
 |
 |
 |
 |
 |
Artigo
|
|
| Green synthesis of FexOy structures using Euterpe oleracea mart extract from the legal Amazon, Brazil |
|
Simara F. BorgesI; Karollyne S. AndradeI; Evilly C. VazI; Manoel D. Morais NetoI; Aluisio A. CabralII; Eledir V. SobrinhoIII; Cláudia Q. da RochaIV; Gilvan P. de FigueredoI,* I. Departamento de Química, Instituto Federal de Educação, Ciência e Tecnologia do Maranhão, 65030-005 São Luís - MA, Brasil Received: 10/28/2023 *e-mail: gilvanfigueredo@ifma.edu.br The synthesis of oxides using plant extracts as precursors has attracted attention as it is an economical, efficient, and ecological route. Euterpe oleracea Mart, available in the Legal Amazon, has great potential due to its composition, rich secondary metabolites. Therefore, this work aimed to investigate the potential of aqueous açaí leaf extract as precursors in synthesizing FexOy-type oxides. The synthesis was developed in two stages to obtain the hematite phases (step 1) and the magnetite phase (step 2). The effect of pH on phase formation and structure was evaluated in step 1. Then, the material was obtained in step 2 by precipitation with pH adjustment. The powders were characterized by thermal analysis, X-ray diffraction, infrared spectroscopy, and scanning electron microscopy. The results showed that pH played a significant role in the thermal behavior, crystallite sizes, and structural parameters of hematite, with neutral or acidic pH being more attractive for obtaining crystallites between 21 and 24 nm. Magnetite with 21.51 nm crystallites was precipitated, indicating possible capping of the Fe3O4 particles and preserving their magnetic characteristics. INTRODUCTION The Brazilian Amazon comprises the largest extension of continuous humid forests on the planet, around 3,700 km2, and covers more than 5 million km2 of the national territory. It is characterized by its extensive river networks, dense vegetation cover, and incomparable biological richness. It is recognized as the most prominent Brazilian biome and one of the most crucial biodiversity reserves in the world. Moreover, the Legal Amazon has an area of 5,217,423 km2, representing 61% of the Brazilian territory. In addition to containing the entire Brazilian Amazon biome, it contains 20% of the Cerrado biome and part of the Mato Grosso Pantanal. It encompasses the whole states of Acre, Amapá, Amazonas, Mato Grosso, Pará, Rondônia, Roraima and Tocantins and part of the state of Maranhão.1,2 The oxides have been previously synthesized through various methods, including sol-gel, hydrothermal, co-precipitation, microwave, and combustion. However, with the increasing population growth rate and industrial factories, recent efforts have been focused on utilizing more eco-friendly synthesis methods to prevent environmental pollution and minimize harmful chemical residues.3 Thus, there has been an increasing interest in the search for ecologically friendly synthesis methods in the literature.4-6 Green chemistry and its 12 principles seek to ensure a process with a low level of waste generation following conscientious and sustainable behavior.7 The biggest obstacle in developing synthetic methods that adhere to these principles is the complete or partial replacement of ecologically hazardous solvents with those that provide little to no risk.8 Within this context, green chemistry is a interdisciplinary scientific field that concentrates on the design, development, and execution of chemical processes. Green chemistry aims to enhance the sustainability of chemical products, reducing their impact on human health and creating a greener future.9-11 Green synthesis uses alternative, less harmful environmental inputs using low-cost, less toxic, and less dangerous products. It consists of using enzymes, amino acids, algae, microorganisms, and plant extracts as organic precursors for synthesizing nanostructures from different categories of materials. In this sense, some compounds extracted from plants can function as chelating, polymerizing, stabilizing, and capping agents for the synthesis mechanisms of metal oxide nanostructures.12 There are several biological sources used in this type of synthesis. However, the use of plant extracts is the most reported in the literature since the biomolecules or phytochemicals products present in these extracts can interact with the metal cations of the precursor salt through chelation and polymerization mechanisms, similar to what happens in the reaction medium of classical methodologies used for the synthesis of metal