
 |
 |
 |
 |
 |
Artigo
| Two novel coumarin compounds: synthesis, in vitro antibacterial anticancer, and in silico docking and molecular dynamics |
|
Le Thi Thanh TrucI; Nguyen Ngoc Huyen TranI; Nguyen Van ThoiI; Nguyen Van HueII; Nguyen Thi Hong AnhIII,* I. Faculty of Chemical Engineering, Industrial University of Ho Chi Minh City, 71456 Ho Chi Minh City, Vietnam Received: 04/20/2024 Coumarin derivatives possess diverse biological activities, encompassing antibacterial, antifungal, anticancer, antidiabetic, anti-inflammatory, and antioxidant effects. This study concentrates on the synthesis of two new coumarins: the first derived from rhizomes of Luvunga scandens and the second from 7-(diethylamino)-4-methyl-2H-chromen-2-one via bromination facilitated by NBS (N-bromosuccinimide) reagent. Their antibacterial and anticancer properties were evaluated in vitro, alongside in silico docking targeting the chromen-2-one group. Bromination of compound 1 yielded novel compound 2, while compound 4 was efficiently synthesized from 7-(diethylamino)-4-methyl-2H-chromen-2-one (3) using NBS. Testing on lung, breast, and liver cancer cell lines indicated that compound 2 effectively inhibited Staphylococcus aureus and Bacillus cereus at concentrations of 13.20 and 6.6 µM, respectively. It also demonstrated significant inhibition against MCF-7 breast cancer cells with a half-maximal inhibitory concentration (IC50) value of 50.37 ± 3.70 µM. Molecular docking revealed compound 2's interaction with 2VF5 enzyme, elucidating its bacterial inhibition mechanism, with Gibbs energy and inhibition constant values of -7.42 kcal mol-1 and 3.62 µM, respectively. Additionally, compound 2 exhibited anticancer activity via alignment with topoisomerase 1 (3TOP): 1T8I, supported by molecular dynamics simulations illustrating strong ligand interactions. Analysis of pharmacophore kinetics highlighted the complex's hydrophilic nature, indicating its potential therapeutic efficacy. INTRODUCTION The natural compound coumarin (2H-1-benzopyran-2-one) exhibits a wide range of biological activities including anti-inflammatory, anticoagulant, antibacterial, antifungal, antiviral, anticancer, antihypertensive, anti-tuberculous, antispasmodic, anti-fat, anti-hyperglycemic, antioxidant, and neuroprotective properties.1,2 Notable strategies for synthesizing coumarin derivatives like furano and pyranocoumarin have been documented. These derivatives have shown anti-cancer properties and are produced from cinnamic acid-based starting materials.3 Six coumarins and limonoids were identified and characterized for the first time in the rhizomes of Luvunga scandens (Roxb.).4 The extracts and bioactive compounds from the P. genus have potential for ginseng-based anti-cancer immunotherapeutic research.5 Coumarin is a heterocyclic molecule that features a benzopyrone structure, which consists of a benzene ring fused to a pyrone ring. The substance was discovered in 1820 after being obtained from the Coumarouna odorata Aubl. tree (Dipteryx odorata). The plant possesses various pharmacological properties beneficial for medicinal purposes, such as antihypertensive, antitubercular, anticonvulsant, antiadipogenic, antihyperglycemic, antioxidant, and neuroprotective effects. The pharmacological potential and molecular mechanisms of action of coumarin derivatives against viral illnesses have been studied by Li et al.6 and Xu et al.7 Recent advancements in coumarin derivatives have been documented regarding their anticancer properties and the development of a quantitative structure-activity relationship (QSAR) model,8-11 by Rawat and Reddy12 as well as the innovative approach to designing potential anticancer drugs using the heterocycle coumarin skeleton by Dorababu.13 Valproic acid and coumarin-3-carboxylic acid boost the decrease in the growth and movement of very aggressive lung cancer cells, along with their biological mechanisms of action.14 Coumarin compounds, both natural and synthesized, have been found to demonstrate interactions with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) proteins and have been investigated for their pharmacokinetics.15 Coumarin-derived compounds exhibited anti-inflammatory properties16 and the antioxidant effects of 4-methyl coumarin were demonstrated.17 Carbazole alkaloids and coumarins found in Claussen plants have shown anticancer properties by causing cell cycle arrest, inducing apoptosis, and enhancing immune responses.18 Chalcone-coumarin derivatives have been recognized as promising anti-cancer drugs with demonstrated efficacy in both laboratory and living organism settings. They exhibit potent inhibition of human breast cancer cell lines, particularly those derived from the coumarin skeleton. Osthole, a coumarin derivative found in the fruit plant Cnidium monnieri, has demonstrated anti-breast cancer properties in women.19,20 Coumarin-derived natural products have demonstrated antihypertensive effects.21 Coumarin and its derivatives have been synthesized and shown to possess a wide range of biologically inhibitory properties, such as antibacterial activity. Structure-activity relationship (SAR) research is utilized to design coumarin derivatives with antibacterial properties by examining specific molecular structures. Xu et al.7 investigated the potential of coumarin derivatives at C-3 and C-4 positions for creating new antibacterial drugs. The antibacterial activity of coumarin derivatives is investigated through SAR analysis.22,23 Various studies22,24,25-29 have shown that 4-hydroxy-7-hydroxy- and 3-carboxycoumarin derivatives, coumarin-imidazole framework, coumarin derivatives synthesized from 8-amino-4,7-dihydroxy-chromen-2-one, coumarin based on thiazole derivatives, and 8-ethoxycoumarin derivatives exhibit significant antibacterial activity. Certain coumarin derivatives have shown antifungal properties.30 Various green synthesis methods have been utilized to produce coumarin derivatives, such as microwave-assisted synthesis of coumarin-thiazolidinone hybrids with docking studies, microwave and montmorillonite K10 catalysis, microwave synthesis of 3-aryl-furo[3,2-c] coumarins with antimicrobial screening, solid acid catalyst-assisted microwave synthesis of coumarin derivatives, TiCl4 catalyst in solvent-free conditions for coumarin derivatives, high-yield anti-cancer products of 3-aminohexahydrocoumarin derivatives under microwave conditions, and synthesis of coumarin via Pechmann reaction using ZrCl4 solid catalyst in solvent-free conditions. The coumarin synthesis in a solvent-free environment using Pechmann and Knoevenagel condensation reactions had been reported by Sugino and Tanaka.31 Usman et al.32 synthesized copper(II) combination of coumarin with attached imidazole as chemotherapeutic drugs for lung cancer: molecular insights using DFT-based vibrational analysis. Aazam et al.33 synthesized a novel series of coumarin-silver complex to evaluate antibacterial activity in vitro. Creaven et al.34 synthesized Cu and Ni-coumarin complex to study antioxidant activity. Patel et al.35 studied the complexes of 3-(pyridin-2-yl) coumarins and CuII against human cancer cell lines in vitro. Pivetta et al.36 synthesized CuII, NiII, CoII, and coumarin complexes to test cytotoxicity activity in vivo. Recent QSAR studies8-11 utilize computational chemistry solutions effectively to predict drug activity, design drugs, and explain biological activities. Several important software tools include Autodock tools.37-42 Mechanized docking is achieved through the utilization of various algorithms such as the Lamarckian genetic algorithm, free energy empirical combination function, Monte Carlo simulation, annealing, traditional genetic algorithm, and Lamarckian genetic algorithm as described by Morris et al.43 The enzymatic active site was identified in a publication by Khan et al.44 in 2020. Docking calculations were conducted using Discovery Studio Visualizer (DSC),45,46 Molegro Molecule Viewer (MMV),47 and PyMOL software48 by Van Vo et al.48 The accuracy of the static docking model for ligand/pose and enzyme/protein interactions is evaluated using the root-mean-square deviation (RMSD) value.40,49,50 If the RMSD value exceeds 2 Å, the interaction between the best docking pose of active compound and one crystal structure of the active enzyme: PDB must be simulated using molecular dynamics (MD) modeling for a duration of 0 to 100 ns. This was achieved through software such as Gromac,51,52 Desmond,53,54 or other relevant tools cited in the literature. Pharmaceutical experts have focused on in silico modeling of absorption, distribution, metabolism, excretion, and toxicity for the rational design of drugs. Several predictive models in the field of ADMET (absorption, distribution, metabolism, excretion, toxicity) have been developed by researchers.55-57 The scientific community thus begins to use in silico models as a possible alternative, developing a large number of models and strategies capable of predicting the toxicological properties of several chemical compounds.58 Recently some useful green chemistry methods was applied to synthesize high yield of heterocyclic derivatives.59,60 This study identified a new coumarin compound from the root of rhizomes of Luvunga scandens, created a new bromocoumarin using N-bromosuccinimide (NBS), synthesized bromocoumarin from 7-(diethylamino)-4-methyl coumarin, assessed antimicrobial and cytotoxic activities, and performed docking studies and molecular dynamics (MD) using Desmond software54 on a Linux platform.
