
 |
 |
 |
 |
 |
Artigo
| Efficient synthesis of 1-(bromomethyl)-3,5-dimethoxybenzene: X-ray structure, hirshfeld surface analysis, DFTs, and molecular modelling investigations as tyrosinase inhibitor |
|
Aamer SaeedI,*; Syeda Abida EjazII,*; Mubashir AzizII; Pervaiz Ali ChannarIII; Laila SumreenIV; Rabail UjanV; Tanveer A. WaniVI; Seema ZargarVII; Tuncer HökelekVIII; Ulrich FlörkeIX
I. Department of Chemistry, Quaid-i-Azam University, 45320 Islamabad, Pakistan Recebido em: 08/06/2023 *e-mail: abida.ejaz@iub.edu.pk; aamersaeed@yahoo.com The synthesis of 1-(bromomethyl)-3,5-dimethoxybenzene, a crucial intermediate in the formation of various natural products, was carried out through a straightforward three-step procedure starting from 3,5-dihydroxybenzoic acid. The structure of the synthesized derivative was fully characterized using a combination of spectroscopic techniques and single crystal X-ray diffraction. The crystalline sample was found to possess monoclinic symmetry with dimensions a = 8.4054(10) Å, b = 4.4245(6) Å, c = 25.016(3) Å, volume V = 928.9(2) Å3 and Z = 4.0. In order to assess the inhibitory potential of the synthesized crystal, a comprehensive in-silico investigation was conducted which involved molecular docking, density function theory (DFT) analysis, and molecular dynamics (MD) simulation studies. The docking analysis was utilized to determine the binding orientation of the compound with respect to the target proteins, while DFT analysis was utilized to evaluate the molecule's reactivity profile. The MD simulation approach was used to evaluate the structural rearrangements and chemical interactions that occurred during the simulation period. The results of these studies suggest that the synthesized crystal could potentially serve as a dual inhibitor of RNR and tyrosinase and could be a suitable candidate for the synthesis of noval anti-cancer agent for the treatment of associated malignancies. INTRODUCTION Ribonucleotide reductase is an essential enzyme that plays a significant role in synthesis of DNA and repair. It is present in all living organisms and is responsible for converting ribonucleotides, the building blocks of RNA, into their respective deoxyribonucleotides, which are the building blocks of DNA. The importance of ribonucleotide reductase lies in its ability to regulate the pool of deoxyribonucleotides, which are used as substrates for DNA synthesis. Defects in ribonucleotide reductase have found to link with various diseases, including cancer and other DNA damage-related disorders.1 Understanding the mechanism and regulation of ribonucleotide reductase is crucial for developing new therapeutic strategies to treat such diseases.2,3 Aberrant expression of ribonucleotide reductase (RNR) protein may lead to development of metastasis. Similarly, tyrosinase enzyme catalyzes the melanin formation and responsible for skin pigmentation.4 Tyrosinase is an enzyme that plays an essential role in the development of skin cancer. It is involved in the production of melanin pigment that imparts color to the skin, hair, and eyes. In normal skin, tyrosinase is regulated and controlled, but in skin cancer, its activity can become unregulated, leading to the overproduction of melanin. This excess melanin results in the formation of dark spots on the skin, also known as age spots, which can progress to more serious forms of skin cancer such as melanoma. Therefore, tyrosinase activity is considered a basic factor in the development of skin cancer and its regulation is an important target for the prevention and treatment of skin cancer.5 Both enzymes can serve as promising targets for treatment of cancer and associated malignancies. The halogenated bromophenols have significant medicinal values including antifungal, microbial, cytotoxicity and enzyme inhibition properties.6 The 1-(bromomethyl)-3,5-dimethoxybenzene has been widely used as a key starting material in the synthesis of various naturally occurring isocoumarins and 3,4-dihydroisocoumarins, having dioxygenation at carbon 6 and 8 due to the fact that they are biogenetically obtained from the acetate-polymalonate pathway. For example, 6-methoxymellein,7 diaporthin,8 fusamarin,9 peniolactol,10 cytogenin,11 scorzocreticin,11 feralolide,12 and dihydroisocoumarins from Ononis natrix,12 are well-known compounds. In addition, it has been used in the total synthesis of a naturally occurring substance denbinobin, which is chemically 1,4-phenanthrenequinone and it inhibits the replication of HIV-1,13 and cytotoxic compounds which include Leucetta-derived 2-aminoimidazoles, naamidine H and naamine G,14 9-tetrahydrocannabinol, which is naturally obtained from the Cannabis sativa (marijuana).15 Although its crystal structure is already known from powder diffraction analysis,16 therefore as we were successful in growing single crystals, we have re-determined the structure from single crystal data. Furthermore, the inhibition profile of the synthesized crystal against RNR and tyrosinase enzymes was determined through comprehensive in-silico investigations including DFT analysis, molecular docking, and MD simulation studies. The DFT studies were performed to evaluate the physicochemical properties and electronic density of the compounds, and the molecular docking approach was adopted to determine the in-silico binding orientation of the compound against the selected targets. The stability of the top-ranked pose was then evaluated through molecular dynamic simulation studies. These findings of current study highlight the potential of the synthesized crystal as a dual inhibitor of RNR and tyrosinase and lay the foundation for further studies towards its development as a therapeutic agent. There are several studies2,3 that have investigated the mechanisms of action for inhibitors of RNR and tyrosinase, which are both potential targets for the treatment of skin cancer. One study proposed a transmetalative approach using a Ti(IV) chelator to inhibit both RNR R1 and R2 subunits, suggesting that this approach could have a dual inhibitory effect on RNR and tyrosinase. Another study4 investigated the mode of action of an antiproliferative compound and found that RNR is considered one of the main targets of TSCs, yet there may be additional targets that contribute to their efficacy. Additionally, some compounds such as monobenzone have been shown to inhibit tyrosinase and induce melanosome autophagy, which could provide a potential mechanism of action for dual inhibition of RNR and tyrosinase. However, further research is needed to fully understand the mechanism of action and potential efficacy of dual inhibition of RNR and tyrosinase for the treatment of skin cancer.