oxides, such as sol-gel and polymeric precursors or modified Pechini.3,13 The green synthesis of nanostructures produced from plant extracts is carried out in an easy and fast reaction, marked by a change in the medium's color. Synthesis can be observed in three phases: activation, growth, and termination. Nanostructures combine in the growth phase, forming a range of morphologies that can influence their properties.14 Iron oxide nanostructures are interesting since they can be found in various environments ranging from aquatic and terrestrial systems to different natural ecosystems. They consist of combinations of the chemical elements iron (Fe) and oxygen (O) and come in various forms, such as magnetite (Fe3O4), maghemite (γ-Fe2O3), and hematite (α-Fe2O3). These iron-based structures have distinct physicochemical properties, are highly available in nature, and have low toxicity. It has been possible to verify an extensive study of iron oxides in recent years for applicability with adjustable anticorrosive, optical, and magnetic properties, in addition to presenting excellent chemical stability and biocompatibility, high surface/volume ratio, large specific surface area, and easy separation.15 Thus, iron oxides are highly valued in several industrial applications, covering both the food and pharmaceutical industries, due to these unique characteristics. Their versatility and special properties make them promising materials for various uses.16 The plant species Euterpe oleracea Mart (Arecaceae) is native to the Amazon's lowland regions and abundant in Maranhão, Brazil. It belongs to the palmaceae group, considered one of the most productive in this ecosystem. Açaí is exploited from it, a fruit much required for food purposes due to its organoleptic and nutritional characteristics and the presence of compounds with antioxidant and anti-inflammatory properties.17 The use of dry straw from Euterpe oleracea leaves is limited, and there are no studies on its use to add value to this abundant biomass. However, recent studies show that several phenolic compounds of hydroxycinnamic acid are found in the leaf extract, as well as apigenin di-C-glycosides (ACGs), a group of flavonoids,18 secondary metabolites such as phenols, tannins, steroids, triterpenes, resins, catechins, saponins and alkaloids, anthocyanins and anthocyanidins.19 Therefore, studies where these biomolecules become necessary and competitive for different applications are required, mainly using the green synthesis of metal oxides assisted by biomolecules from plant extracts. Therefore, we investigated the potential of the aqueous extract of açaí leaves (Euterpe oleracea Mart) to be used as a natural organic precursor in synthesizing FexOy-type structures in this paper. The α-Fe2O3 and Fe3O4 oxides synthesized in this work are of interest in the chemical and pharmaceutical industries, among others.
EXPERIMENTAL Plant material The leaves of Euterpe oleracea plants were collected in São Luís, Maranhão (2º35'57.0"S 44º12'09.1"W), Legal Amazon region, Brazil. The collection followed Brazilian biodiversity protection and registration laws in the National System of Genetic Heritage and Associated Traditional Knowledge (SisGen No. A04BFAE). Obtaining and chemical characterization of the extract The leaves were washed with distilled water, dried in the open air for 72 h, and ground in a knife mill. Then, 3 g of leaf powder was mixed with 300 mL of distilled water and left under constant stirring for 4 h at 60 ºC. After filtration, the aqueous leaf extract was collected and used in green synthesis. The extract previously frozen at –10 ºC was freeze-dried for chemical investigation to remove all the aqueous solvents. For high-performance liquid chromatography (HPLC) analysis, the extract went first through the clean-up stage via solid phase extraction (SPE) to eliminate interfering substances. A Strata C-18 silica cartridge (500 mg 6 mL-1 , Phenomenex, Torrance, USA) activated with 3 mL of methanol (MeOH, Sigma-Aldrich, Munich, Germany) was used. In this case, 0.0060 g of the extract was dissolved in 1.0 mL of MeOH. Next, the dissolved sample was transferred to the SPE cartridge (6 mL), eluted with 1.0 mL of MeOH, and subjected to a cold jet drying process until complete evaporation of the solvent. Then, they were solubilized in 1.0 mL MeOH and filtered through a polypropylene microfilter (Millipak®, Darmstadt, Germany ) with a 0.45 µm pore size. The clean extract was analyzed on a Shimadzu chromatograph (model LC-20AT - Prominence) with a diode array