
 |
 |
 |
 |
 |
Artigo
| Effects of preparation method on catalytic behavior of Ni-Me (Me = Co, Nb)/γ-Al2O3 for oxidative dehydrogenation of ethane |
|
Mario Hurtado CotilloI,II; José M. López NietoII; Benjamín SolsonaIII; Rosario Sun KouIV; Gino PicassoI,* I. Technology of Materials for Environmental Remediation (TecMARA) Research Group, Faculty of Sciences, National University of Engineering, 15333 Lima, Peru Received: 08/21/2023 *e-mail: gpicasso@uni.edu.pe Catalysts based on Ni-Me (Me = Co, Nb) supported on γ-Al2O3 were prepared by the coprecipitation-chemical deposition method via reflux, and hydrothermal method for oxidative dehydrogenation of ethane. For coprecipitation method, ammonia and urea were used as precipitating agents. In both methods, the Ni content was 30 wt.%, whereas that of Me (Co, Ni) was 5 wt.%. The activity and selectivity towards ethylene appeared to depend strongly on the composition of the catalysts and, interestingly, on the preparation conditions. In the present article, it is shown that mixed hydrothermal samples using urea showed the best performance. Specifically, the hydrothermally prepared NiNb/γ-Al2O3 sample was the one that showed the highest values. The introduction of Nb to Ni framework allowed to enrich with nucleophilic oxygen sites displayed via X-ray photoelectronic spectra (XPS) which lead to the formation of low reducible and selective sites. INTRODUCTION The catalytic technology, in particular the oxidative dehydrogenation (ODH) of ethane, seems to be promising due not only to the much lower operating temperatures compared to the thermal option but also to the high selectivities achieved (70 to 90%) with conversion activities higher than 50%.1-4 The ODH process overcomes the shortcomings of thermal cracking such as thermodynamic limitations, intense energy use, coke formation and separation procedures. Despite its advantages, catalytic ODH is rarely applied on an industrial scale due to the difficulties of starting a new industrial technology to replace a well-established process, as well as the need for only a modest improvement in yield to olefins.2-6 Therefore, an important task to explore is to find appropriate catalytic systems with sufficient yield toward ethylene to scale to massive industrial demand. Particularly, an important reserve of natural gas located in the region of Cusco, in Camisea (Peru), has been exploited in the last years. The commercialization of the reserve began in 2004 until nowadays. Since this natural gas reserve is ethane-rich, as it contains circa 10% ethane in its composition, there is a growing interest in giving it an added value to produce ethylene.7,8 The innumerable derivatives from ethylene as synthetic polymers, styrene, ethylene oxide, vinyl chloride, and vinyl acetate monomers, dichloroethane, ethylbenzene, acetaldehyde, ethane, among others, would allow to increase the economics of the country. The aim is to find a suitable catalyst with a selectivity towards ethylene > 70% that justifies a long-term investment. Despite the fact that catalysts with high selectivity are necessary to overcome the drawbacks of ethane ODH, the current process produces unwanted byproducts such as CO and CO2. Additionally, these catalysts should be low cost, thermally stable, and have a high conversion. Hence, the methods for preparing the catalyst are crucial to properly incorporate the active phase and to influence in the final characteristic of the catalyst. Particularly, the hydrothermal synthesis represents some of the most interesting procedures of tailoring catalysts dealing to enhance chemical dispersion and crystal size of the catalyst, especially for multicomponent Mo-V-O based catalysts.9,10 Some characteristics in hydrothermal method are well-studied in the case of forming pure phases with high dispersion of vanadium catalysts.9-13 For instance, the vanadium-containing catalyst MoVNbTe promoted by MnOx, prepared using the hydrothermal method, led to an almost 20% increase in ethane conversion performance at 400 ºC.14 Khalil et al.15 synthesized hematite nanoparticles by hydrothermal reaction with good crystallinity through the formation of goethite nanorods with the formation of hematite crystal providing the control of crystal size and shape at low temperatures. Asghari et al.16 prepared by hydrothermal/impregnation technology a series of chromium oxide catalysts supported over molecular sieves based on Si/Ce for ethane ODH using CO2 as an oxidation reactant, where the results revealed that the catalytic activity depended on the distribution of particle size over the surface and under influence of plasma, the particle size shifted to the smaller and the uniformity improved. Recently, it has been shown that the selectivity towards ethylene in ethane ODH for MoV oxide (M1) based catalysts, prepared hydrothermally, strongly depend on the composition and the operation temperatures.17 In the last years, many catalysts based on NiO have been studied18,19 for the oxidative dehydrogenation (ODH) of ethane due to the high activity of the NiO-based starting material, its easy accessibility but low ethylene selectivity. Many efforts have been made to increase the selectivity towards ethylene such as controlling the morphological properties,17 the electronic surface properties,20 increasing reaction sites via metal support interactions21 as well as doping the structure of NiO with different transition metal oxides as Fe,22,23 Co,24 Nb.18,25 In addition, some boron26 and chloride27 materials were also explored. In a previous paper,22 Ni-Fe/γ-Al2O3 catalysts were explored for ODH of ethane achieving an ethylene selectivity of 60% for the supported sample with Ni/Fe molar ratio of 0.9. This result was attributed to the formation of lower reducibility species with controlled particle size and appropriate dispersion of the active phase. Additionally, there was an increase in the concentration of nucleophilic oxygen surface species. This work explores the influence of preparation method of catalysts based on Ni-Me-O (Me = Co, Nb) supported on γ-Al2O3, using hydrothermal procedure and chemical coprecipitation-deposition by reflux as a reference method. To find the final role of the interaction between a metal oxide and the support and the participation of active phases of Ni under influence of Nb or Co dopants is the main purpose of this work.
