
 |
 |
 |
 |
 |
Artigo
|
|
| Explorando o mecanismo de degradação enzimática do poliuretano (PU) por meio das poliuretanases esterases A e B (PueA E PueB): uma abordagem integrada de docking, Dinâmica Molecular (MD) e QM/MM Exploring the mechanism of enzymatic degradation of polyurethane (PU) by means of polyurethanase esterases A and B (PueA and PueB): an integrated approach of docking, Molecular Dynamics (MD) and QM/MM |
|
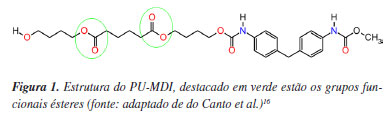
Thiago Antonio Valdez GarciaI,* I. Departamento de Físico-Química, Instituto de Química, Universidade Federal do Rio Grande do Sul, 91501-970 Porto Alegre - RS, Brasil Recebido: 06/08/2024 *e-mail: quimthiagogarcia@gmail.com; netz@iq.ufrgs.br The continuous global production of polyurethane (PU) and its environmental persistence generate negative impacts on ecosystems, driving the search for sustainable handling solutions, specifically the use of enzymes. Therefore, this study seeks to elucidate the understanding of the enzymatic degradation mechanism of polyurethanase esterases A and B (PueA and PueB) applied to PU-MDI (methylene diphenyl diisocyanate). In this sense, previously built structures were used and dockings were carried out for the PueA and PueB complexes. The results indicate binding scores of -10.743 and -9.587 kcal mol-1 for PueA and PueB, respectively. Furthermore, the complexes were then subjected to molecular dynamics simulations (MD) for 500 ns in triplicate, showing a good interaction between the enzymes and the PU-MDI. Finally, a QM/MM (quantum mechanics/molecular mechanics) simulation, starting with an adequate snapshot from classical MD was carried out, and a relaxed scan was performed to obtain the occurrence coordinates, followed by umbrella sampling to obtain the free energy profile of the systems. The results indicate that the PueA and PueB reaction mechanisms occur in two steps, with determining energy barriers of 11.95 and 15.10 kcal mol-1, respectively, indicating that these enzymes may be used to degrade PU. INTRODUÇÃO Na década de 1950 iniciou a produção em grande escala de plásticos. Atualmente, essa expansão já passa de 230 vezes. Esse rápido crescimento dos polímeros se deve às suas propriedades únicas, como, por exemplo: a relação resistência/peso, alta moldabilidade, impermeabilidade, resistência à degradação física e química e baixo custo.1 Os resultados divulgados pelo Plastics Europe 2023 mostraram que a produção anual global de plásticos chegou a 400,3 Mt (milhões de toneladas) em 2022. Dessa massa, 21,2 Mt corresponde somente a poliuretano (PU).2 Nesse mesmo sentido, os resíduos plásticos mais do que dobraram, de 156 Mt em 2000 para 353 Mt em 2019. Ou seja, quase dois terços de todos os resíduos plásticos vêm de aplicações com vida útil inferior a cinco anos, como embalagens, produtos de consumo e têxteis. Somente 55 Mt desses resíduos foram coletados para reciclagem, destes, 9% foram reciclados, 19% foram incinerados e quase 50% foram para aterros sanitários. Já os 22% restantes foram descartados em lixões não controlados, queimados a céu aberto ou vazados para o meio ambiente.3 Além disso, estima-se que os resíduos plásticos atinjam 1.014 Bt (bilhões de toneladas) em 2060. Dessa forma, projeta-se que o vazamento de plásticos para o meio ambiente aumente de 79 Mt em 2019 para 153 Mt em 2060.3 Nessa lógica, conforme outro estudo,4 a acumulação de plásticos e microplásticos em ambiente aquático produz impactos negativos causando efeitos biológicos e ecológicos adversos que consequentemente geram dano econômico global aos ecossistemas marinhos de 13 bilhões de dólares por ano. Ademais, por causa da contínua produção em nível mundial dos polímeros e durabilidade indesejável no ecossistema, a degradação para fins de reciclagem se tornou uma preocupação crescente. Nesse contexto, a busca por soluções sustentáveis para a degradação desses materiais tem recebido, especial, visibilidade e uma das alternativas é a utilização de enzimas para degradá-los.5 Estudos recentes6,7 apontam que os polímeros sintéticos a base de petróleo são o principal alvo. Entre eles destacam-se: PVC (policloreto de vinila), PET (polietileno tereftalato), PE (polietileno), PP (polipropileno), PI (poliimida), PS (poliestireno) e PU. Esse último é um polímero com ligações do tipo uretana (carbamato) obtido pela reação de polimerização entre um diisocianato e um diol ou um poliol poliéster. Desses polímeros, o PET e o PU são suscetíveis ao mecanismo de hidrólise devido a presença de grupos funcionais ésteres que possibilitam a quebra da ligação química entre o carbono e o oxigênio. No entanto, o PET é o polímero mais reciclado de forma mecânica, no Brasil.8 Por outro lado, sua degradação enzimática está sendo desenvolvida em colaboração internacional incluindo grupos de pesquisa brasileiros.9 Todavia, a pesquisa in silico no âmbito da degradação enzimática do PU, no Brasil, ocorre de forma tímida.10 Diante disso, o presente trabalho aborda a degradação enzimática de PU, sistema em relação ao qual o grupo já possui estudos prévios, usando técnicas de docking, dinâmica molecular (MD) e métodos híbridos de mecânica quântica/mecânica molecular (QM/MM) que são amplamente utilizadas no campo da química computacional e biologia estrutural. O docking é uma técnica que prediz a orientação preferencial de uma molécula (ligante) ao se ligar a um receptor (enzima) visando identificar interações e afinidades potenciais. A MD, por sua vez, simula o movimento e comportamento de átomos e moléculas ao longo do tempo, permitindo o estudo detalhado das conformações e flutuações estruturais dos sistemas biomoleculares em um ambiente simulado. O método QM/MM combina a precisão dos cálculos de mecânica quântica, geralmente, aplicada em átomos da região do sítio ativo das enzimas e a mecânica molecular para o restante do sistema, proporcionando uma abordagem balanceada entre acurácia e custo computacional. As esterases A e B, PueA e PueB, foram classificadas pela EC (do inglês, Enzyme Commission) como 3.1.1.1, hidrolases. Elas são secretadas por micro-organismos do gênero Pseudomonas chlororaphis e suas estruturas primárias mostraram a sequência de aminoácidos, no sítio ativo, característica das serina hidrolases, ou seja, GXSXG onde na notação comum usada na bioquímica e na biologia molecular, G representa glicina, X pode representar qualquer aminoácido e S representa a serina.11,12 Por outro lado, os substratos naturais das esterases são, de forma geral, os triglicerídeos que tem grupo funcional éster.13 Nesse sentido, sua atividade catalítica natural é quebrar a ligação éster (C-O) por meio de um mecanismo alternativo de hidrólise formando como produtos, ácidos carboxílicos e álcoois. O PU-MDI (difenilmetano diisocianato) apresenta em sua estrutura química (Figura 1) dois grupos funcionais do tipo éster que o torna susceptível a degradação enzimática por meio das esterases. Dessa forma, o principal fator é a presença de grupos químicos semelhantes, os quais possibilitam a interação dos substratos não naturais com as enzimas.14,15
Essas duas esterases PueA e PueB foram objetos de estudo in silico do grupo de pesquisa17 ao qual o autor está inserido mostrando por meio dos resultados do docking e da MD interações favoráveis e boa estabilidade com o PU-MDI. Por outro lado, essas esterases também foram objeto de estudo in situ em que os resultados apontaram atividade catalítica de ambas na presença do PU.18 Nesse sentido, o objetivo dessa pesquisa, com base nos conhecimentos estruturais e dinâmicos prévios a respeito destes sistemas, é propor e estudar o mecanismo de degradação do PU pelas enzimas PueA e PueB usando as técnicas de química computacional supramencionadas.
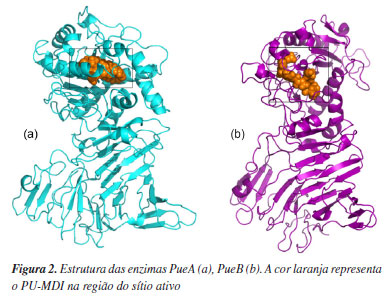
METODOLOGIA Estruturas Os modelos tridimensionais (3D) da PueA e da PueB foram determinados por modelagem comparativa. Essa técnica é baseada na ideia de que as estruturas 3D e a função de duas proteínas ou enzimas são provavelmente semelhantes se suas sequências primárias tiverem pelo menos 25% de identidade.19 Dessa forma, é possível determinar teoricamente as estruturas 3D de enzimas, se houver uma estrutura 3D de uma enzima homóloga já disponível em bancos de dados como o PDB (do inglês, Protein Data Bank).20 Também é possível notar que as estruturas internas, por exemplo: α-hélices e folhas-β, são geralmente conservadas em enzimas homólogas além de estruturas externas, como loops.21,22 As sequências primárias das enzimas PueA e PueB foram obtidas do GenBank, o banco de dados de sequência genética da NCBI (do inglês, National Center of Biotechnology Information).23 A busca de enzimas homólogas a serem usadas como modelos na modelagem por homologia foi realizada pelo servidor BLAST (Basic Local Alignment Search Tool) usando o algoritmo BLASTp.23 Os múltiplos alinhamentos foram obtidos usando o algoritmo Muscle24,25 no programa AliView.26 A partir do melhor alinhamento entre estruturas de modelo e sequências de destino, o Modeller 9.827 foi usado para construir os modelos das enzimas por meio do método de restrições espaciais. O melhor modelo para cada enzima foi selecionado com base nos resultados do PROCHECK28 e Verify-3D.29 A análise de sequências para a determinação dos domínios e locais funcionais foi realizada pelo PROSITE.30,31 A estrutura do monômero de PU-MDI (Figura 1) foi construída no Gauss View32 e otimizada no programa Gaussian 0933 usando o funcional B3LYP34 e as funções de base 6-31G(d,p).35 Após a otimização verificou-se a ausência de frequências negativas indicando que aquela era a geometria molecular de menor energia. Também se otimizou o monômero com solvente implícito, não observando diferenças significativas na energia das duas estruturas. Além disso, foi analisado os estados de protonação em pH neutro usando Marvin Sketch 19.836 o qual é precisamente aquele mostrado na Figura 1. Ainda, este padrão contribui como 100% até o pH 8,40 atingindo 99% em pH 10,80 e 90% em pH 11,80. Somente acima do pH 12,00 que outras formas, como o NH desprotonado, passam a cumprir um papel. Tanto a estrutura das enzimas (Figura 2) quanto a do monômero (Figura 1) foram construídas em trabalhos anteriores16,17 do nosso grupo de pesquisa.
