
 |
 |
 |
 |
 |
Artigo
|
|
| Método multirresíduo de agrotóxicos em polpa de açaí empregando método QuEChERS modificado e UHPLC-MS/MS Multiresidue method for pesticides in açaí pulp using a modified QuEChERS method and UHPLC-MS/MS |
|
Rafael Bel Prestes da Silva; Luana Floriano; Cleusa Fátima Zanchin; Franciele Fátima Machado; Júlia Antunes de Oliveira; Departamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria - RS, Brasil Recebido: 26/08/2024 *e-mail: renato.zanella@ufsm.br The northern region of Brazil, particularly the states of Pará and Amazonas, concentrates the majority of açaí national production. Considering the possible use of pesticides in this species, this study aimed to develop a comprehensive method for the multiresidue determination of pesticides in açaí pulp, using a modified QuEChERS (quick, easy, cheap, effective, rugged, and safe) method with analysis by ultra-high performance liquid chromatography with tandem mass spectrometry (UHPLC-MS/MS). Various parameters of extraction were investigated, and the method was successfully validated. Analysis by UHPLC-MS/MS provided high selectivity and sensitivity. The main modification to the original QuEChERS method was the use of the C18 adsorbent associated with primary secondary amine (PSA) in the clean-up step by dispersive solid phase extraction (d-SPE). The method was validated for 83 compounds, showing adequate accuracy (70 to 120%) and precision (RSD ≤ 20%). The method limits of quantification, established by recovery tests, ranged from 5 to 25 µg kg-1. The method limits of detection were from 1.5 to 7.5 µg kg-1. The method was applied to commercial samples of açaí pulp, and no pesticide residues were detected. The proposed method proved to be fast, efficient and suitable for multiresidue determination of pesticides in açaí pulp in routine analysis. INTRODUÇÃO A flora da Floresta Amazônica possui destaque graças à sua biodiversidade, com espécies muitas vezes endêmicas e outras ainda não identificadas. Dentre as frutas destaca-se o açaí, produzido pelo açaizeiro (Euterpe oleracea Mart. e Euterpe precatoria Mart.). O fruto do açaizeiro possui potencial agroindustrial, tecnológico, nutricional e econômico1,2 graças à versatilidade para aplicação como ingredientes de formulações de alimentos, devido à sua composição nutricional e pelas suas potenciais propriedades bioativas.3 A partir da extração com água da parte comestível do fruto maduro do açaí pode-se obter produtos comerciais. O fruto é pequeno e com pouca polpa, além de uma casca muito dura, que impede seu consumo in natura, e sim na forma de polpa, extraída mecanicamente com a adição de água.4 O produto é um dos principais alimentos das populações ribeirinhas e vem rapidamente ganhando espaço no mercado de bebidas energéticas, por apresentar diversos benefícios nutricionais.3 Para o cultivo do açaizeiro há a possibilidade de uso de vários métodos de controle de plantas invasoras, como o uso de agrotóxicos. Entretanto, o uso indiscriminado e intensivo de agrotóxicos é um fator de risco à saúde do consumidor e ao meio ambiente.5 Considerando-se os riscos apresentados pelo uso inadequado de agrotóxicos, o seu uso e a presença nos alimentos têm sido regulamentados. Entre as técnicas para a determinação de resíduos de agrotóxicos em alimentos destacam-se a cromatografia gasosa e a cromatografia líquida, ambas acopladas à espectrometria de massas.6 Porém, a diversidade dos agrotóxicos utilizados e a ocorrência de níveis muito baixos nos alimentos, representam um desafio considerável quando se trata de supervisionar e avaliar a presença dessas substâncias.7 Atualmente, há 20 agrotóxicos registrados para o cultivo do açaí e, portanto, é importante monitorar a ocorrência de resíduos de agrotóxicos em açaí. Originalmente, o método QuEChERS (do inglês, quick, easy, cheap, effective, rugged, and safe) foi proposto por Anastassiades etal.8 em 2003 para extração de agrotóxicos em frutas e hortaliças. Este método tem sido amplamente explorado para diversas matrizes alimentares, devido à sua capacidade de extrair uma vasta gama de agrotóxicos, com adequada recuperação dos analitos, remoção dos possíveis interferentes da amostra, boa precisão, baixo custo, rapidez, facilidade e segurança.6 O método QuEChERS consiste em uma etapa de extração seguida de limpeza do extrato orgânico destinada à remoção de coextrativos, tais como açúcares, lipídios e ácidos orgânicos. Nestas etapas, fatores como ajuste do pH do solvente de extração e diferentes materiais sorventes podem afetar a eficiência do preparo de amostra.9 Modificações do método QuEChERS permitem adequar as condições para diferentes matrizes e centenas de agrotóxicos.10 Apenas três estudos11-13 apresentam métodos para a determinação de resíduos de agrotóxicos em açaí estão descritos na literatura. O estudo realizado por Tran et al.11 apresenta resultados da determinação de resíduos de agrotóxicos em polpa de açaí a partir do uso de LC-MS/MS (cromatografia líquida com detecção por espectrometria de massas em série) para a detecção de 174 agrotóxicos e GC-MS/MS (cromatografia gasosa com detecção por espectrometria de massas em série) para a detecção de 117 agrotóxicos, com o uso do QuEChERS como preparo de amostra, entretanto não foram apresentados informações sobre a validação dos métodos, bem como em relação aos limites de detecção ou quantificação. O estudo realizado por Paz et al.12 validou um método com o uso de QuEChERS como preparo de amostra para detecção de resíduos de 14 agrotóxicos organoclorados em polpa de açaí com determinação por cromatografia gasosa com detecção por captura de elétrons (GC-ECD), com posterior confirmação por GC-MS/MS. Já o estudo de Froés et al.13 validou um método para a determinação dos agrotóxicos bromuconazol, fenbuconazol, parationa-metílica, cresoxim-metílico e teflubenzurom em polpa de açaí, porém com o uso de dispersão da matriz em fase sólida (MSPD, do inglês matrix solid phase dispersion) e detecção por cromatografia líquida com detecção por arranjo de diodos. Dada a preocupação crescente com os potenciais impactos negativos para a saúde humana e o meio ambiente decorrentes do uso excessivo de agrotóxicos na produção e armazenamento de frutas, foi desenvolvido um método analítico para a determinação multiclasse de resíduos de agrotóxicos em polpa de açaí. Um procedimento de preparo de amostras rápido utilizando a abordagem QuEChERS original e combinação de sorventes para a limpeza do extrato, seguido de análise por UHPLC-MS/MS (cromatografia líquida de ultra-alta eficiência acoplada à espectrometria de massas em série), foi estabelecido após a avaliação de diferentes condições de extração e sorventes na etapa de limpeza do extrato. O método foi validado possibilitando a determinação multirresíduo de 83 agrotóxicos. O método foi aplicado na análise de amostras comerciais provenientes do Estado do Amazonas, Brasil. A implementação deste método contribui para o monitoramento eficaz da segurança do alimento, assegurando a conformidade com os limites máximos de resíduos estabelecidos por regulamentações sanitárias.