EXPERIMENTAL Melting point were determined using a MelTemp MPD apparatus. FTIR spectrum was taken on a single beam Nicolet IR 100 (Fourier transform). 1H NMR and 13C NMR spectra were monitored in CDCl3 using 300 MHz Bruker DPX spectrometer. MS analysis was performed by instrument LC system Agilent 1200 series and LECO 183 CHNS analyzer was utilized for elemental analysis. Synthesis of 1-(bromomethyl)-3,5-dimethoxybenzene (4) Phosphorus tribromide (2.60 mL, 7.44 g, 0.027 mol) was added dropwise to a container having solution of 3,5-dimethoxybenzyl alcohol (3) (0.027 mol, 6.64 g) in 50 mL of dry benzene and the reaction mixture was stirred for 4 h at room temperature. After completion of reaction as monitored by the analytical TLC using solvent system n-hexane ethyl acetate (8:2), the reaction mixture was transferred into ice-cold water, stirred vigorously. Ethyl acetate was added, and the organic layer was separated. The organic phase was successively washed with water, 5% sodium bicarbonate dried using anhydrous magnesium sulphate, and then rotary evaporated to leave a white solid. Recrystallization from petroleum ether afforded the pure product as colorless prisms. (7.2 g, or 88%). Structure determination of 1-(bromomethyl)-3,5-dimethoxy benzene (4) The data were recorded at 130(2) K on a Bruker AXS Smart APEX CCD diffractometer16 utilizing MoK rays; 8110 reflections were recorded at an angle of 1.63 < θ < 27.88°.17 Indirect techniques,18 full-matrix refinement with least squares,17 on F2 and 111 parameters for 2208 specific intensities I > 2σ(I), after excluding hydrogen atoms all the remaining atoms were refined anisotropically, H atoms from method of difference,18 fine-tuned Fourier maps on idealized places with Uiso = 1.2 Ueq(C) or 1.5Ueq(Cmethyl) and C-H distances ranging from 0.95 to 0.99 Å. H(Cmethyl) was allowed to spin but not tipped, according to the rules. Table 1 contains selected bond geometries, while Table 2 has more refinement data on these bond geometries. In the case of CCDC, the deposit number is 1884625.

Hirshfeld surface analysis In this study a high-resolution structural analysis was performed19 for the crystal structure of title compound using Crystal Explorer 17.520 in an attempt to discover the intermolecular association present in the crystal structure. The investigation was performed to find the stacking interactions along with the position of atoms in short contacts having hydrogen bonding potential, as well as the quantity of these interactions. It is possible to identify places that are especially significant for intermolecular interactions using the normalized contact distance, abbreviated as dnorm. The white colored surface on the HS plotted over dnorm, represents interactions with distances equal to the total sum of van der Waals radii, while the red and blue colored surfaces are indication of interactions with comparatively shorter bond lengths (in close contact) or longer (in distinct contact) than the van der Waals radii. Computational studies Density functional theory (DFT) Density functional theory is a computational tool for computing the ground state geometries of the selected compounds. The ground state geometries were optimized with DFT calculations by means of the B3LYP functional including STO-3G basis set.21,22 The STO 3G basis set is a minimal basis set used in quantum chemistry. It is used to represent molecular orbitals in terms of atomic orbitals. The STO-3G basis set uses 3 primitive Gaussian functions to describe the valence electrons of an atom. It is a simple and computationally efficient basis set that is commonly used for small to medium sized molecules. In this current study, Gaussian 09W software23 was used for calculating DFT. The resultant check files were interpreted in Gauss View 6.0.24,25 Molecular docking In order to ascertain the chemical basis of the inhibitory mechanism, it is crucial to interpret the binding orientation of the ligand inside biological targets. A molecular docking technique was used to evaluate the way that produced crystals bound to the intended proteins. AutoDock suite was used for the molecular docking.26 The biological targets were retrieved from the Protein Data Bank using the PDB IDs 2BQ127 and 4OUA28 for the ribonucleotide reductase and the tyrosinase enzyme, respectively (https://www.rcsb.org/). In the beginning, undesirable water molecules and hetero atoms were removed using MOE's preliminary preparation wizard. Then, at the sharpest energy gradient, both biological targets were successively reduced in energy and protonated in 3D. MMFF94x force field was also used to include gasteiger charges. After the protein synthesis has been completed, the MOE site finder utility was used to choose the active site's amino acid residue. After preliminary energy minimization through ChemDraw 3D,29 the synthesized crystal was converted into the desired format for docking purposes. Using London dG as a scoring function, the output structure was docked at specific amino acid residues with triangular matcher placement.30 For each biological target, the rigid docking was employed to enhance the reliability of the docking studies.31 Furthermore, docking protocol was validated, ligand molecule against each protein was redocked within the active pocket. The RMSD values of less than 2 Å between regenerated and native pose were considered as validated protocol. Molecular dynamics simulations Molecular dynamics (MD) simulations were performed using Desmond software on a protein-ligand complex. The simulation spanned 100 ns in the TIP3P solvent model. The OPLS3 forcefield32 was chosen for the simulation under periodic boundary conditions.33 The complex was placed in an orthorhombic solvation box, filled with TIP3P water molecules.34 NaCl counter ions, at 0.15 mol L-1 concentration, were added for neutralization. Steric clashes were removed with 2000 steps of energy minimization using the steepest descent method. System equilibration occurred in an NPT ensemble at 300 K temperature and 1.01 bar pressure. Short-range van der Waals interactions had a 10-angstrom cutoff. To ensure constant conditions, a Martyna-Tobias-Klein barostat and Nose-Hoover thermostat were employed.35 Trajectories of the simulation were saved every 100 ps. The entire MD run for each protein-ligand complex lasted 100 ns. For motion equations, a time step of 2 fs was used. The particle mesh Ewald method36 ensured accurate electrostatic interactions.37 These simulations provided insights into the molecular behavior of the biological target and the ligand molecule.
