
 |
 |
 |
 |
 |
Revisão
|
|
| α-Iminocarbenos metálicos derivados de N-sulfonil-1,2,3-triazóis na síntese de alcaloides α-Imino metal carbenes derived from N-sulfonyl-1,2,3-triazoles in the synthesis of alkaloid |
|
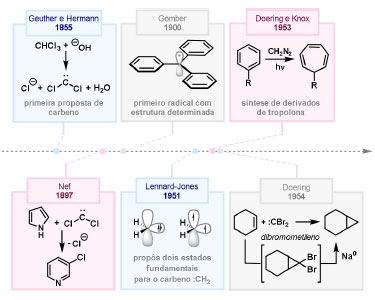
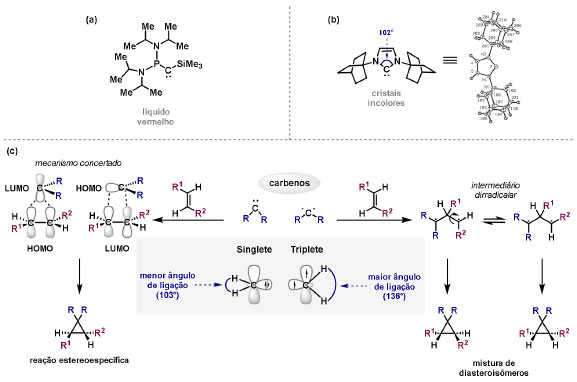
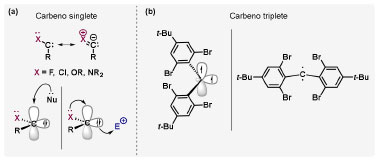
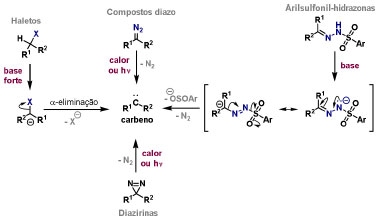
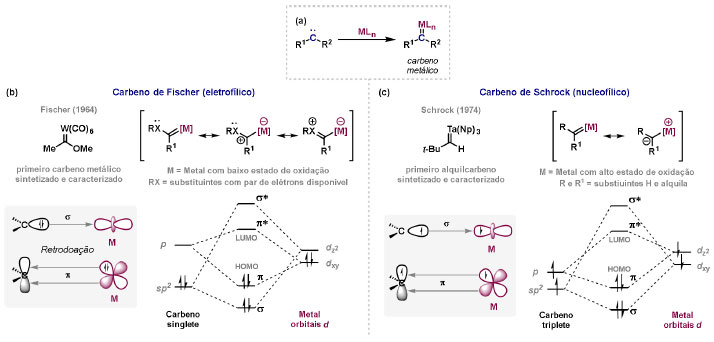
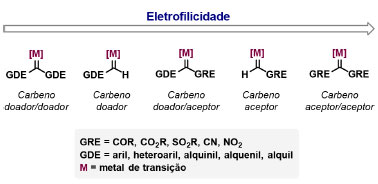
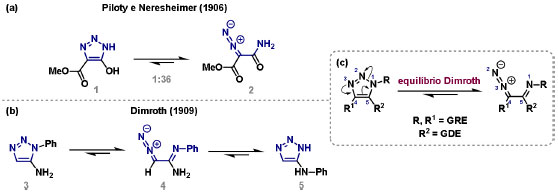
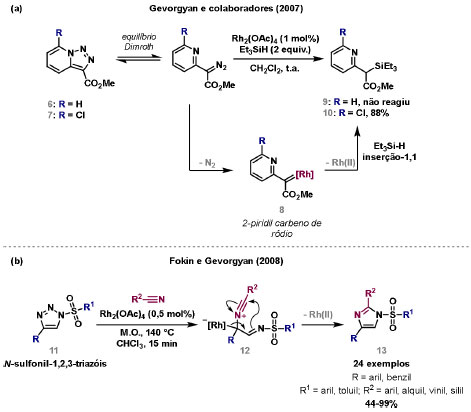
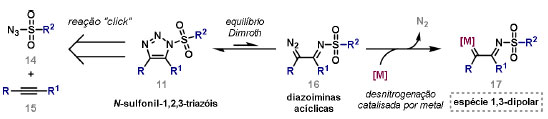
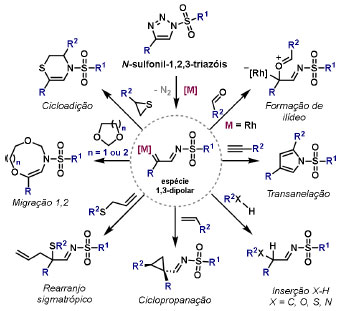
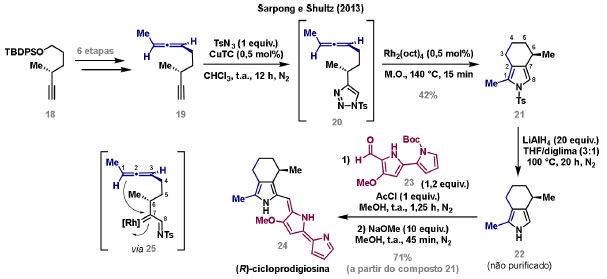
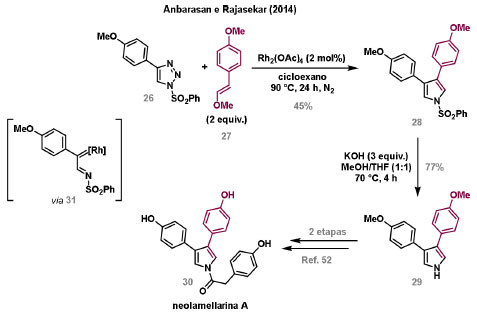
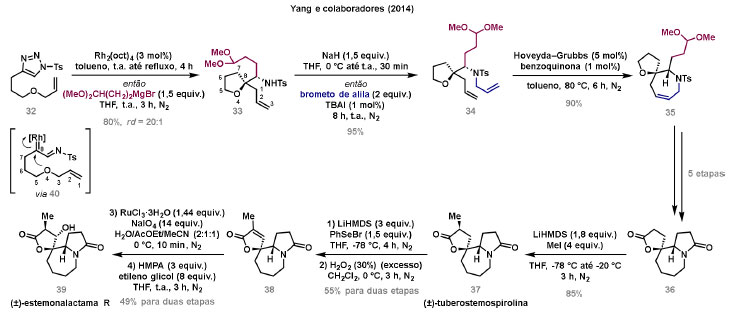
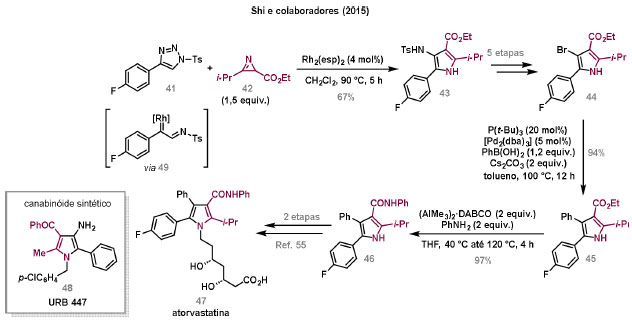
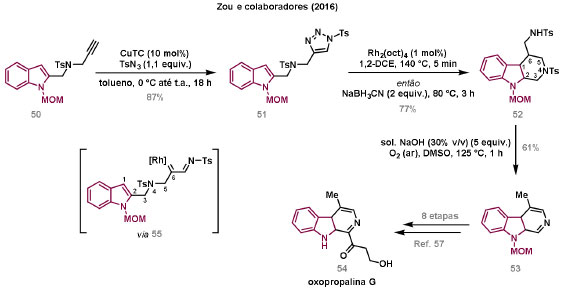
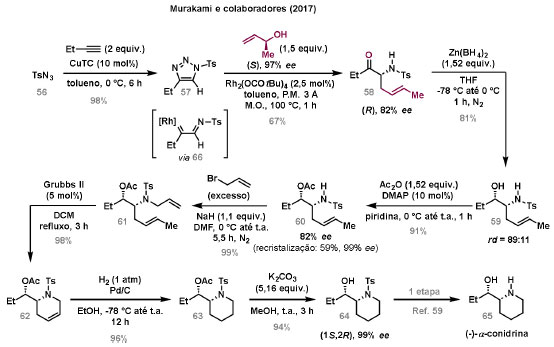
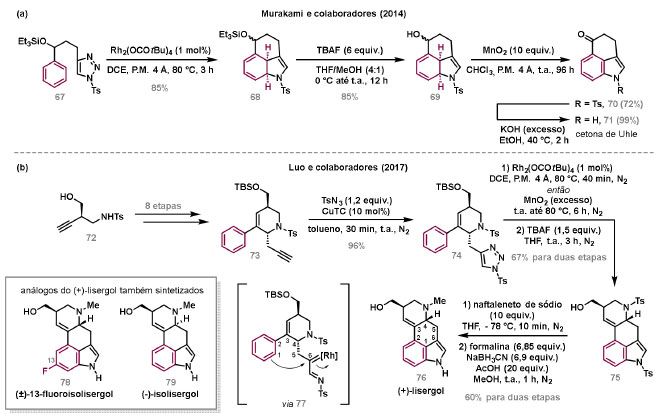
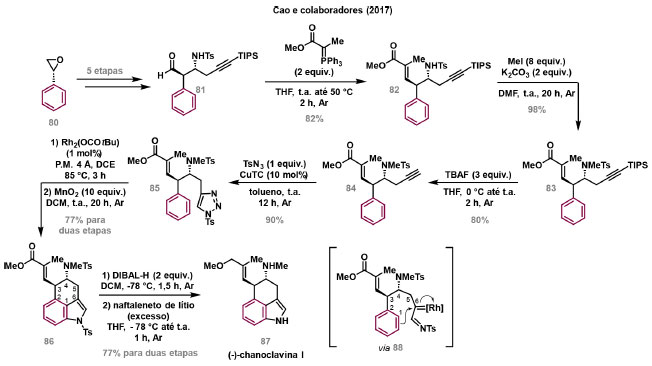
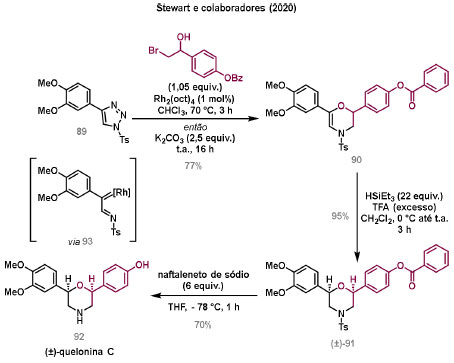
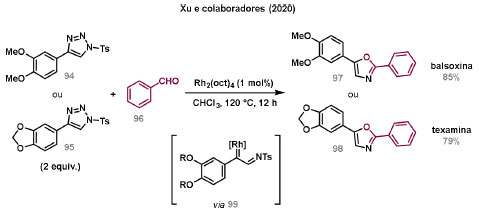
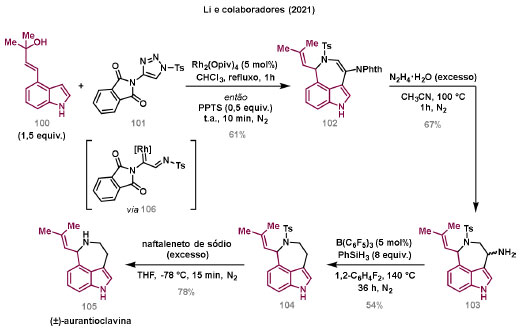
Vinícius V. Souza; Cristiano Raminelli* Departamento de Química, Instituto de Ciências Ambientais, Químicas e Farmacêuticas, Universidade Federal de São Paulo, 09972-270 Diadema - SP, Brasil Recebido: 28/10/2024 *e-mail: raminelli@unifesp.br The study of carbenes and metal carbenes has attracted researchers from different countries due to the versatility of these reactive species in preparative organic chemistry. In this review, we discuss the structural aspects, reactivity, and formation methods of carbenes, which provide theoretical support for understanding the structure/reactivity relationship of metal carbenes, whose classifications and formation methods have been presented. Emphasis is placed on the class of α-imino metal carbenes, which have allowed the construction of various heterocyclic compounds, including alkaloids with complex structures. Total and formal syntheses of nitrogen-containing compounds using α-imino metal carbenes from N-sulfonyl-1,2,3-triazoles have been reviewed. In this regard, an overview of the synthetic routes established in these works, including the key reactions highlighted by the authors, has been provided. INTRODUÇÃO O estudo de carbenos, especialmente na presença de metais de transição, tem atraído pesquisadores de todo mundo, devido à formação de espécies conhecidas como carbenos metálicos ou metal carbenos.1 Neste artigo de revisão, pretendemos abordar características estruturais, a reatividade e métodos de formação de carbenos, para dar suporte teórico para a compreensão da relação entre a estrutura e a reatividade de carbenos metálicos, além de apresentar suas classificações e métodos de geração, visando demonstrar a grande versatilidade desses intermediários organometálicos em química orgânica preparativa. Daremos ênfase à classe dos α-iminocarbenos metálicos, os quais têm permitido a construção de diversos compostos heterocíclicos nitrogenados,2 incluindo alcaloides com estruturas complexas.3 Neste trabalho em particular, apresentaremos 11 (onze) estudos contendo sínteses totais e formais, envolvendo α-iminocarbenos metálicos derivados de compostos N-sulfonil-1,2,3-triazólicos, as quais serão delineadas de maneira a dar uma visão geral das rotas sintéticas desenvolvidas, trazendo as reações chaves destacadas pelos autores. CARBENOS A primeira suposição da existência de um carbeno foi feita em 1855 por Geuther e Hermann.4 Foi sugerido que a hidrólise alcalina do clorofórmio prosseguia através da formação de um intermediário de reação com um carbono divalente chamado diclorocarbeno. Posteriormente, em 1897, Nef5 propôs o mesmo intermediário na transformação entre pirrol e clorofórmio em 3-cloropiridina. Naquela época, a proposta da existência de carbenos como intermediários foi muito ousada, pois ainda não se sabia da existência de radicais livres. Foi somente em 1900 que os radicais livres foram descobertos, ou seja, através de experimentos de análise elementar realizados por Gomberg,6 a estrutura para o radical trifenilmetileno foi determinada. No entanto, o interesse pelos carbenos foi reavivado apenas no início da década de 1950 com Lennard-Jones e Pople,7 que propôs os estados singlete e triplete para carbenos. Em 1953, Doering e Knox8 reportaram a síntese de tropolonas por meio da adição de carbeno metileno a benzenos substituídos com grupos alquila. Entretanto, a contribuição mais significativa de Doering e seus colaboradores9 veio um ano depois, quando relataram a primeira reação de ciclopropanação entre bromofórmio e alcenos via intermediário dibromometileno (Esquema 1). Carbenos são espécies neutras reativas que contêm um átomo de carbono divalente com