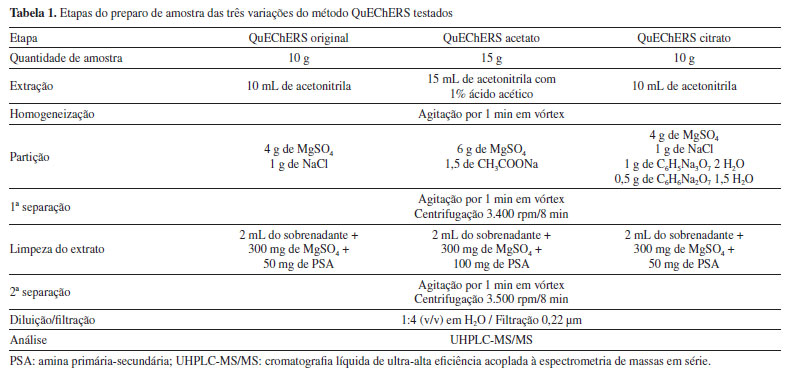
PARTE EXPERIMENTAL Reagentes e materiais O enfoque experimental adotado nesta pesquisa centrou-se na etapa de desenvolvimento e validação de um método abrangente para o preparo de amostras. Especificamente, o método QuEChERS foi empregado para a extração de resíduos de agrotóxicos presentes na polpa de açaí, e posteriormente, a determinação dos analitos foi conduzida pela técnica de UHPLC-MS/MS. Os reagentes, solventes e demais materiais utilizados no desenvolvimento deste estudo foram: acetona, grau HPLC (Sigma-Aldrich, Alemanha); acetonitrila, grau HPLC (Biograde, EUA); ácido acético glacial 100% e ácido fórmico 98% (Sigma-Aldrich, Alemanha); acetato de etila (Mallinckrodt, EUA); acetato de sódio (Avantor, México); citrato de sódio di-hidratado, cloreto de cálcio, cloreto de sódio, formiato de amônio, hidrogenocitrato de sódio sesquiidratado e sulfato de magnésio anidro p.a. (Sigma-Aldrich, EUA); metanol, grau HPLC e hexano grau HPLC (Sigma-Aldrich, Alemanha); Extran® MA neutro (Merck, Brasil); água ultrapurificada em sistema Milli-Q Direct 3UV® com resistividade de 18,2 MΩ cm (Millipore, França). Os seguintes sorventes foram avaliados na etapa de clean-up: alumina neutra (Sigma-Aldrich, EUA); biocarvão obtido do bagaço do malte (Hordeum vulgare) por via hidrotérmica, ativado com ácido fosfórico, durante 14 h a 150 ºC e biocarvão obtido do bagaço do malte (Hordeum vulgare) via pirólise, a 500 ºC por 1 h (Universidade Federal de Viçosa, Brasil); Bond Elut Nexus, Bondesil C18, Bondesil NH2, Bondesil PSA e carvão ativado (Agilent Technologies, EUA); carvão grafitizado (GCB) e Supelclean ENVI-Carb (Supelco, EUA), cortiça extraída da casca de sobreiro, Celite® 521 Terra diatomácea (Sigma-Aldrich, EUA); Florisil® 60-120 mesh (J.T. Baker, EUA); quitosana de casca de caranguejo (Sigma-Aldrich, Japão); quitosana obtida e caracterizada na Universidade Federal do Rio Grande, Brasil, a partir de resíduos de camarão-rosa (Farfantepenaeus brasiliensis); nanotubos de carbono estabilizados em esponja de quitosana (CNT-CS, do inglês carbon nanotube stabilization in chitosan sponge), produzida e caracterizada na Universidade Federal do Rio Grande, a partir de resíduos de camarão (Penaeus brasiliensis) com massa molar de 150,3 ± 5 kDa e grau de desacetilação de 87 ± 1%; Oasis HLB (Waters, Irlanda); sílica (Sigma-Aldrich, EUA); Strata-X (Phenomenex, EUA); Supel QuE Z-Sep+® (Sigma-Aldrich, EUA) e zeólita (Quênia). Também foram utilizados filtros de nylon de 13 mm e porosidade de 0,22 μm (Allcrom, Brasil); frascos de vidro (vial), capacidade de 2 e 4 mL (Agilent Technologies, EUA), tubos de polipropileno (PP), com tampa de rosca, de 50 e 15 mL (Sarstedt, Alemanha), argônio com grau de pureza 5.0 (Air Liquide, Brasil) e vidrarias comuns de laboratório. Instrumentação e condições Os equipamentos utilizados durante a elaboração deste trabalho foram: agitador Vortex Lab Danger (IKA®, EUA); agitador votex modelo 771 (Fisatom, Brasil); balança analítica APX-200 (Denver Instrument Company, Brasil); balanças analíticas de precisão modelos AUX-220, AUW-220D e UX-420H (Shimadzu, Japão); centrífuga FL9-0815B (First Lab, Brasil); centrífuga refrigerada NT 825 (Novatecnica, Brasil); micropipetadores automáticos com capacidade de 20 a 10000 μL (Brand, Alemanha); sistema de ultrapurificação de água, Milli-Q Direct-Q® 3UV (Millipore, França); banho ultrassônico Sonorex RK510 (Bandelin, Alemanha); Sistema UHPLC-MS/MS modelo Xevo TQ (Waters, EUA) equipado com: cromatógrafo a líquido modelo Acquity; Coluna Acquity UPLC™ BEH C18 (50 × 2,1 mm d.i.; 1,7 μm) Waters (EUA); amostrador automático sample manager Acquity (Waters, EUA); detector MS triplo quadrupolo e fonte de ionização por eletronebulização (ESI); sistema gerador de nitrogênio com pureza ≥ 99%, modelo NM30L-MS (PEAK Scientific, Escócia); sistema de aquisição de dados pelo software MassLynx V.4.1 (Waters, EUA); argônio 5.0 usado como gás de colisão no sistema UHPLC-MS/MS; sistema de espectrofotometria de absorção molecular UV-visível modelo 8453 (Agilent Technologies, EUA), equipado com sistema de aquisição de dados pelo software ChemStation (Agilent Technologies, EUA); sistema de cromatografia gasosa acoplada à espectrometria de massas (GC-MS) modelo Intuvo 9000 com analisador de massas tipo triplo quadrupolo modelo 7010 (Agilent Technologies, EUA). A separação cromatográfica foi obtida usando como fase móvel A uma solução de água:metanol (98:2 v/v) e B metanol:água (98:2 v/v), ambas contendo 5 mmol L-1 de