
 |
 |
 |
 |
 |
Assuntos Gerais
| A history of carbonic acid and the central role of bicarbonate in the CO2/H2O system |
|
Claudimir Lucio do Lago* Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo - SP, Brasil Received: 11/10/2023 *e-mail: claudemi@iq.usp.br Carbon dioxide and the related species bicarbonate and carbonate are known for centuries and are deeply involved with modern chemistry knowledge from the beginning. Carbonic acid, by its turn, was intuited more than a century before being synthesized for the first time, which was not obtained, however, without stumbles and mistakes. The equilibrium between HCO3- and CO32- in aqueous medium is free of disputes. However, the interconversion of the other three species is rather controversial. Several papers starting in the beginning of the 19th century until the present days are critically reviewed in order to understand the controversial history of the CO2/H2O system. Finally, experimental evidence and theoretical calculations allow us to place HCO3- as the central species as the conjugate acid for CO32- and conjugate base for two different acids: H2CO3 and CO2. INTRODUCTION Aqueous solutions containing carbon dioxide and related species are ubiquitous. The acid-base properties of CO2 are widely recognized and, in many cases, play a fundamental role in various systems, ranging from the interior of the cells of the most ancient life forms to industrial processes. An examination of a wide range of textbooks and even contemporary scientific articles reveals a well-established sequence of events, which can be summarized as follows: 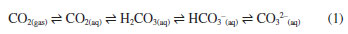 Rocks and salts containing carbonate or bicarbonate have been recognized for centuries, while the solubility of CO2 in water has also been a subject of investigation and understanding over time. However, carbonic acid, the central species in this scheme, was detected and synthesized only a few decades ago. The lack of proof of the existence of H2CO3 was, therefore, an obstacle to understanding this system, which otherwise seemed quite reasonable: the formation of a diprotic acid that dissociates sequentially until the formation of the ionic species with two negative charges. When solid H2CO3 was finally isolated in the 1990s,1 the doubt disappeared and the puzzle finally seemed complete. While there is indeed substantial evidence supporting the existence of all these species and their interconversion, there is also evidence indicating that this transformation does not unfold precisely as depicted in Equation 1. Despite the simple appearance of this system, elucidating the mechanisms is quite challenging and, in fact, remains a work in progress. Nevertheless, its rich historical background and diverse facets provide valuable material for educational discussions.
HISTORICAL BACKGROUND Carbon dioxide has received several names since the first time it was observed until a group of chemists led by Lavoisier included it in their proposition to systematize the nomenclature of the chemical compounds. As a product of charcoal (charbon in French) combustion, its name was associated with it, and the term “carbonic acid” was coined in 1781. At that time, acid was a substance that had a distinctive taste and made litmus red. In a similar way, “sulfuric acid” was the chosen name for SO3; and the element responsible for generating these acids was named “oxygen” using fragments in Greek highlighting this feature. Despite the reluctance of part of the community in adopting the new nomenclature, “carbonic acid” was finally accepted and widely used.2 The decades following the introduction of the term “carbonic acid” by Lavoisier were marked by a great advance in chemical knowledge, and even in the beginning of the 19th century, the chemical composition and the term “carbon dioxide” were already established. Moreover, thanks to the works by Davy and Berzelius, the concept of acid moved to recognize the importance of hydrogen. In 1814, Delametherie3 concluded that “oxygen can no longer be regarded as the generator of acids, hydrogen frequently performing its functions”. From that moment on, it was clear that calling CO2 “carbonic acid” was inappropriate. However, the term “carbonic acid” was not dead, because an acid has been postulated: H2CO3. In fact, carbonates and bicarbonates were known, but not the corresponding acid. However, Laurent4 wrote “... H2CO3 being, under ordinary circumstances, incapable of existing, becomes decomposed immediately into H2O and CO2”. This statement comprehends two underlying concepts: (i) the existence of H2CO3 and that (ii) it is formed as an intermediate. Using modern notation, the following sequence of reactions were assumed to happen when a carbonate is acidified: 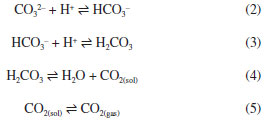 Obviously, H2CO3 was neither isolated nor synthesized in 19th century, and presuming its existence was likely the result of inductive reasoning based on the observation of carboxylic and dicarboxylic acids and their anhydrides, as well as other inorganic oxides, such as SO3. To be fair to the experimental evidence at the time, another reaction should be considered instead of 3 and 4: 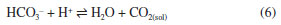 The carbonates and bicarbonates were well known, as well as the products of the reactions with acids. Therefore, reaction 6 dismisses the hitherto undetected H2CO3 keeping adherence to the facts observed so far. In any case, many other scientists continued to use the ambiguous term “carbonic acid” to denote CO2. For instance, Gore and Tyndall5 studied liquid CO2 under the term “liquid carbonic acid”. Arrhenius used the term “carbonic acid” not only