RESULTS AND DISCUSSION Synthesis The compound 1-(bromomethyl)-3,5-dimethoxybenzene (4) was produced in two phases using the technique indicated in Scheme 1 with a modest modification of a previously reported method.
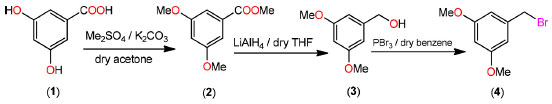 Scheme 1. Synthesis of 1-(bromomethyl)-3,5-dimethoxybenzene
Treatment with dimethylsulfate in dry acetone and anhydrous potassium carbonate as a mild base resulted in the formation of 3,5-dimethoxymethylbenzoate (2) from 3,5-dihydroxybenzoic acid (1). The FTIR spectrum at 1732 cm-1 revealed a distinctive (C=O) stretching absorption, which was confirmed. In order to get 3,5-dimethoxyphenylmethanol, the methyl ester was reduced with lithium aluminum hydride in dry THF (3). In the analysis of the 1H NMR spectrum the signal at 3.06 ppm confirms the presence of hydroxyl group. The reaction of 3,5-dimethoxybenzyl alcohol with phosphorous tribromide in dry benzene at room temperature resulted in the formation of 1-(bromomethyl)-3,5-dimethoxybenzene (4). The lack of a wide signal at 3.06 ppm in the 1H NMR spectrum showed that alcohol had been converted to bromide. Crystal structure of 1-(bromomethyl)-3,5-dimethoxy benzene The aromatic ring is almost perfectly coplanar with the atoms O1, O2, C1, C8, and C9. There are five different displacements from the mean benzene plane, which are 0.0123(17), 0.0173(18), -0.0076(7), -0.0086(15), and -0.0135(16) Å. The torsion angles associated with the methoxy groups are nearly planar. The bond length between C1-C2 was 1.49 Å were as bond angle C6-O2-C9 was 117.83. The detailed bond angle, bond length and torsion angles is tabulated in Table 1. The molecule is connected through various hydrogen bonds forming centro symmetric dimers stacked along the b-axis and forms ring motifs (R22 (8)) around the molecules. Intermolecular C3-H.O1 bonds are formed between the dimers, which results in a two-dimensional network extending down the c-axis (Figure 1).
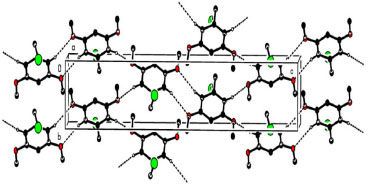 Figure 1. Hydrogen bonds on the a-axis of the crystal packing; hydrogen atoms that are not involved are deleted
Description of crystal structure of 1-(bromomethyl)-3,5-dimethoxy benzene This study conspicuously illustrates the structure of our title compound in single crystal, as established using spectroscopic data, is consistent with the structure of the compound discovered earlier by other means. Table 2 provides experimental characteristics, including crystal data, data collection and data refinement, as well as the findings of the experiment, and is followed by a summary of the experiment's results. It was observed that crystal was colorless having molecular formula of C9H11BrO2. The shape of crystal was monoclinic with P21/c space group. The dimensions of crystal was as follows: a = 8.4054(10), b = 4.4245(6), c = 25.016(3) Å, β = 93.213(2)°, V = 928.9(2) Å3, Z = 4, ρ = 1.652 mg cm-3, μ = 4.38 mm-1, F(000) = 464. In Figure 2, a clear depiction of the crystal structure, along with numbering of atoms that were used to put it together is given. Within the benzene ring A (C2-C7), the atoms O1, O2, C1, C8, and C9 are separated from the mean plane by distances of -0.0473(1), -0.0051(1), -0.0452(1), -0.0132(1), 0.0402(19), -0.0132(1), 0.0402(19), and 0.0402(19) Å. This resulted in the benzene ring A being coplanar with the atoms O1, O2, C1, C8, and C9.
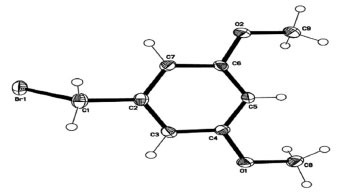 Figure 2. Molecular geometry of 1-(bromomethyl)-3,5-dimethoxybenzene with ellipsoidal displacement at 50%
Table 3 illustrates how intermolecular C-HBnz.OMeth hydrogen bonds (Bnz = benzene and Meth = methoxy), which are formed in the crystal structure between molecules holding all molecules together. All molecules are brought together by centrosymmetric dimers (which include R22 (8) ring motifs) (Figure 1). As a consequence of these dimers a parallel two-dimensional network is created. There is very little amount of interaction present between C and H. The most significant interactions to be mindful of in crystal packing are hydrogen bonding along with van der Waals forces, which are both formed by the crystal packing. Between the two variables, there is no indication of a statistically significant interaction, which is expressed as a correlation.

Surface analysis using the Hirshfeld method The comprehensive Hirshfeld analysis of synthesized crystal was executed on Crystal explorer 17.2. It was observed that the oxygen atoms H3A and H7A play significant role as donors and acceptors in the dominant C-H.O hydrogen bonding and is evident by the sharp red color spots nearby these oxygen atoms; in the same way when electrostatic potential is mapped the positive and negative potential on the Hirshfeld surface (HS) is indicated by the blue and red regions as shown in Figure 3.