EXPERIMENTAL Materials The root rhizomes of Luvunga scandens are harvested in Ninh Hoa ward, Khanh Hoa province, Vietnam in 2023. The chemical reagents such as 7-(diethylamino)-4-methyl coumarin and NBS are supplied from Merck, Germany and Sigma-Aldrich, South Korea, respectively. TLC (thin-layer chromatography), normal phase (230-400 mesh and silica gel for the chromatography column (89943-1KG-F) are bought from Sigma-Aldrich company (South Korea). The analytical TLC was done on silica gel plates (Kieselgel 60 F254, Merck), Singapore. The HR-ESI-MS mass spectra was carried out using a Bruker MicroTOF-QII spectrometer (Germany). The Bruker AM500 FT-NMR was used to record 1H and 13C NMR (nuclear magnetic resonance) spectra, as well as DEPT (distortionless enhancement by polarization transfer), HSQC (heteronuclear single quantum correlation), and HMBC (heteronuclear multiple bond correlation). The FTIR (Fourier transform infrared) spectra were measured with Bruker-German equipment. The software used were AutoDockTools version 1.5.6rc3 (1999-2011),37-42 Discovery Studio Visualizer v21.1.0.20298, copyright 2020,45,46 Molegro Molecular Viewer (MMW), 2012.2.5.0-2012-10-10,47 Desmond in Linux operating system, version 2.0-2018,54 and ChemDraw 12.0.2.1076.61 Methods Extraction and isolation A 1.92 kg of root powder sample had been extracted by Soxhlet method using chloroform solvent, then filtered and dried with Na2SO4. The liquid part had evaporated under vacuum to yield the crude solid of 48 g (2.5%/root). Then, 2.5 g of crude solid have been loaded into the traditional chromatography column with silica gel being stationary phase and mobile phase that is ethyl acetate (EA):n-hexane (He) (2:8) and yield 1.7 g (68%/crude solid) of pure coumarin derivative (1) (Figure 1).

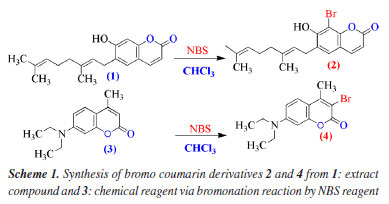
Synthesis compounds One gram of pure coumarin (1) in 10 mL of CHCl3 solvent have been poured into one small 3 neck glass round bottom of 25 mL volumetric then it was added 0.597 g of NBS. Reaction was kept at RT (room temperature) in about 1 h and monitored by TLC with eluent system (EA:He, 4:6). After completing reaction, liquid part has been concentrated in rotary vacuum evaporator until forming crude solid. The crude bromo coumarin is yielded 0.531 g (41.9%). Crude solid has been separated by column chromatography with starting eluent system of EA:He, 9:1, until collecting pure bromo coumarin (2) (Figure 2). Compound 4: 10 g (0.04 mol) of 7-(diethylamino)-4-methyl coumarin (3) has been dissolved in 30 mL of CHCl3 using a magnetic stirrer, then it was added 7.72 g (0.04 mol) NBS in 25 mL CHCl3, and kept reactions at RT by stirring. The reaction was monitored by TLC every 30 min. After completing reaction, 1.5 h, liquid part has been collected in rotary vacuum evaporator until forming crude solid. The solid has been continuously to separate by column chromatography with eluent solvent of EA:He (2:10) (Figure 3 and Scheme 1).


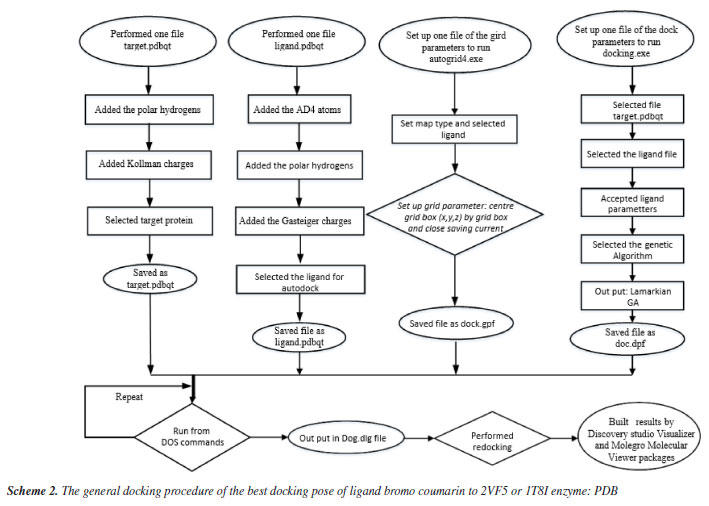
Antimicrobial activity assays in vitro The antibacterial activity of bromocoumarin derivatives was evaluated by disk diffusion method against Staphylococcus aureus (SA) and Bacillus cereus (BC) grown in an incubator at 37 ºC for 24 h. Medium containing solid Muller-Hinton agar (MHA) was prepared for bacterial development. Wells of 6 mm diameter were made in MHA gel disks. It was used a sterile cotton swab to spread each strain evenly onto each plate. The bromo coumarin derivatives have been prepared in concentrations of 5.0, 2.5, 1.25, 0.625, 0.3125 mg mL-1 and gentamicin at a concentration of 1 mg mL-1 for testing antimicrobial activity. Using a sterile micropipette, it was added 30 μL of test compound sample to each well. After 24 h of incubation at 25 ºC, it was used a meter ruler to measure the different levels of inhibition zones. Cytotoxicity Cytotoxicity of bromo coumarin was conducted based on MTT (methyl thiazolyl tetrazolium) assay and followed Shi et al.58 without minor change. The cytotoxicity of the tested compound 2 to MCF-7 cells was assayed using the MTT method. Briefly, MCF-7 cells were collected and seeded in 96-well plates at a density of 4.5 × 105 cells mL-1. After incubation in a humidified atmosphere of 5% CO2 at 37 ºC for 24 h, cells were exposed to fresh medium containing three concentrations (0.1, 1 and 10 µg mL-1) of tested compounds at 37 ºC and allowed to culture for another 24 h. An aliquot of 20 mL MTT tetrazolium salt dissolved in Hank's balanced salt solution at a final concentration of 5 mg mL-1 was added to each well and incubated in the CO2 incubator for 4 h. Finally, the medium was aspirated from each well and 150 mL DMSO (dimethyl sulfoxide) was added to dissolve the formazan crystals. The absorbance of each well was obtained using a Dynatech MR5000 plate counter at wavelength of 570 nm. The viability rate was calculated according to the following formula: viability rate = (OD570 sample/OD570 control) × 100%, where OD stands for optical density at 570 nm. In silico general docking procedure Following Scheme 1, we docked ligands or bromocoumarin derivatives to enzymes 2VF5: PDB and 1T8I, respectively, to understand bacterial inhibition and the reason for the ligand's high cytotoxicity in simulations. (X = 22.670, Y = 1.500, Z = 30.147) or (X = 26.577, Y = 22.697, Z = 8.113) are the active sites on 2VF5 and 1T8I, respectively. The grid parameters for the 1T8I and 2VF5 enzymes are set up in the grid file with 50 elements on the X, Y, and Z axes and 0.5 Å grid point spacing. There were 500 models that were installed.