EXPERIMENTAL Preparation of catalyst Ni-O and Ni-Me-O (Me = Co, Nb) catalysts supported on γ-Al2O3 (Aldrich, < 50 nm-TEM) were prepared as follows: catalysts were prepared using ammonia (NH3(aq) 0.27 M) or urea (CO(NH2)2 0.27 M) as the precipitating agents. Additionally, these catalysts were prepared by two different methods: (i) hydrothermal (using autoclaves), and (ii) coprecipitation-chemical deposition by reflux. In both methods, the Ni content in the single nickel(II) oxide was 30% w/w, and that of Me (Co, Nb) was 5% w/w. Both, 0.14 M of nickel(II) nitrate, and 0.07 M of CTAB (cetyltrimethylammonium bromide) were mixed in 73 mL of water under stirring for 1 h. Later, to the mixture, two grams of γ-Al2O3 were added and stirred for 1 h. In the case of the hydrothermal preparation, the final solution was added into a flask, and Teflon autoclave, with a volume of 73 mL at 135 ºC for 24 h. In the case of the coprecipitation-chemical deposition by reflux, the conditions were similar, except the temperature that was 120 ºC. For both methods, the solid obtained was dried at 100 ºC for 12 h. Eventually, the solids were calcined at 500 ºC for 4 h with a heating ramp of 5 ºC min-1.28 The supported samples prepared by hydrothermal method and urea as precipitating agent were designated as NiMe-H-ur (Me: Nb, Co). Additionally, Ni-H-am (NiO/γ-Al2O3), using ammonia as precipitating agent, was included for comparison purposes. Samples prepared by coprecipitation-chemical deposition by reflux were nominated as NiMe-R-ur (Me: Nb, Co). The only bulk catalyst was designated as Ni-R-Mas. Catalyst characterization The crystalline phases of the samples was identified by powder X-ray diffraction (XRD). This analysis was performed in a Panalytical Cubix Pro diffractometer, supplied with a graphite monochromator operating at 40 kV and 45 mA using CuKα radiation. The scanning speed was 0.04º per second in the range of 5º to 80º. The identification of the patterns was made using ICDD. The crystallite size was estimated by Scherer's equation using X'Pert HighScore software for the most intense peak at 43.29 ºC. The specific surface area of samples was measured by N2 sorption using Micromeritics Gemini VII equipment. The pre-treatment of the samples was made in 10 mL min-1 of He, at 200 ºC for 2 h. The weight of the sample was around 100 mg, and the relative pressure of N2/He was between 0.05 to 0.985 (total 32 points). The distribution of pore diameter of the catalyst was evaluated using BJH method (Barrett-Joyner-Halenda). The temperature programmed reduction of H2 (TPR-H2) profiles were performed by the Micromeritics Autochem 2910 equipment. The mass of the samples was rough 16 mg. The mixed gas of 10% H2/He was used to reduce the samples. The temperature was linearly increasing from 100 to 800 ºC with a heating ramp of 10 ºC min-1, and the flow rate of 50 mL min-1. The TCD detector was calibrated by the reduction of Ag2O reference. The composition on catalyst surface was determined by XPS analysis using a commercial spectrometer UNI-SPECS UHV with a vacuum of 5 × 10-7 Pa. The Mg Kα line was used (hν = 1254.6 eV) as the ionization source and the pass energy of the analyzer was adjusted to 10 eV. The inelastic noise of the high-resolution Ni 2p3/2, Co 2p3/2, Nb 3d, Al 2p, C 1s and O 1s spectra was subtracted using Shirley's method. Composition was determined by the relative proportions of peak areas corrected for Scofield atomic sensitivity factors to an accuracy of ± 5%. The spectra were deconvolved using a Voigtian-like function, with Gaussian (70%) and Lorentzian (30%) combinations. The width at half height varied between 1.4 and 2.5 eV, and the position of the peaks was determined with an accuracy of ± 0.1 eV using the carbon signal. The chemical environment of metal species in the prepared samples was evaluated by Raman spectroscopy, Horiba Scientific equipment, with a wavelength of 532 nm (green light) and potential of 50%. The bulk composition of metal oxide samples was analyzed by elemental analysis by energy-dispersive X-ray spectroscopy (EDX). The analysis was performed using an EDX720 instrument from Shimadzu. The source was an X-ray tube with an Rh target and the detector was a Si (Li) working at 5-15 kV. Catalyst activity The catalytic tests of prepared samples in the dehydrogenation of ethane were carried out at atmospheric pressure in a fixed bed reactor.22 The activity of ethane and selectivity toward ethylene were evaluated at different reaction conditions at temperatures ranging from 300 to 480 ºC, and a constant W/F ratio of 0.48 g s mL-1. Additionally, the feed mixed gas composition was 5% C2H6/5% O2 balance with helium. The selectivity toward ethylene was studied as a function of conversion for a variable W/F ratio of 0.30-1.92 g s mL-1, at a constant reaction temperature of 420 ºC, to find the conversion as a function of the selectivity toward ethylene for each catalyst. The data was collected using a gas chromatograph Varian GC-3800 with a TCD detector, and two columns as Porapack Q, and molecular sieve 5A. This last column allowed to quantify the concentration of oxygen, and carbon monoxide. The accepted balance of the carbon was 95%. Since the fluxes of feed gas were less than 50 mL min-1, the conversion (C) and selectivity towards ethylene (S) were evaluated as follows: 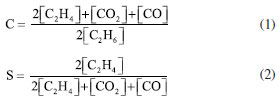 where [C2H6], [C2H4], [CO] and [CO2] are the molar concentrations of chemicals.