detector (DAD), also from the same brand (model SPD-M20A). A Kinetex C18 column (250 mm × 4.6 mm, 5 µm, 100 Å), Phenomenex brand (Torrance, USA) was used. The mobile phase consisted of water acidified with 0.01% formic acid (Sigma-Aldrich, Munich, Germany) (A) and methanol acidified with 0.01% formic acid (B). The exploratory gradient was used as an elution mode, in which the concentration of B varied in the range of 5 to 100% in 95 min, at a flow of 0.9 mL min-1, temperature of 28 ºC, and with an initial pressure of 160 kgf cm-2. The volume of the injected sample was 20 µL. Liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-IT-MS) was performed using a spectrometer (Bruker, Massachusetts, USA). Chromatographic analysis was performed on a Luna 5 µm C18 100 Å column (250 × 4.6 mm, Phenomenex, Torrance, USA). The binary gradient mobile phase consisted of 0.1% formic acid (Sigma-Aldrich, St. Louis, USA) in water (solvent A) and 0.1% formic acid in methanol (Sigma-Aldrich, St. Louis, USA) (solvent B). Compounds were eluted from the analytical column with a 50 min gradient ranging from 5 to 100% solvent B at a constant flow rate of 1 mL min-1 in 30 min. Data acquisition was performed in positive and negative ionization mode, with fragmentation in several stages (MS2 and MS3), according to the following parameters: nebulization gas pressure 50.0 psi; capillary temperature 300 ºC; transfer capillary input voltage –4500 V; desolvation gas N2, flow of 10 L min-1; collision gas He, acquisition range, m/z 50-1200. Raw data were analyzed using Data Analysis 4.3 software (Bruker, Massachusetts, USA). Synthesis of the FexOy structures The FexOy structures were synthesized to obtain the hematite phases (step 1) and the magnetite phase (step 2). The synthesis started in step 1 by adding Fe(NO3)3.9H2O to the Euterpe oleracea extract. The extract reaction with nitrate was performed under constant stirring at 90 ºC until a gel formation occurred. Samples were synthesized at pH 5, 7, and 10 to evaluate the effect of pH on phase formation and particle morphology. To adjust the pH, 0.1 mol L-1 HCl (Synth, Diadema, SP) was used for pH 5, and 1 mol L-1 NH4OH (Synth, Diadema, SP) for pH 7 and 10. The gels were pre-calcined at 250 ºC for 120 min at a rate of 10 ºC min-1 to obtain the organic-inorganic precursors (EAçaí-Fe2O3). Then, they were calcined at 500 ºC for 120 min at a rate of 10 ºC min-1 to form the structure. Otherwise, aqueous solutions of FeCl2.4H2O and FeCl3.6H2O, 0.1 mol L-1 were employed in step 2. The synthesis consisted of mixing the aqueous extract and chloride solutions in a 3:4 ratio (extract/solutions). The extract was slowly dripped into the chloride mixture, and the reaction was conducted under constant stirring for 60 min at 50 ºC. The pH of the mixture was adjusted with 1 mol L-1 NH4OH (Synth, Diadema, SP) until the precipitate formed. The suspension was left to rest for 2 h, centrifuged at 3.000 rpm for 20 min, and vacuum-filtered. The obtained solid was dried at 60 ºC for 24 h (ppt-Fe3O4). Characterization of the oxides Thermogravimetric (TG/DTG) and differential scanning calorimetry (DSC) analyses were simultaneously performed on a Netzsch STA-449C analyzer, starting from room temperature up to 900 ºC at a heating rate of 10 ºC min-1 using a platinum crucible and synthetic air atmosphere. Characterization by X-ray diffraction was performed using a Shimadzu XRD-6100 diffractometer, CuKα radiation source (λ = 1.5406 Å) 30 KV and 30 mA, angular scanning of 20-80º (2θ), scanning speed of 2º min-1 and step of 0.02º. The mean crystallite size was calculated using the Scherrer equation (Equation 1) and Rietveld refinement with the GSAS program. Fourier transform infrared spectroscopy measurements were performed on a Shimadzu IRAffinity-1 analyzer, equipped with a HATR MIRacle module and ZnSe prism, with a scan of 400-4000 cm-1, resolution of 4 cm-1 and ATR mode. The particle morphology was observed by scanning electron microscopy using a Hitachi tabletop microscope TM-3000 microscope.  where Tc is the size of the crystallite (nm), k the Scherrer constant (0.89), λ the wavelength of the radiation used in the equipment (in nm), β the width at half height (FWHM) of the diffraction peak, and θ is the Bragg diffraction angle in degrees.
RESULTS AND DISCUSSION Chemical characterization of the aqueous extract Figure 1 shows the chromatogram obtained by HPLC of the aqueous extract of Euterpe oleracea leaves. Table 1 presents the compounds identified in the chromatogram and elucidated by mass spectrometry.