uma camada de valência de seis elétrons, com dois pares de elétrons formando ligações e dois elétrons não ligantes.10 No entanto, existem alguns carbenos estáveis que podem ser isolados e caracterizados, tais como os estabilizados por substituintes contendo fósforo e silício (Esquema 2a) e carbenos N-heterocíclicos, cuja estrutura foi confirmada em 1991, por cristalografia de raios-X11 (Esquema 2b). Os carbenos podem existir nos estados singlete e triplete. O estado singlete apresenta dois elétrons não ligantes emparelhados em um orbital sp2 e um orbital p vazio, com um ângulo de ligação de aproximadamente 103° para o carbeno metileno.12 O estado triplete, com um ângulo de ligação de aproximadamente 136° para o carbeno metileno, apresenta dois elétrons não ligantes desemparelhados em orbitais distintos, um em orbital sp2 e outro em orbital p.13 Ambos os estados eletrônicos exibem geometrias angulares e reatividades distintas. Um exemplo clássico é a reação de ciclopropanação entre carbenos e alcenos, em que o mecanismo da reação está diretamente relacionado com o tipo de carbeno. Um carbeno singlete, com elétrons emparelhados em orbital sp2 e orbital p vazio, realiza uma reação de cicloadição através da sobreposição de orbitais de fronteira HOMO (em inglês, highest occupied molecular orbital) e LUMO (em inglês, lowest unoccupied molecular orbital), por meio de uma única etapa concertada, favorecendo a formação de um único estereoisômero. Por outro lado, um carbeno triplete atua como um radical e reage via mecanismo por etapas, através da formação de um intermediário dirradicalar, que possui uma rotação livre na ligação C-C, favorecendo a formação de uma mistura de diasteroisômeros14 (Esquema 2c). Estudos experimentais e teóricos dos orbitais moleculares do carbeno metileno indicam que o estado triplete apresenta uma energia aproximadamente 8,5 kcal mol-1 mais baixa do que o estado singlete.15 Contudo, a estrutura e o método empregado para a geração de um determinado carbeno definirá em qual estado ele será formado. A multiplicidade do carbeno depende do solvente em que ele é formado, onde solventes mais polares coordenantes doam densidade eletrônica ao orbital p vazio de carbenos singletes e estabilizam tal forma. Por outro lado, solventes apolares não coordenantes favorecem carbenos tripletes.16 Intramolacularmente, substituintes com pares de elétrons não ligantes (por exemplo, F, Cl, OR e NR2) favorecem a formação de carbenos no estado singlete, através da doação de elétrons por efeito de ressonância para o orbital p vazio do carbeno. Por essa razão, tais carbenos são considerados espécies que atuam tanto como eletrófilos, quanto como nucleófilos17,18 (Esquema 3a). A presença de grupos aromáticos e volumosos favorece a formação do estado triplete, devido à estabilização proporcionada pela doação de elétrons π, principalmente, para o orbital p semipreenchido, e devido aos efeitos estéricos que levam à formação de maiores ângulos de ligação (em comparação com o estado singlete)19 (Esquema 3b). Uma variedade de métodos foi desenvolvida para a geração de carbenos. A grande maioria dessas reações envolve compostos diazo como precursores de carbenos, através da liberação de N2 por decomposição térmica ou fotoquímica.20 No mesmo sentido, a decomposição de arilsulfonil-hidrazonas induzida por base, gera carbenos através de um intermediário "tipo diazo", estabilizado por ressonância, com a eliminação de arilsulfinato e nitrogênio.21 Outro método para a geração de carbenos é a decomposição térmica ou fotoquímica de diazirinas.22 Apesar da importância dos composto diazo, vale mencionar que as α-eliminações induzidas por base em haletos de alquila, constituem o primeiro método desenvolvido para formação de carbenos23 (Esquema 4). CARBENOS METÁLICOS Os carbenos, na presença de alguns metais de transição, podem formar compostos organometálicos relativamente estáveis denominados carbenos metálicos ou metal carbenos24 (Esquema 5a). Existem dois tipos principais de metal carbenos, chamados de carbenos de Fischer ou carbenos de Schrock.25 Em 1964, o primeiro carbeno metálico foi sintetizado e caracterizado por Fischer e Maasböl,26 por meio do tratamento de W(CO)6 com metil-lítio, seguido de reação com CH2N2, a qual resultava na formação de um composto de coordenação de tungstênio, que apresentava a estrutura de um carbeno metálico. Mais tarde, carbenos metálicos com características semelhantes foram definidos como carbenos do tipo Fischer. Eles são constituídos principalmente por metais de baixos estados de oxidação como Cr(0), Mo(0), W(0), Fe(0) e Mn(0), em combinação com carbenos no estado singlete. Neste caso, a ligação é explicada como uma interação entre o carbono carbênico