Docking molecular Depois de obtidas as estruturas dos receptores e do ligante foi realizado o docking molecular para investigar a interação entre eles com o objetivo de prever a conformação mais estável do complexo enzima substrato.37 Nesse sentido, o docking foi realizado pelo DockThor,37 um servidor web, assim submeteu-se os arquivos no formato PDB das enzimas e do PU-MDI. A única modificação foi a troca do estado de protonação da HIS313, um dos aminoácidos da tríade catalítica presente no sítio ativo da PueA, da posição épsilon, padrão em pH neutro, para a posição delta. Esse ajuste no estado de protonação foi necessário para viabilizar a transferência do H da SER207 para o N da posição épsilon, primeiro passo do mecanismo de hidrólise, conforme mostrado na Figura 1Sa, Material Suplementar. Desse modo, configurou-se o centro do grid, em ambas as enzimas, de forma cúbica com arestas de 20 Å, totalizando uma malha de 531.441 pontos, centralizado nas coordenadas do átomo de oxigênio (OG) dos resíduos catalíticos, SER207 no caso da PueA e SER152 na PueB. Dinâmica molecular Após o docking, parametrizou-se o monômero com o programa AcPype,38 por outro lado, a topologia das enzimas foi construída por meio do comando pdb2gmx do GROMACS 2023.2.39 Além disso, usou-se o mesmo comando para adicionar hidrogênios e modificar o estado de protonação da HIS313 para posição delta na estrutura da PueA. Na PueB não foi alterado o estado de protonação padrão em meio neutro de nenhum resíduo. Em seguida, os complexos (enzima-substrato), PueA e PueB, além do PU-MDI livre, foram montados em caixa cúbica de acordo com a Tabela 1.
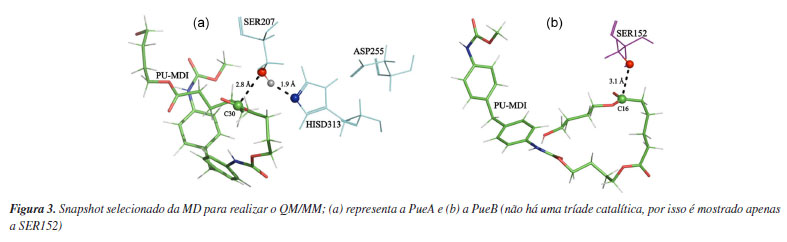
Os dois sistemas foram, primeiramente, relaxados por meio do algoritmo steepest descent com um máximo de 50,000 passos, depois sujeitos a equilibração nos ensembles NVT e NPT por 100 ps em cada um. Por fim a MD de 500 ns (nanossegundos), em triplicata, no ensemble NPT com temperatura de 310 K mantida constante pelo termostato V-rescale40 e a pressão controlada pelo barostato Parrinello-Rahman.41 Além disso, o tempo do passo foi de 2 fs (fentossegundos) e as interações de longo alcance foram calculadas com o algoritmo PME (Particle-Mesh Ewald) usando um corte de 10 Å. Ademais, o modelo de água usado foi o TIP3P42 em concentração fisiológica (0,154 mol L-1 de NaCl), o campo de força escolhido para descrever o sistema foi o AMBER03.43 Utilizou-se de forma análoga, esse procedimento, para a simulação do PU-MDI livre. QM/MM Essa abordagem permite estudar o mecanismo de reação por meio da mecânica quântica, QM (do inglês, quantum mechanics) enquanto os efeitos do meio circundante são modelados usando mecânica molecular, MM (do inglês, molecular mechanics). Com esse objetivo selecionou-se a configuração mais adequada a partir dos frames da trajetória da MD conforme apresentado na Figura 3. No caso do complexo PueA escolheu-se um frame com a menor distância entre o hidrogênio (HG) da SER207 e o nitrogênio (NE2) da HISD313 ao mesmo passo da menor distância do oxigênio (OG) da SER207 e do carbono (C30) do PU-MDI. Já no complexo PueB em que há apenas um resíduo catalítico procurou-se o frame de menor distância entre o oxigênio (OG) da SER152 e o carbono (C16) do PU-MDI, a nomenclatura desses átomos está detalhada nas Figuras 6S e 7S (Material Suplementar). Esse critério de seleção dos snapshots da MD em que envolvem as menores distâncias dos átomos no ataque nucleofílico foi baseado em outro trabalho44 com mecanismo reacional semelhante. Por outro lado, também se selecionou, entretanto, de forma aleatória um frame da MD do ligante livre em meio aquoso para comparar as barreiras energéticas dos mecanismos com e sem catalisador.
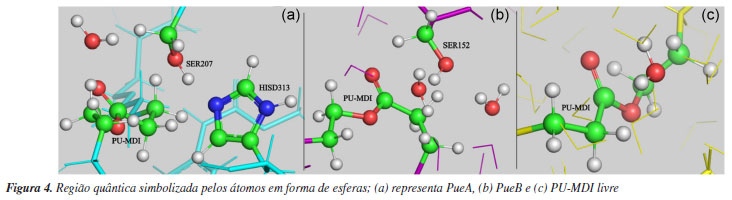
A partir dos snapshots descritos acima realizou-se um corte esférico de cada um dos três sistemas com raio de 20 Å a contar do PU-MDI como mostra a Tabela 2. Na sequência fixou-se os átomos da extremidade da esfera sendo que sua espessura foi de 2 Å, em seguida a otimização de geometria usando o campo de força Amber, depois selecionou-se a região QM conforme Figura 4. Essa parte foi descrita pelo hamiltoniano semi-empírico PM6,45 pois foi o método que melhor descreveu o perfil de energia potencial tanto para a PueA quanto para a PueB no conjunto dos métodos semi-empíricos testados: AM1, AM1dphot, PM3 e PM6 conforme indicam os resultados nas Figuras 8S e 9S (Material Suplementar). Por fim, os sistemas foram otimizados novamente usando na região QM o hamiltoniano semiempírico PM6.