formiato de amônio e 0,1% (v/v) de ácido fórmico. A eluição foi efetuada no modo gradiente, da seguinte forma: 0 a 0,25 min: 5% B; 7,75 min: 100% B, mantido até 8,50 min; 8,51 a 10,00 min: 5% B. A vazão foi de 0,225 mL min-1 com um tempo total de corrida cromatográfica de 10 min. O volume de injeção foi de 10 μL e a temperatura da coluna foi ajustada em 40 ºC. O nitrogênio foi usado como gás de dessolvatação a 600 L h-1 e o gás de colisão a 0,15 mL min-1. O espectrômetro de massas foi operado com ionização por electrospray, em ambos os modos positivo e negativo (ESI+ e ESI-). A detecção foi realizada no modo de monitoramento de reações selecionadas (SRM), onde duas transições foram monitoradas para cada composto analisado. Para a quantificação, foi escolhido o íon com maior abundância, enquanto a segunda transição mais intensa foi utilizada para fins qualitativos, auxiliando na identificação dos analitos. Coleta de amostras branco e de polpa de açaí As amostras de polpa de açaí denominadas branco, empregadas para o desenvolvimento e a validação do método, foram obtidas no comércio dos municípios de Humaitá e Manicoré, no Estado do Amazonas, Brasil. Para a etapa de aplicação do método foi realizada a aquisição de 30 amostras comerciais de açaí provenientes do Estado do Amazonas. As amostras foram coletadas em maio e dezembro de 2022, em dezembro de 2023 e em março de 2024. Após a coleta, as amostras foram armazenadas em freezer a -20 ºC. Compostos selecionados e preparação de soluções analíticas Foram selecionados 83 agrotóxicos de uso corrente e de diferentes classes. Os padrões analíticos foram adquiridos da LGC Standards (Alemanha) com pureza superior a 95%. Preparou-se 10 mL de soluções estoques individuais de cada composto a partir do padrão sólido, com concentração de 1000 mg L-1. A pureza de cada padrão foi levada em consideração no cálculo da massa a ser empregada e diluiu-se em solvente apropriado, que inclui acetonitrila, acetona ou acetato de etila. A partir destas soluções estoques, foi preparada uma solução mistura dos compostos em estudo na concentração de 5 mg L-1 em acetonitrila, que foi armazenada em frasco âmbar a uma temperatura de -4 ºC. Esta solução foi utilizada para a etapa de fortificação das amostras branco e para o preparo das curvas analíticas em extrato da matriz branco e em solvente, nas concentrações 0,5, 1, 2, 5, 10 e 20 μg L-1. Para avaliar o desempenho analítico, o trifenilfosfato (Sigma-Aldrich, EUA) foi adicionado como padrão interno (PI) na concentração de 10 μg L-1 às soluções a serem analisadas. Além disso, para assegurar a eficiência do processo de extração, foi utilizada a atrazina-d5 (Sigma-Aldrich, EUA) como padrão de controle (PC), preparada na concentração 10 mg L-1, adicionada antes da etapa de extração visando uma concentração final de 10 μg L-1. Desenvolvimento da etapa de preparo de amostras Testes iniciais Visando avaliar qual das versões do método QuEChERS proporciona a extração dos analitos de forma mais eficiente, uma série de testes foi conduzida. Esses testes foram baseados nas três variações do método QuEChERS, a saber: o método QuEChERS original,8 bem como suas versões tamponadas, o QuEChERS citrato14 e o QuEChERS acetato.15 Essa etapa de avaliação representa um elemento crucial do processo analítico, visto que tem a finalidade de identificar qual abordagem de extração apresenta o melhor desempenho em termos de eficácia e resultados confiáveis para a matriz em estudo e os agrotóxicos selecionados. O foco central dessa avaliação está na obtenção de recuperações aceitáveis (70 a 120%) e um desvio padrão relativo (RSD) ≤ 20%, de um amplo espectro de resíduos de agrotóxicos, conforme preconizado pela SANTE.16 Cada variação desse método possui particularidades que podem influenciar significativamente os resultados. Nesta avaliação foram utilizadas as amostras sem resíduos de agrotóxicos (branco) de polpa de açaí. Os testes tiveram por objetivo encontrar as melhores condições de extração dos compostos em estudo, sendo que os ensaios de fortificação foram realizados em triplicata (n = 3), na concentração de 50 μg kg-1. Para avaliar a interferência da matriz, a etapa de adição do padrão às amostras foi realizada antes da extração (fortificação na amostra) e após o procedimento de extração (padrão no extrato). Os extratos foram filtrados e diluídos 5 vezes em água ultrapura antes da injeção no sistema UHPLC-MS/MS. Os testes realizados com as três variações do método QuEChERS empregaram as condições descritas na Tabela 1.
