeadas entre o azul (maiores distâncias) e o vermelho (menores distâncias). Os gráficos bidimensionais foram mapeados entre 0,4 e 3,0 Å para cada par de distâncias de e di. As cores dos gráficos bidimensionais foram modificadas com o auxílio do software GIMP 2.10.24®22 para uma melhor distinção das diferentes interações não-covalentes. O software VESTA®23 foi usado para a medida das distâncias das ligações não-covalentes a partir das informações cristalogáficas dos arquivos CIF e os valores foram comparados com a soma dos raios de van der Waals para os halogênios.24
ANÁLISE DOS DADOS As superfícies de Hirshfeld mostram uma tendência similar dentro de cada um dos grupos nos quais os complexos foram separados (Tabela 1). Por isto, serão discutidos neste texto somente os resultados comparativos para os complexos orto-substituídos de Zn2+ (Figura 1). Para os outros complexos, as superfícies de Hirshfeld, os gráficos bidimensionais de impressão digital e os demais dados obtidos podem ser consultados no Material Suplementar (os dados para os complexos orto-, meta- e para-substituídos estão disponibilizados nas Seções 1, 2 e 3, respectivamente).
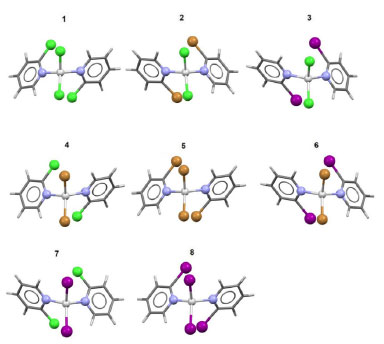 Figura 1. Representações estruturais dos complexos orto-substituídos de Zn2+ (complexos de 1 a 8) geradas a partir dos arquivos CIF correspondentes (números CCDC apresentados no rodapé da Tabela 1). Azul: nitrogênio; verde: cloro; amarelo: bromo; rosa: iodo; cinza-escuro: carbono; cinza-claro: hidrogênio; branco: Zn2+
Nos complexos [MY2(XPy)2] estudados neste trabalho, tanto o doador quanto o aceitador de LX são átomos de halogênios, onde X é o átomo de halogênio eletrófilo (doador de LX) e Y é o átomo de halogênio nucleófilo (aceitador de LX).3,25,26 Sendo assim, as LX intermoleculares estudadas têm a forma MY···XPy. Em uma LX, considera-se que a interação doador-aceitador ocorra devido ao buraco sigma presente no átomo de halogênio doador e, uma vez que este buraco se torna mais positivo com o aumento da polarizabilidade e consequente diminuição da eletronegatividade do halogênio, a sua força segue tipicamente a ordem F < Cl < Br < I.17 Esta característica é observada nas superfícies de Hirshfeld na função dnorm para os complexos de 1 a 8 (Figura 2), onde as áreas em vermelho, relacionadas ao átomo de halogênio doador de LX, aumentam com o aumento da polarizabilidade de X. Esta observação é corroborada pelas distâncias entre os átomos de halogênio Y···X envolvidos nas LX, onde para os complexos de 1 a 3, por exemplo, as menores distâncias são 3,5155(11), 3,4206(12) e 3,4141(12) Å, respectivamente (Tabela 1S).
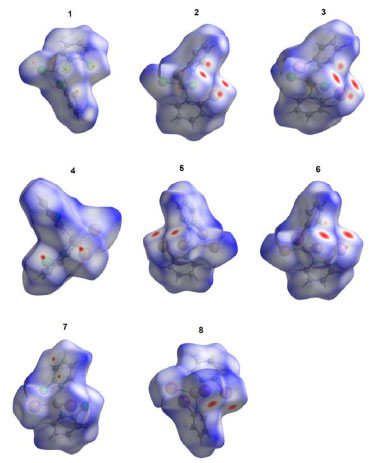 Figura 2. Superfícies de Hirshfeld na função dnorm para os complexos de 1 a 8
As Figuras 3, 4 e 5 mostram os gráficos de impressão digital para os complexos 1, 2 e 3, respectivamente, onde as LX são representadas na cor verde e aparecem como uma "linha" quando X = Y ou como "asas" quando X ≠ Y. 27 Isso ocorre devido à natureza pseudo-espelhada destes gráficos,28 ou seja, quando X = Y temos apenas contatos do tipo Cl···Cl na superfície (complexo 1) e quando X ≠ Y os contatos são do tipo X···Y/Y···X (complexos 2 e 3). Os contatos mais próximos observados nos gráficos, com valores de de + di próximos de 3,6 Å (complexo 1), 3,5 Å (complexo 2) e 3,4 Å (complexo 3) acompanham o aumento da polarizabilidade de X e consequente aumento das áreas em vermelho nas superfícies de Hirshfeld e estão de acordo com os dados cristalográficos listados na Tabela 1S. A porcentagem de contribuição das interações X···Y nas superfícies de Hirshfeld aumenta com o aumento da polarizabilidade de X, sendo os valores encontrados de 4,3, 4,6 e 5,7% para os complexos 1, 2 e 3, respectivamente, como indicado nos gráficos decompostos nas Figuras 3-5. Na Tabela 2S estão listadas todas as contribuições relativas das interações não-covalentes para a superfície de Hirshfeld para todos os complexos orto-substituídos.