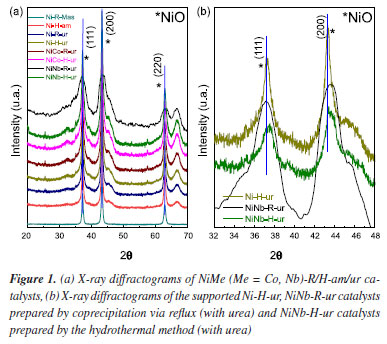
RESULTS AND DISCUSSION X-ray diffraction (XRD) and N2 sorption The X-ray diffraction patterns for the supported samples are shown in Figure 1a. All the diffractograms of samples, no matter the precipitating agent, presented diffraction peaks corresponding to NiO, 2θ = 37.3, 43.4, 63.0, 75.5, and 79.5º (JCPDS 4-835)18 and γ-Al2O3, 2θ = 19.6, 32.6, 37.2, 39.4, 45.7, 60.9, 67.0, and 84.4º (JCPDS 29-63).29 This figure was observed for the samples prepared either by hydrothermal method or coprecipitation method via reflux (Figure 1a). Moreover, no peaks assigned to the presence of Co or Nb were detected, probably due to the presence of possible substitutions of Co for Ni or by the incorporation of Nb5+ that can locate in vacant or interstitial Ni sites. In a larger scale, a shift in the higher angles corresponding to mixed supported samples NiCo-series and NiNb-series (Figure 1b) is observed, related to the compression of the lattice parameter. This reveals that the incorporation of Co or Nb to the lattice of NiO affects the initial crystalline structure, probably influencing the nucleophilic oxygen species present in this catalyst and increasing the pore volume in the mixed catalyst NiNb-H-ur (Figure 2) and with similar picture with NiCo-H-ur.
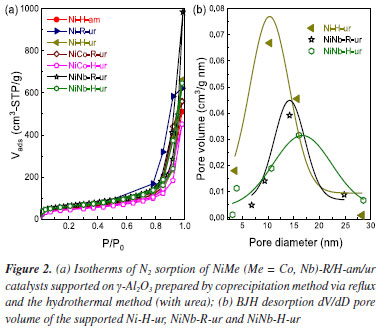
A list of the prepared samples with their physicochemical characteristics is presented in Table 1. As observed from isotherm profiles (Figure 2), regardless of the method of preparation, the samples prepared using urea as a precipitating agent on single supported NiO led to higher total specific surface. The improvement in mesoporosity is important considering that the reactant gases are expected to diffuse better over the external surface. Both samples NiNb-H-ur and NiNb-R-ur present the highest mesoporous surface area, 189 and 199 m2 g-1, respectively, which suggests that the introduction of Nb into Ni sites contributes to improving the contact area of the catalysts with the reactants over the support.
