
 |
 |
 |
 |
 |
Artigo
| Exploring internal standards and matrix matching for lanthanides determination in tea by LA-ICP-MS |
|
Ana Caroline Vedovato RubinI; Vinicius Machado NevesI; Ana Barbosa VianaI I. Departamento de Química, Universidade Federal de Santa Maria, Campus de Camobi, 97105-900 Santa Maria - RS, Brasil Received: 06/20/2024 *e-mail: vdressler@gmail.com Lanthanides (Lns) have been increasingly used in different fields, raising some concerns due to potential adverse effects to human life. In this work, it is proposed a laser ablation-inductively coupled plasma mass spectrometry (LA-ICP-MS) method for Lns determination in tea. Cellulose powder, leave powder and filter paper were evaluated as matrix matched standards and 13C, 103Rh, 115In, 197Au and 205Tl as internal standards (IS). Operating conditions of the LA-ICP-MS system, sample preparation procedures, and spiking samples with IS and standards were optimized. Better conditions of IS mixed with powdered standards or samples was achieved when the powder particles size was lower than 50 μm and 2 mL of IS or standard solutions were added to 500 mg of sample. Best results were obtained using Au as IS and filter paper. The method was applied to Lns determination in mint tea, where Ce, La and Nd concentrations were 580 ± 50, 282 ± 15 and 340 ± 60 ng g-1, respectively. The concentrations of the other Lns were below their limit of quantification (LOQ). The Lns concentrations determined by LA-ICP-MS were in good agreement (95% confidence level, t-test) with those found using ultrasonic nebulization-ICP-MS with a precision better than 20%. INTRODUCTION The lanthanides elements (Lns) are employed in electronic devices, fertilizers, lasers, rechargeable batteries, ceramics, medicine, catalysts, new materials, etc.1-5 However, the Lns effects on the environment and human health is little known and has drawn attention of the scientific community and environmental and health agencies.4-6 It has been observed that Lns application in soil resulted in a considerable growth of roots and aerial parts of plants.7 Ni et al.8 affirmed that the presence of Lns in tea received little attention when compared with other elements, despite suspicion that excessive exposure to Lns oxides can be harmful to human health. A compressive discussion on the effects of the lanthanides can be found in Gómez-Merino et al.9 Beneficial and harmful effects of these elements on plants have been reported and may be element, concentration and time of exposure dependent. So, it is known that La and Ce have a beneficial effect on the growth of certain plant species. In the same sense, it is reported that some lanthanides improve germination if the seed is previously treated with the element. Lanthanides have similar chemical and physical characteristics where the electrons of the outer layer are distributed in the 4f orbital, the ionic radius varies only from 1.06 Å for La3+ to 0.85 Å for Lu3+ and 3+ is the most common valence. These characteristics of the Lns elements make their determination unusually difficult, mainly by classical methods of determination.10 Therefore, in recent years, more appropriate techniques for Lns determination have been employed, mainly inductively coupled plasma optical emission spectrometry (ICP OES), inductively coupled plasma mass spectrometry (ICP-MS), X-ray fluorescence (XRF) and neutron activation analysis (NAA).11 However, drawbacks also exist in the application of such techniques. For instance, NAA requires special instruments, mainly a nuclear reactor, and the limit of detection (LOD) is relatively high, impairing its application in many cases, especially in biological and environmental material analysis. In the case of XRF the LOD is also high for the determination of most of these elements. On the other hand, ICP OES and ICP-MS are currently the main techniques used for Lns determination,11 mainly due to the multielemental analysis feature, the high sample throughput and the low LOD achieved. In addition, ICP-MS allows isotopic analysis. Emission spectra of Lns are complex and spectral lines overlap are prone to occur in ICP OES. Nevertheless, modern instruments are equipped with monochromator capable of good resolution and software with algorithms that allow spectral interferences correction. One of the main difficulties in Lns determination using ICP-MS is the interference by polyatomic ions, mainly oxide and hydroxide ions generated in the plasma. The most remarkable interferent ions are those from Ba and lighter Lns on heavier Lns. These interferences ions are generated by reaction with oxygen present in the plasma gas, atmosphere, and mainly solvent in the analysis of solutions. However, such interferences can be considerably reduced through solvent removal. To this end, nebulizers associated with aerosol desolvation units, electrothermal vaporization (ETV) or laser ablation (LA) can be employed to introduce the sample into the ICP.12 Nevertheless, Lns determination by ETV-ICP-MS is difficult due to refractory carbides formation in the graphite platform, requiring special conditions and sample throughput is very low. In this sense, LA allows direct solid