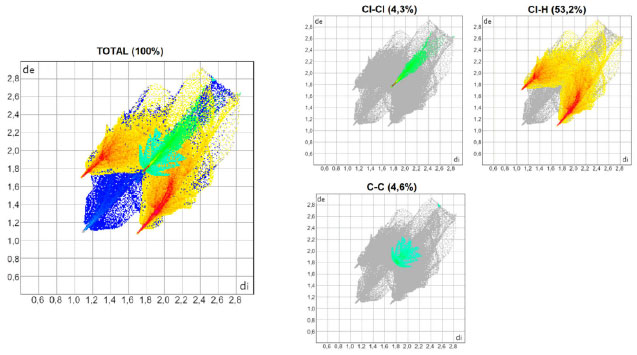 Figura 3. Gráficos de impressão digital para o complexo 1, [ZnCl2(o-ClPy)2]
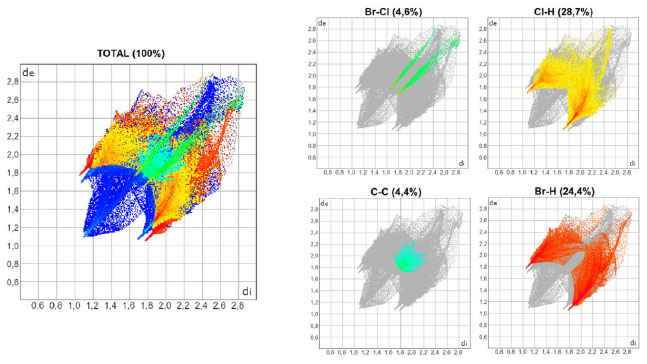 Figura 4. Gráficos de impressão digital para o complexo 2, [ZnCl2(o-BrPy)2]
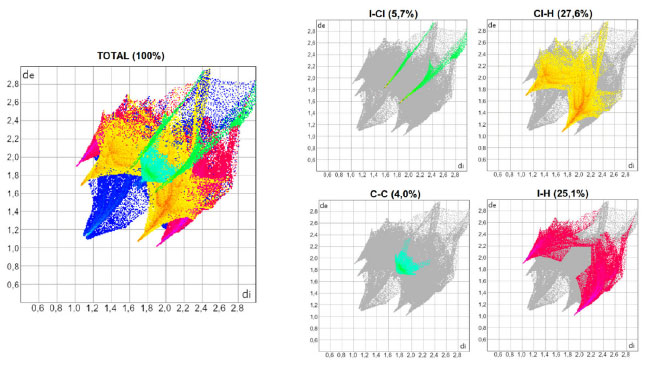 Figura 5. Gráficos de impressão digital para o complexo 3, [ZnCl2(o-IPy)2]
A área bidimensional que é representada pelas distâncias de versus di vai se tornando maior com o aumento da polarizabilidade de X quando se compara os gráficos decompostos das interações do tipo X···H e Y···H nas Figuras 3-5, o que ocorre porque as faixas de distâncias para estas interações vão se ampliando. As pontas alaranjadas indicam as regiões onde as ligações não-covalentes Cl···H são mais próximas, ou seja, os menores valores de de + di, que estão em torno de 2,8 Å para todos os complexos do Grupo A. Nos complexos onde X ≠ Y, há também as interações Br···H (complexo 2) e I···H (complexo 3), onde os valores encontrados de de + di para estas interações são próximos de 2,8 e 2,9 Å para 2 e 3, respectivamente. A superfície de Hirshfeld na função shape index é uma ferramenta usada para visualizar as interações π···π em um arranjo cristalino através da presença de triângulos adjacentes de cores vermelho e azul.29,30,31 Dentre os complexos listados na Tabela 1, todos aqueles orto-substituídos e os complexos 13 e 14 possuem interações π···π do tipo face a face com deslocamento, como exemplificado na Figura 6 para o complexo 8. Os gráficos bidimensionais (Figuras 3-5) refletem a importância desta interação na manutenção do arranjo tridimensional destes sólidos, com porcentagens de contribuição de 4,65, 4,45 e 4,00% para os complexos de 1 a 3, respectivamente. Os valores observados para os demais complexos orto-substituídos de Zn2+ estão listados na Tabela 2S. A observação de que os pré-ligantes orto-substituídos favorecem a presença deste tipo de interação não-covalente demostra como as ferramentas de Hirshfeld podem ser aliadas na escolha das espécies químicas que estarão presentes no meio reacional visando à formação de um tipo específico de arcabouço supramolecular que possa vir a ter propriedades específicas.