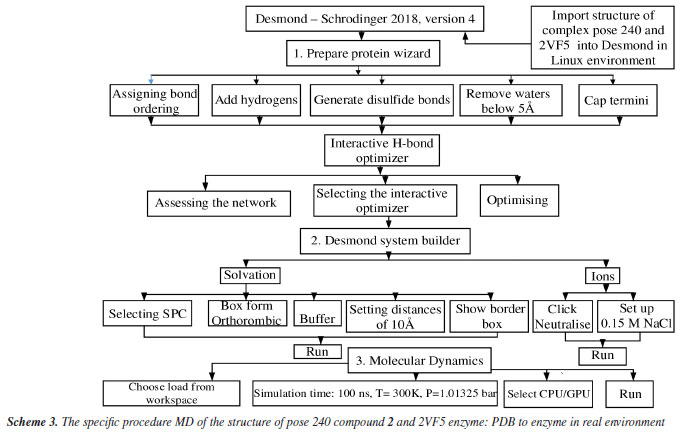
Molecular dynamic simulation The best stable complex between the best docking pose and enzyme 2VF5 in silico MD is implemented based on Scheme 3.
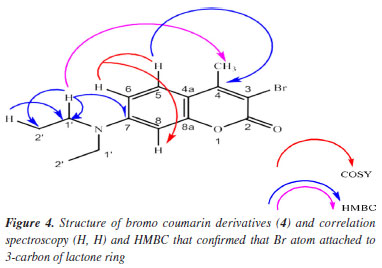
RESULTS AND DISCUSSIONS Compound 1: after extraction and isolation, coumarin is ostruthin (1). The bromination of ostruthin with NBS formed the bromo ostruthin (2) compound in yield of 41.9%. The physical chemistry of 1 was published in our previous article62 and indicated in Table S1 (Supplementary Material); compound 2: (E)-8-bromo-6-(3,7-dimethylocta-2,6-dien-1-yl)-7-hydroxy-2H-chromen-2-one (2): light yellow powder; IR (λmax, cm-1, KBr): 3337 (stretching vibration of OH, phenol), 1722 (stretching vibration of double bond of C=O), 1610 (stretching vibration of C=C double bond, alkene), 579 (stretching vibration of C-Br); 1H, 13C, HSQC, and HMBC, HRMS and FTIR have been indicated in Supplementary Material from Figures 3S to 5S, 7S and 9S: 1H NMR (500 MHz, D6, acetone): d (ppm) 8.89 (s, 1H, OH phenolic), 7.85 (d, J 9.5 Hz, 1H, H-4), 7.38 (s, 1H, H-5), 6.23 (d, J 9.5 Hz, 1H, H-3), 5.36 (t, J 6.8 Hz, 1H, H-12), 5.10 (t, J 7.5 Hz, 1H, H-16), 3.42 (d, J 7.0 Hz, 2H, CH2, H-11), 2.13-2.09 (m, 2H, H-14), 2.06-2.03 (m, 2H, H-15), 1.71 (s, 3H, H-19), 1.63 (s, 3H, H-18), 1.57 (s, 3H, H-20); 13C NMR (125 MHz, D6, acetone): d (ppm) 160.5 (C=O, C-2), 113.6 (C-3), 144.7 (C-4); 128.1 (C-5), 127.4 (C-6), 156.1 (C- 7), 98.5 (C-8), 151.5 (C-9), 113.7 (C-10), 29.1 (C-11), 122.0 (C-12), 137.9 (C-13), 40.4 (C-14), 27.2 (C-15), 125.0 (C-16), 131.8 (C-17), 25.8 (C-18), 16.2 (C-19), 17.7 (C-20); HRMS: theory calculation: [M + H]+ = 377.0752, experiment value: [M + H]+ = 377.0758. All experimental data have proved that it was exactly structure 2. Compound 4: 3-bromo-7-(diethylamino)-4-methyl-2H-chrome-2-one: yellow powder; as indicated in Figures 10S to 15S. The IR, 1H, 13C, DEPT, COSY (H,H), HSQC, HMBC, and HRMS confirmed the structure of bromo coumarin (4) and proved that bromo atom is attached to C-3 carbon of lactone ring of 4 via nucleophilic reaction of nucleophile bromo atom as followed data spectroscopy: IR (νmax, cm-1) 2957 (CH3), 1716, 1621, 1577, 1517 (stretching vibration of C=C double bond), 1410, 1348, 1267, 1213, 1075 (stretching vibration of C-O/C-N single bond), 830, 805 (stretching vibration of aromatic ring bearing substitution group), 744 (stretching vibration of C-Br single bond); 1H NMR (500 MHz, chloroform, D1, TMS): d (ppm) 1.20 (t, J 7.3 Hz, 6H, 2CH2-CH3), 2.51 (s, CH3), 3.40 (q, 4H, J 7.2 Hz, 2CH2-CH3), 6.47 (d, J 2.0 Hz, H8-Ar), 6.60 (dd, J1 9.3, J2 2.3 Hz, H6-Ar), 7.40 (d, J 9.0 Hz, H5-Ar). As shown in Figure 4, the COSY (H,H)-red lines demonstrates that H-5 proton couples with proton H-6 then coupling with H-8 proton, confirming interactions on 1H NMR and protons matching signals that are consistent with the structure of compound 4; 13C NMR (125 MHz, chloroform-d1, TMS), DEPT, and HSQC: d (ppm) 12.4 (CH3, CH3-CH2-N), 19.1 (CH3, CH3-alkenyl), 44.9 (CH2), 97.4 (CH), 105.7 (C-Br), 109.13 (CH), 109.06 (C=C), 126.1 (CH), 150.6 (C-N), 151.5 (C-CH3), 154.5 (C=C), 158.1 (C=O). As shown in Figure 4, Figures 13S and 14S exposed H, C correlation via HMBC (blue and pink colors) of compound 4: H-5 proton is correlated to C-4 and H-1' proton to C-7 and C-2'; HRMS: experimental value of [M + H]+ = 310.0343 and theoretical value of [M + H]+ = 310.0443, respectively, as shown in Figure 15S. All spectroscopy data such as IR, 1H, 13C, DEPT, COSY, HMBC, and HMBC proved that bromo atom is bonded to C-3 as seen in Figure 4.