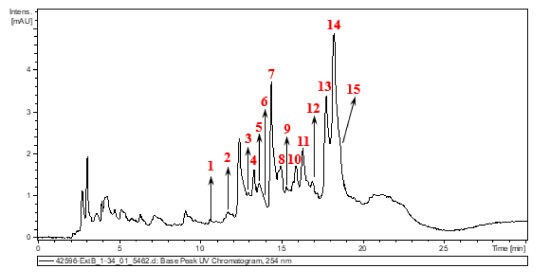 Figure 1. UV-Vis chromatogram at 254 nm of Euterpe oleracea leaf extract
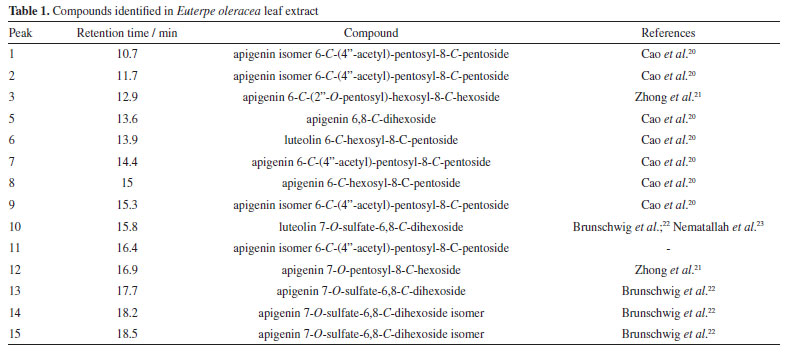
The compounds found agree with the results of Laurindo et al.18 and Sprenger et al.19 The former authors demonstrated that in the aqueous extract of the leaves, several phenolic compounds of hydroxycinnamic acid are found, including 3-O-caffeoylquinic acid, 4-O-cafeoylquinic acid and 5-O-caffeoylquinic acid, in addition, the açaí leaf contains apigenin di-C-glycosides (ACGs), a group of flavonoids including 6,8-di-C-hexosyl apigenin; 6,8-di-C-hexosyl apigenin sulfate; 6-C-hexosyl-8-C-pentosyl apigenin isomers; 6-C-glucosyl luteolin or homoorientin; 6-C-pentosyl-8-C-hexosyl apigenin isomers; 8-C-glucosyl luteolin; and 6-C-glycosyl apigenin. On the other hand, Sprenger et al.19 concluded that açaí fruit extracts present secondary metabolites such as phenols, tannins, steroids, triterpenes, resins, catechins, saponins and alkaloids, anthocyanins, and anthocyanidins. Figure 2 shows an example of a route that the molecules identified in the extract usually go through during fragmentation.
 Figure 2. Fragmentation route of apigenin 6-C-(4''-acetyl)-pentosyl-8-C-pentoside. In blue, the ions of MS3 are expressed, and in black, the ions of MS2
The mechanism of the reaction and the formation of metal oxide structures in plant extract solutions have not yet been entirely determined. Previous results have shown that compounds in the plant extract solutions, such as proteins, phenols, and flavonoids, can bind to the metal as chelating agents, yielding the formation of the metal-phenolate complex. The stability of metal oxide structures can be described based on the interaction between the surface of the metals with the amino group and free carboxylic acid. Additionally, the proteins present in the environment prevent the accumulation and contribute to the stability of FexOy by covering the nanostructures during the green synthesis reaction. A complexing agent functions similarly to a modified sol-gel process, which chelates metal cations in the solution and uniformly mixes them on an atomic scale.3,24 Thermal analysis Figures 3 and 4 present the mass loss measurements and the thermal events related to the precursors (EAçaí-Fe2O3 pH 5, 7, and 10) and the precipitate (ppt-Fe3O4), respectively. Figure 3 shows that the mass loss percentage tends to increase with the increasing pH of the precursor synthesis reaction (EAçaí-Fe2O3 pH 5, 7, and 10). The highest residual mass yield of 96.18% was observed for the synthesis carried out at pH 5, clearly being a consequence of the lower content of free or weakly adsorbed water on the surface of the sample, characterized by the lower percentage of mass loss in event I, as shown in Figure 4 and Table 2. Therefore, when prepared in an alkaline medium with pH adjusted with ammonium hydroxide, the precursors present a more significant layer of water.
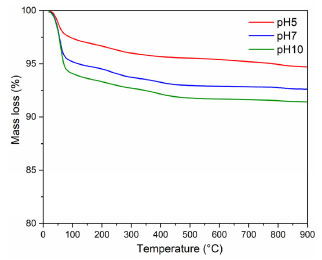 Figure 3. Thermogravimetric curves of the (EAçaí-Fe2O3) precursors obtained at pH 5, 7, and 10
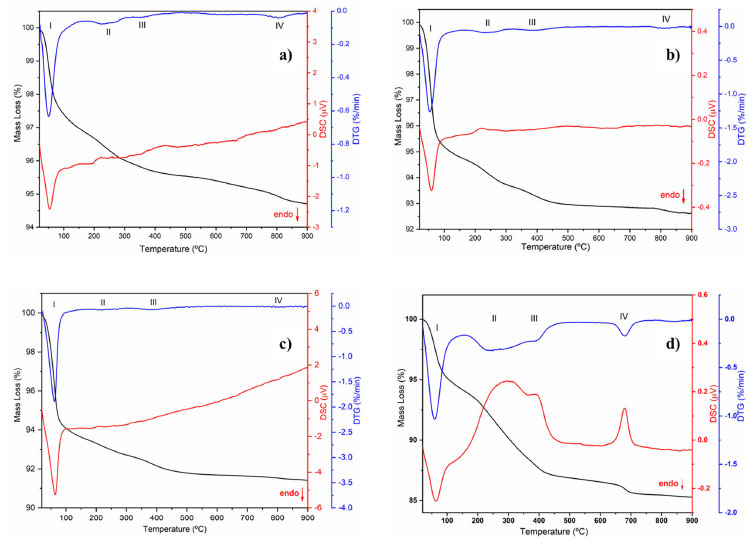 Figure 4. TG/DTG/DSC curves for the EAçaí-Fe2O3 precursors obtained at (a) pH 5, (b) pH 7, (c) pH 10, and for the (d) ppt-Fe3O4 precipitate
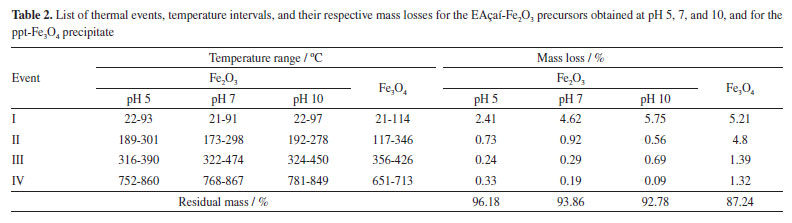