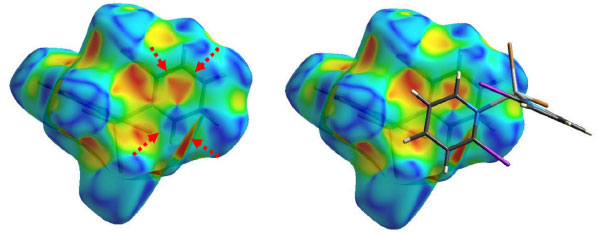 Figura 6. Superfície de Hirshfeld na função shape index para o complexo 8. Esquerda: as setas vermelhas indicam os triângulos adjacentes característicos da presença da interação π···π. Direita: duas moléculas adjacentes, dentro e fora da superfície, demostram o empilhamento dos anéis aromáticos em um arranjo do tipo face a face com deslocamento
Ainda com o objetivo de demonstrar como as ferramentas de Hirshfeld podem ser usadas na comparação de compostos similares, foi verificada a influência da natureza do íon metálico sobre a porcentagem de contribuição da interação X···Y nas superfícies de Hirshfeld, comparando-se os pares de complexos 1-9, 2-10, 3-11 e 5-12. Não foi observada nenhuma mudança significativa nos dados para estes pares de estruturas, indicando que nos complexos estudados, a mudança na acidez do íon metálico não foi suficiente para impor mudanças significativas nas forças das interações não-covalentes. Em outra comparação, analisou-se as estruturas de zinco(II) onde X = Y, ou seja, os complexos 1, 5, 8 e 9, onde observou-se que há um aumento nas porcentagens das contribuições das interações X···Y com o aumento da polarizabilidade de X. No entanto, nesta comparação não se observa uma relação direta com as distâncias de interação X···Y (Tabela 1S), o que indica que não é possível fazer este tipo de correlação entre as estruturas de diferentes grupos. Ainda, quando a comparação é feita mantendo X constante e variando Y, temos as combinações de complexos 1-4-7, 2-5, 3-8 e 10-12; onde para cada uma delas a porcentagem de contribuição aumenta com o aumento da polarizabilidade de Y. Nestes casos, as distâncias de interação X···Y aumentam com o aumento da polarizabilidade de Y (Tabela 1S), ou seja, há uma diminuição na força da LX. Esta última comparação é interessante, visto que mostra que é somente o aumento na polarizabilidade do átomo de halogênio doador de LX que exerce influência na força desta interação não-covalente.
CONSIDERAÇÕES FINAIS O estudo das ligações não-covalentes através das ferramentas da análise da superfície de Hirshfeld para complexos de coordenação correlatos atua como aliado na comparação da força destas interações. A indicação qualitativa e quantitativa de cada uma das contribuições não-covalentes para a manutenção da rede tridimensional é um recurso importante para uma visão completa de todas as forças que mantém o estado sólido. Sendo assim, estas ferramentas auxiliam o químico sintético na interpretação de propriedades químicas e físicas dependentes da organização supramolecular e podem, inclusive, fornecer embasamento na seleção de pré-ligantes, contraíons e solventes para a síntese racional em busca de propriedades e/ou estruturas supramoleculares pré-determinadas. MATERIAL SUPLEMENTAR O material suplementar descrito ao longo deste trabalho está disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem à Universidade Tecnológica Federal do Paraná e ao Programa de Pós-Graduação em Química do campus Curitiba.