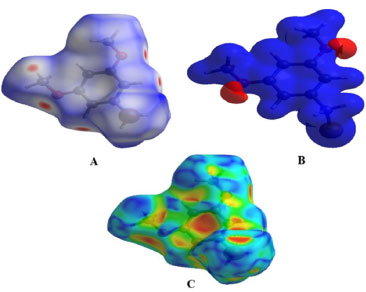 Figure 3. 3D views of the 1-(bromomethyl)-3,5-dimethoxybenzene. The first view (A) shows the 1-(bromomethyl)-3,5-dimethoxybenzene plotted over dnorm ranging from -0.1668 to 1.1141 a.u, while the second view (B) displays the compound V/S electrostatic potential energy using STO-3G basis set using Hatree fock calculations. Finally, the third view (C) shows the 1-(bromomethyl)-3,5-dimethoxybenzene V/S shape-index
The positive electrostatic potential (hydrogen-bond donors) which is represented by the blue region, and the negative electrostatic potential (hydrogen-bond acceptors) is represented by the red areas near the hydrogen atoms H3A and H7A indicating their roles as the negative electrostatic potential (hydrogen-bond acceptors). As long as there are no red or blue triangles, the π.π interactions are very low (HS). An example of the HS shape-index is existence of two adjoining red and blue triangles are present, which allows you to see how stacking is occurring. Because the title chemical has no π.π interactions with other molecules, it is clear from Figure 4 that it does not interact with other molecules. If you look at the two-dimensional plot of the structure, it is also possible to detect the intermolecular interactions that exist between the molecules.
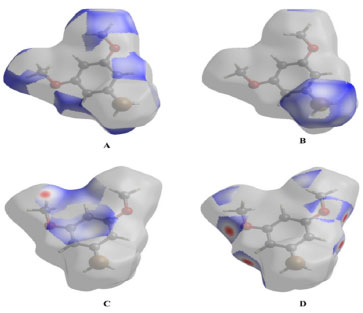 Figure 4. 3D representation of Hirshfeld surface with interatomic parameters
Figure 4A-4D depict the intermolecular interactions between hydrogen and oxygen, illustrating the comprehensive insights gained from the Hirshfeld surface analysis regarding these interactions. The compound's HS analysis data is presented in Table 4. By merging de and di values, a comprehensive overview of the intermolecular interactions within the crystal can be visualized in a two-dimensional fingerprint plot, as illustrated in Figure 5. Figure 5a depicts two-dimensional plot, whereas Figures 5b-5h depict the fingerprint plots divided into, H-Br/BrH, H-O/O-H, C-C, O-O, H-H, H-C/C-H and C-Br/BrC contacts38 also how much each one contributed to the Hirshfeld surface.38 In Figure 5b according to the high hydrogen content of the molecule, the 40.3% benefaction to the HS surface may be observed as widely dispersed regions of high density in the fingerprint plot delineated into H.H interactions because of high hydrogen atoms in the molecule.
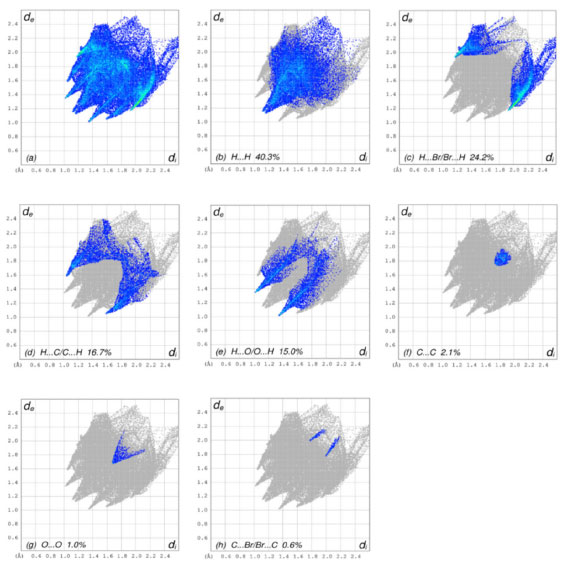 Figure 5. The 2D graphs of 1-(bromomethyl)-3,5-dimethoxybenzene showing interactions and bonding distances (a) which are separated into (b) H.H (40.3%), (c) H.Br/Br.H (24.2%), (d) H.C/C.H (16.7%), (e) H.O/O.H (15.0%), (f) C.C (2.1%), (g) O.O (1.0 %), (h) C.Br/Br.C (0.6%)
The pair of practically splitted spikes with the ends at de + di ~ 2.28 Å due to the short interatomic H.H interactions. The presence of H.Br/Br.H contacts in the structure, which account for 24.2% of the overall HS contribution, have an asymmetric distribution of contact sites, as shown in the Figure below (Figure 5c). Consider the case of the Br.H interactions in Figure 5c, which contribute more (15.8%) than their HBr counterparts (8.4%) and are shown as a spikes pair with the tips at de + di ~ 3.09 Å. The weak interatomic hydrogen bonding between Br and H results in a short interatomic hydrogen bonding between Br and H. In Figure 5d, the weak C-H interaction present within the crystal forms the characteristic wings pair similar to forceps that results in the fingerprint plot describing the H.C/H.C contacts (Figure 5d) to get an asymmetric points distribution, with C.H interactions accounting for a greater proportion of the total (8.9%) compared to the H-C portion (7.8%). As a result, when the molecule tips are present at de + di ~ 2.63 Å, the combined H/C interactions provides 16.7% of the total molecular HS area (Table 4). The H.O/O.H connections in the structure (Figure 5e), which account for 15.0% of the overall HS contribution, exhibit a similar asymmetric distribution of contact sites to the H.O/O.H connections in the structure, as well. H.O/O.H interactions account for a bigger proportion of the total (8.1%) than their H.H counterparts (6.8%), as seen by a spike pair with their tops at de + di ~ 2.34 Å which are the result of weak interatomic H.O/H.H contacts in Figure 5e (Table 4). In the following step, the symmetrical points distribution in the fingerprint plot that has been represented into C.C contacts (Figure 5f) and is used to visualize the contributing 2.1% to overall packing of crystal as a spoon-like spike with the tip at de = di ~ 1.75 Å, which is the resulting from weak interatomic C.C contacts (Table 4). Each of the Hirshfeld surface representations in Figures 5a-5d are plotted with the function dnorm to show how individual intermolecular interactions, as well as their quantitative contributions, are depicted. These representations illustrate the molecular linkages, as well as their quantitative contributions are given above. With the use of the Hirshfeld surface analysis, we can confirm that interactions between the H-atoms are important in the creation of the packing. As a result of several of H/H, H/O/OH, and H/C/CH contacts, it explains the probable importance of hydrogen bonding and van der Waals forces in the packing of crystal. Molecular docking studies Molecular docking employs computational methods to predict how well a ligand binds to its protein target, making it essential for identifying potential inhibitors of bio-targets like RNR and tyrosinase. RNR, for instance, is an enzyme that is vital to DNA synthesis by converting ribonucleotides to deoxyribonucleotides. It is composed of two dissimilar