Figure 4 presents the TG/DTG/DSC curves of the EAçaí-Fe2O3 precursors obtained at pH 5 (Figure 4a), pH 7 (Figure 4b), and pH 10 (Figure 4c), and of the ppt-Fe3O4 precipitate (Figure 4d), where it is possible to observe four well-defined mass loss events for each sample. As can be seen in Table 2, at event I, all the samples presented the highest mass loss percentage. This endothermic event observed for all DTG/DSC curves in Figure 4 may be associated with the loss of free or weakly adsorbed water on the surface of the samples. It is possible to observe that events II, III, and IV for the precursor material (EAçaí-Fe2O3) are exothermic, discrete, and with slow mass loss. Events II and III are associated with: (i) decomposition of organic extract molecules (Table 1) remaining in the precursor; (ii) decomposition of nitrate residues that were not eliminated during pre-calcination; (iii) transformation from the amorphous to the crystalline phase of γ-Fe2O3. Event IV corresponds to the γ-Fe2O3 to α-Fe2O3 (hematite) phase transformation.25 Notably, the sample synthesized at pH 10 showed a higher mass loss percentage in event I (5.57%) and lower in event IV (0.09%), indicating the high thermal stability of the final phase at temperatures higher than 500 ºC. According to the literature,26 the pH of the green synthesis reaction plays a crucial role in the morphology of metal oxide particles due to the different activation modes of the biomolecules present in the extract and their interactions with metal cations in solution. These interactions may explain the differences observed in the thermal behavior of the samples synthesized in this work. In addition, it is possible to observe a mass loss equivalent to 12.76% for the precipitate (ppt-Fe3O4), with more evident exothermic events II, III, and IV and more significant mass loss than those observed for the hematite precursors (EAçaí-Fe2O3). This result indicates that the magnetite precipitate particles (ppt-Fe3O4) may be capped or encapsulated by the molecules of the extract (Table 2). However, it presents a yield of approximately 87% in terms of magnetite (Fe3O4), similar to that observed in recent work published in the literature that used an extract from Rhus coriaria.27 X-ray diffraction Figure 5 presents the X-ray diffractograms of the samples synthesized in step 1 to obtain the hematite-type FexOy structure. It is possible to observe narrow and well-defined diffraction lines for all samples, with a principal plane (104) at 2θ ~ 33º, characteristic of the rhombohedral α-Fe2O3 phase, according to crystallographic file (Joint Committee on Powder Diffraction Standards) JCPDS No. 01-079-1741. The mean crystallite sizes (CS) of the α-Fe2O3 phase obtained at pH 5 (23.76 nm) and pH 7 (21.39 nm) were smaller than those commonly observed in the literature.27-29 The crystallite size at pH 10 achieved 42.74 nm. This behavior indicates a strong effect of the pH of green synthesis on the crystallization mechanism of the hematite, making it more attractive to use pH 7 or 5, depending on the desired yield in terms of hematite mass (Table 2).
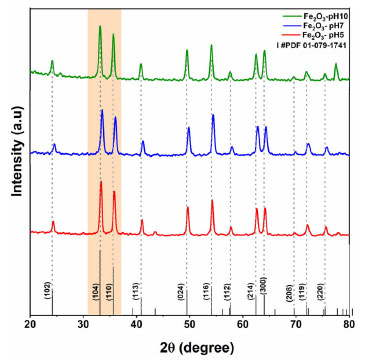 Figure 5. X-ray diffraction patterns of samples provided at pH 5, 7, and 10 and calcined at 500 °C for 120 min
Figure 6 presents the X-ray diffractogram of the sample synthesized in step 2 to provide the magnetite-type FexOy structure. It can be observed that the diffraction lines correspond to the crystallographic planes of magnetite (Fe3O4) with cubic structure and Fd-3m symmetry, according to crystallographic file JCPDS No. 00-088-0866, with evidence for the crystallographic plane (311) at 2θ ~ 35.5º. The identified phase has an average crystallite size of 21.51 nm, indicating the high potential of synthesis via precipitation with Euterpe oleracea extract in obtaining small Fe3O4 particles when compared to those obtained with Rhamnidium elaeocarpum extract,30 potato extract31 and Moringa oleifera extract.32 The synthesis is simple and relatively low cost, mainly due to energy savings, as there is no need for heat treatment of the material.