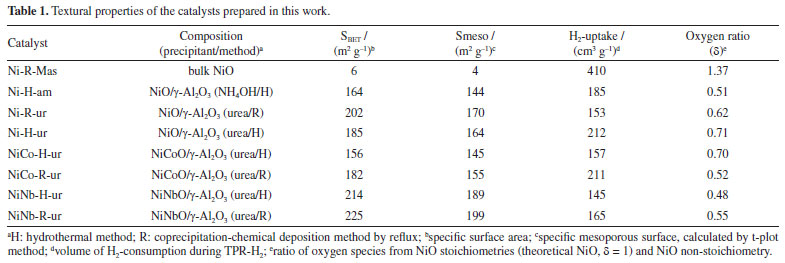
As is shown in the Figure 2a, isotherms of all the catalysts are type-IV corresponding to micro-mesoporous surface for the presence of hysteresis loop with shape type I assigned to the enrichment of cylindrical or conical porous over the surface. The introduction of Co or Nb (5% w/w) onto the support surface allowed to obtain a pore diameter ranging from 10 to 18 nm (BJH method). This is related to the decrease in pore volume observed in the Nb mixed catalysts, NiNb-H-ur and NiNb-R-ur (Figure 2b), compared to single Ni-H-ur, contributing to better saturation of the pores. The main parameters of XRD studies are shown in Table 1S (Supplementary Material). The crystallite sizes could play a significant role in the selectivity toward ethylene according to the literature,1,7,28,30 especially if the crystallite size decreases, which is a factor of enhancement of the selectivity toward ethylene. This can be related to the depression of electrophilic species as the crystallite size is falling down.21,22 However, for the samples Ni-H-ur, NiCo-H-ur, and NiNb-H-ur, the particle size was very similar with a slightly difference in lattice parameter (Table 1S). The lattice parameter of nickel oxide in samples of Ni-H-ur, NiCo-H-ur and NiNb-H-ur was less than the corresponding to bulk NiO no matter the precipitating agent. However, when metals such as Co and Nb are incorporated to NiO, a slight displacement of the peak at 43.62º of (200) plane is observed, which suggests the possible incorporation (partial or total) of metals into the NiO lattice and consequently in the intermetal interaction. Temperature programmed reduction of H2 (TPR-H2) The TPR-H2 profiles of the supported catalysts are shown in Figure 3. For comparison purposes, the results obtained for bulk Ni-R-Mas are also included. The reduction of bulk NiO to Ni metallic is according to the Equation 3. 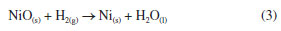
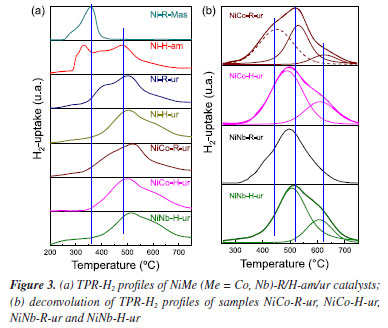
In the supported Ni-H-am and Ni-R-ur catalysts, the reduction to Ni metallic is also observed, but the main peak of reduction is shifted to the right compared with the bulk sample, probably due to the metal-support interaction, according to Equation 4. This shifting was also observed in other supported samples (Ni-H-ur, NiCo-H-ur and NiNb-H-ur). 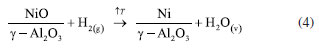 A redistribution of reducible species in all supported samples is clearly observed in the Figure 3a. For the single Ni-H-ur sample, compared to the Ni-R-ur sample, the formation of reducible species occurs at higher temperatures, even when compared to the single Ni-H-am sample. The introduction of Co and Nb into the NiO framework further increased the reduction temperatures, probably due to Ni-synergy effects and metal-support interactions. In addition, differences are also observed in mixed supported samples depending on the preparation (Figure 3b), that shows the deconvolution display of TRP profile. For instance, in the catalyst prepared by reflux (NiCo-R-ur), three peaks of reducible deconvolution peaks can be clearly observed, whereas in the catalyst prepared hydrothermally (NiCo-H-ur) only two peaks are detected, but with a major formation of more reducible species. The mixed sample NiNb-H-ur (Figure 3b) presents a TPR pattern very similar to NiCo-H-ur. The sample prepared from reflux NiNb-R-ur presents only one peak in deconvolution without the formation of a peak at high temperatures, probably due to the formation of one oxygen species. The particular characteristics in the TPR profiles of both NiNb-H-ur and NiCo-H-ur will be further discussed in the XPS analysis and during the catalytic reaction test. Referring to H2-uptake, the Co- and Nb-doped samples consumed less hydrogen than supported single NiO (Ni-H-ur). Thus, the hydrogen consumption for the NiCo-H-ur sample was 157 and NiNb-H-ur 145 cm3-STP/g (STP: standard conditions of T and P) compared with supported NiO 212 cm3-STP/g (Table 1). This is possibly assigned to the lower extent of oxidation of Ni species in the mixed samples. X-ray photoelectronic spectroscopy (XPS) The spectra of O 1s for all of the samples (Figure 4b) show 2 types of oxygen: nucleophilic oxygen (On) species corresponding to O2- (lattice oxygen), and electrophilic oxygen (Oe) species corresponding to O2-, O- or hydroxyl species.31,32 The nature of the oxygen species has been related to the selectivity towards ethylene, in a way that the predominance of nucleophilic species are usually linked to a higher formation of partial oxidation or dehydrogenation products, such as ethylene. For these samples, the higher is the O1s binding energy (BE), the higher selectivity toward ethylene is expected.22 In fact, the maximum value of BE of O 1s (530.5 eV) is achieved by the sample NiNb-H-ur, which is the one that presents the highest selectivity to ethylene, as it will be shown later. On the other hand, the relationship between On and Oe species (On/Oe) shows an interesting behavior in Nb-containing mixed sample with a magnitude of 2.04, which is the maximum obtained by all the catalysts studied in this work. This is probably due to the incorporation of Nb atoms in the NiO framework, as observed in the XRD and TPR analysis.