in the title of his seminal paper “On the Influence of Carbonic Acid in the Air upon the Temperature of the Ground”, but also 145 times throughout the whole text without mentions to “carbon dioxide” or CO2.6 Certainly, Tyndall and Arrhenius had no doubt about the chemical composition of what they called “carbonic acid” and used the term simply because it was usual. The problem is that others did not have this understanding so clear in mind. For instance, Tolman7 stated about H2CO3: “this acid has never been separated as such, but doubtless exists”. He supported this statement by citing Bunsen's book Gasometry: Comprising the Leading Physical and Chemical Properties of Gases.8 Bunsen had indeed used consistently the term “carbonic acid” throughout the book, but one can conclude by checking the experimental section that Bunsen was writing about CO2 and not H2CO3. Fortunately, the inappropriate nomenclature for CO2 was eventually abandoned by the end of the 19th century. For instance, Jones9 edited a collection of papers with the then modern theory of solutions. At the end of the chapter about Arrhenius' work, his background and achievements are described and there the term “carbonic acid” was deliberately replaced by “carbon dioxide” in the title of the previously cited paper about the greenhouse effect. Although a number of studies were published in the first decades of the 20th century on equilibrium and kinetics related to the CO2/H2O system, the first somewhat successful attempts to synthesize H2CO3 were carried out in the 1960's. Over that decade, carbonic acid was claimed to be synthesized as ether adducts H2CO3·O(CH3CH2)210 and H2CO3·O(CH3)2.11 Although, in hindsight, the routes used may indeed have been successful, the products were not pure, and the spectral confirmation was dubious. However, in 1987, the production of H2CO3 by pyrolysis of NH4HCO3 in the gas phase and its mass spectrometric identification was achieved.12 In 1991, H2CO3 was obtained by proton irradiation of a 1:1 H2O + CO2 ice mixture at 20 K, as in the work of Moore and Khanna.1 Carbonic acid was also synthesized through the use of aqueous solutions containing HBr and KHCO3. This process involved a unique cryogenic technique where water was subsequently removed through sublimation at 200 K, as in the work of Hage et al.1 This last route is a more compelling demonstration, because despite the use of a modern cryogenic approach, it is essentially the well-known reaction between bicarbonate and an acid. Surprisingly, along with the convincing demonstrations of the existence of H2CO3 came a hasty conviction about how it behaves in water. The model assumed to be true in most of the works encompasses the reactions 2, 3, and 4. However, there is another plausible model, in which reaction 4 is replaced by 6. In addition, there is a conciliatory model that encompasses 2, 3, 4, and 6. In the following sections, experimental evidence and theoretical reasoning will be used in the elucidation of which model should we adopt. In a nutshell, we have to look for evidence that either the first or the second model occurs; if both are true, the conciliatory model would automatically be proven.
KINETIC AND EQUILIBRIUM CONSTANTS AND THE MECHANISM OF REACTION The already cited Bunsen8 quantitative study about solubility of CO2 in different solvents was followed by other ones using different approaches and different conditions in order to describe the CO2/H2O system. For instance, in 1916, Johnston13 demonstrated the determination of the concentrations of the different species in a solution adopting not only the set of reactions, but also a similar notation used today. Four years before, McBain14 demonstrated the slow kinetics of the CO2 hydration reaction using an alkaline solution with phenolphthalein. Through these quantitative studies, it was possible to observe that the kinetic and thermodynamic behavior was compatible with the existence of H2CO3. Thus, the set of reactions 2-6 seemed to be correct after all. In 1960, Kern15 revised the achievements from the first decades of the 20th century and published an instructive paper about the CO2 hydration. Although new methods and approaches have been used to improve the accuracy of the values of the equilibrium and kinetic constants since then,16 the order of magnitude of these constants still holds. At that point, it was already clear that there was a slow hydration step and a fast acid dissociation step. The dissociation of inorganic and organic acids is generally fast and, thus, the equilibrium between H2CO3 and HCO3– can be accepted without further reservations. However, what exactly is the hydration step: reverse reaction 4 or 6? This intriguing question is still debated today. First, let's evaluate what the initial kinetic and thermodynamic studies reveal. It is straightforward to show that equilibria 3 and 4, as well as their equilibrium constants (K3 and K4) can be combined to represent equilibrium 6, whose equilibrium constant is given by K6 = K4/K3. Conversely, whether we assume that equilibrium 6 is the real one, then K4 can be numerically obtained by using the same equation. In other words, one cannot argue which is the right hydration mechanism based on the values obtained experimentally to the concentration of the species. Figure 1 shows the concentration profiles obtained by numeric calculation based on equilibria from 2 to 5. However, if equilibrium 4 is replaced by 6, exactly the same values are obtained. Moreover, since the hydration is the only slow reaction in both models, monitoring the concentration of the species is not also a way to doubtless determine the mechanism of reaction.