subunits i.e., R1 containing polythiols and R2 containing free tyrosyl radical and non-heme iron site. Overexpression of RNR has been observed in many cancers, which has led to the development of RNR inhibitors as potential anti-cancer agents. Similarly, tyrosinase is responsible for melanin synthesis, which is essential for skin pigmentation. Overexpression of tyrosinase has been observed in melanoma, a type of skin cancer, and tyrosinase inhibitors have been developed as potential anti-cancer agents for melanoma. Choosing both RNR and tyrosinase as biotargets for molecular docking has a rationale since their inhibition may have synergistic effects in cancer treatment. For instance, melanoma has been found to be resistant to chemotherapy. By combining a tyrosinase inhibitor with an RNR inhibitor, it may increase the efficacy of treatment. Molecular docking studies targeting both biotargets can help identify potential drug candidates that can be tested for their effectiveness in combination therapy for cancer treatment. In the current study, the focus was on the molecular docking of 1-(bromomethyl)-3,5-dimethoxybenzene (4) within the active site of both RNR and tyrosinase enzymes, which could potentially lead to the discovery of a novel drug candidate with dual activity against these two biotargets. Interpretation of molecular interactions Hydrogen bonds and hydrophobic contacts were prominent in the docked conformation of (4)'s interactions with the RNR protein. Through hydrogen bonds, compound 4 was interacting with significant amino acid residues. A hydrogen bond was observed between the electronegative oxygen atom of (4) and SER606 with a bond length of 2.99 Å. Additional interactions include hydrophobic associations with SER217, ARG293, ALA605, THR604, THR607, ALA201, MET602, and SER202. The best conformation yielded a docking score of -20 kJ mol-1. The putative interactions are illustrated in Figure 6.
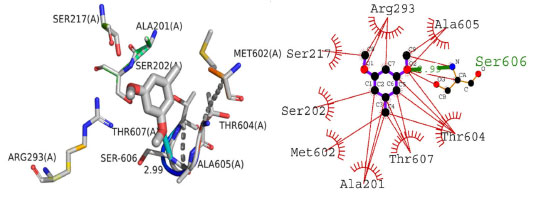 Figure 6. The presumed 2D and 3D binding orientation of 1-(bromomethyl)-3,5-dimethoxybenzene with in active pocket of RNR protein. The grey colored compound in 3D orientation is representing 1-(bromomethyl)-3,5-dimethoxybenzene (4)
The binding orientation of compound 4 was also investigated inside active pocket of tyrosinase enzyme. Tyrosinase plays important role in melanogenesis and ultimately lead to skin disorders. The current study delved into the binding mechanism of (4) within the active pocket of the tyrosinase enzyme. The docked posture of (4) revealed significant molecular interactions with the enzyme, encompassing both hydrophilic and hydrophobic associations. The stability of the complex was reinforced by a hydrogen bond between the oxygen atom of (4) and SER394 of the enzyme. This bond, with a length of 2.69 Å, indicates its strong nature. Hydrophobic interactions involved residues such as PHE400, HIS381, THR391, LEU382, GLY389, ASN378, HIS215, TYR362, GLU360, HIS377, and HIS192. The docking score for the complex stood at -24 kJ mol-1. Figure 7 displays these anticipated interactions.
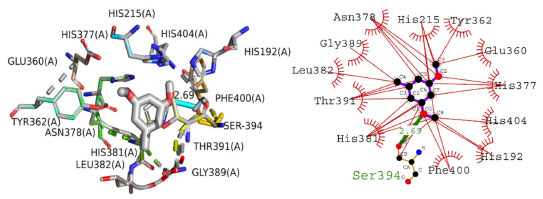 Figure 7. The presumed 2D and 3D binding orientation of 1-(bromomethyl)-3,5-dimethoxybenzene with in active pocket of tyrosinase enzyme. The grey colored compound in 3D orientation is representing 1-(bromomethyl)-3,5-dimethoxybenzene (4)
Density functional theory Density functional theory (DFT) is an extensively used technique of quantum mechanics for determination of electronic densities of the compounds. It is used to analyze the electronic configuration, reactivity, efficiency and energy surface simulation of a compound. All DFT calculations in this research were conducted using Gaussian-09, utilizing the RHF calculation method and the STO 3G basis set. The primary analysis involved determining energies and the energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Compound 4's structural geometry was designed to have the steepest energy gradient, and no phantom frequencies were detected that would have pointed to the precise local minima of the optimized geometry. The compound's polarizability and dipole moment were 64.83 and 1.06 debye, respectively. Frontier molecular orbitals (FMOs) investigation also demonstrated the compound's chemical reactivity. The low energy gap between HOMO/LUMO orbitals were predictive of high chemical reactivity of the compound. The HOMO/LUMO orbitals with their energies along with optimized structure are shown in Figure 8.
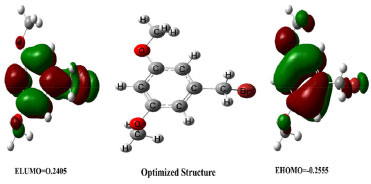 Figure 8. HOMO and LUMO orbitals and optimized structure of compound
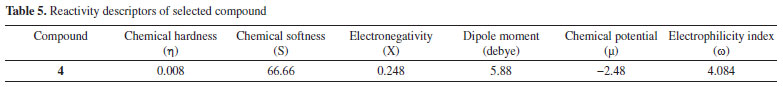
Compound 4 is a highly reactive compound with a low chemical hardness of 0.008, indicating it can easily undergo chemical changes, and a high chemical softness of 66.66, indicating it is very easy for electrons to flow into or out of the compound. The compound has a relatively low electronegativity of 0.248, which suggests it does not strongly attract electrons towards itself in a chemical bond. The dipole moment of the compound is 5.88 debye, indicating a significant separation of electrical charges within the molecule. The chemical potential is -2.48, indicating that it requires a relatively low amount of energy to add a molecule to the system, while keeping the temperature and volume constant. The electrophilicity index of compound 4 is 4.084, suggesting that it has a tendency to accept electrons. The properties of compound 4 could make it useful in various applications, depending on the specific context and desired outcomes. The detailed reactivity descriptors are provided in Table 5.