sample analysis, avoiding solvent introduction in the plasma, decreasing oxides and hydroxides generated from solvent in the ICP. In addition, LA allows elements mapping in the sample. Mapping in 2D and 3D dimensions with spatial resolution in the range of μm can be achieved.12-16 In laser ablation-inductively coupled plasma mass spectrometry (LA-ICP-MS), vapor and fine particles of the sample containing the analytes are formed in the sample ablation process and then transported with the aid of a carrier gas to the ICP-MS instrument for analyte detection. The ablation process, vapor/particles transport to the ICP and ionization in the ICP are affected by the sample matrix and inaccurate results are obtained if the standards and samples matrices are not similar.17,18 For that reason, calibration with certified reference materials (CRMs) whose matrix is similar to the sample, in-house prepared matrix matched standards, standard addition calibration, and solution-based calibration have been proposed.14,17,19,20 In relation to matrix matching calibration for biological material analysis, different materials are proposed and prepared in-house.21 In this case, standards are adjusted to the specific tissue under study by spiking homogenized tissue material with the elements of interest. In this way, spiked matrix-matched tissue resembles the actual matrix of the sample under investigation. Another commonly accepted form is the use of spiked material that mimics the composition of the biological sample. For plant tissues, this implies the use of pure cellulose paper, e.g. filter paper, hydrocolloid gels, and other materials.22-24 However, regardless the calibration approach, internal standardization in LA-ICP-MS analysis is usually necessary to achieve satisfactory precision and accuracy;25,26 internal standardization compensates several effects that occurs in sample ablation, aerosol transport and ionization processes in the ICP. For better compensation, similar mass to charge ratio (m/z), ionization energy and volatilization temperature of analyte and the internal standards (IS) are required. The IS must also be free of spectral interferences and homogenously distributed in the sample.25 As such, any element that does not fulfil all these requirements cannot be used as an IS. Usually, the IS must be chosen experimentally. Isotopes of elements naturally present in sample and standards (for example, 12C and 13C in plants) or spiked to them have been used as IS.12,17 Carbon has the advantage of being uniformly distributed in the plant tissues,26,27 but the low m/z of 12C and 13C and high ionization potential of C might compromise precision and accuracy for some elements determined in plants. Another drawback is related to the ablation process, in which carbon in gas phase or in smaller particles can be released from the solid sample or standards. As such, the behavior of IS and analyte in the ICP will be different.28,29 Even so, calibration in LA-ICP-MS is not well consolidated and is a current research field. Calibration with matrix matched standards for Lns determination in mint tea was evaluated in the present work. Pellets of cellulose powder, leave powder, and filter paper were evaluated for matrix matched standards preparation and 13C, 103Rh, 115In, 197Au, 205Tl as IS. Accuracy was accessed through sample digestion and analyte determination by ultrasonic nebulization-ICP-MS (USN-ICP-MS). Mint tea was chosen to show the performance of the method, since the concentration of the lanthanides in this plant is relatively high.
EXPERIMENTAL Reagents, solutions and materials Ultrapure water (resistivity of 18 MΩ cm) was produced by a Milli-Q system (Millipore corp., USA) and was used for solutions preparation and materials cleaning. Concentrated HNO3 (65% m m-1, Merck, Germany) was purified by sub-boiling distillation (Milestone, Model Duopur, Italy) and used for solutions and samples preparation. Calibration solutions were obtained by serial dilution of a multielement solution containing 10 mg L-1 (CLMS 1, SPEX CertiPrep, USA) of Lns. For internal standardization, Rh, In, Au and Tl solutions were prepared by serial dilution of 1000 mg L-1 (Certipur, Merck IV, Germany) monoelement solutions of these elements. Argon with purity of 99.996% (White Martins-Praxair, Brazil) was used for plasma generation. Instrumentation Laser ablation was done using an LSX-266 Nd:YAG laser system (CETAC Technologies, Inc., USA), with quadrupled frequency at 266 nm. A quadrupole ICP-MS instrument (PerkinElmer Sciex, ELAN DRCII, Canada), equipped with a quartz torch fitted to a quartz injector-tube of 2 mm internal diameter, was employed for elements detection. An ultrasonic nebulizer (CETAC, 6000 UT, USA) was employed for solution introduction in the ICP. This system was used to reduce oxides and hydroxides formation in the ICP due to partial water removal. The aerosol generated by laser ablation in the ablation cell was transported to the ICP through a Tygon tube (0.5 cm i.d. × 100 cm length) internally covered with fluorinated ethylene propylene (FEP). Operating conditions of LA-ICP-MS and USN-ICP-MS, and monitored isotopes are summarized in Table 1.