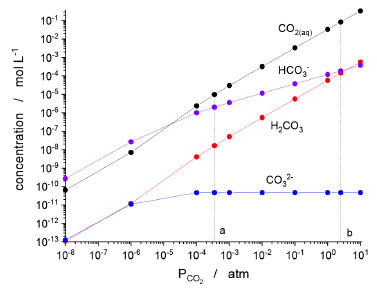 Figure 1. Concentration profiles of the carbon-containing species in the aqueous phase of a CO2/H2O system as a function of the CO2 partial pressure. Equilibrium constants: K2 = 2.13 × 1010 L mol-1,17 K3 = 4.0 × 103 L mol-1,15K4 = 588,15 and K5 = 30 atm L mol-1.18 The same plot is obtained if reaction 4 is replaced by 6 (K6 = 2.24 × 106 L mol-1).15 Dashed line a emphasizes the regular atmospheric condition, and line b emphasizes the threshold pressure above which H2CO3 is more abundant than HCO3-
In 1961, Eigen et al.19 proposed a model to explain the systems SO2/H2O and CO2/H2O, which essentially is the conciliatory model including reactions 4 and 6, i.e., the hydration/dehydration reaction could take place alternatively through one or other way. This model including equations 3, 4, and 6 is often accepted today. In this seminal paper, the authors paid attention to something important: the reaction mechanism. They suggested that reaction 6 takes place according to the formation of a zwitterionic intermediate: 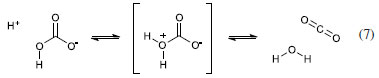 The alternative path (reactions 3 and 4) would demand a less plausible concerted reaction involving water molecules from the neighborhood. The authors stated that the relaxation measurements they obtained were actually related to reaction 6 (through mechanism shown in 7), and that the kinetic constants for reaction 4 that were presented in that paper were only of a formal nature, i.e., they were calculate based on the results for reaction 6 and the pKa for H2CO3. It is rather strange that the authors proposed a conciliatory model, which is not supported by their own experimental results, but even so the model is maintained. Obviously, there is no way to elucidate this point, but apparently the authors understood the limitation of available instrumental techniques and decided to maintain the possibility of an alternative mechanism. In 1977, Pocker and Bjorkquist16 investigated the kinetic isotope effect on CO2 hydration/dehydration in H2O and D2O. Fundamentally, the idea is to distinguish the mechanism based on deuterium isotope effect (kH2O/kD2O) for the reaction involving the possible transition states shown in Scheme 1.
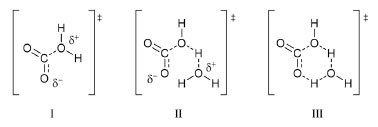 Scheme 1. Possible transition states for CO2 hydration. States I and II are related to reaction 6, while III is related to reaction 4. State I precedes the zwitterion proposed by Eigen et al.,19 while II precedes the ion pair H3O+ HCO3-, which then dissociates and diffuses away
The first transition state would be the precursor of the zwitterion proposed by Eigen et al.19 The second one would be similar to the first, but allowing the formation of HCO3– and H3O+ without necessarily generating a zwitterionic species. The last transition state would be one formed in the concerted rearrangement leading to H2CO3 instead HCO3–. Their results for the deuterium isotope effect for hydration and dehydration reactions suggest that the first two transition states are more likely. Recent studies about kinetic isotope effects taking into account not hydrogen, but the natural isotopes of carbon and oxygen are not conclusive.20 A fundamental problem with the approach used in these studies is that only the isotope ratios for the tangible species CO2 and HCO3– can be measured; H2CO3 remains elusive. In any way, the results are in agreement with the conclusion by Pocker and Bjorkquist.16 An investigation about the protonation reaction of HCO3– was carried out by Adamczyk et al.21 using a photoacid that is optically triggered to transfer a proton to HCO3– on an ultrafast time scale. The monitoring of IR bands allowed demonstrating that not only H2CO3 is formed in aqueous medium on a nanosecond time scale, but also that it acts like an ordinary carboxylic acid. In addition, the pKa was also obtained, and the value 3.45 ± 0.15 is similar to the value 3.36 obtained by Thiel and Strohecker (cited by Buytendyk et al.)22 and other obtained by different methods. Transient signals in the region around 2364 cm-1 were not detected, which suggest that CO2 is not formed by dehydration of H2CO3 in the nanosecond time scale, i.e., the acid has a lifetime extending beyond 1 ns. It is important to note that this result does not contradict the models studied here, as reaction 3 is considered in both. The authors also stated that as the vibrational marker is located at a frequency typical for carbonyl stretching, the zwitterionic structure suggested by Eigen et al.19 can be excluded. This conclusion, however, does not consider the possibility that the zwitterion is not an intermediate, but a transition state. As an intermediate, it is reasonable to assume that its presence can be detected through spectroscopic methods. However, a transition state would not be detected. Moreover, the results are compatible with the second transition state shown in Scheme 1. Once again, the three models under investigation deny neither the protonation of HCO3– forming H2CO3, nor its reverse reaction. It is noteworthy that some studies consider a mechanism for the H2CO3 decomposition involving an intramolecular transition state: 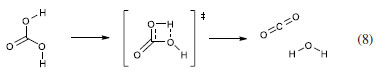 For instance, Terlouw et al.12 consider that the calculated activation energy higher than 40 kcal mol-1 for reaction 8 explains why H2CO3 is a stable species in the gas phase and supports the detection of it in their study about formation of H2CO3 in gas phase. However, the authors pointed out that the mechanism could, in fact, be different in solution.