Molecular dynamics simulation Molecular dynamics modeling is employed to predict the thermodynamic properties of living systems under physiological settings. The Desmond MD simulation software was utilized to analyze the best-docked conformation. These 100 ns simulations provided pivotal insights into the stability of the protein-ligand complex. From the MD trajectories, multiple analytical measures such as RMSD, contact profile, RMSF, and ligand interaction profile were derived. Figure 9 depicts the RMSD plot for the protein's C-alpha, the protein-ligand complex, and the ligand atoms. Protein C-alpha atoms' RMSD patterns remained noticeably constant. The structural changes were barely noticeable at the start of the trajectory but showed structural reorganizations by the end of the MD simulations. These changes, which had an overall range of 1.1 to 2.9 Å, were kept within acceptable bounds. Protein C-alpha atoms have an average RMSD of 2.5 Å.
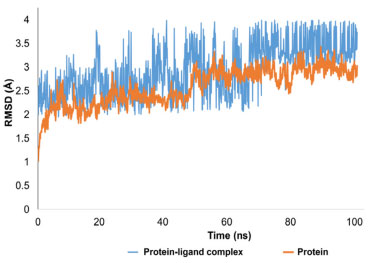 Figure 9. RMSD trajectory analysis for protein (C alpha), protein ligand complex (blue trajectory) and ligand atoms (orange colored trajectory)
Interpretation of protein-ligand stability The protein-ligand complex's RMSD pattern showed a steady pattern. At first, the reference frame was used to align all 1000 frames, and an RMSD pattern was generated. The protein-ligand complex's average RMSD was determined to be 2.8 Å. The compound first oscillated at 2.5 Å and was firmly anchored to the active pocket's amino acid residues. Inside the binding region, there were a few minor structural changes, although these variations were within acceptable bounds. A ligand molecule was in interaction with the amino acid residues SER202, PRO294, ALA605, and TYR625 in both hydrophilic and hydrophobic ways. 3.3 Å separated the atoms of SER202 and the ligand molecule. The interatomic distances between the other residues specified varied from 2.6 to 3.7 Å as well. The overall RMSD pattern of the trajectory was stable, and the ligand made adequate contact with the targeted protein. When the ligand displayed minor rearrangements and the RMSD pattern increased to 3.5 Å after 70 ns, a little strange behavior was seen. The trajectory's equilibration, which stayed stable and equilibrated for the remainder of the trajectory, was encouraging. The root mean square fluctuations (RMSF) analysis is vital for pinpointing local structural alterations in proteins. In this research, a residue-specific RMSF was computed for every residue. While most residues showed optimal fluctuations, certain residues, namely HIS108, ARG12, GLY264, THR265, ASN266, and ASN109, displayed significant structural changes, varying between 3 and 4.3 Å. Notably, these residues are part of the C and N terminals, which are typically less dense than the central backbone and alpha strand regions of the protein. It was observed that amino acid residues including ASN144, TYR145, PHE146, THR150, ARG153, TYR155, ALA201, SER202, PRO203, SER217, SER244, ALA245, LYS292, ARG293, PRO294, PHE329, MET602, PRO603, THR604, ALA605, SER606, THR607, ALA608, GLN609, LEU611, GLY612, ASN613, GLU615, SER622, TYR625 and ARG740 were in contact with ligand molecule. All these residues exhibited very few fluctuations ranges from 0.6 to 1.2 Å. The overall protein displayed an average RMSF of 1.07 Å, which is considered within acceptable limits. Figure 10 depicts the RMSF progression, with green lines signifying interactions between the ligand and specific amino acid residues.
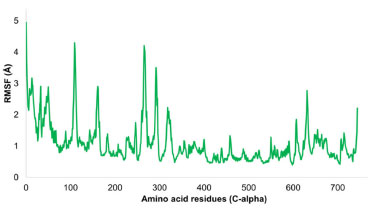 Figure 10. Evolution of RMSF for amino acid residues of targeted protein
The connections between a ligand and protein are heavily influenced by their contact interactions. The nature and duration of these interactions directly impact the stability of their combined structure. In the studied complex, a variety of interactions, including hydrogen bonds, water bridges, and hydrophobic connections, were identified. Key hydrogen bonds were notably formed with SER202 and SER606 from the target protein. Hydrophobic contacts, particularly with PRO294, TYR625, ALA605, LEU428, and PRO603, were sustained for over 10% of the simulation period, enhancing the complex's stability. Figure 11 displays the interaction profile of the simulated ensemble.