De posse das informações obtidas no teste, avaliou-se o efeito matriz (EM) através da comparação entre a média das triplicatas da área obtida para a concentração 10 μg kg-1 em extrato da matriz branco com a média das triplicatas da área da mesma concentração em solvente, utilizando a Equação 1: 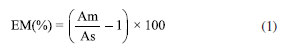 onde: Am é a área da solução analítica preparada no extrato da matriz (açaí), e As é a área da solução analítica preparada no solvente (acetonitrila). Avaliação do uso de sorventes na etapa de limpeza Após a fase inicial de testes, o método que proporcionou os resultados mais promissores foi selecionado para uma avaliação adicional, com foco específico na etapa de limpeza do extrato obtido. Nesta etapa, um conjunto de 20 sorventes foi submetido a testes individuais, a fim de avaliar a eficácia de cada um deles em limpar o extrato, eliminando interferências indesejadas. Todos os procedimentos foram avaliados em termos de recuperação e RSD, mas também em relação à eficiência na remoção de coextrativos, através de avaliação visual, por espectrofotometria de absorção molecular UV-Vis, por GC-MS no modo full scan, que permite avaliar de forma ampla os compostos orgânicos presentes no extrato, e por gravimetria. A partir do extrato obtido pelo método QuEChERS original, procedeu-se com a transferência de uma alíquota de 2 mL do extrato (sobrenadante) resultante da etapa de extração para um tubo de PP de 15 mL contendo 300 mg de MgSO4 e as seguintes quantidades de sorvente: 10 mg para os sorventes a base de biocarvão, carvão ativado e GCB, e 50 mg para os demais sorventes. Para garantir a interação efetiva entre os componentes, o conteúdo do tubo foi submetido a uma agitação vigorosa por 1 min para garantir a distribuição homogênea dos componentes, promovendo uma interação completa entre os coextrativos e o sorvente selecionado. Após a etapa de agitação, o tubo foi centrifugado a 3.500 rpm, por 8 min a 20 ºC. Essas condições foram selecionadas para assegurar a separação eficiente das fases. O sobrenadante resultante desse processo de centrifugação foi cuidadosamente coletado para as etapas posteriores. A partir dos resultados, realizou-se uma avaliação visual, selecionando-se os oito extratos que apresentavam maior eficiência na remoção dos pigmentos para serem avaliados por espectrofotometria de absorção molecular UV-Vis. A partir disso, foram selecionados os dois extratos que apresentaram melhores resultados de limpeza e realizou-se teste com as quantidades definidas de sorventes, combinando-os com C18. Nessa etapa, realizou-se uma análise do tipo varredura (full scan) em sistema GC-MS a fim de verificar qual sorvente apresentaria melhores resultados de limpeza. Comparou-se os resultados do extrato sem limpeza com as duas melhores condições de limpeza do teste anterior, bem como com estas mesmas condições acrescidas de 250 mg de C18. A avaliação gravimétrica constitui uma técnica analítica fundamental que pode ser empregada para avaliar a eficácia na remoção dos coextrativos.17,18 Este método envolve a evaporação do solvente para a quantificação precisa das massas dos componentes antes e após a aplicação do processo de limpeza.19 Tais testes foram realizados para avaliar a quantidade de coextrativos presentes no extrato após o processo de limpeza, avaliando a eficiência desta nos diferentes sorventes testados. Para os testes gravimétricos utilizou-se os sorventes considerados mais eficazes, conforme identificado pelas análises por GC-MS. Com isso, após as etapas prévias de limpeza, 1 mL de extrato de cada teste foi transferido para tubos de vidros com massas previamente aferidas. Estes tubos foram então colocados em uma estufa, onde a temperatura foi inicialmente mantida a 40 ºC por 30 min. Em seguida, a temperatura foi aumentada em incrementos de 10 ºC até atingir 110 ºC, sendo mantida nesse patamar até que o extrato estivesse completamente seco. Após a completa secagem do extrato, determinou-se a massa do material residual e realizou-se o cálculo para cada teste de limpeza. A variação na massa dos coextrativos, determinada antes e depois da etapa de purificação, forneceu uma medida quantitativa da eficácia do método de limpeza empregado. Este método permite uma avaliação rigorosa da eficiência de remoção dos coextrativos, assegurando a confiabilidade dos resultados experimentais obtidos. Após estes testes, utilizou-se o nível de fortificação de 50 μg kg-1 na amostra branco, em triplicata, para avaliar o número de compostos com resultados aceitáveis. Para escolher o melhor sorvente de limpeza do extrato considerou-se aquele que proporcionou um maior número de compostos recuperados adequadamente. Validação do método O método de extração que demonstrou os melhores resultados na etapa de testes foi validado conforme as diretrizes estabelecidas nos documentos orientativos do INMETRO20 e do guia SANTE.16 Durante a validação do método analítico foram avaliados os parâmetros: seletividade, linearidade, efeito matriz, limite de detecção, limite de quantificação, precisão e exatidão, parâmetros estes geralmente utilizados para validação desse tipo de estudo.11-13 Cada um desses parâmetros foi cuidadosamente avaliado para assegurar a confiabilidade do método empregado, garantindo assim a sua aplicabilidade em análises de rotina e a sua conformidade com padrões internacionais de qualidade.
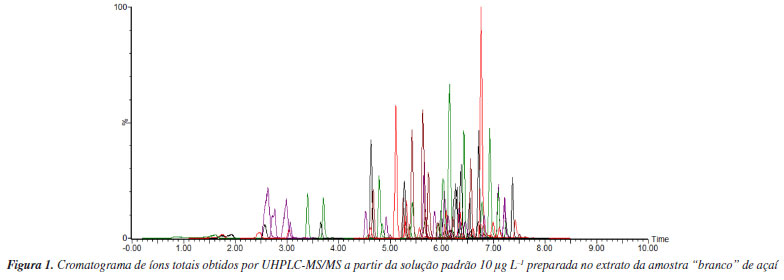
RESULTADOS E DISCUSSÃO Determinação realizada por UHPLC-MS/MS A Tabela 1S (Material Suplementar) apresenta os compostos analisados por UHPLC-MS/MS, utilizando ionização ESI- para fipronil e fluxapiroxade e ESI+ para os demais compostos, e modo de aquisição por monitoramento de reações selecionadas (SRM), tempos de retenção (tR), voltagem de cone (CV), energias de colisão (CE) e íons precursores e produtos para quantificação e identificação de cada analito em estudo. A Figura 1 apresenta um cromatograma obtido por UHPLC-MS/MS a partir da transição de quantificação de cada analito a partir de uma solução padrão da mistura dos analitos em 10 µg L-1, preparada em extrato da amostra branco. O sistema UHPLC-MS/MS permitiu a identificação e quantificação dos agrotóxicos em um tempo de análise de 10 min.
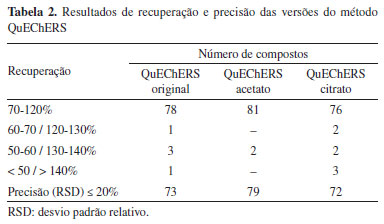
Avaliação da etapa de extração A Tabela 2 apresenta os resultados dos testes com as diferentes versões do método QuEChERS. A avaliação foi feita em triplicata fortificando a matriz branco com 83 agrotóxicos na concentração de 50 μg kg-1. Ao comparar as três versões do método QuEChERS foram observados resultados de recuperação e de precisão (RSD) consistentes e similares. Os testes apresentaram resultados satisfatórios, com recuperação entre 70 e 120% e RSD ≤ 20%, para 78 compostos com o QuEChERS original, 76 compostos com a versão citrato e 81 compostos com o acetato. Supõe-se que alguns compostos não foram recuperados por apresentarem menor caráter ácido, devido ao uso de acetonitrila como solvente de extração no QuEChERS acetato.