THE CARBONIC ANHYDRASE Another term coined for CO2 in the 19th century was “carbonic anhydride”, which follows directly from the assumption that CO2 would be the dehydration product of H2CO3. For instance, in 1860, Miller23 suggested that, in the absence of a definite “hydrate of carbonic acid” (H2CO3), “carbonic acid gas” (CO2) should be called “carbonic anhydride”. In 1933, when finally the enzyme that catalyzes the reaction between CO2 and H2O was isolated from ox blood and characterized, Meldrum and Roughton24 heeded their colleague Phillip Eggleton's suggestion and called it “carbonic anhydrase”.25 The term not only alludes to the dehydration of H2CO3, but this acid was then used to explain how the enzyme works (reactions 3 and 4). That enzyme – actually a member of a whole class of enzymes that catalyze the same reaction – was studied over the next decades, and by the end of the century, the mechanism of action of carbonic anhydrases was already elucidated.26 The catalysis mechanism uses to be represented as a set of 4 to 6 steps involving the central zinc atom, but it can also be depicted, in its most simplified form, by two elementary steps:27 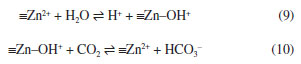 It is noteworthy that at no time – even in the detailed version of the mechanism – it is proposed that H2CO3 is formed as a free molecule or a complex with the enzyme. Therefore, carbonic anhydrase actually catalyzes reaction 6 instead 4. Of course, without the enzyme, the path through reaction 4 could be indeed the correct one. However, despite the suggestive name of the enzyme, there is no support for reaction 4 here.
THE NUCLEOPHILIC ATTACK OF CO2 Since the first quantitative studies on the CO2/H2O system, it has been established that CO2 can also react in an aqueous medium in a distinctive way through the following reaction: 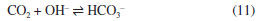 The importance of this reaction is, of course, higher in alkaline solutions, because of the low availability of OH– in neutral or acid environments. This reaction helps to understand, for instance, the kinetics of bubbling of CO2 in alkaline solutions. The mechanism of reaction is quite straightforward: the nucleophilic attack of CO2 by OH–, as cited by Pocker and Bjorkquist,16 and Stirling.28 One can see the similarity between this reaction and the reverse reaction 7, in which H2O would be the nucleophile. In this case, however, there must be a base to recover the H+; ordinarily, water also would play this role. Considering the competition between H2O and OH– as nucleophiles, the action of carbonic anhydrase presented in the previous section can be considered as a strategy to boost the mechanism via OH– at low pH values.27 There is another important nucleophilic attack of CO2 in aqueous medium: the carbamate formation. Carbamates are formed for primary and secondary amines including amine moiety in proteins.29 In an aqueous medium containing the two previous nucleophiles plus amines, one can think of the reactions and the products as resulting of a competition guided by availability and nucleophilicity of all of them. Said et al.30 proposed a unified approach to the reaction mechanisms based on experimental data and density functional theory (DFT) calculations. They conclude that the zwitterionic form of the carbamate (R-NH2+-COO–) is energetically unflavored, which is indeed correct. However, once again, the result must be taken carefully, because this form should be considered as either an intermediate or a transition state, and R-NH-COO– remains the stable species in solution. Anyway, this similarity in behavior between these nucleophiles shows how it is possible to understand and support a model including reaction 7.