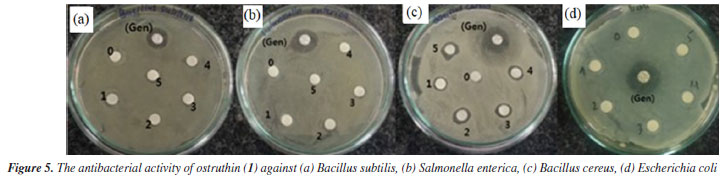
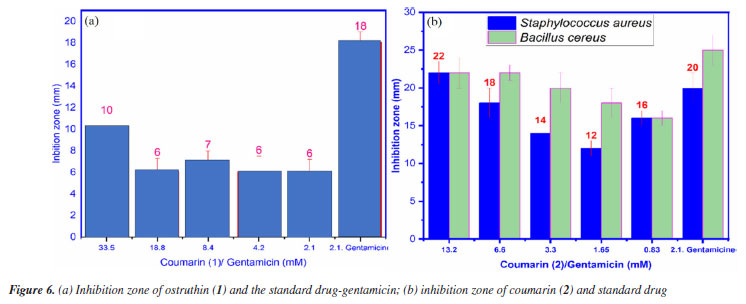
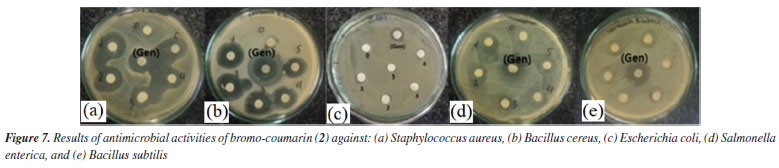
Antimicrobial activity Compound 1, also known as ostruthin, was tested for antimicrobial activity against Bacillus subtilis, Salmonella enterica, Bacillus cereus, and Escherichia coli, as depicted in Figures 5a-5d. Ostruthin (1) does not exhibit any antibacterial activity based on the results. Gentamicin exhibited significant activity at a concentration of 2.1 µM, resulting in an inhibition zone diameter of 18 mm, as illustrated in Figure 6a. The diameter of the inhibition zone of ostruthin (1) was measured at concentrations of 33.5, 18.8, 8.4, 4.2, and 2.1 mm, resulting in values of 10, 6, 7, 6, and 6 mm, respectively. These values correspond to the labels 0, 1, 2, 3, and 4, as depicted in Figure 6a. Figure 7 displays the bromination of ostruthin (1) to yield bromo ostruthin (2), as seen in Scheme 1. Compound 2 was tested for antimicrobial activity against Staphylococcus aureus, Bacillus cereus, Escherichia coli, Salmonella enterica, and Bacillus subtilis. Compound 2 demonstrated the excellent inhibition against Staphylococcus aureus (SA) and Bacillus cereus (BC) but only moderate inhibition against Salmonella enterica. It did not show any inhibition against Escherichia coli and Bacillus subtilis. Compound 2 displayed strong antimicrobial activity against both SA and BC bacteria, with larger inhibition zones observed for BC bacteria compared to SA, as shown in Figure 6b. The compound effectively inhibited BC bacteria at a concentration of 6.6 mM and SA bacteria at a concentration of 13.2 mM.
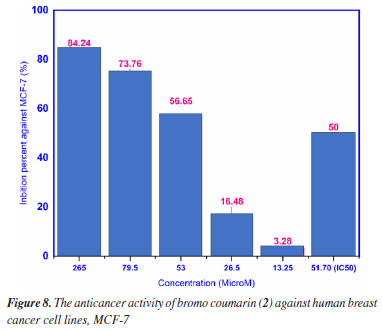
Anticancer activity The cytotoxicity of compound 2 against human breast cancer cell line had performed by MTT method as shown in Figure 8. As seen in Figure 8, compound 2 had demonstrated the potential ability of cytotoxicity against human breast cancer cell line (MCF-7) by the value of half-maximal inhibitory concentration (IC50) has been calculated 51.70 mM that had compared to drug camptothecin with value of 3.28 µM.
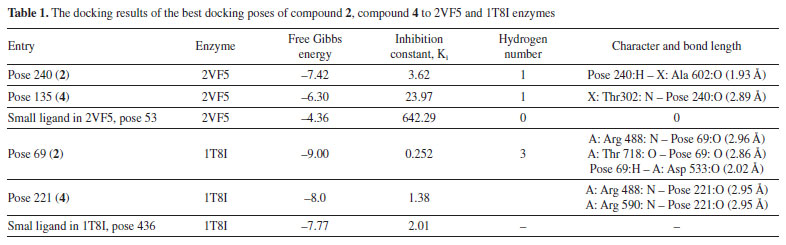
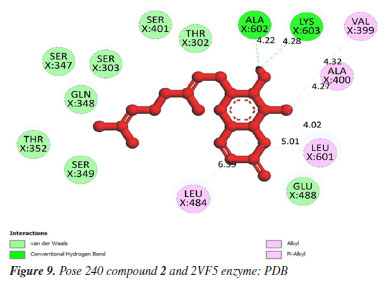
In silico docking and MD simulations Docking study Anti-bacteria against 2VF5 enzyme: PDB Compounds 2 and 4 exhibited a strong interaction with the 2VF5: PDB enzyme, leading to potent effects against SA and BC in vitro. The utilization of this enzyme is elucidated according to the general mechanism outlined by Mai et al.45 and Ece.53 Pose 240 (compound 2): pose 240 is the best docking pose among 500 docking conformations. It was docked to the active center of the 2VF5 enzyme, resulting in a free Gibbs energy value of -7.42 kcal mol-1 and an inhibition constant of 3.62 µM, as shown in Table 1. The value of estimated free energy of binding (ΔGº) is calculated based on AutoDock software:37-42 final intermolecular energy (1), final total internal energy (2), torsional free energy (3), and unbound system's energy (4), ΔGº (kcal mol-1) = (1) + (2) + (3) - (4). The inhibition constant is calculated based on the formula, . The number of hydrogen bondings are determined based on the signification interaction model by Discovery Studio visualizer software.45,46 Pose 240 exhibited a hydrogen bond between the hydrogen atom of hydroxyl phenolic and the oxygen atom on Ala 602 on 2VF5, as indicated in Table 1. The hydrogen bond is strong due to its short bond length. Figure 9 illustrates the significant ligand interaction model on a 2D diagram, showing that pose 240 has strong interactions with 2VF5 enzymes as three parts of this pose interact effectively.45 Pose 240's functional group forms two hydrogen bonds with the hydrogen atom of hydroxyl phenolic to Ala 602 and Lys 603 in the X chain. The capping group of pose 240 is determined by alkyl or pi-alkyl interactions from Leu 484 and Leu 601 to the lactone ring. The linkage of this pose is identified through alkyl or pi-alkyl interactions between Ala 400 and Leu 601 with the bromine atom of the pose. Figure 10 illustrates a ligand map displaying secondary interactions like hydrogen bonding, steric, and overlap interactions. Three hydrogen bonds are formed by Ala 602, Lys 603, and Val 399 with the hydrogen atom of hydroxyl phenolic. The residual amino acids involved in steric interactions in enzymes are Ser 303, Ser 347, Ser 349, Gln 348, Leu 484, and Glu 488. Steric interactions around pose indicate a strong interaction between pose 240 and the enzyme in overall conformational processing. Interactions that overlap are depicted as purple circles on 240 atoms. Increased diameter of the purple circle results in intensified overlapping interaction.