O efeito matriz (EM) foi avaliado de acordo com o guia SANTE16 comparando as inclinações das curvas preparadas no extrato branco da matriz e no solvente (acetonitrila). O EM foi calculado para cada versão do método QuEChERS e os resultados são mostrados na Tabela 3. Pode-se observar que, para o método QuEChERS original (T1), dos 83 agrotóxicos avaliados, 74 apresentaram resultados de efeito matriz entre -20 e +20%, considerado insignificante.
O método QuEChERS citrato revelou uma menor capacidade de recuperar os agrotóxicos quando comparado às outras variações, concluindo-se que o QuEChERS citrato não seria a escolha mais adequada para as necessidades analíticas do estudo em questão. Entre as versões QuEChERS original e acetato, no método original não é necessária a utilização de ácido acético e do sal acetato de sódio, o que segue os princípios da Química Verde que preconiza que a utilização de substâncias auxiliares (solventes, agentes de separação etc.) deve ser evitado sempre que possível.21 Comparando-se com estudos anteriores, pode-se perceber que o método QuEChERS original já foi utilizado para determinação multirresíduo em polpa de açaí para 174 agrotóxicos11 e o QuEChERS citrato já foi validado para a mesma matriz, porém, para um método com apenas 14 agrotóxicos, sendo este utilizado por conta das características físico-químicas específicas de alguns analitos desejados para estudo.12 Considerando tais fatores, bem como a perspectiva da Química Verde, que promove práticas sustentáveis e de menor impacto ambiental, a escolha do método QuEChERS original emergiu como uma decisão estratégica, embasada tanto nos resultados obtidos quanto nas considerações econômicas e ecológicas associadas aos reagentes empregados. Avaliação dos sorventes utilizados na etapa de limpeza do extrato A polpa de açaí é caracterizada como uma matriz complexa devido à variabilidade dos componentes presentes em suas matérias-primas. Os principais nutrientes encontrados neste alimento incluem carboidratos, lipídios, proteínas, vitaminas e minerais. Dentre estes, os macronutrientes proteicos e lipídicos são os mais relevantes no que diz respeito à interferência na etapa de preparo de amostras. Portanto, foi avaliada a etapa de limpeza mais adequada com o objetivo de remover esses interferentes, sem eliminar os compostos de interesse. Optou-se por realizar testes com 20 diferentes materiais para avaliar a remoção dos coextrativos presentes na matriz, sem comprometer a recuperação dos compostos. A Figura 2 apresenta os extratos obtidos após a realização de cada um dos 20 testes de limpeza, em ordem decrescente de remoção do coextrativo.

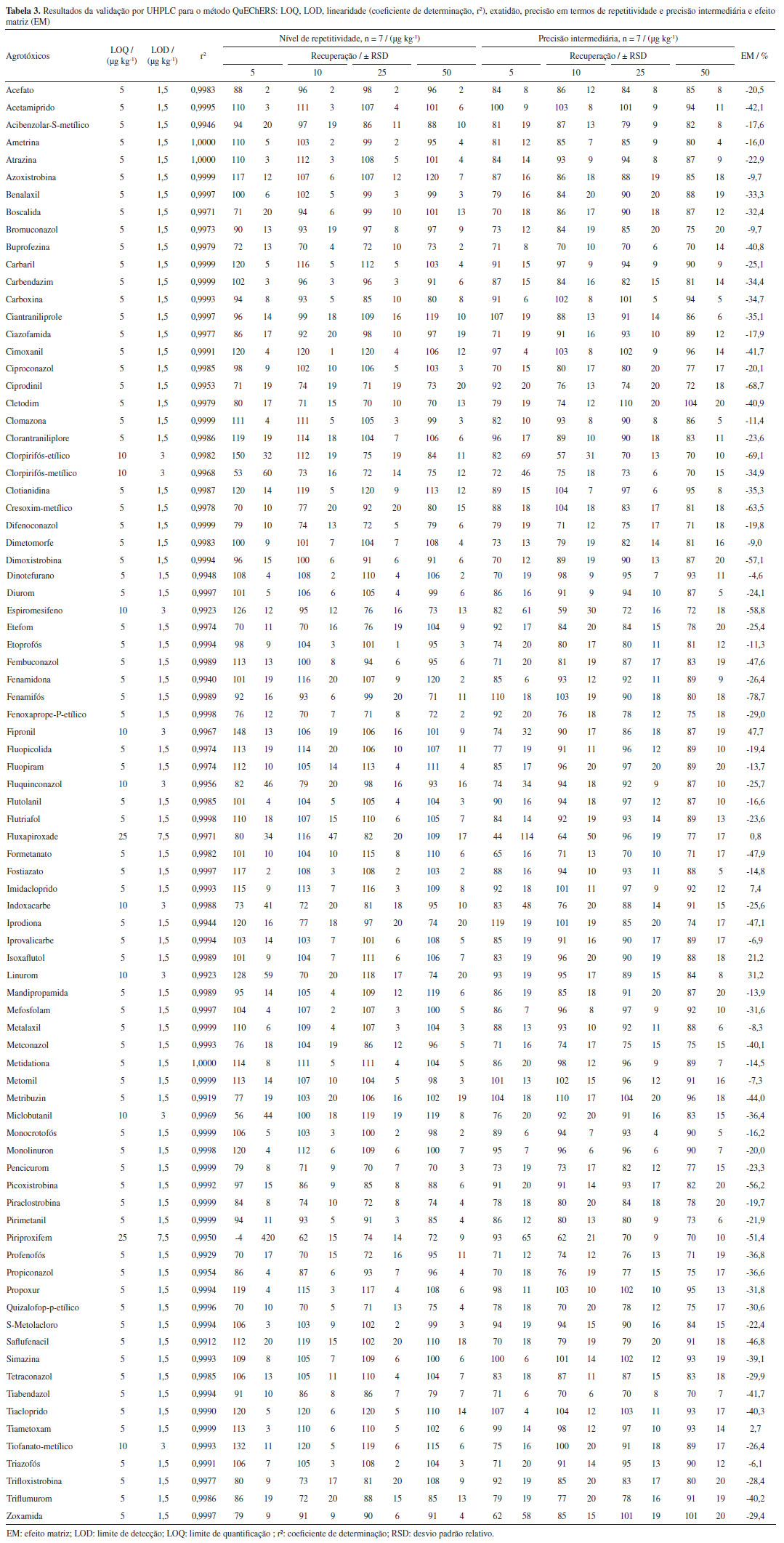
Com base nesses resultados, foram escolhidos os oito extratos que demonstraram maior eficácia na etapa de limpeza, de forma visual: propilamino, PSA, Strata-X, Oasis HLB, GCB, carvão ativado, quitosana de camarão-rosa e quitosana de casca de caranguejo. Tais extratos foram posteriormente analisados por espectrofotometria de absorção molecular UV-Vis, entre 200 e 800 nm, a partir da qual foram obtidos os espectros de absorção apresentados na Figura 3. Pela análise dos espectros UV-Vis dos extratos é possível observar a predominância de compostos que exibem absorção na região do ultravioleta. Provavelmente os compostos presentes nos extratos pertencem a mesma classe de moléculas, tendo bandas no UV (entre 217 e 309 nm) muito semelhantes às dos espectros de compostos fenólicos e de seus derivados, que são compostos que ocorrem em açaí.22 Examinando os espectros, é possível constatar que os sorventes PSA e o propilamino foram os sorventes que proporcionaram a maior eficácia na limpeza de interferentes. Isso se evidenciou pela redução da intensidade do sinal observada em todos os picos, ao se fazer a comparação dos sinais obtidos dos outros extratos com a adição dos diferentes sorventes com o sinal obtido do extrato sem processo de limpeza.