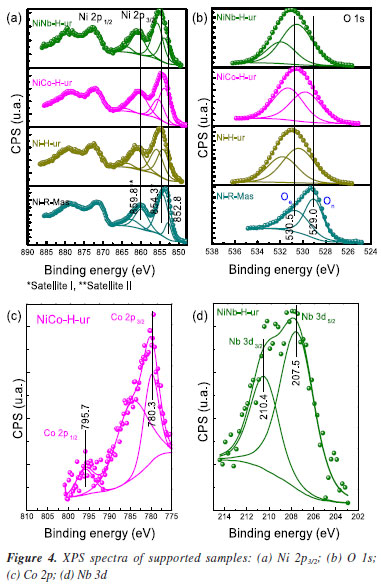
On the other hand, the deconvolution of the XPS spectra of NiCo-containing sample shows two peaks corresponding to Co 2p3/2 and Co 2p1/2 at 780.3 and 795.7 eV, respectively (Figure 4c). These peaks indicate the presence of cobalt oxides with oxidation states of +2 or +3 species.33 Similarly to the NiCo-based catalyst, the NiNb-containing sample also presents two peaks in position at 207.5 and 210.4 eV attributed to Nb 3d5/2 and Nb 3d3/2, respectively (Figure 4d). It is necessary to mention that the Nb is only found as Nb5+ species, as a consequence of the calcination procedure in the preparation of catalysts. Furthermore, the analysis of the atomic composition of Nb and Co content in the bulk of supported sample was determined by EDX analysis (Table 2S, Supplementary Material). Compared with an analogous ratio of Nb and Co content over the surface results between 0.0375 and 0.0486, and in the bulk, it was obtained from the Table 2S, 0.032 for Nb and 0.058 for Co so the metallic dopants are well dispersed over the surface. As observed in Table 2, the sample Ni-R-Mas presents 49.7% of surface Ni while in the supported samples Ni-H-ur, NiCo-H-ur and NiNb-H-ur present 9.9, 11.7, and 8.1%, respectively. This explains the metallic charges deposited over the alumina support, roughly 30 wt.% related to the total mass. The main peak position of Ni 2p3/2 in the bulk NiO sample is located at 852.8 eV, which is different to those in the supported samples (in the 853.7 to 854.2 eV range). This trend is confirmed by the position of satellites peaks Sat I and Sat II. This could be attributed to different chemical environment of nickel sites in the bulk and supported catalysts (Figure 4a). The origin of satellite peaks was discussed these by Solsona et al.34 relating them to the presence of Ni2+, Ni2+-OH species or Ni2+ vacancies. It is noteworthy the shift to higher binding energies of the main peak of Ni 2p3/2 in the case of supported nickel catalysts, probably due to the metal-support interaction. Moreover, the difference ΔSat I-Main peak is higher than 1.5 eV in supported samples with a maximum in 1.8 eV in NiNb-H-ur which represents the extent of electronic repulsion in the last sample.35 The position of Nb is likely octahedral, as it is the most exposed atom in the catalyst, which can play a very important role in the selectivity behavior towards ethylene.
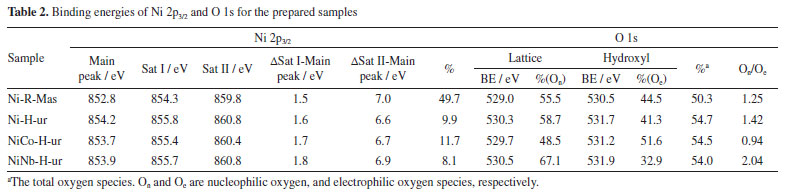
The oxygen species of samples were recalculated to evaluate the effect of preparation condition on the stoichiometry of solid sample. With this aim, the ratio of oxygen species (d) coming from stoichiometric NiO (theoretical NiO) and non-stoichiometric NiO has been quantified from TPR analysis and shown in Table 1. For more details see Supplementary Material. The oxygen ratio of samples Ni-H-ur, NiCo-H-ur and NiNb-H-ur were 0.71, 0.52 and 0.48, respectively (Table 1), so the structure composition of mixed system Ni-Me plays a crucial role in the oxygen distribution in the supported samples. Thus, the lower content of bulk oxygen species, the higher is the expected selectivity toward ethylene. However, this trend is only partially observed in the supported NiO oxide catalyst prepared by the hydrothermal method with ammonium hydroxide (sample Ni-H-am), since the ammonia as a precipitating agent led to lower δ. Catalytic performance for ODH of ethane The catalytic behavior of these catalysts was studied for the ODH of ethane. Firstly, the catalytic activity was evaluated as the variation of the ethane conversion (%) with the temperature of the samples (Figure 5). For comparison purpose, all samples in the Figure 5 are supported with the same surface density (ds = 14.4 at nm-2). As it can be observed, the samples prepared from coprecipitation via reflux were more active than those hydrothermal. Indeed, the sample NiCo-R-ur appeared to be the most active, even more active than Ni-H-ur since the temperature to achieve the 15% of conversion increases from 420 to 435 ºC. The enrichment of oxygen species in NiCo-R-ur respect to Ni-R-ur (Table 1) and the higher H2-consumption in TPR profiles of the mixed sample relative to the single, 211 vs. 153 cm3-STP/g (Table 1) allowed to enhance the activity of the reaction. In contrast, the sample promoted with niobium (NiNb-H-ur) presented a lower activity, probably due to the presence of the less reactive oxygen species in the TPR-H2 experiments (Table 1). The supported samples prepared from hydrothermal compared with coprecipitation via reflux present less reactive oxygen species and, accordingly, a higher reduction temperature is observed (Figure 3b). As a matter of fact, the TPR profiles of NiCo-H-ur and NiNb-H-ur depict a shoulder at higher temperatures meanwhile in NiCo-R-ur a shoulder is present at lower temperatures. On the other hand, the selectivity to the olefin obtained with mixed supported samples is presented in Figure 5b. As it is shown, the selectivity toward ethylene dropped with the reaction temperature and the sample NiNb-H-ur appeared to be the most selective toward ethylene, probably due to the formation of less reducible species (Figure 3b) and the higher value of ratio of On/Oe, as presented in Table 2. For this sample, the selectivity toward CO2, among all the samples, is the lowest (Figure 5c), revealing the formation of selective oxygen species over the surface which led to the partial oxidation of ethane to ethylene.