no estado singlete com doação σ para o orbital d vazio do metal e retrodoação π do metal para o carbeno.27 Embora essas estruturas apresentem grupos contendo heteroátomos como substituintes, por exemplo, grupos alcóxi ou amino, a ligação carbono-metal (relativamente longa) é polarizada no sentido do metal, tornando o carbono carbênico eletrofílico28 (Esquema 5b). Em 1974, Shrock29 relatou a primeira síntese de um alquilcarbeno de tântalo com alto estado de oxidação estável. Neste contexto, carbenos com características semelhantes são classificados como carbenos do tipo Shrock. Esses compostos possuem metais com altos estados de oxidação, como W(VI) e Mo(VI), em combinação com carbenos no estado triplete. Neste caso, o carbeno no estado triplete interage com orbitais d do metal para formar uma ligação dupla covalente (relativamente curta), que torna o carbono carbênico nucleofílico26,30 (Esquema 5c). Em linhas gerais, os grupos funcionais presentes no carbeno são responsáveis por modular sua reatividade e aplicabilidade através das diferentes propriedades eletrônicas. Grupos retiradores de elétrons (GRE) aumentam o caráter eletrofílico do átomo de carbono e grupos doadores de elétrons (GDE) atuam com o efeito oposto. Considerando a eletrofilicidade, metal carbenos podem ser diferenciados e classificados da seguinte forma: doador/doador; somente doador; doador/aceptor; somente aceptor e aceptor/aceptor31 (Esquema 6). Embora existam muitas vantagens no uso de compostos diazo como precursores de carbenos metálicos, também existem algumas desvantagens. Os compostos diazo são potencialmente explosivos e tóxicos, além de apresentar baixa estabilidade térmica (eles se decompõem tipicamente acima de 80 °C). Como resultado, enormes esforços têm sido dedicados para encontrar substitutos adequados para tais compostos,32 dentre os quais, destacamos alcinos em combinação com Au e ilídeos de sulfoxônio.32 Neste contexto, 1,2,3-triazóis surgiram como substâncias sintéticas versáteis para geração in situ de compostos α-imino diazo sob condições brandas, pois são estáveis possibilitando armazenamento, além de serem mais seguros para o manuseio. Em 1906, Piloty e Neresheimer33 prepararam o 5-oxi-1,2,3-triazol (1) e observaram que se apresentava em equilíbrio com o composto diazodicarbonílico 2 (de anel aberto) (Esquema 7a). Após três anos, Dimroth34 preparou o 5-amino-1,2,3-triazol (3) e relatou que com o tempo, ocorria um rearranjo intramolecular espontâneo, através da formação de uma diazoimina acíclica 4, gerando 5-(N-fenilamino)-1,2,3-triazol (5) (Esquema 7b). O processo de isomerização anel-cadeia em 1,2,3-triazóis ficou conhecido como rearranjo de Dimroth e foi estudado posteriormente por diversos grupos, incluindo Grünanger et al.,35 Hermes e Marsh,36 Harmon et al.37 e Habraken et al.38 Esses estudos e outros tornaram claro que o deslocamento do equilíbrio Dimroth era altamente dependente da natureza dos substituintes presentes no anel. Descobriu-se que a presença de grupos retiradores de elétrons nas posições N-1 e C-4, bem como grupos doadores de elétrons na posição C-5 dos triazóis deslocam o equilíbrio em direção às diazoiminas de anel aberto. Os carbenos gerados podem ser classificados como aceptores quando R1 é um GRE ou como doadores/aceptores quando R1 é um GRE e R2 é um GDE (Esquema 7c). O potencial dos 1,2,3-triazóis como precursores de carbenos metálicos foi percebido pela primeira vez em 2007 por Gevorgyan e colaboradores.39 O 2-piridil carbeno de ródio 8 foi gerado a partir de 7-cloropiridotriazol (7) e sofreu inserção em ligação Si-H na presença de trietilsilano, para gerar o produto sililado 10 em 88% de rendimento. O substituinte cloro foi crucial, uma vez que o análogo não clorado não reagiu, demonstrando a sutil relação entre a estrutura e a reatividade do derivado 1,2,3-triazólico39 (Esquema 8a). Após o trabalho de Gevorgyan e colaboradores,39 foram feitos esforços para descobrir outros triazóis que promovessem a formação de carbenos metálicos, impulsionando o emprego de 1,2,3-triazóis como precursores desses organometálicos e suas aplicações em síntese orgânica. Em 2008, Fokin et al.40 revelaram os N-sulfonil-1,2,3-triazóis 11 como importantes precursores de carbenos metálicos, sendo submetidos à transanelação na presença de nitrilas catalisada por Rh(II), para gerar imidazóis substituídos 13. Nessa transformação, o N-sulfonil-1,2,3-triazol 11 gera um carbeno metálico, que é atacado por uma nitrila para gerar o intermediário polar 12, que sofre ciclização com a liberação do catalisador, para gerar o imidazol substituído 1340 (Esquema 8b). Os N-sulfonil-1,2,3-triazóis 11 podem ser facilmente preparados a partir de alcinos terminais e sulfonil azidas