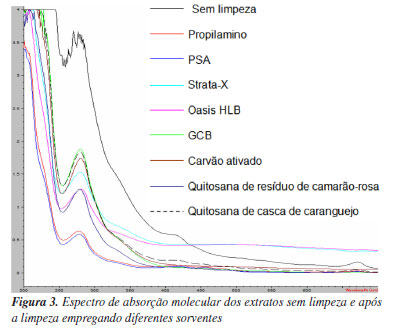
Com isso, os extratos nos quais foi realizada a etapa de limpeza com estes dois sorventes foram selecionados para a realização das análises por GC-MS full scan, comparando seu uso puro com a associação de 250 mg de C18, sendo este acrescentado na limpeza devido sua eficiência em amostras que contenham grande quantidade de lipídios.10,15 A Figura 4 apresenta os resultados dessa análise.
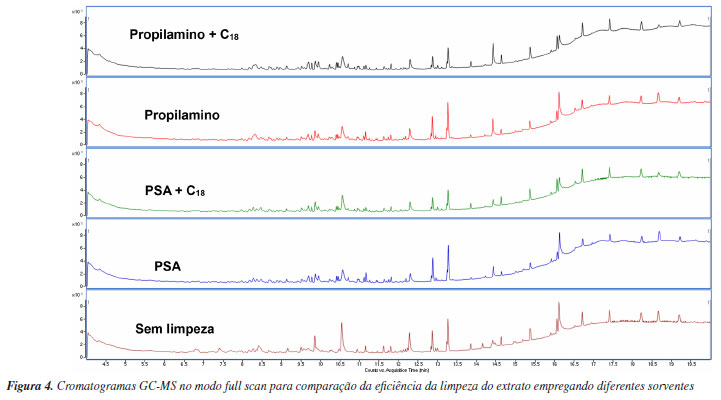
Os resultados obtidos a partir da técnica GC-MS full scan indicam que a associação dos sorventes PSA e C18 promove uma limpeza mais eficiente do extrato de açaí. Para fins comprobatórios, realizou-se o teste gravimétrico, em triplicata, visando verificar quantitativamente a eficiência de diferentes abordagens de limpeza. Notavelmente, foi constatada uma massa ligeiramente maior nas amostras submetidas às etapas de limpezas com PSA e C18 (1,82 mg), em comparação à limpeza feita com uso de propilamino e C18 (1,71 mg). Ademais, os extratos que não foram submetidos a nenhum processo de limpeza apresentaram uma massa de 5,62 mg. Com isso, a limpeza com PSA e C18 apresenta uma porcentagem de cerca de 67,6% de diminuição de massa, enquanto a limpeza com propilamino e C18 apresenta uma diminuição de cerca de 69,6%. A análise comparativa dos resultados revela que a utilização do sorvente propilamino resultou em uma limpeza mais efetiva, notavelmente evidenciada pelos menores valores de interferentes mostrados no teste de gravimetria. Este resultado sugere que a escolha do sorvente propilamino contribui para a obtenção de extratos mais limpos e, portanto, mais adequados para a subsequente determinação de resíduos de agrotóxicos em polpa de açaí. Porém, ao verificar os resultados de recuperação dos 83 agrotóxicos, associados ao C18, percebe-se que o propilamino apresentou o menor número (73) de compostos com recuperação satisfatória. O PSA apresentou recuperação satisfatória para 75 agrotóxicos. Ao se comparar com os demais trabalhos que também utilizaram QuEChERS como preparo de amostra,11,12 pode-se perceber que o uso do PSA é utilizado em ambos, tendo, ainda a presença do C18 em um deles, o que indica que tais sorventes são adequadamente utilizados para este tipo de matriz para a determinação por UHPLC-MS/MS. Validação de método A validação do método analítico para os agrotóxicos listados na Tabela 3 foi realizada em conformidade com as diretrizes estabelecidas pelos guias INMETRO20 e SANTE.16 As soluções analíticas utilizadas neste estudo foram preparadas tanto em solvente (acetonitrila) quanto no extrato obtido a partir das amostras branco de polpa de açaí. O método validado envolve a pesagem de 10 g da amostra de polpa de açaí, à qual são adicionados 10 mL de acetonitrila, seguidos de agitação em vórtex por 1 min. Em seguida, os sais de extração QuEChERS original (4 g de MgSO4 e 1 g de NaCl) são adicionados, e a mistura é novamente agitada em vórtex por 1 min e submetida a centrifugação (8 min, 3400 rpm a 20 ºC). Na etapa de limpeza do extrato, 1 mL do sobrenadante é adicionado ao sorventes (300 mg MgSO4, 50 mg de PSA e 250 mg de C18). Após agitação por 1 min em vortex e centrifugação (8 min, 3500 rpm a 20 ºC), o sobrenadante (extrato de açaí) é diluído 5 vezes em água ultrapura e em seguida é filtrado através de filtros de nylon de 13 mm com porosidade de 0,2 µm, e então analisado por UHPLC-MS/MS. Dessa maneira, foram avaliados os parâmetros de validação do método analítico para os 83 agrotóxicos em estudo, com base nos seguintes critérios: seletividade, linearidade, efeito matriz, limite de detecção (LOD), limite de quantificação (LOQ), exatidão e precisão (repetibilidade e precisão intermediária). O método proposto demonstrou uma seletividade adequada, conforme evidenciado pela análise comparativa dos cromatogramas do extrato da amostra branco de açaí, do branco dos reagentes e do extrato da amostra branco de açaí fortificado com uma solução de 10 µg L-1. Nessas análises, não foram detectados interferentes no mesmo tempo de retenção dos agrotóxicos selecionados, confirmando assim a seletividade do método. A linearidade do procedimento analítico foi verificada por meio da avaliação do coeficiente de determinação (r²), que foi obtido a partir de curvas analíticas preparadas no extrato do açaí. Inicialmente, a curva analítica foi elaborada em acetonitrila e, subsequentemente, no extrato de açaí. As concentrações dos pontos da curva que