BICARBONATE AS A MEMBER OF THE MONOALKYL CARBONATE CLASS Analogy and inductive reasoning are helpful in human attempts to understand the world, and it is no different in chemistry. In the case of H2CO3, the analogy with other inorganic acids could be useful to predict its behavior. Another approach is to classify H2CO3 as a dicarboxylic acid. Starting from, for example, adipic acid (C6) and going down to oxalic acid (C2), we have a set of dicarboxylic acids that are well known. What if carbonic acid (C1) is the first member of this family? Although, at first glance, the structural similarity can be highlighted, there is no advantage in such a classification. For example, the intramolecular anhydrides down to C4 were known. The tense 4-atom ring malonic anhydride (C3) was only synthesized in 1978,31 and oxalic anhydride (C2) is still unknown. Therefore, carbonic anhydride (C1) would be definitely an outlier in this series. The same could be said about the acidity, because the trend for the first and second pKa values starting from adipic acid is disrupted from oxalic to carbonic acid. Therefore, there is no reason to consider H2CO3 a special case of a carboxylic acid as sometimes it is presented. There is, however, another class in which H2CO3 could be advantageously included: the alkyl carbonic acids, or hemiesters of carbonic acid (HECAs). Their salts, the monoalkyl carbonates (MACs), are long known. One of the first citations to a MAC is from 1886. Habermann32 claimed to have obtained monomethyl carbonate during electrochemical oxidation of potassium acetate in a mixture of water and methanol. However, the MACs were synthesized and systematically studied years after that.33 In the series H-(CH2)n-OCO2–, monomethyl carbonate is the first member (n = 1), or alternatively, bicarbonate would be the first one (n = 0). The classic MAC synthesis approach consists of bubbling CO2 into a solution of an alkali or alkaline earth metal alkoxide in the corresponding alcohol. One can observe the similarity with the reaction 11, being just the case of exchanging the nucleophiles alkoxide and hydroxide. The most studied reaction of MACs is their hydrolysis. It was soon realized that, in many cases, the hydrolysis was not complete, with a balance between MAC and HCO3– remaining. Even so, investigations involving MACs remained basically in the direction of hydrolysis. Only recently, the interconversion between HCO3– and MACs was demonstrated to occur freely in aqueous solutions of alcohols and sugars,34 and monoethyl carbonate was demonstrated to be present in alcoholic beverages.35 Not only the MACs are of interest, but also the HECAs: the counterpart of H2CO3 in that series. The synthesis of methylcarbonic acid was described in 1972 by Gatow and Behrendt;36 not surprisingly one of the groups that proposed the synthesis of H2CO3 in the 1960's.11 They used a similar approach adding HCl to a methanol solution of sodium methyl carbonate at –50 ºC. Similar to what was observed for the synthesis of H2CO3 adducts with methyl and ethyl ether,10,11 the product decomposes into CH3OH and CO2 above –36 ºC. The similarity between the members of this class is apparent, but what is actually gained by using this classification? A particularly important case is the α-H2CO3. The synthesis of H2CO3 from HBr and KHCO3 by using the cryogenic approach, as studied by Hage,1 was preceded by an attempt in which methanol, and not water, was chosen as the solvent.37 The product was different physically and spectroscopically from that obtained subsequently in the absence of methanol. As the products were solid, these differences were attributed to differences in the crystal forms, and they were named α-H2CO3 and β-H2CO3 according to the chronology, i.e., using methanol and water, respectively, as the solvent. At this point, it should seem obvious to the attentive reader that the putative α-H2CO3 was actually methyl carbonic acid, as the use of methanol as the solvent was sufficient to convert bicarbonate to monomethyl carbonate. However, seen in retrospect, this failure is even natural, given our little practice in associating both HCO3– with MACs and H2CO3 with HECAs. The fact is that even after the authors38 have revisited the so-called α-H2CO3 in a subsequent paper and explained in detail the actual origin of the differences in the products, sometimes we still find citations to the forms α-H2CO3 and β-H2CO3. Another contribution from MAC to the present topic was to the elucidation of the hydration/dehydration mechanism. Pocker et al.39 investigated the decomposition of CH3OCO2–, C2H5OCO2–, and sec-C4H9OCO2– in H2O and D2O, and concluded that a transition state similar to the second one in Scheme 1 in involved in the reactions.
THEORETICAL CALCULATIONS By now, it should be clear that elucidating the mechanism of CO2 hydration is not straightforward. In addition to the set of experimental strategies used for elucidation, theoretical calculations and computational simulations have emerged as a complementary tool. The stability of solid H2CO3 is one of the questions that have been addressed. The synthesis and study of solid H2CO3 had revealed its surprising stability as a solid when compared to its fast decomposition in aqueous solution. This topic was addressed by Loerting et al.40 They calculated the energies of the transition states for the decomposition of H2CO3 in the presence of a growing number (n) of water molecules according to the schemes shown in Figure 2.
 Figure 2. Decomposition of pure H2CO3 and in presence of one and two water molecules. The energy barriers are shown beneath the transition states (source: adapted from Loerting et al.)40
The first reaction shown in Figure 2 is the same as Equation 8, and the calculated energy was similar to the previous result. The decrease in energy as the number of water molecules increases is evident, and the estimated half-life for n = 2 is 119 s at 300 K. In contrast, the estimated half-life for pure H2CO3 is 0.18 million years, which suggests that H2CO3 is a quite stable species. The half-life of 119 s for n = 2 is considerably greater than the experimental value of 0.056 s. Obviously, compared to the half-life of pure H2CO3, these values for n = 2 may seem, at first glance, to be compatible. However, there are flaws in this approach. The first one is that the first reaction in Figure 2 represents the behavior of a single molecule, which would be the case of H2CO3 in the gas phase, but not in liquid or solid phase. Kumar et al.41 and de Marothy42 investigated the behavior of dimers of H2CO3 and concluded that the energetic barrier can be even smaller than the one for the cluster involving H2O. The second one is that it is assumed that H2CO3 would remain intact in aqueous phase given the opportunity for the concerted mechanism to take place. In other words, no calculation was carried out starting from HCO3– and going through the Eigen's zwitterion, and, thus, no comparison is possible. In 2014, Galib and Hanna43 investigated the mechanisms and energetics for the decomposition of H2CO3 in water using Car-Parrinello molecular dynamics (CPMD). They found that, in fact, the concerted mechanism may occur, but only in small cluster of water: up to 9 molecules in their simulations. For bigger clusters – and, thus, in bulk –, the decomposition occurs through the formation of what the authors called a solvent-separated H3O+HCO3– ion pair intermediate. Quantum chemistry calculations by Zeebe20 suggest that this second mechanism prevails from four water molecules. Four years before, Stirling and Pápai44 also had used CPMD to investigate the hydration of CO2 and conclude that H2CO3 forms via HCO3–. They have detected the formation of the zwitterion H2O+CO2– proposed by Eigen et al.,19 which dissociates to form HCO3–. In a second step, HCO3– is protonated to form H2CO3. Figure 3 shows the calculated free energy profile of the involved species.