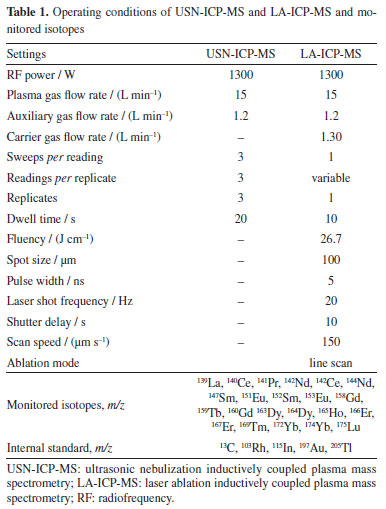
A planetary ball grinder (Retsch, PM 200, Germany) was employed for mint tea and leaves grinding. Pellets of the sample, leaves or cellulose were obtained using a manual hydraulic press (Specac, UK). For Lns determination by USN-ICP-MS, the mint tea sample was acid digested in closed vessels assisted by microwave radiation (Berghof, Speedwave Four, Germany). Sample preparation and analysis Mint tea (Mentha spicata) sample (in sachets) was purchased in a local market. The tea was taken from several sachets and ground in zirconia flasks with zirconia balls, at 500 rpm for one hour to obtain particles size lower than 50 µm. For LA-ICP-MS analysis, 500 mg of ground tea were transferred to a glass flask (10 mm height × 5 mm i.d.) in which the IS elements were added to obtain a final concentration of 1.0 µg g-1 Rh, In, Au, and Tl. The mixture was homogenized manually for 5 min and left drying at room temperature for 48 h in a class 100-laminar flow hood until constant weight. The dry mixture was then homogenized again, transferred to the 10 mm-diameter device of the hydraulic press, and pressed at 5.0 ton for 2.0 min to obtain a pellet. Pellets of approximately 10 mm diameter and approximately 2 mm thickness were obtained. By using this condition of drying, the pellet has a good physical consistency, where it is not brittle either during pressing or when analyzed by LA. The pellet was fixed using a double-sided adhesive tape on a glass slide and subsequently placed on the laser ablation chamber, followed by LA-ICP-MS analysis. Three pellets of the sample were prepared and analyzed. For analysis using USN-ICP-MS, aliquots of the ground tea sample were acid digested in the microwave oven system, under the conditions cited in Table 2. For acid digestion, 250 mg of sample was transferred to TFM flask that accompanies the microwave oven system, followed by addition of 6.0 mL of concentrated HNO3. After digestion, the solution was transferred to polypropylene flask and the volume was made up to 30 mL with purified water. Further dilution was carried out if necessary. Blanks were prepared in the same way as the sample using only HNO3. Sample was prepared in triplicate. For accuracy evaluation of the USN-ICP-MS method, the certified reference material (CRM) BCR-668 (mussel tissue, Institute for Reference Materials and Measurements, Belgium) was analyzed. For this, 250 mg of the CRM were decomposed by using the same conditions as described for the other samples. The accuracy of the LA-ICP-MS method was evaluated by comparing the results with those obtained by USN-ICP-MS.