foram avaliadas incluíram os seguintes níveis de concentração: 0,5; 1; 2; 5; 10 e 20 µg L-1. Tal intervalo escolhido para análise permitiu que 83 compostos avaliados apresentasse um coeficiente de determinação (r2) superior a 0,990, conforme demonstrado na Tabela 3. Além disso, a faixa de trabalho estabelecida para o método foi de 0,5 a 20 µg L-1, garantindo a precisão e a exatidão das quantificações realizadas nesse intervalo de concentrações. O efeito matriz (EM) foi avaliado pela comparação dos coeficientes angulares das curvas analíticas preparadas tanto na acetonitrila quanto no extrato do branco de açaí, utilizando a Equação 1. A faixa de concentração avaliada variou de 0,5 a 20 µg L-1. Como muitos agrotóxicos apresentaram efeito matriz superior a ±20%, o efeito matriz foi compensado por meio de uma curva no extrato branco da matriz. O limite de quantificação (LOQ) do método foi determinado a partir dos ensaios de fortificação. Este limite foi considerado como o menor nível de fortificação (µg kg-1) que apresentou uma razão sinal/ruído superior a 10, onde a precisão e a exatidão dos compostos de interesse estavam dentro dos níveis aceitáveis de recuperação e desvio padrão relativo (70 a 120%; RSD ≤ 20%) conforme as diretrizes preconizadas pela SANTE.16 Já o valor do limite de detecção (LOD) do método foi calculado a partir do LOQ, dividindo este valor por 3,33. Conforme a Tabela 3, os valores de LOQ do método variaram entre 5 e 25 µg kg-1, enquanto os valores de LOD ficaram entre 1,5 e 7,5 µg kg-1, valores estes considerados satisfatórios para o método desenvolvido para determinação de resíduos de agrotóxicos em alimentos. Ao comparar com o método desenvolvido por Froés et al.13 empregando a técnica MSPD,13 que apresentou LOQ entre 50 e 100 µg kg-1 e LOD entre 20 e 50 µg kg-1, verifica-se que o método proposto apresentou valores de LOQ e LOD bem mais baixos. Além de tais valores, dos 5 agrotóxicos analisados por MSPD, apenas bromuconazol, fenbuconazol e cresoxim-metílico foram bem recuperados. Comparando-se com os resultados obtidos pelos métodos QuEChERS descrito por Tran et al.,11 que obteve valores de LOQ entre 2 e 120 µg kg-1, os valores são semelhantes aos obtidos pelo método proposto neste trabalho. No estudo desenvolvido por Paz etal.,12 os valores de LOQ encontram-se entre 5,16 e 17,45 µg kg-1, muito próximos aos obtidos neste estudo, porém os valores foram estabelecidos com base na relação sinal-ruído igual a 10 e não a partir de ensaios de recuperação. Com o intuito de avaliar a exatidão e a precisão do método, foram considerados os valores de recuperação e de desvio padrão relativo recomendados pela legislação internacional, especificamente as diretrizes SANTE.16 Esses valores aceitáveis são definidos entre 70 e 120% para recuperação e um desvio padrão relativo (RSD) ≤ 20%. Para avaliar a exatidão do método, foram realizados ensaios de recuperação dos compostos selecionados em quatro níveis de fortificação: 5, 10, 25 e 50 µg kg-1, com cada nível sendo testado em sete replicatas. Como resultado obteve-se que 72 dos 83 agrotóxicos avaliados apresentaram valores de recuperação dentro do intervalo aceitável no nível de fortificação mais baixo (5 µg kg-1). Destes, 9 agrotóxicos demonstraram recuperações adequadas no nível de 10 µg kg-1, e apenas 2 agrotóxicos teve uma recuperação aceitável a partir do nível de 25 µg kg-1. Em relação aos valores de RSD, 75 dos 83 agrotóxicos apresentaram valores adequados para os 4 níveis selecionados (Tabela 3). Além da precisão avaliada com base nas replicatas, a precisão intermediária foi examinada ao longo de um intervalo de sete dias em relação ao primeiro teste. Esse teste de validação foi repetido por um analista distinto, utilizando os mesmos lotes e fabricantes de reagentes e solventes e mantendo o mesmo local, condições e equipamento. Aplicação do método desenvolvido O método validado neste estudo foi aplicado em 30 amostras comerciais do estado do Amazonas, Brasil. Após a análise de 30 amostras de açaí coletadas no Amazonas, não houve detecção de nenhum dos 83 agrotóxicos validados neste estudo. As aplicações dos outros métodos descritos na literatura11-13 para a mesma matriz também não detectaram a presença de resíduos de agrotóxicos em polpa de açaí. Esses resultados refletem o fato da maioria dos açaizeiros do Amazonas estarem localizados em áreas extrativistas,3 onde a prática do cultivo sustentável e sem uso de agrotóxicos predomina.