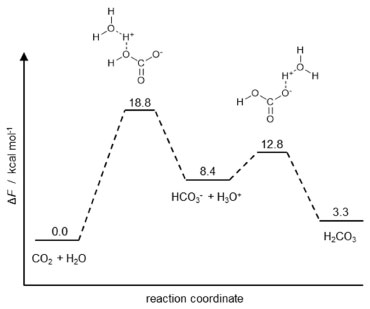 Figure 3. Calculated free energy profile according to Stirling and Pápai44
Based on the ΔF of H2CO3 and HCO3– + H+, the authors calculated the pKa as 3.7, which is in good agreement with experimental results. Although the authors did not make the same calculation for the other branch, one can calculate the other pKa as 6.2. This value is in good agreement with the so-called apparent pKa for carbonic acid, which brings us to a more appropriate model to describe the CO2/H2O system. There are actually two acids: the stronger Brønsted (or Arrhenius) acid H2CO3 (pKa ca. 3.7) and the weaker Lewis acid CO2 (pKa ca. 6.2). For both of them, the conjugate base is the same HCO3–. By its turn, the protonation of HCO3– can be seen as a simple and elegant example of thermodynamic versus kinetic reaction control. The protonation of HCO3– that results in H2CO3 is a fast reaction just like any other carboxylate protonation. However, H2CO3 is only the kinetic product, because there is a more stable product: CO2. Therefore, the preparation of H2CO3 by protonation of HCO3– at low temperature as shown previously can be seen as a typical case of kinetic control of a reaction.
CONCLUSIONS Carbon dioxide has been known and studied for centuries. It is intimately connected to the history of chemistry and its nomenclature. However, surprisingly its behavior in water remains under study and debate even today, because of the controversial species H2CO3. Initially, H2CO3 had its existence intuited at a time with very limited instrumental techniques. Later, it became fundamental for understanding the equilibria and kinetics of the CO2/H2O system. After it was finally synthesized, part of the mystery was solved, but its role in aqueous media remained controversial. Most of the time, H2CO3 is seen as a central species in the conversion of CO2(aq) to HCO3–. However, experimental data and computer simulations suggest that the conversion takes place directly without the participation of H2CO3. In fact, the calculations suggest that H2CO3 may indeed be the intermediate, but under conditions of great restriction of water molecules that could solvate a transition state containing ionic groups. However, what would this environment look like? For instance, the droplets in cloud, fog, or mist have a diameter typically between 10 to 15 µm. The smallest droplets have ca. 1 µm in diameter, which corresponds to 1010 water molecules – way above the limit of less than 10 water molecules. Even the inner side of a mitochondrion or a mycoplasma bacterium are environments with a pretty big number of water molecules. Therefore, although possible, the hydration through the formation of H2CO3 before HCO3– seems to be quite rare. It is important to emphasize that, despite the non-participation of H2CO3 in the CO2 hydration process, the species can be significant in some cases. For instance, as shown in Figure 1, for CO2 partial pressure above 3 atm, H2CO3 becomes more abundant than HCO3–. In addition, calculations suggest that at the Earth's upper mantle, H2CO3 can be the most abundant carbon species in aqueous CO2 solutions.45 Moreover, on a short time scale and without catalysis in aqueous environments, this is still the acid to be considered. The elucidation of the mechanism of reaction is an important topic in chemistry. For instance, the action mechanism of a carbonic anhydrase helps the development of drugs and the studies on CA-related diseases. Catalyzed and non-catalyzed hydration of CO2 is a relevant topic not only in biochemistry, but also in carbon capture and storage (CCS) processes. In this sense, a better understanding of the reactions and the similarity and dissimilarities between water and other nucleophiles is equally important. In pursuit of these objectives, there is room for the development of new instrumental techniques capable of helping to elucidate existing structures, as well as computer systems allowing increasingly realistic simulations. In this course, understanding of natural and biological processes, as well as technological development for CCS and other industrial processes would benefit from the new knowledge. Until now, experimental evidence and theoretical calculations are not absolutely conclusive about the intermediates or transition states involving the Eigen's zwitterion or an ion pair, but both of them eventually result in HCO3–, which position it as the central species in aqueous media. By protonation, HCO3– can form either CO2 or H2CO3, or CO32– by deprotonation. Bicarbonate is, therefore, a curious case of being the conjugate base for two different acids: H2CO3– – the elusive, but correctly named carbonic acid – and CO2, which formerly was also called carbonic acid. Despite the ambiguous nomenclature and confusions that permeated the 19th century, the evidence suggests that Lavoisier and his colleagues were not wrong after all in calling CO2 and acid.