por reação "click" catalisada por Cu(I).41 Os compostos 11 encontram-se em equilíbrio com suas correspondentes diazoiminas acíclicas 16, que na presença de metais de transição, levam à formação de α-iminocarbenos metálicos 17, através da liberação de N2. O caráter eletrofílico do carbono carbênico e o nitrogênio da sulfonilimina nucleofílico explicam a natureza 1,3-dipolar dessas espécies. Ademais, o equilíbrio pouco deslocado na direção dos produtos proporciona uma baixa concentração de espécies reativas permitindo uma alta quimiosseletividade, sem a necessidade de uma adição lenta42 (Esquema 9). A geração lenta dos α-iminocarbenos metálicos, em condições relativamente brandas, permitiu suas aplicações em uma variedade de transformações úteis em síntese orgânica, por exemplo, em ciclopropanações,43 inserções em ligações X-H,44 transanelações,45 rearranjos sigmatrópicos,46 migrações 1,2,47 formações de ilídeos48 e cicloadições49 (Esquema 10), podendo ser considerados intermediários versáteis na síntese de compostos N-heterocíclicos e produtos naturais. APLICAÇÃO DE CARBENOS METÁLICOS EM SÍNTESE TOTAL DE ALCALOIDES Em 2013, Schultz e Sarpong50 relataram uma rota sintética eficiente para compostos contendo núcleo pirrólico obtidos a partir de alenilalcinos, através de duas etapas reacionais one-pot, envolvendo α-iminocarbenos metálicos produzidos por catalisador de ródio(II). Em seguida, realizaram a síntese estereosseletiva do alcino terminal 18 (não apresentada neste texto), o qual forneceu o alenilalcino 19, em seis etapas reacionais. O composto 19 foi submetido à reação de cicloadição com tosilazida catalisada por tiofeno-2-carboxilato de cobre(I) (CuTC), levando à formação do N-sulfonil-1,2,3-triazol 20 (não isolado), que na presença de octanoato de ródio(II) como catalisador, resultou no intermediário 21, através do α-iminocarbeno metálico 25 e eliminação de Rh(II) (ver intermediário 12 no Esquema 8), em 42% de rendimento. Posteriormente, o composto 21 teve o grupo tosila removido por tratamento com LiAlH4, promovendo a formação do intermediário 22, que após condensação com o aldeído 23 forneceu (R)-cicloprodigiosina (24) em 71% de rendimento (para as três últimas etapas)50 (Esquema 11). A rota sintética apresentada no Esquema 11 corresponde à primeira síntese assimétrica para o produto natural (R)-cicloprodigiosina (24), que tem despertado grande interesse farmacológico pelo seu potencial anticancerígeno.50 Em 2014, Rajasekar e Anbarasan51 relataram a reação de cicloadição (3 + 2) entre N-sulfonil-1,2,3-triazóis e éteres vinílicos catalisada por ródio, que resultou em pirróis polissubstituídos via α-iminocarbenos metálicos. Esse método foi utilizado para a síntese formal do alcaloide neolamellarina A (30) com ação antitumoral (Esquema 12). A cicloadição (3 + 2) entre o N-sulfonil-1,2,3-triazol 26 e o éter vinílico 27 catalisada por acetato de ródio(II), que ocorre via intermediário 31, resultou no composto sulfonilado 28 em 45% de rendimento. Em seguida, a remoção do grupo sulfonila promoveu a formação do pirrol 29 em rendimento de 77%. A conversão do intermediário 29 em neolamellarina A (30) pode ser realizada através de duas etapas reacionais relatadas na literatura.52 Ainda em 2014, Yang e colaboradores53 reportaram a reação intramolecular em substâncias contendo uma porção N-sulfonil-1,2,3-triazólica e um grupo éter alílico catalisada por ródio, seguida por rearranjo sigmatrópico [2,3] e adição de reagente de Grignard, as quais promoveram a formação de diidroisobenzofuranos e tetraidrofuranos de maneira diastereosseletiva. Esse método foi utilizado na síntese de dois produtos naturais bioativos, (±)-tuberostemospirolina (37) e (±)-estemonalactama R (39). A síntese foi iniciada a partir da ciclização intramolecular do N-sulfonil-1,2,3-triazol contendo éter alílico 32 na presença de catalisador de ródio(II), através do α-iminocarbeno metálico 40, seguida por rearranjo sigmatrópico [2,3] e adição de brometo de (3,3-dimetoxipropil)magnésio, para fornecer o tetraidrofurano 33 em rendimento de 80% e razão diastereoisomérica de 20:1. Em seguida, o composto 33 foi tratado com NaH e brometo de alila, fornecendo o intermediário 34 em 95% de rendimento. O composto 34 foi submetido a uma metátese de fechamento de anel utilizando catalisador de Hoveyda-Grubbs, para fornecer o oxa-espirocíclo 35 em 90% de rendimento, que após cinco etapas reacionais, resultou na lactona 36. A metilação da lactona 36 com LiHMDS e iodeto de metila, promoveu a formação de (±)-tuberostemospirolina (37) em rendimento de 85%, a qual foi convertida após quatro etapas reacionais em (±)-estemonalactama R (39) (Esquema 13). Em 2015, Shi e colaboradores54 relataram uma reação de cicloadição (3 + 2) entre N-sulfonil-1,2,3-triazóis e 2H-azirinas catalisada por ródio, para a formação de pirróis polissubstituídos. As reações prosseguiam através da formação de α-iminocarbeno metálico, seguida de um ataque nucleofílico da azirina e posterior expansão do anel para fornecer os 3-aminopirróis correspondentes. Esse método foi aplicado à síntese formal da atorvastatina (47), fármaco usado principalmente para regular os níveis de colesterol no sangue e prevenir eventos associados a doença cardiovascular (Esquema 14). Reação de cicloadição (3 + 2) entre o N-sulfonil-1,2,3-triazol 41 e a 2H-azirina 42 catalisada por Rh2(esp)2, resultou no composto pirrólico 43, via intermediário 49, em rendimento de 67%, que foi convertido após cinco etapas reacionais no brometo 44. O composto 44 foi submetido ao acoplamento de Suzuki-Miyaura, resultando no intermediário 45 em rendimento de 94%. Subsequente amidação do composto 45 resultou no intermediário chave 46 em 97% de rendimento. A conversão do composto 46 em atorvastatina (47) pode ser realizada através de duas etapas reacionais reportadas na literatura.55 No mesmo trabalho, o método desenvolvido foi empregado na síntese de URB 447 (48), canabinoide sintético que atua como inibidor de apetite.55 Em 2016, Zou e colaboradores56 estabeleceram um método de anelação intramolecular em indóis contendo uma porção N-sulfonil-1,2,3-triazólica catalisado por ródio, para a preparação de tetraidro-β-carbolinas (após finalização com NaBH3CN), via α-iminocarbenos metálicos. A utilidade sintética do método desenvolvido foi avaliada na síntese formal do alcalóide oxopropalina G (54). O alcino terminal 50 foi submetido à reação de cicloadição com tosilazida catalisada por tiofeno-2-carboxilato de cobre(I) (CuTC), que resultou no N-sulfonil-1,2,3-triazol 51 em rendimento de 87%. O triazol 51 foi submetido à ciclização na presença de octanoato de ródio(II) em quantidade catalítica, através do α-iminocarbeno metálico 55, seguida de redução com NaBH3CN, resultando no intermediário 52 em 77% de rendimento. Após tratamento com solução de NaOH na presença de oxigênio (O2) do ar, o composto 53 foi obtido em rendimento de 61%, completando assim a síntese formal da oxopropalina G (44), que poderia ser alcançada em oito etapas reacionais relatadas na literatura57 (Esquema 15). Em 2017, Murakami e colaboradores58 desenvolveram uma reação entre N-sulfonil-1,2,3-triazóis e álcoois alílicos catalisada por ródio, que ocorre através da inserção de um α-iminocarbeno metálico ao grupo hidroxila, formando um intermediário que sofre rearranjo sigmatrópico [3,3], para fornecer 2-aminocetonas N-sulfoniladas de maneira estereosseletiva. Este método foi aplicado com sucesso à síntese formal da (-)-α-conidrina (65), um alcalóide piperidínico, isolado da planta Conium maculatum L., através de nove etapas reacionais. Por reação de cicloadição entre 1-butino e tosilazida (56) catalisada por tiofeno-2-carboxilato de cobre(I) (CuTC), o triazol 57 foi preparado em 98% de rendimento. O composto 57 foi submetido à reação de inserção empregando (S)-but-3-en-2-ol [97% de excesso enantiomérico (ee)] catalisada por Rh2(OCOtBu)4 (via intermediário 66), que após rearranjo sigmatrópico [3,3], resultou na 2-aminocetona N-sulfonilada 58 em 67% de rendimento e 82% de ee. O composto 58 foi reduzido com Zn(BH4)2 para formar o intermediário 59 em rendimento de 81%, com razão diastereoisomérica (rd) de 89:11. O composto 59 foi acetilado com anidrido acético (Ac2O), promovendo a formação do acetato 60 em rendimento de 91%, com 82% de ee, que passou a 99% de ee após recristalização. O composto 60 foi submetido à alilação resultando no intermediário 61 em rendimento de 99%, que após metátese de fechamento de anel, utilizando o catalisador de Grubbs de 2ª geração, formou a 3,4-desidropiperidina 62 em 98% de rendimento. Após reações de hidrogenação utilizando H2 e Pd/C e hidrólise com K2CO3, o intermediário 64 foi obtido em 90% de rendimento para as duas etapas reacionais. A conversão do composto 64 em (-)-α-conidrina (65), pode ser realizada através de uma etapa reacional reportada na literatura59 (Esquema 16). Em 2014, Murakami e colaboradores2 divulgaram uma transanelação intramolecular em compostos aromáticos, contendo uma porção N-sulfonil-1,2,3-triazólica, catalisada por ródio, que resultava na formação de esqueletos indólicos, envolvendo α-iminocarbenos metálicos. A reação desenvolvida promoveu a transformação do material de partida 67 no derivado indólico 68 em 85% de rendimento, que após desproteção com TBAF (fluoreto de tetrabutilamônio), oxidação com MnO2 e desproteção com KOH, levou à formação da cetona de Uhle (71) em 61% de rendimento para as três etapas, a qual pode ser