Calibration For calibration in USN-ICP-MS, calibration solutions with Lns concentration ranging from 0.25 to 10 µg L-1 were prepared in 5% v v-1 HNO3, without addition of internal standard. For calibration in LA-ICP-MS, filter paper, cellulose powder pellet, and powdered leaves pellet were tested as support for Lns spiking. Cellulose powder was used as supplied by the manufacturer (Synth, Brazil). Filter paper was used without treatment. The leaves were already dried and grounded as described in "Sample preparation and analysis" sub-section. Concentrations of Lns in the leaves were previously determined and are lower than Lns concentration in mint tea or lower then LOD of the LA-ICP-MS method. The leaves sample was digested as described for mint tea and Lns determined by USN-ICP-MS. The pellets were prepared as above described in "Sample preparation and analysis" sub-section. The procedure of filter paper spiked with standards was adapted from that reported by Nunes et al.27 and Neves et al.30 In short, 40 μL of solution containing the Lns and IS elements (Rh, In, Au and Tl) in 5% v v-1 HNO3 were transferred with the aid of a micropipette on the center of a 17 nm diameter-filter paper disc (Whatman quantitative filter paper, ashless, grade 42) which was then slowly dried under an infrared lamp at temperature around 80 ºC. This filter paper was used without additional treatment. The volume of solution added to the filter paper has been optimized to avoid chromatographic effect, which can lead to heterogeneous distribution of the elements added to the filter paper.27 After drying, the paper disc was fixed on a quartz slide using a double-sided adhesive tape before proceeding with the LA-ICP-MS analysis. Five filter paper discs were spiked to obtain a calibration curve, where the concentrations of spiked Lns in the dried disks were 0.0 (blank), 0.5, 1.0, 2.5 and 5.0 µg g-1, and that of the spiked IS elements (Rh, In, Au and Tl) was 1.0 µg g-1. To prepare standards in the form of pellets, 500 mg of powdered leaves or cellulose were transferred to a glass flask (10 mm height × 5 mm i.d.) and mixed with 2 mL of solution containing Lns and IS elements (Au, In, Rh and Tl) in 5% v v-1 HNO3. Only 2 mL of 5% v v-1 HNO3 (without adding Lns) were used for preparing the blanks. The mixtures were left drying at room temperature in a class 100-room. Then, the spiked leaves or cellulose were pressed into pellets in the same way as the tea sample (see "Sample preparation and analysis" sub-section). Each pellet was fixed on a quartz slide using a double-sided adhesive tape before proceeding with the LA-ICP-MS analysis. Instruments performance For USN-ICP-MS, RF (radiofrequency) power, carrier gas flow rate and lens voltage were adjusted to achieve the highest 115In signal and lowest oxides and double charge ions signal intensity. The ratios 140Ce16O+/140Ce+ and 138Ba++/138Ba+ were lower than 0.3 and 2%, respectively. The working conditions for LA-ICP-MS were adjusted to achieve the highest signal-to-background ratio (SBR) and lower relative standard deviation (RSD). The 156CeO+/140Ce+ and 138Ba++/138Ba+ ratios were also monitored and kept lower than 0.5 and 2%, respectively. The carrier gas flow rate influence was also evaluated, and the best sensitivity achieved at 1.30 L min-1. The mass spectrometer dwell time was set to 10 ms to obtain high number of readings per m/z. The total analysis time of the mass spectrometer per ablated line has been adjusted through the number of readings per replicate, and according to the length of each ablated line, which was about 5 mm. Laser power (fluence), spot size, laser shot frequency and scan speed of the LA system were adjusted to achieve the best sensitivity. The conditions set (Table 1) were similar with those reported by Nunes et al.27 and Neves et al.30
RESULTS AND DISCUSSION LA-ICP-MS has been widely employed for quantitative analysis and elements mapping in plants and other biological tissues.13-15,26,27,30-35 The main advantage of using LA-ICP-MS for Lns determination is that interferences by oxides and hydroxides are markedly reduced.12 In addition, it is not necessary to digest the sample, as observed in the analysis by ICP-MS with nebulization systems for introducing the sample into the plasma. By LA-ICP-MS, depending on the purpose of the analysis or the kind of sample, the sample can be analyzed directly. However, calibration for quantitative analysis using LA-ICP-MS is still difficult,14,35 whereas solid matrix matched standards have been generally used. Nevertheless, solid standards prepared in house may be inhomogeneous, making calibration difficult and affecting precision and accuracy.12,17 Particles size for the standards should be as small as possible and internal standardization used to improve the calibration and analysis performance. Filter paper, cellulose and powdered leaves were evaluated in the present study by considering the similarity of their matrices to that of the sample; 103Rh, 115In, 197Au and 205Tl were tested for internal standardization by considering the proximity of ionization potential and m/z of these isotopes and those of the Lns, and low spectral interference. 13C has also been investigated as IS because it is naturally present and homogeneously distributed in the investigated matrices (paper, cellulose, and leaves). It is worth citing that 13C has been the most exploited IS in biological tissue analysis using LA-ICP-MS.12,28,29 Evaluation of internal standards and matrices Internal standardization can compensate for changes in laser interaction with the solid matrix, inhomogeneous analyte distribution in the solid matrix, plasma drift and other matrix effects.17,19 The IS choice is critical because the ionization potential and m/z of analyte and IS element should be ideally similar, and both free of spectral interferences.36 In addition, the IS element must be homogeneously distributed in samples and standards. Figures 1 and 2 show the signal profiles of 13C naturally present and those of the spiked IS elements (in cellulose, powdered leaves and filter paper), and the mean and standard deviation (SD) of the signal's intensities, respectively. According to Figure 1, only the 13C signal profile is similar in the three matrices, whereas Figure 2 reveals the more homogeneous 13C distribution. In contrast, the signals profiles of the spiked IS elements exhibit more variation. Even though the cellulose and powdered leaves pellets had been prepared in the same way, the distribution of the spiked IS elements was not similar. The reason for the higher inhomogeneous distribution of IS elements in cellulose than in powdered leaves has not been studied, but it is an issue that needs further investigation. Indium was the IS element most heterogeneously distributed in cellulose and powdered leaves, as demonstrated by the higher SD in Figure 2. When compared to In, Tl was more homogeneously distributed in cellulose followed by Au and Rh. Gold and Rh distribution was more homogeneous than was Tl in powdered leaves. The lower signal intensities for filter paper are probably due to the lower mass ablated, given that the filter paper is thinner than the pellets of cellulose or powdered leaves. All paper in the ablated line of filter paper was ablated using 30% laser energy. The energy of the laser was similar to those used by Nunes et al.27 and Neves et al.30 that corresponds to a fluency of 26.7 J cm-1. All other parameters are described in Table 1.