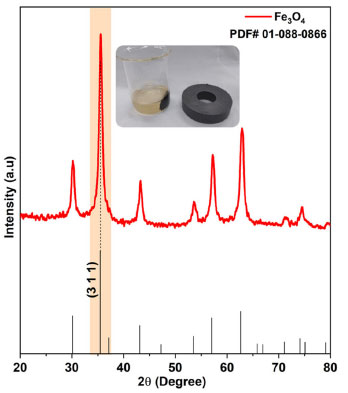 Figure 6. X-ray diffraction pattern of the sample obtained via precipitation in step 2
A quick test with a magnet was performed, and as can be seen in Figure 6, the particles were attracted by the applied magnetic field. The Rietveld refinement results obtained from XRD data (Figures 5 and 6) are presented in Table 3. The refinement was convergent, with acceptable parameters for metallic oxides, indicating that a single crystalline material formed with 100% volumetric fraction and nanometric crystallites. According to the data in Table 3, it is possible to notice that all values referring to the lattice parameters and unit cell volume of the hematite samples are higher than those observed for the standard sample in JCPDS No. 01-079-1741, indicating increasing atomic distances during the crystallization process. On the other hand, an opposite behavior was observed for the magnetite sample with more excellent atomic approximation, which the decrease of the lattice parameters and unit cell volume can justify. The considerations made based on the Rietveld refinement results developed in this work have a significant role in elucidating the microstructural behavior of iron-based oxides that are synthesized by alternative green synthesis strategies with plant extracts based on the sol-gel principle and co-precipitation, as applied in step 1 and 2, respectively.
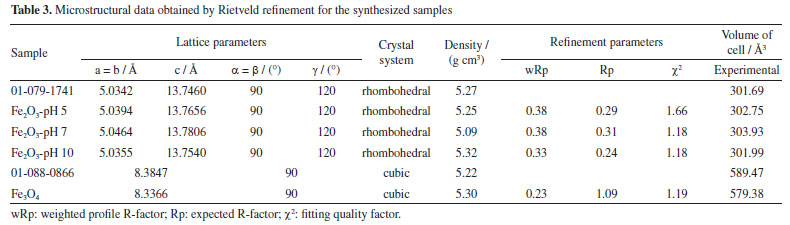
Fourier-transform infrared spectroscopy (FTIR) The vibrational analysis in the infrared region presented in Figure 7 for the α-Fe2O3 samples highlights the calcination effects. Samples of pre-calcined precursors present bands between 3900-3200 and 1700-1200 cm-1, attributed to the O–H stretching of alcohols, phenols, and adsorbed water, C=O stretching (carboxylic acids), and vibration of the C–O group (alcohols and esters).33 The absence of these bands in the calcined samples is noticeable, as well as the appearance of bands between 800-400 cm-1, which can be attributed to metal-oxygen bond34 vibrations of the hematite structure, mainly for the synthesized samples at pH 5 and 7.
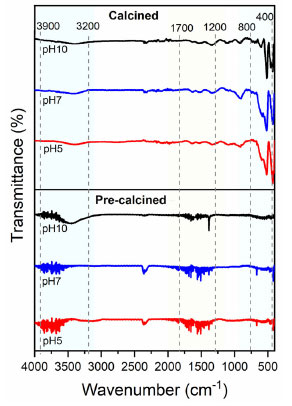 Figure 7. Infrared spectra of hematite samples obtained at pH 5, 7 and 10 (pre-calcined and calcined)
Figure 8 presents the FTIR spectrum of the Fe3O4 sample, where the presence of bands between 3700-2500 and 1775-1300 cm-1 correspond to the O–H stretching of alcohols, phenols, and adsorbed water, stretching C=O (carboxylic acids), and vibration of the C–O group (alcohols and esters).33 Bands between 800-400 cm-1 can also be seen, indicating the formation of Fe–O bonds and confirming the achievement of the magnetite structure. These results corroborate the thermal analysis data, indicating possible compounds from Euterpe oleracea extract capping or encapsulating the magnetite particles. Metal oxide capping has been identified as an essential strategy for stabilizing nanostructures and maintaining their performance in a given application.14
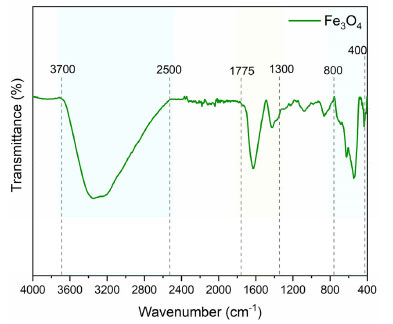 Figure 8. Infrared spectra of the magnetite sample (Fe3O4)
Scanning electron microscopy (SEM) Micrographs of the samples are shown in Figures 9a-9h. All hematite samples (Figures 9a-9f) present morphology as blocks containing irregular surfaces, voids, and agglomerates. It was impossible to observe the effect of pH on the morphology of the synthesized particles. Otherwise, micrographs of magnetite reveal massive blocks with regular surfaces (Figures 9g-9h).