considerada um intermediário sintético útil para síntese de compostos ergolínicos de origem natural (Esquema 17a). Em 2017, Luo e colaboradores60 empregaram a abordagem do Esquema 17a na síntese do (+)-lisergol (76) e seus análogos. A síntese foi iniciada a partir do álcool propargílico quiral 72, preparado a partir do (R)-aldeído de Garner (não apresentada no trabalho). Após oito etapas reacionais, incluindo proteção com TBS (terc-butildimetilsilano), acoplamento de Suzuki e homologação de Seyferth-Gilbert, o alcino 73 foi alcançado e submetido à reação de cicloadição com tosilazida catalisada por tiofeno-2-carboxilato de cobre(I) (CuTC), para gerar o N-sulfonil-1,2,3-triazol 74. Uma transanelação intramolecular através de reação de cicloadição (3 + 2) catalisada por Rh2(OCOtBu)4, que ocorre via α-iminocarbeno metálico 77, seguida de oxidação com MnO2 e posterior desproteção do grupo TBS com TBAF, resultaram no intermediário ergolínico 75 em 67% para as duas etapas reacionais. (+)-Lisergol (76) foi obtido após desproteção dos grupos tosila e reação de metilação em 60% para as duas etapas reacionais. Além do (+)-lisergol (76), dois análogos [(±)-13-fluorolisergol (78) e (-)-isolisergol (79)] foram obtidos usando a mesma estratégia sintética (Esquema 17b). Ainda em 2017, Cao e colaboradores61 relataram a síntese do alcaloide ergolínico (-)-chanoclavina I (87) usando uma estratégia semelhante a de Luo e colaboradores.60 A transformação chave envolveu uma anelação intramolecular catalisada por ródio, do intermediário aromático com porção N-sulfonil-1,2,3-triazólica 85, através da formação do carbeno metálico 88, seguida de oxidação com MnO2, as quais proporcionaram o composto tricíclico 86 em 77% de rendimento para as duas etapas. A redução do intermediário 86 com DIBAL-H (hidreto de diisobutilalumínio), seguida por desproteção do grupo tosila, resultaram na (-)-chanoclavina I (87) em rendimento de 77% (para duas etapas) (Esquema 18). Em 2020, Stewart e colaboradores62 desenvolveram uma reação entre N-sulfonil-1,2,3-triazóis e haloidrinas catalisada por ródio, que ocorre através da inserção de um α-iminocarbeno metálico ao grupo hidroxila, seguida de ciclização intramolecular promovida por base, para formação de oxazinas. A versatilidade sintética do método foi explorada na síntese do produto natural antimicrobiano (±)-quelonina C (92). A reação de inserção via intermediário 93, seguida de anelação após adição de base, entre o N-sulfonil-1,2,3-triazol 89 e 3-bromo-2-hidroxipropil benzoato catalisada por octanoato de ródio(II), resultou na 3,4-diidro-2H-1,4-oxazina 90 em rendimento de 77%. O intermediário 90 foi submetido a redução com HSiEt3, resultando no composto 91 em rendimento de 95%. Reação de desproteção dos grupos benzoíla e tosila com naftaleneto de sódio, forneceu a (±)-quelonina C (92) em 70% de rendimento (Esquema 19). Em 2020, Xu e colaboradores63 desenvolveram a reação de transanelação entre N-sulfonil-1,2,3-triazóis e aldeídos arílicos catalisada por ródio, que promoveu a formação de uma variedade de 2,5-diariloxazóis, via α-iminocarbenos metálicos 99. O desenvolvimento do método resultou nos produtos naturais balsoxina (97) e texamina (98) em rendimentos de 85 e 79%, respectivamente (Esquema 20). No ano de 2021, Li e colaboradores64 estabeleceram um método de ciclização entre N-sulfonil-1,2,3-triazóis e indóis catalisado por ródio, na preparação de azepino[5,4,3-cd]indóis, através de α-iminocarbenos metálicos. Essa estratégia foi utilizada na síntese total do alcaloide (±)-aurantioclavina (105), que foi realizada em apenas quatro etapas reacionais. A reação de cicloadição (4 + 3) entre o indol 100 e o triazol 101 catalisada por Rh2(Opiv)4, via α-iminocarbeno metálico 106, proporcionou o azepino 102 em rendimento de 61%. A reação do intermediário 102 com excesso de hidrazina, resultou no composto 103 em 67% de rendimento. Os grupos amino e tosila foram removidos em rendimentos de 54 e 78%, respectivamente, resultando no produto desejado 105 (Esquema 21). CONSIDERAÇÕES FINAIS Neste trabalho reunimos um conteúdo que trata de aspectos estruturais, reatividade e métodos de formação de carbenos, que dão suporte teórico para a compreensão da relação estrutura/reatividade de carbenos metálicos, que tiveram as suas classificações e os seus métodos de geração abordados. A versatilidade desses intermediários organometálicos em química orgânica preparativa foi demonstrada através de α-iminocarbenos metálicos, que têm permitido a construção de diversos heterociclos, incluindo alcaloides com estruturas complexas. Sínteses totais e formais de compostos nitrogenados, que fazem uso de α-iminocarbenos metálicos derivados de compostos N-sulfonil-1,2,3-triazólicos, foram