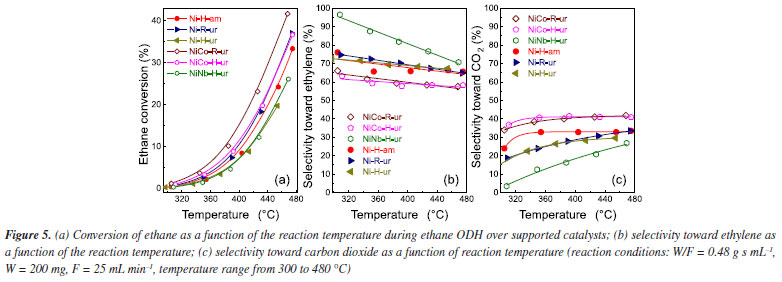
The conversion of ethane and selectivity toward ethylene at 380 and 420 ºC are presented in Table 3S (Supplementary Material). As expected, the ethane to ethylene transformation increased with the increasing temperature meanwhile the selectivity slightly decreased. For undoped NiO supported oxide the application of the urea as a precipitating agent meant a more selective sample towards ethylene. The introduction of Nb5+ dopant enhanced even more the selectivity to ethylene. For instance, in the case of NiNb-H-ur catalyst the selectivity was 76.7% and for Ni-H-ur 68.3%. These results agreed with previous results21-23,31,36 in which a positive effect of the presence of niobium in this type of catalysts has been observed. This positive effect on ethylene selectivity is possibly due to the compression in the crystallite size of the active phase, NiO. As a matter of fact, as it was suggested by some authors,22,31 the selectivity toward ethylene increases when the NiO crystal size diminishes. Moreover, this selectivity is tightly related to metal-support interaction of the active phase. Although there are similar reduction temperatures for the NiCo-H-ur and NiNb-H-ur samples, the last one presented a better behavior in confront of the selectivity toward ethylene. From the XPS spectra, the distance between the main peak and the satellite I can be determined (Table 2). This distance value increases from 1.5 for the bulk NiO sample to 1.8 eV for the NiNb-H-ur sample and this is probably related to the presence of less vacancies, Ni3+ and Ni2+-OH species exposed, in the Nb-containing samples. This favored the increasing of Ni2+-species content respect to Ni3+-species24 and for this reason NiNb-H-ur sample appeared to have less activity but higher selectivity towards ethylene. The ethane conversion at 420 ºC starts to increase in average 10% in relation to the observed at 380 ºC as is expected (Table 3S). The yield to ethylene at 420 ºC is almost three times higher than that observed at 380 ºC. However, the single supported sample (Ni-H-ur) and NiCo-H-ur presented a clear trend to the formation of CO2 meanwhile the NiNb-H-ur appeared to have more preference for ethylene since the quantity of CO2 is a half respect to the remaining samples, as is observed in Figures 5c and 6.
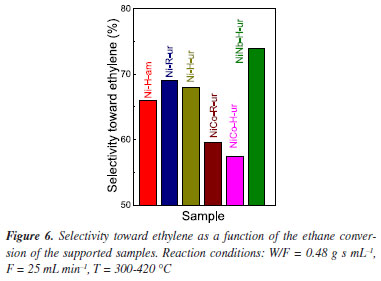
In order to study with a deeper insight of metal atom surroundings, Raman spectroscopy has been employed. As observed in Figure 7, two Raman bands at 460 and 500 cm-1 are identified (Ni-R-Mas), which probably correspond to bulk crystalline NiO.37 In the spectra of Ni-H-ur either the position of the bands or the corresponding intensity changed under the influence of the support attributed to metal-support interaction.38 With the introduction of Co and Nb to NiO, some slightly chemical shifts are observed (for instance at 554 cm-1 for NiCo-H-ur, 507 cm-1 for Ni-R-Mas and 545 cm-1 for NiNb-H-ur), revealing the influence of different chemical surroundings in each case. The peak at 717 cm-1 correspond to stretching oxygen atoms affected by tetrahydric or octahedral of metal atoms (Co or Nb). UV visible-DRS spectroscopy was applied to confirm the atom position. A shift in the band position to lower energy was observed with NiNb-H-ur and Ni-R-ur in relation to NiCo-H-ur and NiCo-R-ur which means a change from tetrahedral to octahedral position39 (Figure 2S).