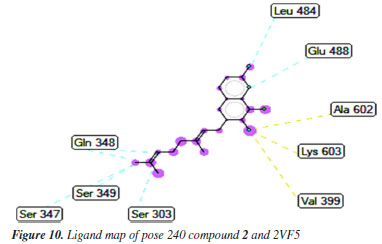
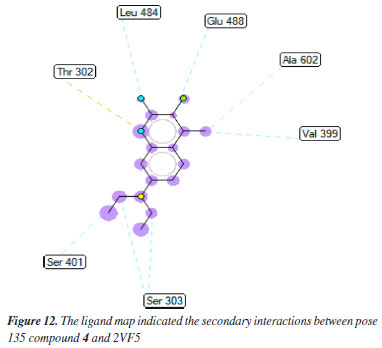
Pose 135 (compound 4): the best docking pose of compound 4 is docked to 2VF5 enzyme with the values of values of ΔGº and Ki of -6.30 and 23.97 µM, respectively. This pose involved one hydrogen bonding interaction between the oxygen atom of a lactone group and the N atom of Thr 302 amino acid, with a bond length of 2.89 Å, as illustrated in Table 1 and Figure 11. As seen in Figure 11, pose 135 interacted with 2VF5 via one hydrogen bond between the oxygen atom of the lactone group and Thr 302, which is the functional group of this pose. The capping group of this pose is identified through a pi-anion interaction or non-polar interaction between Glu 488 and the lactone ring, as well as a carbon-hydrogen interaction (nonpolar interaction) between Gly 301 and the oxygen atom of the C=O double bond. The pose linker is determined by an alkyl interaction between Ala 404 and the carbon atom of the methyl group in the ethyl chain. This demonstrated that pose 135 exhibited good interaction with enzymes. Figure 12 shows a ligand map revealing a secondary interaction between pose 135 and 2VF5. It highlights that pose 135 does not interact effectively with 2VF5 due to the limited secondary interactions in that area. Pose 240 exhibited a stronger interaction with 2VF5 compared to pose 135 in the ligand interaction model, as indicated by the free Gibbs energy values.
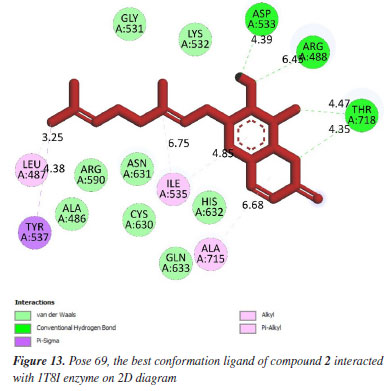
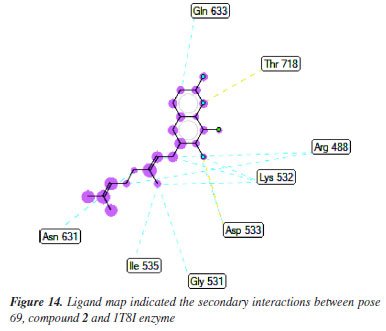
Anti-cancer against 1T8I enzyme: PDB in silico Pose 69 (compound 2): the 1T8I: PDB enzyme was chosen to demonstrate the potential inhibitory effects of the compound in vitro through a general mechanism.63 The optimal docking pose for compound 2, pose 69 that anchored to the active site of 1T8I:PDB with the values of ΔGº and Ki of -9.00 kcal mol-1 and 0.252 µM, respectively. Pose 69 formed three hydrogen bonds to the active center on 1T8I, as shown in Table 1. As seen in Figure 13, one ligand interaction model illustrated the significant interactions between pose 69 and residual amino acid on 1T8I enzyme. This pose exhibited full interactions through three components: hydrogen bonding from Asp 533, Arg 488, and Thr 718 to specific atoms in the ligand; capping group interactions with alkyl or pi-alkyl from Ile 535 and with Ala 715; and a linker part formed by pi-sigma interaction from Tyr 537 and Leu 487. Pose 69 demonstrated effective interaction with the enzyme-ligand interaction model. Figure 14 shows a ligand map displaying secondary interactions developing around this specific pose. The steric interactions occur around pose 69 is influenced by residual amino acids.
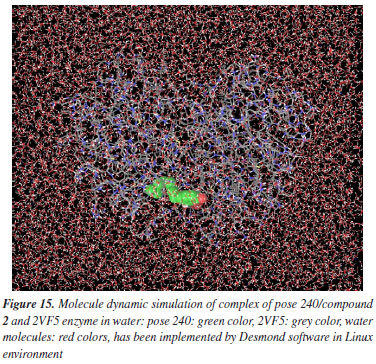
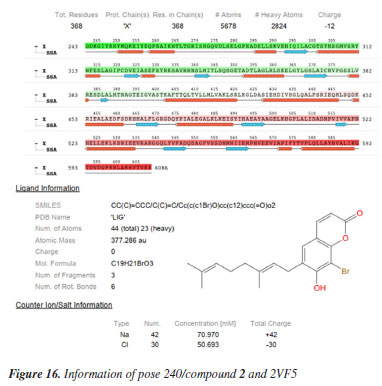
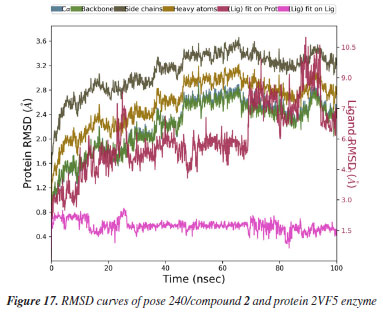
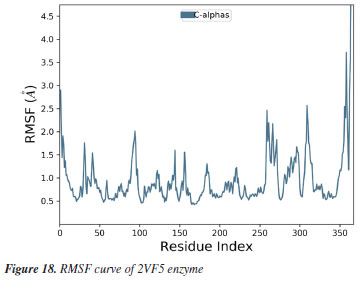
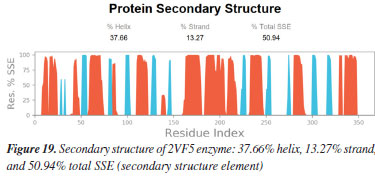
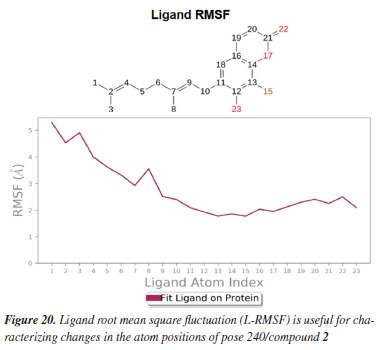
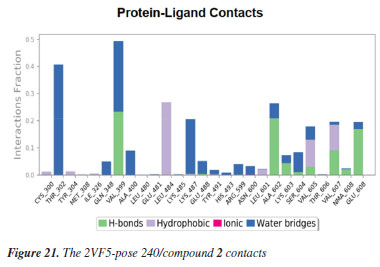
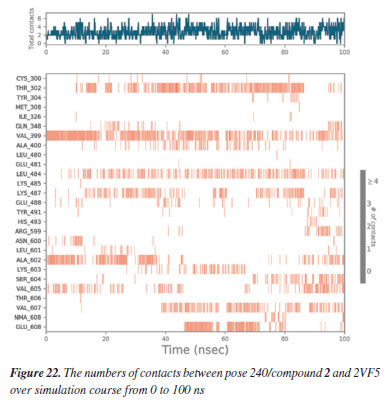
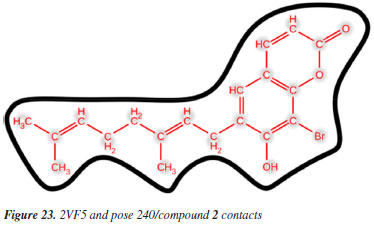
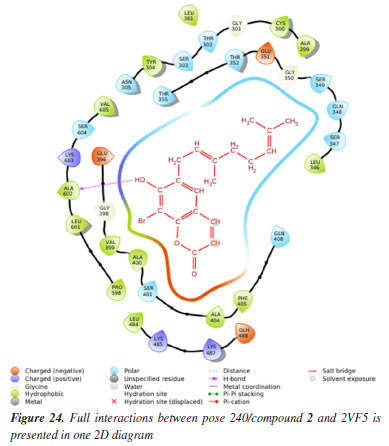
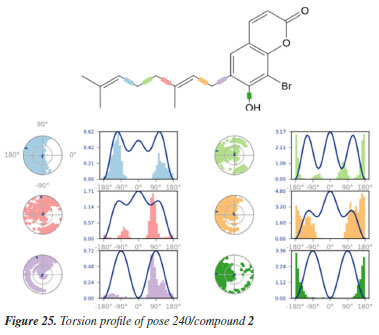
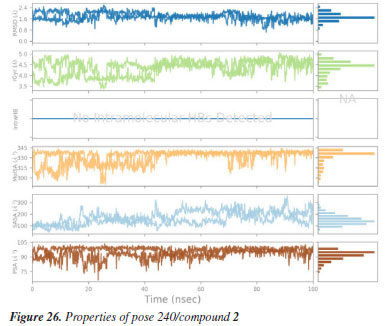
MD simulation A molecular dynamic simulation model was created to analyze the interaction between pose 240 (compound 2) and enzyme 2VF5 to explain why compound showed good activity against Staphylococcus aureus and Bacillus cereus in vitro. The significant simulation results of complex_pose 240_2VF5 are displayed in Figures 15 to 26 that control the stability of complex of pose 240 and 2VF5 throughout one simulation course from 0 to 100 ns. As seen in Figure 15, the conformation of pose 240 and the 2VF5 enzyme is illustrated and surrounded by water molecules. Figure 16 displays the details of enzyme 2VF5, pose 240, and the concentration of NaCl. The enzyme consists of 368 residues, 5678 atoms, 2824 heavy atoms, and a -12 charge. Pose 240, comprises of 44 atoms, 23 heavy atoms, 0 charge, and 6 rotational bonds. Sodium chloride is present at concentrations of 42 and 30 mM, respectively. The total charges of sodium (Na) and chlorine (Cl) ions are +42 and -30, respectively. Figure 17 displays the RMSD values of 2VF5 (left Y-axis) and pose 240 (right Y-axis) plotted against simulation times ranging from 0 to 100 nm during the simulation course. The number of simulation frames from 0 to 100 ns are 1000 frames. The frames of the 2VF5 enzyme were aligned to the reference frame backbone, and the RMSD value was calculated using the selected atoms. The RMSD values of the protein backbone in Figure 17 range from 1.2 to 2.8 Å, falling within the acceptable range of 1.0-3.0 Å. They discovered a counterpart within 40 to 80 ns of simulation time. The RMSD curve for the α-carbon of the 2VF5 enzyme fluctuates between 1.0 to 3.0 Å and stabilizes after 40 to 80 ns. The side chain conformation of 2VF5 has altered the root mean square deviation (RMSD) values from 2 to 3.2 Å. The simulation demonstrated convergence, with RMDS values stabilizing at a constant level. Pose 240 has been aligned with RMSD values ranging from 0.4 to 0.8 Å, showing minimal conformational changes throughout the simulation time (pink curve below). The RMSD values of pose 240 are calculated after aligning the 2VF5-pose 240 complex with the reference protein backbone. The large atoms in pose 240 are then analyzed using the red curve. The RMSD curve for pose 240 (dark red) is lower than that of the