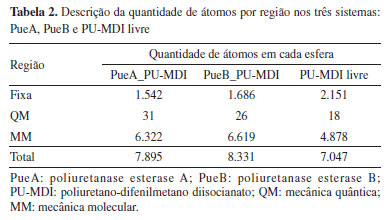
Com cada sistema otimizado e suas regiões QM e MM definidas, iniciou-se a varredura de coordenadas de reação com o objetivo de calcular a energia potencial em função da posição dos átomos. As varreduras dos três sistemas, PueA, PueB e PU-MDI livre, foram realizadas em uma dimensão (1D) sendo um total de 53 passos com tamanho de 0,08 Å para a PueA, 55 passos e 0,08 Å para PueB e 32 passos e 0,05 Å para o PU-MDI livre, sendo que para os três sistemas usou-se uma constante de força equivalente a 4.000 kJ mol-1 Å-2. Finalmente, a partir dos resultados dessa varredura procedeu-se com o umbrella sampling (US)46,47 (também em 1 D), sendo que a constante de força foi de 1.300 kJ mol-1 Å-2 e 20 ps em cada janela de simulação. Posteriormente, aplicou-se a metodologia WHAM (weighted histogram analysis method)48,49 para se obter o perfil de energia livre. Todo procedimento QM/MM foi realizado com a interface gráfica EasyHybrid 3.050 e os cálculos rodados com o software pDynamo3.51
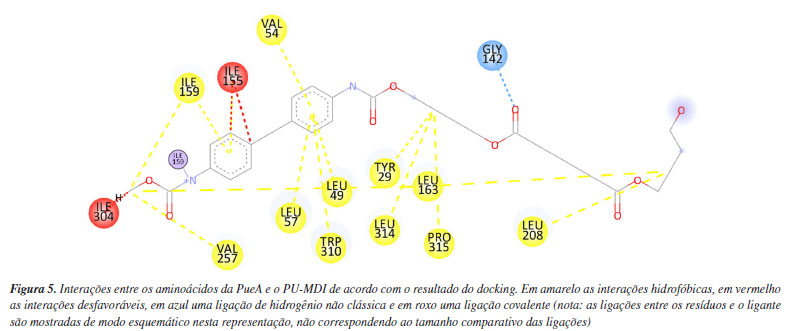
RESULTADOS E DISCUSSÃO O docking do PU-MDI na PueA apresentou as seguintes interações com o ligante, conforme Figura 5. Nesse sentido, observa-se que a maioria das interações foram hidrofóbicas alquil, pi-alquil e entre anéis aromáticos, seguido de uma interação covalente e uma ligação de hidrogênio não clássica entre o oxigênio da carbonila do PU-MDI e o hidrogênio ligado covalentemente ao carbono (sp3) da GLY142. Essa interação intermolecular é considerada um pouco mais fraca do que as ligações de hidrogênio clássicas conforme estudado por Johnston e Cheong.52 Além disso, nota-se três interações desfavoráveis com os resíduos ILE155 e ILE304, possivelmente, causadas por sobreposições espaciais ou colisões estéricas.
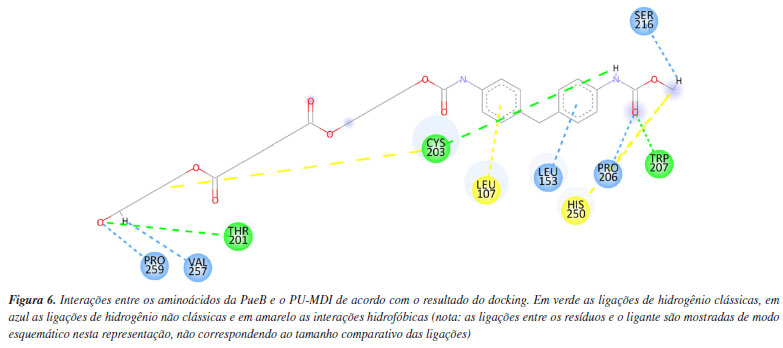
Por outro lado, o resultado do docking da PueB (Figura 6) mostrou mais interações polares, sendo três ligações de hidrogênio clássicas e cinco não clássicas. Também se formaram interações apolares do tipo alquil e pi-alquil. Dessa forma, ao se comparar as duas enzimas quanto às interações com o PU-MDI, percebe-se que a PueA embora tenha interações desfavoráveis, há um número maior de resíduos interagindo com o substrato. Ademais, em termos numéricos a função score da PueA e da PueB foram, respectivamente, -10,743 e -9,587 kcal mol-1. Ou seja, o PU-MDI, aparentemente, se ajusta melhor em um sítio ativo mais hidrofóbico (como da PueA) do que hidrofílico (como da PueB).