 Figure 9. Micrographs of the α-Fe2O3 samples obtained at pH 5 (a) 120×, (b) 1000×; pH 7 (c) 120×, (d) 1000×; pH 10 (e) 120×, (f) 1000×; and the Fe3O4 sample (g) 120×, (h) 1000×
CONCLUSIONS Nanocrystalline hematite (α-Fe2O3) and magnetite (Fe3O4) structures were obtained through green synthesis using aqueous Euterpe oleracea Mart leaf extract containing phenolic compounds and secondary metabolites. Rietveld's refinement confirmed the formation of single crystalline phases, in which unit cells were higher than the standard for hematite, while they had lower volume than the standard for magnetite. The results showed that pH played a significant role on the thermal behavior and structure of oxides, especially the crystallite size of hematite particles, with neutral or acidic pH being more attractive to obtain smaller particles. The obtained magnetite also presented a small crystallite size and magnetic characteristics without needing heat treatment, validating a simple synthesis with relatively low cost and high energy savings. Therefore, the results demonstrated that is possible to synthesize oxides with excellent structural properties using a more environmentally friendly method.
ACKNOWLEDGMENTS To IFMA for the infrastructure and financial support (PRPGI notices No. 89/2021, No. 186/2021 and No. 287/2022). To the Department of Materials Engineering (DeMat-UFRN) and Tomaz R. de Araújo for the scanning electron microscopy analyses. CAPES for the scholarship granted in notice/CAPES No. 13/2020 (Support Program for the Development of Postgraduate Studies in the Legal Amazon). The co-authors thank FAPEMA, CNPq, and IFMA for the scientific initiation grants.
REFERENCES 1. Prestes, F. F.; Brazilian Journal of Animal and Environmental Research 2021, 4, 3848. [Crossref] 2. Paolino, C. C.; Amaral, F. G.; Cruz, C. B. M.; Meio Ambiente (Brasil) 2021, 3, 17. [Crossref] 3. Lachini, S. A.; Eslami, A.; Int. J. Hydrogen Energy 2024, 51, 51. [Crossref] 4. Mahmoud, M. E.; Amira, M. F.; Abouelanwar, M. E.; Salam, M. A.; J. Environ. Chem. Eng. 2021, 9, 104697. [Crossref] 5. Kunoh, T.; Takeda, M.; Matsumoto, S.; Suzuki, I.; Takano, M.; Kunoh, H.; ACS Sustainable Chem. Eng. 2018, 6, 364. [Crossref] 6. Nadagouda, M. N.; Hoag, G.; Collins, J.; Varma, R. S.; Cryst. Growth Des. 2009, 9, 4979. [Crossref] 7. Marco, B. A.; Rechelo, B. S.; Tótoli, E. G.; Kogawa, A. C.; Salgado, H. R. N.; Saudi Pharm. J. 2019, 27, 1. [Crossref] 8. Lenoir, D.; Schramm, K. W.; Lalah, J. O.; Sustainable Chem. Pharm. 2020, 18, 100313. [Crossref] 9. Juibari, N. M.; Eslami, A.; J. Therm. Anal. Calorim. 2017, 130, 1327. [Crossref] 10. Ahmad, S.; Munir, S.; Zeb, N.; Ullah, A.; Khan, B.; Ali, J.; Bilal, M.; Omer, M.; Alamzeb, M.; Salman, S. M.; Ali, S.; Int. J. Nanomed. 