backbone (dark green) between 0 and 70 ns, indicating that pose 240 has remained close to its initial binding site and exhibited oscillations beyond 70 to 90 ns. The peaks on Figure 18 represent the regions of the 2VF5 that experienced the highest fluctuations during the simulation. The N- and C-terminal tails of the protein have exhibited the highest degree of fluctuation compared to other regions. Alpha helices and beta strands, which are secondary structure elements, tend to be more rigid than the unstructured regions of a protein and therefore exhibit less fluctuation compared to loop regions. As depicted in Figure 19, the enzyme's secondary structure consists of 37.66% helix, 13.27% strand, and 50.54% secondary structure elements (SSE). The plot has revealed the distribution of secondary structure elements (SSE) by residue index across the protein structure. Figure 20 illustrates how the ligand root mean square fluctuation (L-RMSF) can be utilized to explain variations in the positions of the ligand atoms. It was possible to explore the interaction of ligand fragments with the 2VF5 and their entropic role in the binding process by examining the ligand RMSF. The ‘fit ligand on protein' line for the 2VF5 enzyme shows the ligand fluctuations in the lower panel. The RMSF values in pose 240 have been altered from 2 to 5 Å. The contacts between the 2VF5 enzyme and Pose 240 are shown in Figure 21 within the time range of 0 to 100 ns. Enzyme interactions with pose 240 can be monitored during the simulation and classified into three types: hydrogen bonds, hydrophobic bonds, and water bridges. The stacked bar charts are standardized throughout the orbital period. The amino acid residue Val 399 in 2VF5 engages in hydrogen bonding with water bridges, persisting for 50% of the simulation duration. Hydrogen bonds have been established between Ala 602 and the water bridge, contributing to stability for 25% of the simulation duration. Residue 484 of leucine has established hydrophobic interactions that persist throughout less than 30% of the simulation duration. The hydrogen bonds, depicted in light green, are crucial in ligand binding. Hydrogen-bonding properties play a crucial role in drug design by significantly affecting drug specificity, metabolism, and absorption. Figure 22 displays the interactions and contacts, which are summarized in Figure 21. The total number of interactions that 2VF5 had with pose 240 throughout the orbital period is displayed at the top. The bottom displayed the residues that interacted with pose 240 in every trajectory frame. Several residues, including Thr 302, Val 399, Leu 484, Lys 487, and Ala 602, are associated with multiple interactions in pose 240. In Figure 23, pose 240's atom interactions with the amino acid residues of the 2VF5 enzyme occurred for over 30.0% of the simulation time in the selected trajectory. All atoms in pose 240 have engaged with water molecules, represented by the grey color. Figure 24 displays the complete interaction system between pose 240 and the 2VF5 enzyme in an aqueous environment. The pharmacokinetics display a single boundary delineating areas around position 240 as polar (light blue), positively charged (violet), hydrophobic (light green), and negatively charged. A hydrogen bond is created between Ala 602 and the hydrogen atom of hydroxyl phenolic on pose 240. It interacted with Ala 602 through a water bridge, maintaining stable interactions for approximately 30% of the simulation time, as shown in Figure 24. Additionally, it had multiple interactions with Ala 602, as depicted in Figure 21. The enzyme's residual amino acids involved in polar interactions are Ser 347, Gln 348, Thr 352, Thr 355, Gln 408, Ser 401, Thr 302, Ser 303, Asn 305, and Ser 604. The amino acids comprising 2VF5 are Leu 346, Gly 398, Val 399, Ala 400, Ala 404, Phe 405, Ala 299, Cys 300, Tyr 301, Val 605, Ala 602, Leu 601, Pro 598, and Leu 488. The amino acids in 2VF5 include positively charged residues like Lys 487, Lys 485, and Lys 603, as well as negatively charged residues like Glu 488, Glu 396, and Glu 351. Pose 240's pharmacophores are characterized by being more hydrophilic (light blue), less hydrophobic, negatively charged, and positively charged. Figure 25 displays the torsion profile of pose 240, consisting of six torsion bonds in various colors. Each rotatable bond torsion is accompanied by a dial plot and a bar plot of matching color. Radial charts display the evolution of torsion over the course of the simulation. The time evolution is graphed outward from the center of the radial plot, starting at the simulation's initial point. The information displayed on the dial plots is condensed in the bar plots, revealing the probability density of torsion. The plot demonstrates the potential of the rotatable bond by summing the potential of the related torsions, provided that torsional potential information is accessible. The potential values are shown in kilocalories per mole and can be found on the left Y-axis of the chart. Torsion potential relationships can provide insights into the conformational strain experienced by pose 240 to sustain an enzyme-bound conformation. Figure 26 displays the characteristics of pose 240 that includes the RMSD, radius of gyration, intermolecular hydrogen bonds, molecular surface area, solvent accessible surface area (SASA), and polar surface area vary between 1.6 and 2.0 Å2, 4.0 and 4.5 Å2, 0.315 and 340 Å2, 150 to 250 Å2, and 90 to 100 Å2.