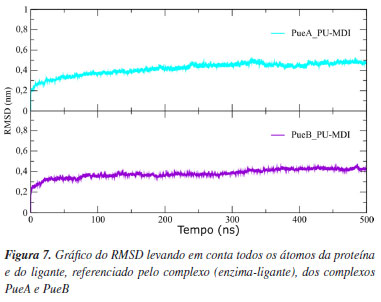
As simulações MD foram analisadas do ponto de vista da estabilidade, flexibilidade (Figura 4S) e interação (Figura 5S) com o ligante. Dessa maneira, a partir do gráfico da raiz quadrada do desvio quadrático médio (RMSD, do inglês root mean square deviation), apresentado na Figura 7, observou-se que os dois complexos, PueA e PueB, mantiveram boa estabilidade ao longo da simulação, no caso da PueA, aproximadamente, após 150 ns a sua oscilação ficou entre 0,3 e 0,5 nm (nanômetro) enquanto que a PueB se estabilizou depois dos 100 ns, girando em torno de 0,4 nm.
Reação de acilação O mecanismo proposto para degradar o PU teve como base o mecanismo teórico de reação da hidrólise de ligações éster catalisada pelas esterases.53 Nesse sentido, a Figura 8 ilustra as etapas da degradação que ocorrem no sítio ativo da PueA bem como as correspondentes variações de energia livre.
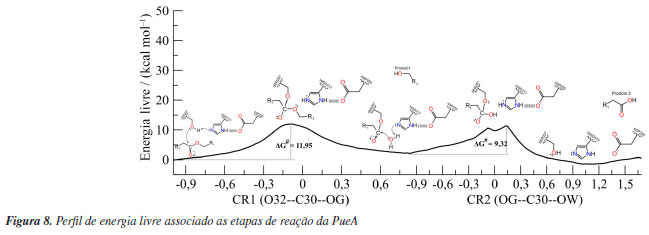
Os resultados do US sugerem um mecanismo de duas etapas, acilação e desacilação. Na primeira, há o ataque nucleofílico do OG da SER207 ao C30 do PU-MDI e, simultaneamente, a transferência do HG da SER207 para o NE2 da HISD313, ressalta-se que a desprotonação da SER207 ocorreu sem a necessidade de realizar uma varredura em duas dimensões (2D). Recentemente, Moliner e colaboradores54 estudaram esse mesmo mecanismo usando US em 2D e seus resultados, também, apontaram que o ataque nucleofílico e a desprotonação ocorrem ao mesmo tempo. Analisando as cargas parciais desses átomos conforme Tabela 1S (Material Suplementar) nota-se que a carga do OG diminuiu de -0,60 para -0,64 no TS1 (estado de transição, em inglês, transition state) aumentando a nucleofilicidade do SER207 enquanto que a carga do NE2 aumenta de -0,38 para -0,20 aumentando seu caráter ácido. A carga do C30 aumenta de 0,66 para 0,77 durante a formação da ligação química com o OG, por outro lado a carga do O31 passa de -0,56 a -0,82 pois esse oxigênio durante a formação do intermediário tetraédrico (TI), do inglês, tetrahedral intermediate, passa a fazer uma ligação simples e o O32 diminui sua carga parcial levando a quebra da ligação éster (C30--O32) do PU-MDI. Essa etapa tem uma barreira energética de 11,95 kcal mol-1. A energia de ativação calculada recentemente para esse mecanismo de degradação do PU usando, também, a PueA por outro grupo de pesquisa54 aplicando uma metodologia semelhante de QM/MM encontrou uma barreira de 22 kcal mol-1. Essa diferença nos valores pode ser atribuída aos diferentes ligantes, nesse e naquele estudo, além da escolha do método semi-empírico e a quantidade de átomos incluídos na região quântica. Depois, da transferência do HG para o O32 regenerando a HISD313, consequentemente, formando um intermediário acil-enzima (AE) e o primeiro produto, um álcool. Na PueB, a fase de acilação pode ser observada na Figura 9. Essa enzima tem apenas um resíduo catalítico (SER152), enquanto a PueA tem uma tríade catalítica (SER207, HISD313 e ASP255). Nessa primeira fase o mecanismo de degradação da PueB foi desenvolvido com base nos trabalhos de Ekici et al.,55 Lee e colaboradores,56 Hol e colaboradores.57 Esses autores investigaram a família das serina hidrolases, não comum, as quais tem no sítio ativo apenas a serina como resíduo catalítico. Nesse sentido, seus estudos sugerem que uma molécula de água pode desempenhar a função equivalente à histidina durante a catálise. Dessa forma, o ataque nucleofílico do oxigênio (OG) da SER152 ao C16 do PU-MDI e a transferência do hidrogênio (HG) para o oxigênio (OW) da água ocorrem concomitantemente formando o TI1. Analisando as cargas formais desses átomos na Figura 2S observa-se uma leve diminuição da carga do OG de -0,59 para -0,47 indicando que a SER152 tem um menor caráter nucleofílico do que a SER207. Já as cargas parciais dos átomos da molécula de água nessa fase se mantêm, praticamente, sem alterações, enquanto que a carga do C16 aumenta de 0,65 para 0,68 e a carga do O15 aumenta de -0,47 para -0,61 levando a quebra da ligação éster (O15--C16) do PU-MDI. Essa análise das cargas mostra que a densidade eletrônica se concentra no O15 e não no oxigênio da carbonila como ocorre na PueA, isso pode ser devido a um maior número de moléculas de água nas proximidades do mecanismo da PueB. Dessa forma, a fase de acilação ocorre com um custo energético de 15,10 kcal mol-1. Nota-se que a PueB precisou de 3,15 kcal mol-1 a mais para formar o TS1 em comparação com a PueA. Outrossim, observa-se nas Figuras 8 e 9 que a barreira energética dessa fase de acilação das duas enzimas é a própria etapa determinante da reação. Nesse sentido, a tríade catalítica da PueA se mostrou mais eficiente do que um único resíduo catalítico (da PueB), esse resultado vai ao encontro dos obtidos experimentalmente por Crookes-Goodson e colaboradores.18 Essa etapa é finalizada com a formação do intermediário AE e do álcool.