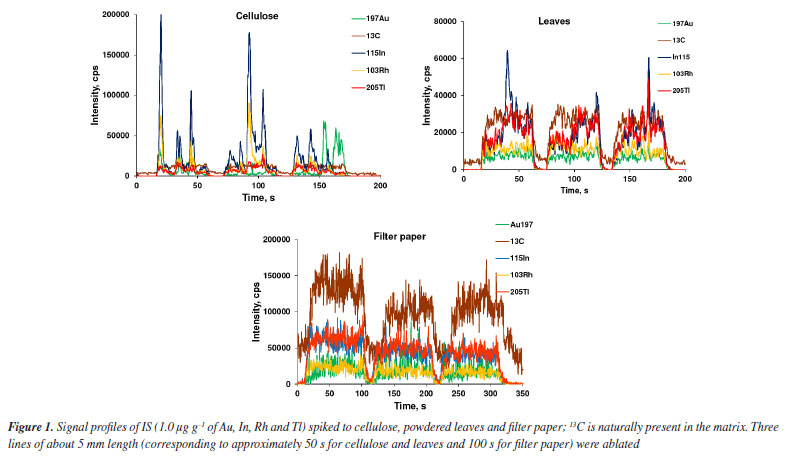
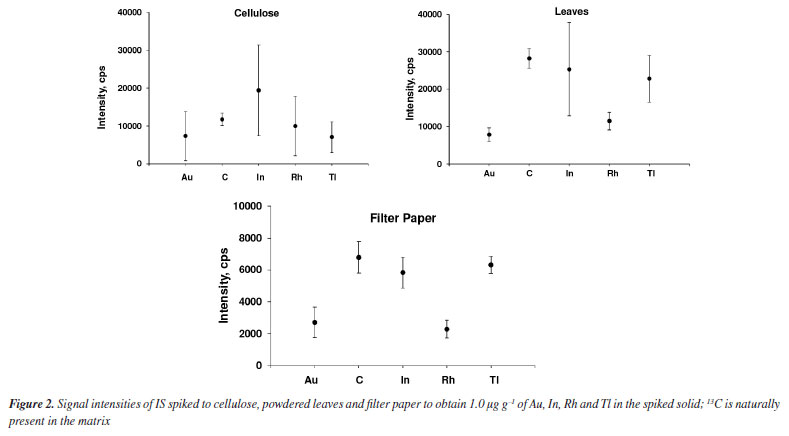
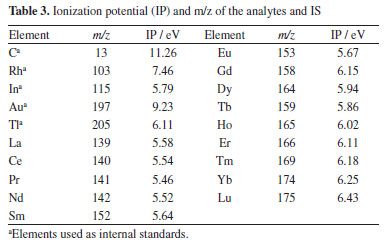
Despite the widespread use of 13C as IS,12,26,27,29 it should be considered the matrix-dependent partitioning of carbon into carbon-containing gaseous species and carbon containing particles transported to the ICP while other elements are exclusively transported as particles. The generation of carbon-containing gaseous species depends on oxygen and matrix constituent's affinity towards oxygen.26,29 An inhomogeneous distribution of carbon in certain matrices would be an additional disadvantage of using 13C as IS.33 In the case of Lns determination, the large difference among the ionization potential (11.26 eV for C and 5-6 eV for Lns) and m/z of 13C and that of monitored Ln isotopes ions (ranges from 139 to 175) might also compromise accuracy. Conversely, the Rh, In, Au and Tl ionization potentials (7.46, 5.79, 9.23 and 6.11 eV, respectively) as well as the isotopes m/z are closer to those of Lns. The ionization potential and m/z of the analytes and IS are shown in Table 3. In addition, the baseline signal of the isotopes of these elements are lower when compared to 13C. In general, the signal intensities of the elements are lower for filter paper that is due to the lower mass ablated, given that the filter paper is thinner than the pellets of cellulose or powdered leaves. However, these elements must be added to samples and standards and may result in inhomogeneous distribution as can be observed in Figures 1 and 2. Addition of IS have generally been carried out through solution spiking, coating the samples or standards surface with metallic films or organic polymers doped with the IS elements.25,37-39 Although it is more complex than spiking a solution of IS elements, surface coating enables homogeneous distribution of IS elements. However, the surface and thickness of the solids (standards and samples) to be ablated must be very regular; the IS cannot compensate for irregular surface or thickness.