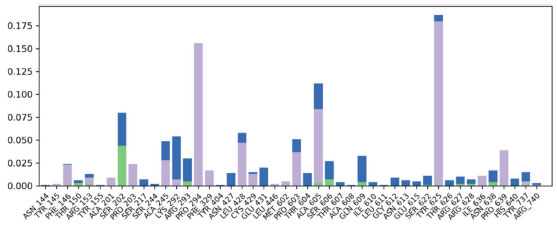 Figure 11. Contact profile of the protein-ligand complex. Green peaks indicate hydrogen bonds, while purple highlights hydrophobic interactions
Interpretation of protein-ligand stability Through MD simulations, the stability of tyrosinase in complex with 1-(bromomethyl)-3,5-dimethoxybenzene was also assessed. A further confirmation of putative molecular interactions in the initial docked conformation of the ligand with the tyrosinase came from MD simulations. Following a 100 ns simulation of the complex, it was discovered that the C-alpha atoms of the protein and tyrosinase-ligand combination had an important stability pattern. The stability pattern of the evolution of protein RMSD was examined, and it was found that the protein remained stable and equilibrated with little variations. The protein's average RMSD was found to be 1.8 Å, which is completely acceptable. The protein-ligand complex displayed a stable and equilibrated trajectory, with fluctuations of about 0.8 Å. The minute fluctuations showed that the ligand was still adequately bound to the amino acid residues in the active site. Significant contact was made by the ligand molecule with HIS215, TYR362, HIS377, ASN378, LEU382, and HIS381. Initial ligand rearrangements were visible in the RMSD pattern, but these rearrangements became stable as the RMSD pattern shrank to 2.8 Å. The average RMSD of the protein-ligand combination was discovered to be 3.2 Å. The results of the molecular dynamics simulation of the complex showed that the HIS215, GLU216, ARG321, GLU360, TYR362, HIS377, ASN378, HIS381, LEU382, GLY388, GLY389, GLN390, and HIS404 were all in contact with the ligand molecule. Thankfully, all of these residues belonged to the active site and showed minimal variation. The typical separation between the ligand and these residues was between 2.5 and 3.3 Å. These patterns represent the complexes with the fewest structural variations and sufficient stability under physiological conditions. Figure 12 is illustrating the evolution of RMSD pattern for protein and protein ligand complex.
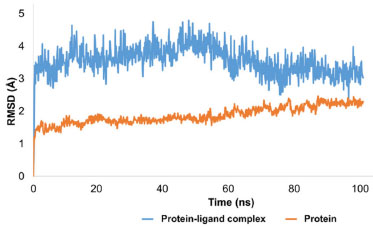 Figure 12. Evolution of RMSD pattern for tyrosinase and protein-ligand complex
Variability in the tyrosinase enzyme by individual residues was examined. Apart from a few amino acids in the C and N terminals, the C-alpha atoms of the selected protein maintained a prominent stability pattern. Noteworthy fluctuations exceeding 3 Å were identified in residues like HIS81, GLY202, VAL203, GLN205, GLU206, and ASP82. In contrast, most residues showed minor structural variations and consistently remained stable throughout the simulation. Ligand-bound residues such as HIS215, GLU216, ARG321, GLU360, TYR362, HIS377, ASN378, HIS381, LEU382, GLY388, and GLY389 registered slight fluctuations, ranging between 0.4 and 1.5 Å. The entire protein's average RMSF was calculated as 1.20 Å. Figure 13 visualizes the RMSF values for the tyrosinase enzyme.
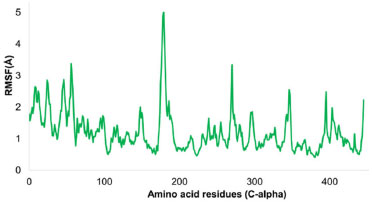 Figure 13. Illustration of RMSF for tyrosinase enzyme
Figure 14 shows the contact profile of the chosen ligand. Significant hydrophobic and hydrogen bonding interactions were found to be stabilizing the protein ligand complex. In particular, hydrophobic contact with TYR362 persisted for 40% of simulation time while hydrophobic bonding with ASN378 persisted for more than 50% of simulation duration. Additionally, for 10% of the simulation time, hydrophobic interaction was also seen with LEU382.
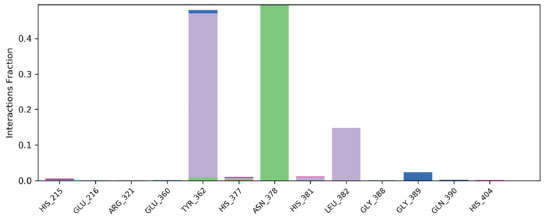 Figure 14. Contact profile of ligand with amino acid residues of tyrosinase enzyme
CONCLUSIONS Conclusively, based on the desire of the potential candidate for the treatment of skin cancer in this study, a title compound 1-(bromomethyl)-3,5-dimethoxybenzene (4), an intermediate towards a range of natural products, was successfully synthesized and its structure was confirmed through different spectroscopic methods. The synthesized crystal was subjected to a thorough analysis, including Hirshfeld analysis and comprehensive in-silico studies, in order to determine its properties, reactivity and potential of the title compound against the tyrosinase and RNR. The results of molecular docking, density functional theory (DFT), and molecular dynamics (MD) simulation studies revealed that the compound has significant inhibitory potential and chemical reactivity, particularly as a dual inhibitor of the RNR and tyrosinase enzymes. The further research is needed to fully understand the mechanism of action and potential efficacy of dual inhibition of RNR and tyrosinase for the treatment of skin cancer. Based on the in-silico findings, the title compound was found as a promising candidate for the synthesis of more potential candidate which can be explored at molecular level in addition to the computational investigations.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
ACKNOWLEDGEMENTS All the authors want to extend their appreciation to researchers supporting project (project number RSP2024R357), King Saud University, Riyadh Saudi Arabia for funding this research. The authors declare no conflict of interest.