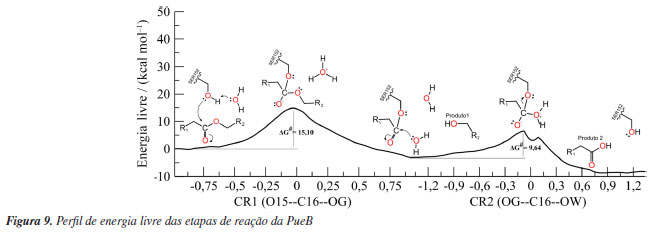
Reação de desacilação Nessa segunda etapa, o mecanismo da PueA segue com o ataque nucleofílico do OW da molécula de água ao C30 do PU-MDI simultâneo a transferência do HW1 para o NE2 da HISD313 formando o segundo intermediário tetraédrico (TI2) com uma barreira energética de 9.32 kcal mol-1. Observa-se um aumento da carga do C30 (de 0,68 para 0,78) e um aumento da carga do OW (de -0,63 para -0,69) bem como a diminuição da carga do ND1 (de -0,20 para -0,11). Posteriormente, ocorre a quebra da ligação entre os átomos C30 e OG da SER207 e a transferência do HW1 da HISD313 para o OG, dessa forma, regenerando a SER207 e formando o segundo produto, um ácido carboxílico. Esses produtos, álcool e ácido, também foram previstos por Kemona e Piotrowska.58 A desacilação da PueB inicia-se com o ataque nucleofílico do OW da molécula de água ao C16 do PU-MDI formando o segundo intermediário tetraédrico seguindo para a transferência do HW2 para o OG da SER152 e a quebra da ligação entre os átomos OG--C16 regenerando a SER152 e formando o segundo produto semelhante a PueA. Essa etapa mostrou que a barreira energética de ambas enzimas foi praticamente igual, mesmo com a PueB tendo apenas um resíduo catalítico. Outro fator que interfere nos mecanismos das hidrolases é a formação da cavidade oxiânion, que pode ser formada por moléculas de água ou por resíduos da enzima. Nos sítios de ligação, tanto da PueA e da PueB, essa região é formada por resíduos das cadeias laterais, sendo a LEU208 e a GLY142 na PueA e a LEU153 e a CYS203 na PueB. Eles estabilizam a carga gerada no oxigênio da carbonila durante a formação dos intermediários tetraédricos. O principal objetivo das enzimas é diminuir a energia de ativação, para isso elas propõem um mecanismo alternativo de menor energia conforme detalhado para PueA e PueB nas Figuras 1S e 2S. Desse modo, com a finalidade de comparar a energia de ativação da hidrólise com e sem biocatalisador também simulou-se a hidrólise do PU-MDI livre (Figura 3S). A Figura 10 mostra a barreira energética do PU-MDI.
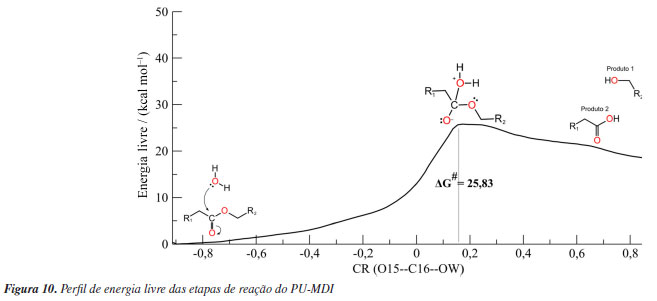
Diante disso, nota-se que a barreira energética da hidrólise sem catalisador é maior. Ou seja, tanto o mecanismo da PueA quanto o da PueB diminuem a energia de ativação indicando que podem biocatalisar a degradação do PU-MDI. Ainda se observou que a barreira energética da PueA é 3,15 kcal mol-1 menor do que a da PueB corroborando com os resultados experimentais do Crookes-Goodson e colaboradores.18
CONCLUSÕES O presente trabalho, por meio de ferramentas computacionais, estudou o mecanismo de degradação do PU-MDI catalisado pelas serina hidrolases PueA e PueB. O resultado do docking mostrou que a PueA teve mais resíduos interagindo com o PU-MDI em seu sítio ativo do que a PueB, indicando maior afinidade de ligação no complexo PueA. Já a MD demonstrou que o PU-MDI permaneceu no sítio ativo ao longo dos 500 ns de simulação em todas as réplicas, sugerindo boa interação e estabilidade na conformação dos complexos. Por outro lado, com a abordagem QM/MM foi possível explorar o mecanismo da reação com e sem catalisador. Nesse sentido, a parte QM descrita pelo hamiltoniano semi-empírico PM6 indicou barreiras energéticas favoráveis para a ocorrência da reação indicando a eficiência catalítica das duas enzimas. Ainda se notou que a energia da ativação da PueA foi reduzida em, aproximadamente, 54% e da PueB em 41% quando comparadas com a barreira energética da reação em meio aquoso. A partir desses resultados estudos futuros podem vir a abordar dímeros ou oligômeros, visando uma descrição mais próxima da realidade experimental. Por fim, a pesquisa pode contribuir para o desenvolvimento de tecnologias sustentáveis de gestão de resíduos, promovendo alternativas biológicas para a degradação de PUs, que são notoriamente resistentes à degradação ambiental. A identificação de micro-organismos e enzimas eficazes, como PueA e PueB, pode levar à criação de processos de tratamento de resíduos mais ecológicos e economicamente viáveis, reduzindo a dependência de métodos físicos e químicos tradicionais, que muitas vezes são caros e poluentes.