ACKNOWLEDGMENTS This work was supported by FAPESP, Brazil (grant 2017/13137-5) and CNPq, Brazil (fellowship 304415/2013-8).
REFERENCES 1. Moore, M. H.; Khanna, R. K.; Spectrochim. Acta, Part A 1991, 47, 255 [Crossref]; Hage, W.; Hallbrucker, A.; Mayer, E.; J. Chem. Soc., Faraday Trans. 1995, 91, 2823. [Crossref] 2. Borvon, G.; Histoire du Carbone et du CO2, 1st ed.; Vuibert: Paris, 2013. 3. Delametherie, J. C.; Philos. Mag.(1798-1977) 1814, 44, 25. [Crossref] 4. Laurent, A.; Chemical Method, Notation, Classification, & Nomenclature; Cavendish Society: London, 1855. [Link] accessed in March 2024 5. Gore, G.; Tyndall, J.; Philos. Trans. R. Soc. London 1861, 151, 83. [Crossref] 6. Arrhenius, S.; Philos. Mag. (1798-1977) 1896, 41, 237. [Crossref] 7. Júnior Tolman, C. F.; The Journal of Geology 1899, 7, 585. [Crossref] 8. Bunsen, R.; Gasometry: Comprising the Leading Physical and Chemical Properties of Gases; Walton and Maberly: London, 1857. 9. Jones, H. C.; The Modern Theory of Solution; Harper and Brothers: New York, 1899. 10. Galinos, A. G.; Carotti, A. A.; J. Am. Chem. Soc. 1961, 83, 752. [Crossref] 11. Gattow, G.; Gerwarth, U.; Angew. Chem., Int. Ed. 1965, 4, 149 [Crossref]; Gattow, G.; Gerwarth, U.; Z. Anorg. Allg. Chem. 1968, 357, 78. [Crossref] 12. Terlouw, J. K.; Lebrilla, C. B.; Schwarz, H.; Angew. Chem., Int. Ed. Engl. 1987, 26, 354. [Crossref] 13. Johnston, J.; J. Am. Chem. Soc. 1916, 38, 947. [Crossref] 14. McBain, J. W.; J. Chem. Soc. 1912, 101, 814. [Crossref] 15. Kern, D. M.; J. Chem. Ed. 1960, 37, 14. [Crossref] 16. Gibbons, B. H.; Edsall, J. T.; J. Biol. Chem. 1963, 238, 3502 [Link] accessed in March 2024; Ho, C.; Sturtevant, J. M.; J. Biol. Chem. 1963, 238, 3499 [Link] accessed in March 2024; Garg, L. C.; Maren, T. H.; Biochim. Biophys. Acta 1972, 261, 70 [Crossref]; Patel, R. C.; Boe, R. J.; Atkinson, G.; J. Solution Chem. 1973, 2, 357 [Crossref]; Pocker, Y.; Bjorkquist, D. W.; J. Am. Chem. Soc. 1977, 99, 6537 [Crossref]; Johnson, K. S.; Limnol. Oceanogr. 1982, 27, 849 [Crossref]; Soli, A. L.; Byrne, R. H.; Mar. Chem. 2002, 78, 65 [Crossref]; Wang, X. G.; Conway, W.; Burns, R.; McCann, N.; Maeder, M.; J. Phys. Chem. A 2010, 114, 1734 [Crossref]; Pines, D.; Ditkovich, J.; Mukra, T.; Miller, Y.; Kiefer, P. M.; Daschakraborty, S.; Hynes, J. T.; Pines, E.; J. Phys. Chem. B 2016, 120, 2440. [Crossref] 17. Van Cappellen, P.; Charlet, L.; Stumm, W.; Wersin, P.; Geochim. Cosmochim. Acta 1993, 57, 3505. [Crossref] 18. Dean, J. A.; Lange's Handbook of Chemistry, 15th ed.; McGraw-Hill: New York, 1999. 19. Eigen, M.; Kustin, K.; Maass, G.; Zeitschrift für Physikalische Chemie 1961, 30, 130. [Crossref] 20. Zeebe, R. E.; Geochim. Cosmochim. Acta 2014, 139, 540 [Crossref]; Yumol, L. M.; Uchikawa, J.; Zeebe, R. E.; Geochim. Cosmochim. Acta 2020, 279, 189 [Crossref]; Sade, Z.; Halevy, I.; Geochim. Cosmochim. Acta 2017, 214, 246. [Crossref] 21. Adamczyk, K.; Premont-Schwarz, M.; Pines, D.; Pines, E.; Nibbering, E. T. J.; Science 2009, 326, 1690. [Crossref] 22. Buytendyk, F. J. J.; Brinkman, R.; Mook, H. W.; Biochem. J. 1927, 21, 576. [Crossref] 23. Miller, W. A.; Elements of Chemistry: Theoretical and Practical, 4th ed.; John W. Parker: London, 1860. 