CONCLUSIONS The novel synthetic compounds 2 and 4 via bromination reactions are performed. Compound 2 inhibited well both Staphylococcus aureus and Bacillus cereus bacteria than compound 1. The complex of pose 240 and 2VF5 indicated the strongest stability in simulation time from 0 to 100 ns. The full interaction simulation course, MD of 2VF5:PDB and compound 2 indicated the hydrophilic property of compound (2) or pose 240.
SUPPLEMENTARY MATERIAL Supplementary material is available at http://quimicanova.sbq.org.br, in the form of PDF file, with free access.
REFERENCES 1. Venugopala, K. N.; Rashmi, V.; Odhav, B.; BioMed Res. Int. 2013, article ID 963248. [Crossref] 2. Garg, S. S.; Gupta, J.; Sharma, S.; Sahu, D.; Eur. J. Pharm. Sci. 2020, 152, 105424. [Crossref] 3. Gray, A. I.; Waterman, P. G.; Phytochemistry 1978, 17, 845. [Crossref] 4. Nguyen, T. P.; Minh, N. P.; Dat, T. B.; Le, T. D.; Nguyen, M. A. T.; Pham, T. N. T.; Tri, D. M.; Nat. Prod. Res. 2017, 31, 2281. [Crossref] 5. Baptiste, S. J.; Le, T. H. Y.; Le, T. K. V.; Vu, D. N.; Nguyen, D. D.; Food Rev. Int. 2022, 38, 1461. [Crossref] 6. Li, Z.; Kong, D.; Liu, Y.; Li, M.; Genes Dis. 2022, 9, 80. [Crossref] 7. Xu, Z.; Chen, Q.; Zhang, Y.; Liang, C.; Fitoterapia 2012, 150, 104863. [Crossref] 8. Srivastav, V. K.; Tiwari, M.; Arabian J. Chem. 2017, 10, S1081. [Crossref] 9. Osman, E. A.; Abdalla, M. A.; Abdelraheem, M. O.; Ali, M. F.; Osman, S. A.; Tanir Y. M.; Abdelrahman, M.; Ibraheem, W.; Alzain, A. A.; Inf. Med. Unlocked 2021, 26, 100765. [Crossref] 10. Kasmi, R.; Hadaji, E.; Chedadi, O.; Aissouq, A.E.; Bouachrine, M.; Ouammou, A.; Heliyon 2020, 6, e04514. [Crossref] 11. Shukla, A.; Tyagi, R.; Meena, S.; Datta, D.; Srivastava, S. K.; Khan, F.; J. Biomol. Struct. Dyn. 2020, 38, 168. [Crossref] 12. Rawat, A.; Reddy, A. V. B.; Eur. J. Med. Chem. Rep. 2022, 5, 100038. [Crossref] 13. Dorababu, A.; Eur. J. Med. Chem. Rep. 2021, 2, 100006. [Crossref] 14. Liu, X.; Chen, L.; Sun, F.; Zhang, G.; Cytotechnology 2013, 65, 597. [Crossref] 15. Chidambaram, S.; El-Sheikh, M. A.; Alfarhan, A. H.; Radhakrishnan, S.; Akbar, I.; Saudi J. Biol. Sci. 2021, 28, 1100. [Crossref] 16. Lin, H.; Yang, H.; Huang, S.; Wang, F.; Wang, D.-M.; Liu, B.; Tang, Y.-D.; Zhang, C.-J.; ACS Appl. Mater. Interfaces 2018, 10, 12173. [Crossref] 17. Pedersen, J. Z.; Oliveira, C.; Incerpi, S.; Kumar, V.; Fiore, A. M.; De Vito, P.; Prasad, A. K.; Malhotra, S. V.; Parmar, V. S.; Saso, L.; J. Pharm. Pharmacol. 2007, 59, 1721. [Crossref] 18. Whang, W. K.; Park, H. S.; Ham, I.; Oh, M.; Namkoong, H.; Kim, H. K.; Hwang, D. W.; Hur, S. Y.; Kim, T. E.; Park, Y. G.; Kim, J.-R.; Kim, J. W.; Exp. Mol. Med. 2005, 37, 436. [Crossref] 19. Yang, D.; Gu, T.; Wang, T.; Tang, Q.; Ma, C.; Biosci., Biotechnol. Biochem. 2010, 74, 1430. [Crossref] 20. Batran, R. Z.; Dawood, D. H.; El-Seginy, S. A.; Ali, M. M.; Maher, T. J.; Gugnani, K. S.; Rondon-Ortiz, A. N.; Arch. Pharm. 2017, 350, 1700064. [Crossref] 21. Bai, R.-R.; Wu, X.-M.; Xu, J.-Y.; Chin. J. Nat. Med. 2015, 13, 721. [Crossref] 22. Feng, D.; Zhang, A.; Yang, Y.; Yang, P.; Arch. Pharm. 2020, 353, 1900380. [Crossref] 23. Dekić, V.; Radulović, N.; Vukicevic, R.; Dekić, B.; Stojanović-Radić, Z.; Palić, R.; Afr. J. Pharm. Pharmacol. 2011, 5, 371. [Crossref] 24. Sahoo, C. R.; Sahoo, J.; Mahapatra, M.; Lenka, D.; Sahu, P. K.; Dehury, B.; Padhy, R. N.; Paidesetty, S. K.; Arabian J. Chem. 2021, 14, 102922. [Crossref] 25. Qin, H.-L.; Zhang, Z.-W.; Ravindar, L.; Rakesh, K. P.; Eur. J. Med. Chem. 2020, 207, 112832. [Crossref] 26. Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.-X.; Eur. J. Med. Chem. 2018, 143, 958. [Crossref] 27. Lin, P.-Y.; Yeh, K.-S.; Su, C.-L.; Sheu, S.-Y.; Chen, T.; Ou, K.-L.; Lin, M.-H.; Lee, L.-W.; Molecules 2012, 17, 10846. [Crossref] 28. Mamidala, S.; Peddi, S. R.; Aravilli, R. K.; Jilloju, P. C.; Manga, V.; Vedula, R. R.; J. Mol. Struct. 