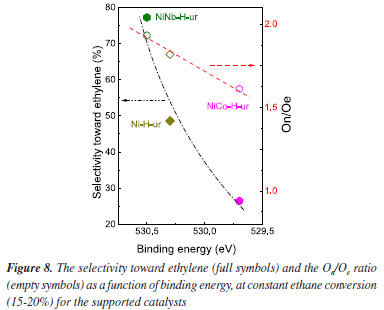
The nature of oxygen was very well distinguished via XPS spectra. In fact, as it is observed in the Figure 8, a direct relation between the On/Oe ratio and the selectivity is clearly observed, revealing different oxygen surroundings in each catalyst. The most selective catalyst, NiNb-H-ur, is the one with the highest value of On/Oe ratio, whereas for NiCo-H-ur and Ni-H-ur, higher concentration of electrophilic oxygen, which are more active for total oxidation reactions, can be observed.
CONCLUSIONS The synthesis of NiO supported on alumina by a hydrothermal method using urea has been shown to be highly effective for the oxidative dehydrogenation of ethane to ethylene. Moreover, the addition of Nb or Co as promoters meant modifications in the physicochemical properties, leading to changes in the catalytic activity and on the selectivity to ethylene. Modifications in the lattice parameters that suggest incorporation of the promoters into the NiO lattice, variations in the reducibility and changes in the characteristics of the surface were observed when the promoters were added. Depending on the element incorporated a different catalytic behavior was observed. Then, the addition of cobalt led to a drop of the selectivity to ethylene whereas the addition of Nb meant an enhancement compared to the unpromoted supported catalyst. These catalytic results can be explained on the basis of the nature of the surface oxygen species. Then, the catalyst with Nb presented the highest concentration of nucleophilic oxygen whereas the catalyst with Co presents the highest concentration of electrophilic oxygen.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
ACKNOWLEDGMENTS The authors thank the following institutions for funding the projects: FONDECYT: contract 138-2019, project No. 237-2015-FONDECYT, Project 219-2015-FONDECYT; and Spanish Ministry of Economy and Competitiveness, MINECO/FEDER (PID2021-126235OB-C31, PID2021-126235OB-C33).
REFERENCES 1. Bortolozzi, J. P.; Weiss, T.; Gutierrez, L. B.; Ulla, M. A.; Chem. Eng. J. 2014, 246, 343. [Crossref] 2. Gärtner, C. A.; vanVeen, A. C.; Lercher, J. A.; ChemCatChem 2013, 5, 3196. [Crossref] 3. Najari, S.; Saeidi, S.; Concepción, P.; Dionysiou, D. D.; Bhargava, S. K.; Lee, A. F.; Wilson, K.; Chem. Soc. Rev. 2021, 50, 4564. [Crossref] 4. Ivan, S.-B.; Fechete, I.; Papa, F.; Marcu, I.-C.; Catal. Today 2021, 366, 133. [Crossref] 5. Ding, W.; Zhao, K.; Jiang, S.; Zhao, Z.; Cao, Y.; He, F.; Appl. Catal. A 2021, 609, 117910. [Crossref] 6. Jin, F.; Cheng, X.; Wan, T.; Gong, J.; Liang, T.; Wu, G.; Catal. Commun. 2022, 172, 106531. [Crossref] 7. Leung, L.; Jenkins, G. P.; Energy Policy 2014, 74, 643. [Crossref] 8. Balvin, L.; Gomez, S.; León, C.; Alvarez, J. C.; Energy Rep. 2020, 6, 256. [Crossref] 9. de Arriba, A.; Solsona, B.; Dejoz, A. M.; Concepción, P.; Homs, N.; de la Piscina, P. R.; López Nieto, J. M.; J. Catal. 2022, 408, 388. [Crossref] 10. Obunai, R.; Tamura, K.; Ogino, I.; Mukai, S. R.; Ueda, W.; Appl. Catal. A 2021, 624, 118294. [Crossref] 11. Le, D.; Chaidherasuwet, N.; Hinchiranan, N.; Appl. Catal. A 2022, 638, 118624. [Crossref] 12. Khanna, H. S.; Mirich, A.; Twombly, A.; Dang, Y.; Shirazi-Amin, A.; Perera, I.; Liu, Y.; Posada, L. F.; Deshlahra, P.; Suib, S. L.; Appl. Catal. A 2022, 635, 118557. [Crossref] 13. Faizan, M.; Zhang, R.; Liu, R.; J. Ind. Eng. Chem. 2022, 110, 27. [Crossref] 14. Xin, C.; Dan, D.; Hang, A.; Bozhao, C.; Yi, C.; J. Taiwan Inst. Chem. Eng. 2019, 95, 103. [Crossref] 15. Khalil, M.; Yu, J.; Liu, N.; Lee, R. L.; J. Nanopart. Res. 2014, 16, 2362. [Crossref] 16. Asghari, S.; Haghighi, M.; Taghavinezhad, P.; Microporous Mesoporous Mater. 