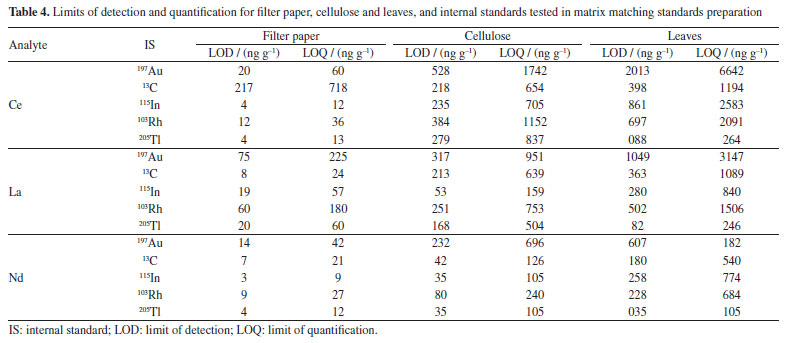
Analytical characteristics of the method and element quantification External calibration is common in solution nebulization-ICP-MS determinations, differently of LA-ICP-MS. As already mentioned, differences in laser interaction between standards and samples, aerosol transport and plasma drift occur in LA-ICP-MS and should be compensated. Matrix matching calibration using solid standards obtained from CRMs or prepared in house is a feasible way to circumvent such interference. A CRM with certified Lns concentrations was not available and, therefore, matrix matching standards were prepared in the present study. The parameters of the lanthanides calibration curves obtained by means of the prepared standards are given in Supplementary Material (Table 1S). The coefficient of determination (R2) was at least 0.98 for all matrices tested and all standards could be used for calibration, except for some elements. However, in general, precision was better for filter paper standards (as also shown in Figure 2); the relative standard deviation (RSD) of signals generated in the ablation of three lines (each with about 5 mm) was better than 20%. On the other hand, the RSD of signals generated in the ablation of three lines over the pellets of cellulose or powdered leaves was typically higher than 20%. The RSD values observed for filter paper are consistent with those reported for LA-ICP-MS analysis.21,27,31 The better precision for filter paper is also demonstrated by the lower LODs and limit of quantification (LOQs) values (Table 4). The LOD and LOQ were estimated considering the standard deviation of the Lns signals of the ablation of 10 lines (each with about 5 mm length) of the blanks. The standard deviation values were multiplied by 3 and 10 to calculate the LOD and LOQ, respectively. These values were then divided by the slope (S) of the respective calibration curves.
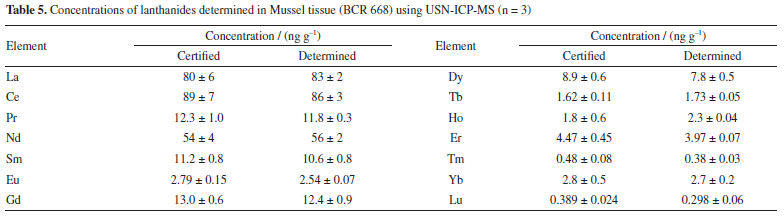
Accuracy of the USN-ICP-MS method was evaluated by the analysis of the CRM BCR-668 (Mussel tissue) and results are shown in Table 5, while the accuracy of the LA-ICP-MS method was evaluated by comparing the results with those obtained by USN-ICP-MS.