REFERENCES 1. Huff, S. E.; Winter, J. M.; Dealwis, C. G.; Biomolecules 2022, 12, 815. [Crossref] 2. Shao, J.; Zhou, B.; Chu, B.; Yen, Y.; Curr. Cancer Drug Targets 2006, 6, 409. [Crossref] 3. Ding, Y.; Zhong, T.; Wang, M.; Xiang, X.; Ren, G.; Jia, Z.; Lin, Q.; Liu, Q.; Dong, J.; Li, L.; Li, X.; Front. Oncol. 2019, 9, 956. [Crossref] 4. Di Petrillo, A.; Santos-Buelga, C.; Era, B.; González-Paramás, A. M.; Tuberoso, C. I. G.; Medda, R.; Pintus, F.; Fais, A.; Food Sci. Biotechnol. 2018, 27, 139. [Crossref] 5. Chen, J.; Ye, Y.; Ran, M.; Li, Q.; Ruan, Z.; Jin, N.; Front. Pharmacol. 2020, 11, 81. [Crossref] 6. Akbaba, Y.; Balaydın, H. T.; Göksu, S.; Şahin, E.; Menzek, A.; Helv. Chim. Acta 2010, 93, 1127. [Crossref] 7. Mercier, J.; Arul, J.; Julien, C.; J. Phytopathol. 1993, 139, 17. [Crossref] 8. Lee, Y. C.; Online Inf. Rev. 2006, 30, 517. [Crossref] 9. Lawrence, M.; Afzal, M.; Kenrick, J.; Heredity 1978, 40, 239. [Crossref] 10. Saeed, A. I.; Bhagabati, N. K.; Braisted, J. C.; Liang, W.; Sharov, V.; Howe, E. A.; Li, J.; Thiagarajan, M.; White, J. A.; Quackenbush, J.; Methods Enzymol. 2006, 411, 134. [Crossref] 11. Diaz, S.; Hodgson, J. G.; Thompson, K.; Cabido, M.; Cornelissen, J. H.; Jalili, A.; Montserrat-Mart, G.; Grime, J. P.; Zarrinkamar, F.; Asri, Y.; Band, S. R.; J. Veg. Sci. 2004, 15, 295. [Crossref] 12. Saeed, A.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; Sturn, A.; Snuffin, M.; Rezantsev, A.; Popov, D.; Ryltsov, A.; Kostukovich, E.; Borisovsky, I.; Liu, Z.; Vinsavich, A.; Trush, V.; Quackenbush, J.; BioTechniques 2003, 34, 374. [Crossref] 13. Koswatta, P. B.; Lovely, C. J.; Chem. Commun. 2010, 46, 2148. [Crossref] 14. Márquez, N.; Calzado, M. A.; Sánchez-Duffhues, G.; Pérez, M.; Minassi, A.; Pagani, A.; Appendino, G.; Diaz, L.; Muñoz-Fernández, M. A.; Muñoz, E.; Biochem. Pharmacol. 2008, 75, 1370. [Crossref] 15. Karlsen, M.; Liu, H.; Johansen, J. E.; Hoff, B. H.; Molecules 2014, 19, 13526. [Crossref] 16. Selak, M. A.; Armour, S. M.; MacKenzie, E. D.; Boulahbel, H.; Watson, D. G.; Mansfield, K. D.; Pan, Y.; Simon, M. C.; Thompson, C. B.; Gottlieb, E.; Cancer Cell 2005, 7, 77. [Crossref] 17. Pao, W.; Wang, T. Y.; Riely, G. J.; Miller, V. A.; Pan, Q.; Ladanyi, M.; Zakowski, M. F.; Heelan, R. T.; Kris, M. G.; Varmus, H. E.; PLoS Med. 2005, 2, e17. [Crossref] 18. Sheldrick, G. M.; Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112. [Crossref] 19. Vanitha, U.; Elancheran, R.; Manikandan, V.; Kabilan, S.; Krishnasamy, K.; J. Mol. Struct. 2021, 1246, 131212. [Crossref] 20. Khera, M.; Goel, N.; J. Mol. Graphics Modell. 2020, 95, 107503. [Crossref] 21. Hirshfeld, J.; Granatstein, V.; IEEE Trans. Microwave Theory Tech. 1977, 25, 522. [Crossref] 22. Spackman, M. A.; Jayatilaka, D.; CrystEngComm 2009, 11, 19. [Crossref] 23. Turner, S.; Endres, A.; International Journal of Applied Management Technology 2017, 16, 3. [Crossref] 24. Graus, F.; Titulaer, M. J.; Balu, R.; Benseler, S.; Bien, C. G.; Cellucci, T.; Cortese, I.; Dale, R. C.; Gelfand, J. M.; Geschwind, M.; Glaser, C. A.; Lancet Neurol. 2016, 15, 391. [Crossref] 25. Spackman, M. A.; McKinnon, J. J.; Jayatilaka, D.; CrystEngComm 2008, 10, 377. [Crossref] 26. Goodsell, D. S.; Sanner, M. F.; Olson, A. J.; Forli, S.; Protein Sci. 2021, 30, 31. [Crossref] 27. Uppsten, M.; FärnegÅrdh, M.; Domkin, V.; Uhlin, U.; J. Mol. Biol. 2006, 359, 365. [Crossref] 28. Mauracher, S. G.; Molitor, C.; Al-Oweini, R.; Kortz, U.; Rompel, A.; Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 2301. [Crossref] 29. Brown, T.; The Science Teacher 2014, 81, 67. [Crossref] 30. McKinnon, J. J.; Jayatilak, D.; Spackman, M. A.; Chem. Commun. 2007, 37, 3814. [Crossref] 31. Li, Y.; Han, L.; Liu, Z.; Wang, R.; J. Chem. Inf. Model. 2014, 54, 1717. [Link] accessed in October 2023 32. Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W.; J. Chem. Theory Comput. 2010, 6, 1509. [Crossref] 33. Barclay, P. L.; Zhang, D. Z.; J. Comput. Phys. 2021, 435, 110238. [Crossref] 34. Bowers, K. J.; Chow, E.; Xu, H.; Dror, R. O.; Eastwood, M. P.; Gregersen, B. A.; Klepeis, J. L.; Kolossvary, I.; Moraes, M. A.; Sacerdoti, F. D.; Salmon, J. K.; Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, United States, 2006. [Crossref] 35. Martyna, G. J.; Tobias, D. J.; Klein, M. L.; J. Chem. Phys. 1994, 101, 4177. [Crossref] 36. Luty, B. A.; Davis, M. E.; Tironi, I. G.; Van Gunsteren, W. F.; Mol. Simul. 1994, 14, 11. [Crossref] 37. Humphreys, D. D.; Friesner, R. A.; Berne, B. J.; J. Phys. Chem. A 1994, 98, 6885. [Crossref] 38. Chang, J.; Zhang, S. Z.; Wu, Y.; Zhang, H. J.; Sun, Y. X.; Transition Met. Chem. 2020, 45, 279. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access