2019, 279, 165. [Crossref] 17. de Arriba, A.; Solsona, B.; García-González, E.; Concepción, P.; López Nieto, J. M.; Appl. Catal. A 2022, 643, 118780. [Crossref] 18. Heracleous, E.; Lemonidou, A. A.; J. Catal. 2010, 270, 67. [Crossref] 19. Abdelbaki, Y.; de Arriba, A.; Solsona, B.; Delgado, D.; García-González, E.; Issaadi, R.; López Nieto, J. M.; Appl. Catal. A 2021, 623, 118242. [Crossref] 20. Li, M.; Gao, Y.; Zhao, K.; Li, H.; He, F.; Lv, P.; Huang, Z.; Fuel Process. Technol. 2021, 216, 106771. [Crossref] 21. Delgado, D.; Sanchís, R.; Cecilia, J. A.; Rodríguez-Castellón, E.; Caballero, A.; Solsona, B.; López Nieto, J. M.; Catal. Today 2019, 333, 10. [Crossref] 22. Cotillo, M. H.; Unsihuay, D.; Santolalla-Vargas, C. E.; Doig, A. P.; Kou, R. S.; Picasso, G.; Catal. Today 2020, 356, 312. [Crossref] 23. Cancino-Trejo, F.; Santes, V.; Alcantara Cardenas, J. A.; Gallardo, M.; Maldonado, Y. G.; Lopéz Miranda, A.; Valdes, O.; de los Reyes, J. A.; Santolalla-Vargas, C. E.; Chem. Eng. J. Adv. 2022, 12, 100404. [Crossref] 24. Boukhlouf, H.; Barama, A.; Benrabaa, R.; Guerrero Caballero, J.; Lofberg, A.; Bordes-Richard, E.; C. R. Chim. 2017, 20, 30. [Crossref] 25. Qiao, A. L.; Kalevaru, V. N.; Radnik, J.; Martin, A.; Catal. Today 2016, 264, 144. [Crossref] 26. Wu, Z.; Zhou, Y.; Ying, H.; Lin, J.; Han, W.-Q.; Chem. Phys. Lett. 2020, 746, 137294. [Crossref] 27. Ayari, F.; Charrad, R.; Asedegbega-Nieto, E.; Mhamdi, M.; Delahay, G.; Farhat, F.; Ghorbel, A.; Microporous Mesoporous Mater. 2017, 250, 65. [Crossref] 28. Justin, P.; Meher, S. K.; Rao, G. R.; J. Phys. Chem. C 2010, 114, 5203. [Crossref] 29. Xu, L.; Lin, X.; Xi, Y.; Lu, X.; Wang, C.; Liu, C.; Mater. Res. Bull. 2014, 59, 254. [Crossref] 30. Li, C.; Chen, Y.-W.; Thermochim. Acta 1995, 256, 457. [Crossref] 31. Biesinger, M. C.; Payne, B. P.; Lau, L. W. M.; Gerson, A.; Smart, R. St. C.; Surf. Interface Anal. 2009, 41, 324. [Crossref] 32. Biesinger, M. C.; Payne, B. P.; Grosvenor, A. P.; Lau, L. W. M.; Gerson, A. R.; Smart, R. St. C.; Appl. Surf. Sci. 2011, 257, 2717. [Crossref] 33. Cabrera-German, D.; Gomez-Sosa, G.; Herrera-Gomez, A.; Surf. Interface Anal. 2016, 48, 252. [Crossref] 34. Solsona, B.; López Nieto, J. M.; Concepción, P.; Dejoz, A.; Ivars, F.; Vázquez, M. I.; J. Catal. 2011, 280, 28. [Crossref] 35. Van Veenendaal, M. A.; Sawatzky, G. A.; Phys. Rev. Lett. 1993, 70, 2459. [Crossref] 36. Botella, P.; García-González, E.; Dejoz, A.; López Nieto, J. M.; Vázquez, M. I.; González-Calbet, J.; J. Catal. 2004, 225, 428. [Crossref] 37. Peña, J. P.; Bouvier, P.; Isnard, O.; J. Solid State Chem. 2020, 291, 121607. [Crossref] 38. Chan, S. S.; Wachs, I. E.; J. Catal. 1987, 103, 224. [Crossref] 39. Qiao, A.; Kalevaru, V. N.; Radnik, J.; Kumar, A. S.; Lingaiah, N.; Prasad, P. S. S.; Martin, A.; Catal. Commun. 2013, 30, 45. [Crossref]
Associate Editor handled this article: Marcela M. Oliveira |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access