apresentadas neste artigo de revisão. Atualmente, o emprego de α-iminocarbenos metálicos constitui uma estratégia estabelecida para a síntese de substâncias orgânicas. O desenvolvimento de catalisadores de metais mais abundantes e, consequentemente, mais acessíveis economicamente seria fundamental para difundir a química destes intermediários organometálicos em síntese orgânica. AGRADECIMENTOS Agradecemos à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Processo No. 2023/03611-2) e o Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Processo No. 304907/2021-9) pelo apoio financeiro. V. V. S. agradece o Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pela bolsa de doutorado. REFERÊNCIAS 1. Doyle, M. P.; Chem. Rev. 1986, 86, 919 [Crossref]; Doyle, M. P.; Forbes, D. C.; Chem. Rev. 1998, 98, 911 [Crossref]; Jia, M.; Ma, S.; Angew. Chem., Int. Ed. 2016, 55, 9134 [Crossref]; Xia, Y.; Qiu, D.; Wang, J.; Chem. Rev. 2017, 117, 13810 [Crossref]; Akter, M.; Rupa, K.; Anbarasan, P.; Chem. Rev. 2022, 122, 13108 [Crossref]; Epping, R. F.; Vesseur, D.; Zhou, M.; de Bruin, B.; ACS Catal. 2023, 13, 5428. [Crossref] 2. Spangler, J. E.; Davies, H. M. L.; J. Am. Chem. Soc. 2013, 135, 6802 [Crossref]; Miura, T.; Funakoshi, Y.; Murakami, M.; J. Am. Chem. Soc. 2014, 136, 2272 [Crossref]; Parr, B. T.; Green, S. A.; Davies, H. M. L.; J. Am. Chem. Soc. 2013, 135, 4716 [Crossref]; Chen, C.; Chen, J.; Wang, H.; Xu, Z.-F.; Duan, S.; Li, C.-Y.; J. Org. Chem. 2023, 88, 9543 [Crossref]; Caiuby, C. A. D.; de Jesus, M. P.; Burtoloso, A. C. B.; J. Org. Chem. 2020, 85, 7433. [Crossref] 3. Li, Y.; Yang, H.; Zhai, H.; Chem. - Eur. J. 2018, 24, 12757. [Crossref] 4. Hermann, M.; Justus Liebigs Ann. Chem. 1855, 95, 211 [Crossref]; Geuther, A.; Justus Liebigs Ann. Chem. 1862, 123, 121. [Crossref] 5. Nef, J. U.; Justus Liebigs Ann. Chem. 1897, 298, 202. [Crossref] 6. Gomberg, M.; J. Am. Chem. Soc. 1900, 22, 757. [Crossref] 7. Hine, J.; J. Am. Chem. Soc. 1950, 72, 2438 [Crossref]; Urry, W. H.; Eiszner, J. R.; J. Am. Chem. Soc. 1951, 73, 2977 [Crossref]; Lennard-Jones, J.; Pople, J. A.; Discuss. Faraday Soc. 1951, 10, 9. [Crossref] 8. Doering, W. V. E.; Knox, L. H.; J. Am. Chem. Soc. 1953, 75, 297. [Crossref] 9. Doering, W. V. E.; Hoffmann, A. K.; J. Am. Chem. Soc. 1954, 76, 6162. [Crossref] 10. Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F.; Nature 2014, 510, 485. [Crossref] 11. Arduengo III, A. J.; Harlow, R. L.; Kline, M.; J. Am. Chem. Soc. 1991, 113, 361 [Crossref]; Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G.; Chem. Rev. 2000, 100, 39. [Crossref] 12. Herzberg, G.; Johns, J. W. C.; J. Chem. Phys. 1971, 54, 2276. [Crossref] 13. Wasserman, E.; Kuck, V. J.; Hutton, R. S.; Anderson, E. D.; Yager, W. A.; J. Chem. Phys. 1971, 54, 4120 [Crossref]; Bernheim, R. A.; Bernard, H. W.; Wang, P. S.; Wood, L. S.; Skell, P. S.; J. Chem. Phys. 1971, 54, 3223. [Crossref] 14. Skell, P. S.; Garner, A. Y.; J. Am. Chem. Soc. 1956, 78, 5430 [Crossref]; Woodworth, R. C.; Skell, P. S.; J. Am. Chem. Soc. 1959, 81, 3383 [Crossref]; Skell, P. S.; Tetrahedron 1985, 41, 1427 [Crossref]; Keating, A. E.; Merrigan, S. R.; Singleton, D. A.; Houk, K. N.; J. Am. Chem. Soc. 1999, 121, 3933. [Crossref] 15. Harrison, J. F.; Acc. Chem. Res. 1974, 7, 378 [Crossref]; Saxe, P.; Schaefer III, H. F.; Handy, N. C.; J. Phys. Chem. 1981, 85, 745 [Crossref]; Hayden, C. C.; Neumark, D. M.; Shobatake, K.; Sparks, R. K.; Lee, Y. T.; J. Chem. Phys. 1982, 76, 3607 [Crossref]; McKellar, A. R. W.; Bunker, P. R.; Sears, T. J.; Evenson, K. M.; Saykally, R. J.; Langhoff, S. R.; J. Chem. Phys. 1983, 79, 5251. [Crossref] 16. García-Garibay, M. A.; Theroff, C.; Shin, S. H.; Jernelius, J.; Tetrahedron Lett. 1993, 34, 8415 [Crossref]; Wang, J.; Kubicki, J.; Peng, H.; Platz, M. S.; J. Am. Chem. Soc. 2008, 130, 6604. [Crossref] 17. Pauling, L.; J. Chem. Soc., Chem. Commun. 1980, 688 [Crossref]; Irikura, K. K.; Goddard III, W. A.; Beauchamp, J. L.; J. Am. Chem. Soc. 1992, 114, 48. [Crossref] 18. Moss, R. A.; Perez, L. A.; Wlostowska, J.; Guo, W.; Krogh-Jespersen, K.; J. Org. Chem. 1982, 47, 4177 [Crossref]; Moss, R. A.; Kmiecik-Lawrynowicz, G.; Krogh-Jespersen, K.; J. Org. Chem. 1986, 51, 2168 [Crossref]; Moss, R. A.; Shen, S.; Hadel, L. M.; Kmiecik-Lawrynowicz, G.; Wlostowska, J.; Krogh-Jespersen, K.; J. Am. Chem. Soc. 1987, 109, 4341 [Crossref]; Mieusset, J. L.; Brinker, U. H.; J. Org. Chem. 2008, 73, 1553. [Crossref] 19. Tomioka, H.; Watanabe, T.; Hattori, M.; Nomura, N.; Hirai, K.; J. Am. Chem. Soc. 2002, 124, 474 [Crossref]; Iwamoto, E.; Hirai, K.; Tomioka, H.; J. Am. Chem. Soc. 2003, 125, 14664 [Crossref]; Itoh, T.; Nakata, Y.; Hirai, K.; Tomioka, H.; J. Am. Chem. Soc. 2006, 128, 957. [Crossref] 20. Ye, T.; McKervey, M. A.; Chem. Rev. 