2021, 1225, 129114. [Crossref] 29. Behrami, A.; Orient. J. Chem. 2014, 30, 1747. [Crossref] 30. Al-Amiery, A. A.; Kadhum, A. A. H.; Mohamad, A. B.; Molecules 2012, 17, 5713. [Crossref] 31. Sugino, T.; Tanaka, K.; Chem. Lett. 2001, 30, 110. [Crossref] 32. Usman, M.; Zaki, M.; Khan, R. A.; Alsalme, A.; Ahmad, M.; Tabassum, S.; RSC Adv. 2017, 7, 36056. [Crossref] 33. Aazam, E.; EL Husseiny, A.; Hitchcock, P.; Alshehri, J.; Cent. Eur. J. Chem. 2008, 6, 319. [Crossref] 34. Creaven, B. S.; Egan, D. A.; Kavanagh, K.; McCann, M.; Noble, A.; Thati, B.; Walsh, M.; Inorg. Chim. Acta 2006, 359, 3976. [Crossref] 35. Patel, J.; Dholariya, H.; Patel, K.; Bhatt, J.; Patel, K.; Med. Chem. Res. 2014, 23, 3714. [Crossref] 36. Pivetta, T.; Valletta, E.; Ferino, G.; Isaia, F.; Pani, A.; Vascellari, S.; Castellano, C.; Demartin, F.; Cabiddu, M. G.; Cadoni, E.; J. Inorg. Biochem. 2017, 177, 101. [Crossref] 37. El-Hachem, N.; Haibe-Kains, B.; Khalil, A.; Kobeissy, F. H.; Nemer, G. In Neuroproteomics - Methods and Protocols, 2nd ed.; Kobeissy, F. H.; Stevens Jr., S. M., eds.; Humana: New York, 2017, p. 391. [Crossref] 38. Schüttelkopf, A. W.; van Aalten, D. M. F.; Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355. [Crossref] 39. Syamsul, E. S.; Umar, S.; Wahyuni, F. S.; Martien, R.; Hamidi, D.; Maced. J. Med. Sci. 2022, 10, 1471. [Crossref] 40. Aamir, M.; Singh, V. K.; Dubey, M. K.; Meena, M.; Kashyap, S. P.; Katari, S. K.; Upadhyay, R. S.; Umamaheswari, A.; Singh, S.; Front. Pharmacol. 2018, 9, 1038. [Crossref] 41. Lam, P. C.-H.; Abagyan, R.; Totrov, M.; J. Comput.-Aided Mol. Des. 2018, 32, 187. [Crossref] 42. Kostal, J.; Adv. Mol. Toxicol. 2016, 10, 139. [Crossref] 43. Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J.; J. Comput. Chem. 1998, 19, 1639. [Crossref] 44. Khan, S. U.; Ahemad, N.; Chuah, L.-H.; Naidu, R.; Htar, T. T.; Progress in Drug Discovery & Biomedical Science 2020, 3, a0000054. [Crossref] 45. Mai, T.-C.; Tran, N.-T.; Mai, D.-T.; Mai, T. T. N.; Duyen, N. H. T.; An, T. N. M.; Alam, M.; Dang, C.-H.; Nguyen, T.-D.; RSC Adv. 2022, 12, 25962. [Crossref] 46. Nguyen, N.-H.; Vu, Y. T.; Nguyen, T.-D.; Cao, T.-T.; Nguyen, H. T.; Le, T.-K.-D.; Sichaem, J.; Mai, D.-T.; An, T. N. M.; Duong, T.-H.; RSC Adv. 2023, 13, 35408. [Crossref] 47. Nguyen, H. T.; Nguyen, T.-T.; Duong, T.-H.; Tran, N.-M.-A.; Nguyen, C. H.; Nguyen, T.-H.-A.; Sichaem, J.; Molecules 2022, 27, 2574. [Crossref] 48. Van Vo, G.; Nguyen, T.-H.-T.; Nguyen, T.-P. ; Do, T.-H.-T.; Tran, N.-M.-A.; Nguyen, H. T.; Nguyen, T. T.; Saudi Pharm. J. 2022, 30, 1301. [Crossref] 49. Bell, E. W.; Zhang, Y.; J. Cheminf. 2019, 11, 40. [Crossref] 50. Agrawal, P.; Singh, H.; Srivastava, H. K.; Singh, S.; Kishore, G.; Raghava, G. P. S.; BMC Bioinf. 2019, 19, 426. [Crossref] 51. Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A. E.; Berendsen, H. J. C.; J. Comput. Chem. 2005, 26, 1701. [Crossref] 52. Nguyen, N.-H.; Tran, N.-M.-A.; Duong, T.-H.; Vo, G. V.; RSC Adv. 2023, 13, 8190. [Crossref] 53. Ece, A.; BMC Chem. 2023, 17, 26. [Crossref] 54. D. E. Shaw Research, Desmond Tutorial, version 3.0, https://www.deshawresearch.com/downloads/download_desmond.cgi/Desmond_Tutorial-0.6.pdf, accessed in September 2024 55. Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; Zheng, M.; Jiang, H.; Q. Rev. Biophys. 2015, 48, 488. [Crossref] 56. Xiong, Y.; He, X.; Zhao, D.; Tian, T.; Hong, L.; Jiang, T.; Zeng, J.; Nucleic Acids Res. 2021, 49, W5. [Crossref] 57. Bitew, M.; Desalegn, T.; Demissie, T. B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R.; PLoS One 2021, 16, e0260853. [Crossref] 58. Shi, F.; Dai, A.-X.; Zhang, S.; Zhang, X.-H.; Tu, S.-J.; Eur. J. Med. Chem. 2011, 46, 953. [Crossref] 59. Saeeda, A.; Ejazb, S. A.; Aziz, M.; Channar, P. A.; Sumreen, L.; Ujan, R.; Wani, T. A.; Zargar, S.; Hökelek, T.; Flörke, U.; Quim. Nova 2024, 47, e-20230131. [Crossref] 60. Carlos, M. F. L. P.; Echevarria, A.; Quim. Nova 2024, 47, e-20230131. [Crossref] 61. Kimberley, R. C.; J. Am. Chem. Soc. 2011, 133, 8388. [Crossref] 62. Minh, A. T. N.; Van, C. N.; Minh, Q. N.; Dinh, T. M.; Mahboob, A.; Rev. Chim. 2020, 71, 220. [Crossref] 63. Nguyen, H.-H.; Tran, N.-M.-A.; Nguyen, T.-H.-T.; Vo, H.-C.; Nguyen, C. H.; Nguyen, T.-H.-A.; Nguyen, N.-H.; Duong, T. H.; J. Saudi Chem. Soc. 2022, 26, 101489. [Crossref]
|

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access