24. Meldrum, N. U.; Roughton, F. J. W.; The Journal of Physiology 1933, 80, 113. [Crossref] 25. Davenport, H. W.; Ann. N. Y. Acad. Sci. 1984, 429, 4. [Crossref] 26. Lindskog, S.; Silverman, D. N. In The Carbonic Anhydrases: New Horizons; Chegwidden, W. R.; Carter, N. D.; Edwards, Y. H.; Birkhäuser Basel: Basel, 2000, p. 175-195 [Crossref]; Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M.; Chem. Rev. 2008, 108, 946. [Crossref] 27. Tripp, B. C.; Smith, K.; Ferry, J. G.; J. Biol. Chem. 2001, 276, 48615. [Crossref] 28. Stirling, A.; J. Phys. Chem. B 2011, 115, 14683. [Crossref] 29. Linthwaite, V. L.; Janus, J. M.; Brown, A. P.; Wong-Pascua, D.; O'Donoghue, A. C.; Porter, A.; Treumann, A.; Hodgson, D. R. W.; Cann, M. J.; Nat. Commun. 2018, 9, 3092. [Crossref] 30. Said, R. B.; Kolle, J. M.; Essalah, K.; Tangour, B.; Sayari, A.; ACS Omega 2020, 5, 26125. [Crossref] 31. Perrin, C. L.; Arrhenius, T.; J. Am. Chem. Soc. 1978, 100, 5249. [Crossref] 32. Habermann, J.; Monatshefte für Chemie und verwandte Teile anderer Wissenschaften 1886, 7, 529. [Crossref] 33. Hempel, W.; Seidel, J.; Ber. Dtsch. Chem. Ges. 1898, 31, 2997 [Crossref]; Allpress, C. F.; Haworth, W. N.; J. Chem. Soc., Trans. 1924, 125, 1223 [Crossref]; Faurholt, C.; Zeitschrift Fur Physikalische Chemie, Stochiometrie Und Verwandtschaftslehre 1927, 126, 72 [Crossref]; Miller, N. F.; Case, L. O.; J. Am. Chem. Soc. 1935, 57, 810. [Crossref] 34. Cieslarova, Z.; dos Santos, V. B.; do Lago, C. L.; Int. J. Greenhouse Gas Control 2018, 76, 142 [Crossref]; do Lago, C. L.; Vidal, D. T. R.; Rossi, M. R.; Hotta, G. M.; da Costa, E. T.; Electrophoresis 2012, 33, 2102 [Crossref]; Vidal, D. T. R.; Nogueira, T.; Saito, R. M.; do Lago, C. L.; Electrophoresis 2011, 32, 850 [Crossref]; Vidal, D. T. R.; Santana, M. A.; Hotta, G. M.; Godoi, M. N.; Eberlin, M. N.; do Lago, C. L.; RSC Adv. 2013, 3, 18886. [Crossref] 35. Rossi, M. R.; Vidal, D. T. R.; do Lago, C. L.; Food Chem. 2012, 133, 352. [Crossref] 36. Gattow, G.; Behrendt, W.; Angew. Chem., Int. Ed. 1972, 11, 534. [Crossref] 37. Hage, W.; Hallbrucker, A.; Mayer, E.; J. Am. Chem. Soc. 1993, 115, 8427. [Crossref] 38. Kock, E. M.; Bernard, J.; Podewitz, M.; Dinu, D. F.; Huber, R. G.; Liedl, K. R.; Grothe, H.; Bertel, E.; Schlogl, R.; Loerting, T.; Chem. - Eur. J. 2020, 26, 285. [Crossref] 39. Pocker, Y.; Davison, B. L.; Deits, T. L.; J. Am. Chem. Soc. 1978, 100, 3564. [Crossref] 40. Loerting, T.; Tautermann, C.; Kroemer, R. T.; Kohl, I.; Hallbrucker, A.; Mayer, E.; Liedl, K. R.; Angew. Chem., Int. Ed. 2000, 39, 892. [Crossref] 41. Kumar, M.; Busch, D. H.; Subramaniam, B.; Thompson, W. H.; J. Phys. Chem. A 2014, 118, 5020. [Crossref] 42. de Marothy, S. A.; Int. J. Quantum Chem. 2013, 113, 2306. [Crossref] 43. Galib, M.; Hanna, G.; J. Phys. Chem. B 2014, 118, 5983. [Crossref] 44. Stirling, A.; Pápai, I.; J. Phys. Chem. B 2010, 114, 16854. [Crossref] 45. Stolte, N.; Pan, D.; J. Phys. Chem. Lett. 2019, 10, 5135. [Crossref] |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access