MATERIAL SUPLEMENTAR Algumas figuras e tabelas dos mecanismos utilizados neste trabalho estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS À Universidade Federal do Rio Grande do Sul (UFRGS) e à Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA). Ao Centro Nacional de Supercomputação (CESUP) pelo apoio operacional, à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo apoio financeiro.
REFERÊNCIAS 1. Organisation for Economic Co-operation and Development (OECD); Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options; OECD Publishing: Paris, 2022. [Crossref] 2. Plastics Europe, Plastics - the Fast Facts 2023, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/, acessado em Setembro 2024. 3. Organisation for Economic Co-operation and Development (OECD); Global Plastics Outlook: Policy Scenarios to 2060; OECD Publishing: Paris, 2022. [Crossref] 4. Yusuf, A. A.; Yusuf, D. A.; Jie, Z.; Bello, T. Y.; Tambaya, M.; Abdullahi, B.; Muhammed-Dabo, I. A.; Yahuza, I.; Dandakouta, H.; Atmos. Pollut. Res. 2022, 13, 101258. [Crossref] 5. Costa, C. Z.; de Albuquerque, M. D. C. C.; Brum, M. C.; de Castro, A. M.; Quim. Nova 2015, 38, 259. [Crossref] 6. Baldera-Moreno, Y.; Pino, V.; Farres, A.; Banerjee, A.; Gordillo, F.; Andler, R.; Polymers 2022, 14, 375. [Crossref] 7. Flores-Castañón, N.; Sarkar, S.; Banerjee, A.; J. Hazard. Mater. Lett. 2022, 3, 100063. [Crossref] 8. Romão, W.; Spinacé, M. A. S.; de Paoli, M. A.; Polim.: Cienc. Tecnol. 2009, 19, 121. [Crossref] 9. Austin, H. P.; Allen, M. D.; Donohoe, B. S.; Rorrer, N. A.; Kearns, F. L.; Silveira, R. L.; Pollard, B. C.; Dominick, G.; Duman, R.; El Omari, K.; Mykhaylyk, V.; Wagner, A.; Michener, W. E.; Amore, A.; Skaf, M. S.; Crowley, M. F.; Thorne, A. W.; Johnson, C. W.; Woodcock, H. L.; McGeehan, J. E.; Beckham, G. T.; Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4350. [Crossref] 10. Alves, L. R.; Carriello, G. M.; Pegoraro, G. M.; Rezende, M. L.; de Menezes, A. J.; Disciplinarum Scientia 2022, 23, 99. [Crossref] 11. Stern, R. V.; Howard, G. T.; FEMS Microbiol. Lett. 2000, 185, 163. [Crossref] 12. Howard, G. T.; Crother, B.; Vicknair, J.; Int. Biodeterior. Biodegrad. 2001, 47, 141. [Crossref] 13. Vaz, M.; Choupina, A.; Revista Eletrônica de Biologia 2012, 5, 42. [Link] acessado em Setembro 2024 14. Howard, G. T.; Blake, R. C. B.; Int. Biodeterior. Biodegrad. 1998, 42, 213. [Crossref] 15. Howard, G. T.; Int. Biodeterior. Biodegrad. 2002, 49, 245. [Crossref] 16. do Canto, V. P.; Thompson, C. E.; Netz, P. A.; J. Mol. Model. 2021, 27, 46. [Crossref] 17. do Canto, V. P.; Thompson, C. E.; Netz, P. A.; J. Mol. Graphics Modell. 2019, 89, 82. [Crossref] 18. Nadeau, L. J.; Barlow, D. E.; Hung, C. S.; Biffinger, J. C.; Crouch, A. L.; Hollomon, J. M.; Ecker, C. D.; Russell, J. N.; Crookes-Goodson, W. J.; Int. Biodeterior. Biodegrad. 2021, 156, 105121. [Crossref] 19. Sali, A.; Blundell, T. L.; J. Mol. Biol. 1993, 234, 779. [Crossref] 20. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. [Crossref] 21. Webb, B.; Sali, A.; Curr. Protoc. Bioinf. 2014, 47, 1. [Crossref] 22. Santos Filho, O. A.; de Alencastro, R. B.; Quim. Nova 2003, 26, 253. [Crossref] 23. Benson, D. A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D. J.; Ostell, J.; Sayers, E. W.; Nucleic Acids Res. 2013, 41, D36. [Crossref] 24. Edgar, R. C.; Nucleic Acids Res. 2004, 32, 1792. [Crossref] 25. Edgar, R. C.; BMC Bioinf. 2004, 5, 113. [Crossref] 26. Larsson, A.; Bioinformatics 2014, 30, 3276. [Crossref] 27. Sali, A.; Modeller, 9v4, r6262; University of California, San Francisco, USA, 2008. 28. Laskowski, B. A. R.; Macarthur, M. W.; Thornton, J. M.; J. Appl. Crystallogr. 