2019, 14, 5087. [Crossref] 11. Ying, S.; Guan, Z.; Ofoegbu, P. C.; Clubb, P.; Rico, C.; He, F.; Hong, J.; Environ. Technol. Innovation 2022, 26, 102336. [Crossref] 12. Rocha, M. B. C.; Araújo, T. R.; Medeiros, R. L. B. A.; Oliveira, M. M.; Figueredo, G. P.; Research, Society and Development 2021, 10, e399101623406. [Crossref] 13. Aarthye, P.; Sureshkumar, M.; Mater. Today: Proc. 2021, 47, 907. [Crossref] 14. Shamaila, S.; Sajjad, A. K. L.; Najam-ul-Athar, R.; Farooqi, S. A.; Jabeen, N.; Majeed, S.; Farooq, I.; Applied Materials Today 2016, 5, 150. [Crossref] 15. Pallela, P. N. V. K.; Ummey, S.; Ruddaraju, L. K.; Gadi, S.; Cherukuri, C. S. L.; Barla, S.; Pammi, S. V. N.; Heliyon 2019, 5, e02765. [Crossref] 16. Duarte, G. M.; Guilherme, L. R.; Rev. Virtual Quim. 2021, 13, 1069. [Crossref] 17. Rogez, H.; Akwie, S. N. L. T.; Moura, F. G.; Larondelle, Y.; J. Food Sci. 2012, 77, 1299. [Crossref] 18. Laurindo, L. F.; Barbalho, S. M.; Araújo, A. D.; Guiguer, E. L.; Mondal, A.; Bachtel, G.; Bishayee, A.; Nutrients 2023, 15, 989. [Crossref] 19. Sprenger, L. K.; Giese, E. G.; Santos, J. N.; Molento, M. B.; Archives of Veterinary Science 2016, 21, 21. [Crossref] 20. Cao, J.; Yin, C.; Qin, Y.; Cheng, Z.; Chen, D.; J. Mass Spectrom. 2014, 49, 1010. [Crossref] 21. Zhong, L.; Wu, G.; Fang, Z.; Wahlqvist, M. L.; Hodgson, J. M.; Clarke, M. W.; Junaldi, E.; Johnson, S. K.; Food Res. Int. 2019, 116, 1153. [Crossref] 22. Brunschwig, C.; Leba, L. J.; Saout, M.; Martial, K.; Bereau, D.; Robinson, J. C.; Int. J. Mol. Sci. 2017, 18, 61. [Crossref] 23. Nematallah, K. A.; Ayoub, N. A.; Abdelsattar, E.; Meselhy, M. R.; Elmazar, M. M.; El-Khatib, A. H.; Linscheid, M. W.; Hathout, R. M.; Godugu, K.; Adel, A.; Mousa, S. A.; J. Funct. Foods 2018, 49, 401. [Crossref] 24. Cao, C.; Yu, L.; Xie, Y.; Wei, W.; Jin, H.; Int. J. Hydrogen Energy 2022, 47, 8716. [Crossref] 25. Mirzaei, A.; Janghorban, K.; Hashemi, B.; Hosseini, S. R.; Bonyani, M.; Leonardi, S. G.; Bonavita, A.; Neri, G.; Process. Appl. Ceram. 2016, 10, 209. [Crossref] 26. Badnore, A. U.; Pandit, A. B.; Chemical Engineering and Processing: Process Intensification 2017, 122, 235. [Crossref] 27. Piro, N. S.; Hamad, S. M.; Mohammed, A. S.; Barzinjy, A. A.; IEEE Trans. NanoBiosci. 2023, 22, 308. [Crossref] 28. Selvaraj, R.; Pai, S.; Murugesan, G.; Pandey, S.; Bhole, R.; Gonsalves, D.; Varadavenkatesan, T.; Vinayagam, R.; Appl.Nanosci. 2021, 11, 2227. [Crossref] 29. Diniz, P. A. F.; Cavalcante, K. S. B.; Souza, J. C.; Marques, G. N.; Santos, F. H. S.; Cutrim, F. M.; Henriques, R. B.; Mendonça, L. T. B.; Nascimento, U. M.; Mater. Res. 2023, 26, e20230016. [Crossref] 30. Jacinto, M. J.; Silva, V. C. P.; Prescilio, I. C.; Correa, A. K.; Ferreira, L. F.; Razini, S. S.; Anais Eletrônicos do 59º Congresso Brasileiro de Química; João Pessoa, Brasil, 2019. [Link] accessed in March 2024 31. Buazar, F.; Baghlani-Nejazd, M. H.; Badri, M.; Kashisaz, M.; Khaledi-Nasab, A.; Kroushawi, F.; Starch-Starke 2016, 68, 796. [Crossref] 32. Darmawan, M. Y.; Istiqomah, N. I.; Adrianto, N.; Tumbelaka, R. M.; Nugraheni, A. D.; Suharyadi, E.; Results Chem. 2023, 6, 100999. [Crossref] 33. Mayedwa, N.; Mongwaketsi, N.; Khamlich, S.; Kaviyarasu, K.; Matinise, N.; Maaza, M.; App. Surf. Sci. 2018, 446, 266. [Crossref] 34. Karpagavinayagam, P.; Prasanna, A. E. P.; Vedhi, C.; Mater. Today: Proc. 2020, 48, 136. [Crossref] |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access