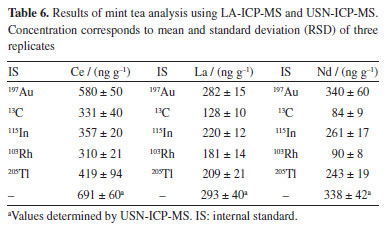
Sample analysis Pellets of the mint tea sample were analyzed by LA-ICP-MS, whereas filter paper spiked with the Lns and IS elements were used for matrix matching calibration. The Ce, La and Nd concentrations are given in Table 6, together with those obtained using USN-ICP-MS. The concentrations of the other Lns were below the respective LOQs and for that reason they were not included in Table 6. According to t-test at 95% confidence level, the Ce, La and Nd concentrations found using LA-ICP-MS and 197Au as IS were in good agreement with those found using USN-ICP-MS. Therefore, 197Au could be selected for internal standardization when Lns are determined in mint tea using LA-ICP-MS and matrix matching calibration with filter paper.
CONCLUSIONS Laser ablation-ICP-MS was evaluated for Lns determination in mint tea by using filter paper, cellulose and powdered leaves for matrix matching calibration combined with C, Rh, In, Au and Tl as IS. Sensitivity, precision and accuracy were particularly good using filter paper for matrix matching and internal standardization with 197Au. In such conditions, Ce, La and Nd in mint tea were accurately determined. Concentration of the other Lns were below the LOQ. It can be stated that through such calibration and sample preparation approaches, LA-ICP-MS is an appropriate method for Lns determination in leaves.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
ACKNOWLEDGMENTS The author would like to thank CNPq (Proc. No. 480929/2011-4 and 302276/2017-3) and CAPES for funding and support.
REFERENCES 1. Zhan, W.; Guo, Y.; Gong, X.; Guo, Y.; Wang, Y.; Lu, G.; Chin. J. Catal. 2014, 35, 1238. [Crossref] 2. Balaram, V.; Geosci. Front. 2019, 10, 1285. [Crossref] 3. Shigematsu, T.; Nakashima, Y.; Ohya, M.; Tatsuta, K.; Koreeda, D.; Yoshimoto, W.; Yamanaka, S.; Sakaguchi, T.; Hanba, Y.; Mima, T.; Negi, S.; Int. J. Nephrol. Renovasc. Dis. 2012, 2012, 81. [Crossref] 4. Panichev, A. M.; Achievements in the Life Sciences 2015, 9, 95. [Crossref] 5. Elias Junior, J.; Santos, A. C.; Koenigkam-Santos, M.; Nogueira-Barbosa, M. H.; Muglia, V. F.; Radiologia Brasileira 2008, 41, 263. [Crossref] 6. Rim, K.-T.; Toxicol. Environ. Health Sci. 2016, 8, 189. [Crossref] 7. Emmanuel, E. S. C.; Anandkumar, B.; Natesan, M.; Maruthamuthu, S.; Aust. J. Crop Sci. 2010, 4, 289. [Link] accessed in November 2024 8. Ni, Z.; Ren, C.; Cheng, J.; Tang, F.; J. Braz. Chem. Soc. 2017, 28, 1960. [Crossref] 9. Gómez-Merino, F. C.; Gómez-Trejo, L. F.; Ruvalcaba-Ramírez, R.; Trejo-Téllez, L. I. In Beneficial Chemical Elements of Plants: Recent Developments and Future Prospects; Pandey, S.; Tripathi, D. K.; Singh, V. P.; Sharma, S.; Chauhan, D. K., eds.; John Wiley & Sons Ltd.; New Jersey, USA, 2023, p. 349-369. [Crossref] 10. Zawisza, B.; Pytlakowska, K.; Feist, B.; Polowniak, M.; Kita, A.; Sitko, R.; J. Anal. At. Spectrom. 2011, 26, 2373. [Crossref] 11. Gorbatenko, A. A.; Revina, E. I.; Inorg. Mater. 2015, 51, 1375. [Crossref] 12. Pozebon, D.; Scheffler, G. L.; Dressler, V. L.; J. Anal. At. Spectrom. 