1993, 26, 283. [Crossref] 29. Lüthy, R.; Bowie, J. U.; Eisenberg, D.; Nature 1992, 356, 83. [Crossref] 30. Sigrist, C. J. A.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P.; Briefings Bioinf. 2002, 3, 265. [Crossref] 31. de Castro, E.; Sigrist, C. J. A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P. S.; Gasteiger, E.; Bairoch, A.; Hulo, N.; Nucleic Acids Res. 2006, 34, W362. [Crossref] 32. Dennington, R.; Keith, T. A.; Millam, J. M.; GaussView, version 6; Semichem Inc., Shawnee Mission, USA, 2016. 33. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, P. G.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.; Gaussian 09, rev. D. 01; Gaussian Inc., Wallingford, USA, 2013. 34. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. [Crossref] 35. Rassolov, V. A.; Ratner, M. A.; Pople, J. A.; Redfern, P. C.; Curtiss, L. A.; J. Comput. Chem. 2001, 22, 976. [Crossref] 36. Marvin Sketch, versão 19.8; ChemAxon, Cambridge, 2019. 37. Santos, K. B.; Guedes, I. A.; Karl, A. L. M.; Dardenne, L. E.; J. Chem. Inf. Model. 2020, 60, 667. [Crossref] 38. Silva, D.; Vranken, B. F.; BMC Res. Notes 2012, 5, 367. [Crossref] 39. Abraham, M.; Alekseenko, A.; Bergh, C.; Blau, C.; Briand, E.; Doijade, M.; Fleischmann, S.; Gapsys, V.; Garg, G.; Gorelov, S.; Gouaillardet, G.; Gray, A.; Irrgang, M. E.; Jalalypour, F.; Jordan, J.; Junghans, C.; Kanduri, P.; Keller, S.; Kutzner, C.; Lemkul, J. A.; Lundborg, M.; Merz, P.; Miletić, V.; Morozov, D.; Páll, S.; Schulz, R.; Shirts, M.; Shvetsov, A.; Soproni, B.; Spoel, D. V. D.; Turner, P.; Uphoff, C.; Villa, A.; Wingbermühle, S.; Zhmurov, A.; Bauer, P.; Hess, B.; Lindahl, E.; Gromacs, version 2023.2; Institute of Technology and Uppsala University, Sweden, 2023. 40. Bussi, G.; Donadio, D.; Parrinello, M.; J. Chem. Phys. 2007, 126, 14101. [Crossref] 41. Parrinello, M.; Rahman, A.; J. Appl. Phys. 1981, 52, 7182. [Crossref] 42. Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L.; J. Chem. Phys. 1983, 79, 926. [Crossref] 43. Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M. C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; Caldwell, J.; Wang, J.; Kollman, P.; J. Comput. Chem. 2003, 24, 1999. [Crossref] 44. Lima, M. C. P.; Seabra, G. M.; Phys. Chem. Chem. Phys. 2016, 18, 30288. [Crossref] 45. Stewart, J. J. P.; J. Mol. Model. 2007, 13, 1173. [Crossref] 46. Kästner, J.; Senn, H. M.; Thiel, S.; Otte, N.; Thiel, W.; J. Chem. Theory Comput. 2006, 2, 452. [Crossref] 47. Kästner, J.; Comput. Mol. Sci. 2011, 1, 932. [Crossref] 48. Kumar, S.; Rosenberg, J. M.; Bouzida, D.; Swendsen, R. H.; Kollman, P. A.; J. Comput. Chem. 1992, 13, 1011. [Crossref] 49. Roux, B.; Comput. Phys. Commun. 1995, 91, 275. [Crossref] 50. Bachega, J. F. R.; Timmers, L. F. S. M.; Assirati, L.; Bachega, L. R.; Field, M. J.; Wymore, T.; J. Comput. Chem. 2013, 34, 2190. [Crossref] 51. Field, M. J.; J. Chem. Inf. Model. 2022, 62, 5849. [Crossref] 52. Johnston, R. C.; Cheong, P. H. Y.; Org. Biomol. Chem. 2013, 11, 5057. [Crossref] 53. Rauwerdink, A.; Lunzer, M.; Devamani, T.; Jones, B.; Mooney, J.; Zhang, Z. J.; Xu, J. H.; Kazlauskas, R. J.; Dean, A. M.; Mol. Biol. Evol. 2016, 33, 971. [Crossref] 54. Świderek, K.; Marti, S.; Arafet, K.; Moliner, V.; Faraday Discuss. 2024, 252, 323. [Crossref] 55. Ekici, Ö. D.; Paetzel, M.; Dalbey, R. E.; Protein Science 2008, 17, 2023. [Crossref] 56. Yoon, J.; Oh, B.; Kim, K.; Park, J.; Han, D.; Kim, K. K.; Cha, S. S.; Lee, D.; Kim, Y.; J. Biol. Chem. 2004, 279, 341. [Crossref] 57. Kim, Y.; Kim, S.; Earnest, T. N.; Hol, W. G. J.; J. Biol. Chem. 2002, 277, 2823. [Crossref] 58. Kemona, A.; Piotrowska, M.; Polymers 2020, 12, 1752. [Crossref]
Editor Associado responsável pelo artigo: Eduardo H. S. Sousa |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access