CONCLUSÕES O presente estudo teve como objetivo desenvolver e validar um método baseado no método QuEChERS para a determinação simultânea de múltiplos resíduos de uma vasta gama de agrotóxicos em polpa de açaí, utilizando UHPLC-MS/MS. Para isso, foram avaliadas diversas versões do método QuEChERS, assim como a eficiência de diferentes sorventes na etapa de limpeza da amostra, visando otimizar a remoção de interferentes. Os parâmetros de validação demonstraram que o método desenvolvido é adequado para a determinação de 83 agrotóxicos na polpa de açaí, apresentando excelente linearidade (com r2 > 0,9912), exatidão satisfatória (com recuperações entre 70 e 120%) e precisão consistente (com RSD ≤ 20%). O limite de quantificação (LOQ) do método variou entre 5 e 25 µg kg-1. A detecção dos compostos por meio de UHPLC-MS/MS garantiu alta detectabilidade, seletividade, exatidão e precisão durante a etapa de validação. O método validado foi aplicado à análise de 30 amostras comerciais de polpa de açaí, nas quais não foram detectados resíduos de agrotóxicos. Dessa forma, o método desenvolvido, utilizando a versão original do QuEChERS com etapa de limpeza utilizando PSA associado ao C18 e quantificação por UHPLC-MS/MS, mostrou-se simples, rápido e eficaz para a determinação multirresíduos de agrotóxicos em polpa de açaí. Além disso, o método proposto possui potencial para ser amplamente utilizado em análises de rotina, proporcionando resultados confiáveis e de alta qualidade.
MATERIAL SUPLEMENTAR Tabela 1S relacionada aos resultados obtidos neste trabalho estão disponíveis em http://quimicanova.sbq.org.br, como arquivo PDF, com livre acesso.
AGRADECIMENTOS Os autores agradecem o apoio financeiro e a concessão de bolsas das agências de fomento brasileiras CNPq e Fundação de Amparo à Pesquisa do Estado do Amazonas (01.02.016301.001624/2021-10-FAPEAM).
REFERÊNCIAS 1. Yuyama, L. K. O.; Aguiar, J. P. L.; Silva Filho, D. F. S.; Yuyama, K.; Varejão, M. J.; Fávaro, D. I. T.; Vasconcellos, M. B. A.; Pimentel, S. A.; Caruso, M. S. F.; Acta Amazônica 2011, 41, 545. [Crossref] 2. do Nascimento, R. J. S.; Couri, S.; Antoniassi, R.; Freitas, S. P.; Rev. Bras. Frutic. 2008, 30, 498. [Crossref] 3. Guimarães, C. S. S.; Vaz, M. A. B.; da Silva, R. B. P.; Revista Ibero-Americana de Ciências Ambientais 2021, 12, 591. [Link] acessado em Janeiro 2025 4. Matta, V. M.; Cruz, A. P. G.; Cabral, L. M. C.; Donângelo, C. M.; Comunicado Técnico 165,Açaí Clarificado por Microfiltração; Embrapa Amazônia Oriental: Belém, 2010. [Link] acessado em Janeiro 2025 5. Gomes, H. O.; Menezes, J. M. C.; da Costa, J. G. M.; Coutinho, H. D. M.; Teixeira, R. N. P.; do Nascimento, R. F.; Ecotoxicol. Environ. Saf. 2020, 197, 110627. [Crossref] 6. da Silva, R. B. P.; de Oliveira, J. A.; Zanchin, C. F.; dos Santos, P. J.; Prestes, O. D.; Zanella, R.; Quim. Nova 2024, 47, e-20240066. [Crossref] 7. Prestes, O. D.; Zanella, R.; dos Santos, P. J. Em Manual de Práticas em Análises Toxicológicas; Peixe, T. S.; Bairros, A. V., orgs.; Brazil Publishing: Curitiba, 2021, p. 301-317. 8. Anastassiades, M.; Lehotay, S. J.; Štajnbaher, D.; Schenck, F. J.; J. AOAC Int. 2003, 86, 412. [Crossref] 9. Li, M.; Dai, C.; Wang, F.; Kong, Z.; He, Y.; Huang, Y. T.; Fan, B.; Sci. Rep. 2017, 7, 42489. [Crossref] 10. Santana-Mayor, A.; Rodríguez-Ramos, R.; Herrera-Herrera, A. V.; Socas-Rodríguez, B.; Rodríguez-Delgado, M. A.; TrAC, Trends Anal. Chem. 2023, 169, 117375. [Crossref] 11. Tran, K.; Eide, D.; Nickols, S. M.; Cromer, M. R.; Sabaa-Srur, A.; Smith, R. E.; Food Chem. 2012, 134, 2398. [Crossref] 12. Paz, M.; Correia-Sá, L.; Vidal, C. B.; Becker, H.; Longhinotti, E.; Domingues, V. F.; Delerue-Matos, C.; J. Environ. Sci. Health, Part B 2017, 52, 48. [Crossref] 13. Froés, M. B. R.; Santos, L. F. S.; Navickiene, S.; Food Anal. Methods 2013, 6, 328. [Crossref] 14. European Committee for Standardization (CEN), Technical Committee CEN/TC 275; EN 15662:2008: Foods of Plant Origin - Determination of Pesticide Residues Using GC-MS and/or LC-MS/MS Following Acetonitrila Extraction/Partitioning and Clean-Up by Dispersive SPE - QuEChERS-Method; European Union, Brussels, 2008. [Link] acessado em Janeiro 2025 15. Lehotay, S. J.; Maštovská, K.; Lightfield, A. R.; J. AOAC Int. 2005, 88, 615. [Crossref] 16. SANTE, European Comission; Document No. SANTE/11312/2021 v2: Guidance Document in Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed; 2024. [Link] acessado em Janeiro 2025 17. Prata, R.; López-Ruiz, R.; Nascimento, L. E. S.; Petrarca, M. H.; Godoy, H. T.; Frenich, A. G.; Arrebola, F. J.; J. Food Compos. Anal. 2024, 129, 106062. [Crossref] 18. Zenebon, O.; Pascuet, N. S.; Métodos Físico-Químicos para Análise de Alimentos, 4ª ed.; Instituto Adolfo Lutz: São Paulo, 2005. 19. Sapozhnikova, Y.; Lehotay, S. J.; Anal. Chim. Acta 2013, 758, 80. [Crossref] 20. Instituto Nacional de Metrologia, Qualidade e Tecnologia (INMETRO); DOQ-CGCRE-008: Orientação sobre Validação de Métodos Analíticos: Documento de Caráter Orientativo - Revisão 09 - Jun/2020; INMETRO, Coordenação Geral de Acreditação: Duque de Caxias, 2020. [Link] acessado em Janeiro 2025 21. Prado, A. G. S.; Quim. Nova 2003, 26, 738. [Crossref] 22. Amorim, I. S.; Amorim, D. S.; Godoy, H. T.; Mariutti, L. R. B.; Chisté, R. C.; Pena, R. S.; Bogusz Junior, S.; Chim, J. F.; Heliyon 2024, 10, e24054. [Crossref]
Editor Associado responsável pelo artigo: Eduardo M. Richter |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access