REFERÊNCIAS 1. Lehn, J. M.; Science 1993, 260, 1762. [Crossref] 2. Devadiga, D.; Ahipa, T. N.; J. Mol. Liq. 2021, 333, 115961. [Crossref] 3. Teyssandier, J.; Mali, K. S.; de Feyter, S.; ChemistryOpen 2020, 9, 225. [Crossref] 4. Tepper, R.; Schubert, U. S.; Angew. Chem., Int. Ed. 2018, 57, 6004. [Crossref] 5. Lu, T. X.; Wheeler, S. E.; Org. Lett. 2014, 16, 3268. [Crossref] 6. Bai, L. Y.; Bose, P.; Gao, Q.; Li, Y. X.; Ganguly, R.; Zhao, Y. L.; J. Am. Chem. Soc. 2017, 139, 436. [Crossref] 7. Liu, X. X.; Liu, Y. H.; Feng, S. S.; Lu, L. P.; J. Mol. Struct. 2023, 1274, 134570. [Crossref] 8. Lindman, B.; Medronho, B.; Alves, L.; Norgren, M.; Nordenskiöld, L.; Q. Rev. Biophys. 2021, 54, e3. [Crossref] 9. Rowe, R. K.; Ho, P. S.; Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2017, 73, 255. [Crossref] 10. Hendricks, M. P.; Sato, K.; Palmer, L. C.; Stupp, S. I.; Acc. Chem. Res. 2017, 50, 2440. [Crossref] 11. Su, Z.; Zhang, R.; Yan, X. Y.; Guo, Q. Y.; Huang, J.; Shan, W.; Liu, Y.; Liu, T.; Huang, M.; Cheng, S. Z. D.; Prog. Polym. Sci. 2020, 103, 101230. Crossref] 12. Desiraju, G. R.; Chem. Commun. 1997, 16, 1475. [Crossref] 13. Hirshfeld, F. L.; Theor. Chim. Acta 1977, 44, 129. [Crossref] 14. Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A.; J. Appl. Crystallogr. 2021, 54, 1006. [Crossref] 15. Thakuria, R.; Nath, N. K.; Saha, B. K.; Cryst. Growth Des. 2019, 19, 523. [Crossref] 16. Mahmudov, K. T.; Kopylovich, M. N.; da Silva, M. F. C.; Pombeiro, A. J. L.; Coord. Chem. Rev. 2017, 345, 54. [Crossref] 17. Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G.; Chem. Rev. 2016, 116, 2478. [Crossref] 18. Pourayoubi, M.; Bayraq, S. S.; Tarahhomi, A.; Necas, M.; Fejfarova, K.; Dusek, M.; J. Organomet. Chem. 2014, 751, 508. [Crossref] 19. Tavassoli, M.; Montazerozohori, M.; Naghiha, R.; Sadeghi, H.; Masoudiasl, A.; Joohari, S.; Lopez, E. V.; Mahmoudi, G.; Mater. Sci. Eng., C 2020, 110, 110642. [Crossref] 20. Badr, A. M. A.; Barakat, A.; Albering, J. H.; Sharaf, M. M.; Ul-Haq, Z.; Soliman, S. M.; Appl. Sci. 2020, 10, 4853. [Crossref] 21. Web of Science, https://www.webofknowledge.com/, acessado em Outubro 2023. 22. Gimp, https://www.gimp.org/, acessado em Outubro 2023. 23. Vesta, https://jp-minerals.org/vesta/en/, acessado em Outubro 2023. 24. Mantina, M.; Chamberlin, A. C.; Valero, R.; Cramer, C. J.; Truhlar, D. G.; J. Phys. Chem. A 2009, 113, 5806. [Crossref] 25. Desiraju, G. R.; Ho, P. S.; Kloo, L.; Legon, A. C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K.; Pure Appl. Chem. 2013, 85, 1711. [Crossref] 26. Gurbanov, A. V. ; Mertsalov, D. F.; Zubkov, F. I.; Nadirova, M. A.; Nikitina, E. V. ; Truong, H. H.; Grigoriev, M. S.; Zaytsev, V. P.; Mahmudov, K. T.; Pombeiro, A. J. L.; Crystals 2021, 11, 112. [Crossref] 27. Öztürkkan, F. E.; Özdemir, M.; Akbaba, G. B.; Sertçelik, M.; Yalçın, B.; Necefoğlu, H.; Hökelek, T.; J. Mol. Struct. 2022, 1250, 131825. [Crossref] 28. Kuriakose, D.; Kurup, M. R. P.; Inorg. Chim. Acta 2020, 505, 119472. [Crossref] 29. Suresh, M.; Srinivasan, K.; J. Mol. Struct. 2022, 1250, 131721. [Crossref] 30. Shikhaliyev, N. Q.; Çelikesir, S. T.; Akkurt, M.; Bagirova, K. N.; Suleymanova, G. T.; Toze, F. A. A.; Acta Crystallogr., Sect. E: Crystallogr. Commun. 2019, 75, 465. [Crossref] 31. Granifo, J.; Gavino, R.; Suarez, S.; Baggio, R.; Acta Crystallogr., Sect. C: Struct. Chem. 2019, 75, 1299. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access