2017, 32, 890. [Crossref] 13. Sussulini, A.; Becker, J. S.; Becker, J. S.; Mass Spectrom. Rev. 2017, 36, 47. [Crossref] 14. Pessôa, G. S.; Lopes Júnior, C. A.; Madrid, K. C.; Arruda, M. A. Z.; Talanta 2017, 167, 317. [Crossref] 15. Marillo-Sialer, E.; Black, J. R.; Paul, B.; Kysenius, K.; Crouch, P. J.; Hergt, J. M.; Woodhead, J. D.; Hare, D. J.; J. Anal. At. Spectrom. 2020, 35, 671. [Crossref] 16. Hare, D. J.; Fryer, F.; Paul, B.; Bishop, D. P.; Doble, P. A.; Anal. Methods 2016, 8, 7552. [Crossref] 17. Miliszkiewicz, N.; Walas, S.; Tobiasz, A.; J. Anal. At. Spectrom. 2015, 30, 327. [Crossref] 18. Lin, J.; Liu, Y.; Yang, Y.; Hu, Z.; Solid Earth Sci. 2016, 1, 5. [Crossref] 19. Martinez, M.; Baudelet, M.; Anal. Bioanal. Chem. 2020, 412, 27. [Crossref] 20. Augusto, A. S.; Sperança, M. A.; Andrade, D. F.; Pereira-Filho, E. R.; Food Anal. Methods 2017, 10, 1515. [Crossref] 21. Limbeck, A.; Galler, P.; Bonta, M.; Bauer, G.; Nischkauer, W.; Vanhaecke, F.; Anal. Bioanal. Chem. 2015, 407, 6593. [Crossref] 22. Šala, M.; Šelih, V. S.; van Elteren, J. T.; Analyst 2017, 142, 3356. [Crossref] 23. Li, Y.; Guo, W.; Hu, Z.; Jin, L.; Hu S.; Guo, Q.; J. Agric. Food Chem. 2019, 67, 935. [Crossref] 24. Doble, P. A.; Gonzalez de Veja, R.; Bishop, D. P.; Hare D. J.; Clases, D.; Chem. Rev. 2021, 121, 11769. [Crossref] 25. Konz, I.; Fernández, B.; Fernández, M. L.; Pereiro, R.; González, H.; Álvarez, L.; Coca-Prados, M.; Sanz-Medel, A.; Anal. Bioanal. Chem. 2013, 405, 3091. [Crossref] 26. Chacón-Madrid, K.; Arruda, M. A. Z.; J. Anal. At. Spectrom. 2018, 33, 1720. [Crossref] 27. Nunes, M. A. G.; Voss, M.; Corazza, G.; Flores, E. M. M.; Dressler, V. L.; Anal. Chim. Acta 2016, 905, 51. [Crossref] 28. Frick, D. A.; Günther, D.; J. Anal. At. Spectrom. 2012, 27, 1294. [Crossref] 29. Deiting, D.; Börno, F.; Hanning, S.; Kreyenschmidt, M.; Seidl, T.; Otto, M.; J. Anal. At. Spectrom. 2016, 31, 1605. [Crossref] 30. Neves, V. M.; Heidrich, G. M.; Rodrigues, E. S.; Enders, M. S. P.; Muller, E. I.; Nicoloso, F. T.; de Carvalho, H. W. P.; Dressler, V. L.; Environ. Sci. Technol. 2019, 53, 10827. [Crossref] 31. Egger, A. E.; Theiner, S.; Kornauth, C.; Heffeter, P.; Berger, W.; Keppler, B. K.; Hartinger, C. G.; Metallomics 2014, 6, 1616. [Crossref] 32. Diniz, A. P.; Kozovits, A. R.; Lana, C. C.; de Abreu, A. T.; Leite, M. G. P.; Int. J. Mass Spectrom. 2019, 435, 251. [Crossref] 33. Becker, J. S.; Matusch, A.; Palm, C.; Salber, D.; Morton K. A.; Becker, J. S.; Metallomics 2010, 2, 104. [Crossref] 34. Cizdziel, J.; Bua, K.; Nowinski, P.; Anal. Methods 2012, 4, 564. [Crossref] 35. Francischini, D. S.; Arruda, M. A. Z.; Anal. Bioanal. Chem. 2024, 416, 2737. [Crossref] 36. Austin, C.; Fryer, F.; Lear, J.; Bishop, D.; Hare, D.; Rawling, T.; Kirkup, L.; McDonagh, A.; Doble, P.; J. Anal. At. Spectrom. 2011, 26, 1494. [Crossref] 37. Neves, V. M.; Heidrich, G. M.; Hanzel, F. B.; Muller, E. I.; Dressler, V. L.; Chemosphere 2018, 198, 409. [Crossref] 38. Bonta, M.; Lohninger, H.; Marchetti-Deschmann, M.; Limbeck, A.; Analyst 2014, 139, 1521. [Crossref] 39. Bonta, M.; Hegedus, B.; Limbeck, A.; Anal. Chim. Acta 2016, 908, 54. [Crossref]
Associate Editor handled this article: Eduardo M. Richter |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access