1994, 94, 1091 [Crossref]; Zhang, Z.; Wang, J.; Tetrahedron 2008, 64, 6577 [Crossref]; Ciszewski, Ł. W.; Rybicka-Jasińska, K.; Gryko, D.; Org. Biomol. Chem. 2019, 17, 432 [Crossref]; Zhang, Z.; Yadagiri, D.; Gevorgyan, V.; Chem. Sci. 2019, 10, 8399. [Crossref] 21. Bamford, W. R.; Stevens, T. S.; J. Chem. Soc. 1952, 4735. [Crossref] 22. Frey, H. M. Em Advances in Photochemistry, vol. 4; Noyes Jr., W. A.; Hammond, G. S.; Pitts Jr., J. N., eds.; John Wiley & Sons, Inc., 1966, p. 225 [Crossref]; Liu, M. T.; Chem. Soc. Rev. 1982, 11, 127 [Crossref]; Moss, R. A.; Acc. Chem. Res. 2006, 39, 267. [Crossref] 23. Hine, J.; Dowell Jr., A. M.; J. Am. Chem. Soc. 1954, 76, 2688 [Crossref]; Kirmse, W.; Angew. Chem., Int. Ed. 1965, 4, 1. [Crossref] 24. Caballero, A.; Pérez, P. J.; Chem. - Eur. J. 2017, 23, 14389. [Crossref] 25. Schrock, R. R.; Chem. Rev. 2009, 109, 3211 [Crossref]; Feliciano, A.; Vázquez, J. L.; Benítez‐Puebla, L. J.; Velazco‐Cabral, I.; Cruz Cruz, D.; Delgado, F.; Vázquez, M. A.; Chem. - Eur. J. 2021, 27, 8233. [Crossref] 26. Fischer, E. O.; Maasböl, A.; Angew. Chem., Int. Ed. 1964, 3, 580. [Crossref] 27. Taylor, T. E.; Hall, M. B.; J. Am. Chem. Soc. 1984, 106, 1576 [Crossref]; Vyboishchikov, S. F.; Frenking, G.; Chem. - Eur. J. 1998, 4, 1428 [Crossref]; Frenking, G.; Solà, M.; Vyboishchikov, S. F.; J. Organomet. Chem. 2005, 690, 6178 [Crossref]; Montgomery, C. D.; J. Chem. Educ. 2015, 92, 1653. [Crossref] 28. Cardin, D. J.; Cetinkaya, B.; Lappert, M. F.; Chem. Rev. 1972, 72, 545 [Crossref]; Santamaría, J.; Aguilar, E.; Org. Chem. Front. 2016, 3, 1561. [Crossref] 29. Schrock, R. R.; J. Am. Chem. Soc. 1974, 96, 6796. [Crossref] 30. Schrock, R. R.; Acc. Chem. Res. 1979, 12, 98 [Crossref]; Schrock, R. R.; Hoveyda, A. H.; Angew. Chem., Int. Ed. 2003, 42, 4592 [Crossref]; Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H.; Nature 2008, 456, 933. [Crossref] 31. Davies, H. M.; Beckwith, R. E.; Chem. Rev. 2003, 103, 2861 [Crossref]; Santiago, J. V.; Machado, A. H. L.; Beilstein J. Org. Chem. 2016, 12, 882 [Crossref]; He, Y.; Huang, Z.; Wu, K.; Ma, J.; Zhou, Y.-G.; Yu, Z.; Chem. Soc. Rev. 2022, 51, 2759. [Crossref] 32. Green, S. P.; Wheelhouse, K. M.; Payne, A. D.; Hallett, J. P.; Miller, P. W.; Bull, J. A.; Org. Process Res. Dev. 2020, 24, 67 [Crossref]; Wang, Y.; Muratore, M. E.; Echavarren, A. M.; Chem. - Eur. J. 2015, 21, 7332 [Crossref]; Burtoloso, A. C. B.; Dias, R. M. P.; Leonarczyk, I. A.; Eur. J. Org. Chem. 2013, 2013, 5005. [Crossref] 33. Piloty, O.; Neresheimer, J.; Ber. Dtsch. Chem. Ges. 1906, 39, 514. [Crossref] 34. Dimroth, O.; Justus Liebigs Ann. Chem. 1909, 364, 183. [Crossref] 35. Grünanger, P.; Finzi, P. V.; Scotti, C.; Chem. Ber. 1965, 98, 623. [Crossref] 36. Hermes, M. E.; Marsh, F. D.; J. Am. Chem. Soc. 1967, 89, 4760. [Crossref] 37. Harmon, R. E.; Stanley Jr., F.; Gupta, S. K.; Johnson, J.; J. Org. Chem. 1970, 35, 3444. [Crossref] 38. Habraken, C. L.; Erkelens, C.; Mellema, J. R.; Cohen-Fernandes, P.; J. Org. Chem. 1984, 49, 2197. [Crossref] 39. Chuprakov, S.; Hwang, F. W.; Gevorgyan, V.; Angew. Chem., Int. Ed. 2007, 46, 4757. [Crossref] 40. Horneff, T.; Chuprakov, S.; Chernyak, N.; Gevorgyan, V.; Fokin, V. V.; J. Am. Chem. Soc. 2008, 130, 14972. [Crossref] 41. Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B.; Angew. Chem., Int. Ed. 2002, 41, 2596 [Crossref]; Tornøe, C. W.; Christensen, C.; Meldal, M.; J. Org. Chem. 2002, 67, 3057. [Crossref] 42. Yoo, E. J.; Ahlquist, M.; Kim, S. H.; Bae, I.; Fokin, V. V.; Sharpless, K. B.; Chang, S.; Angew. Chem., Int. Ed. 2007, 46, 1730 [Crossref]; Raushel, J.; Fokin, V. V.; Org. Lett. 2010, 12, 4952. [Crossref] 43. Schultz, E. E.; Lindsay, V. N.; Sarpong, R.; Angew. Chem. 2014, 126, 10062 [Crossref]; Pan, X.-H.; Jiang, P.; Jia, Z.-H.; Xu, K.; Cao, J.; Chen, C.; Shen, M.-H.; Xu, H.-D.; Tetrahedron 2015, 71, 5124 [Crossref]; Kwok, S. W.; Zhang, L.; Grimster, N. P.; Fokin, V. V.; Angew. Chem. 2014, 126, 3520 [Crossref]; Xu, H.-D.; Xu, K.; Jia, Z.-H.; Zhou, H.; Jiang, P.; Lu, X.-L.; Pan, Y.-P.; Wu, H.; Ding, Y.; Shen, M.-H.; Pan, X.-H.; Asian J. Org. Chem. 2014, 3, 1154. [Crossref] 44. Miura, T.; Tanaka, T.; Biyajima, T.; Yada, A.; Murakami, M.; Angew. Chem., Int. Ed. 2013, 52, 3883 [Crossref]; Chuprakov, S.; Worrell, B. T.; Selander, N.; Sit, R. K.; Fokin, V. V.; J. Am. Chem. Soc. 2014, 136, 195 [Crossref]; Yang, J.-M.; Zhu, C.-Z.; Tang, X.-Y.; Shi, M.; Angew. Chem. 2014, 126, 5242 [Crossref]; Lindsay, V. N.; Viart, H. M.-F.; Sarpong, R.; J. Am. Chem. Soc. 2015, 137, 8368 [Crossref]; Meng, J.; Wen, M.; Zhang, S.; Pan, P.; Yu, X.; Deng, W.-P.; J. Org. Chem. 2017, 82, 1676. [Crossref] 45. Chattopadhyay, B.; Gevorgyan, V.; Org. Lett. 2011, 13, 3746 [Crossref]; Miura, T.; Yamauchi, M.; Murakami, M.; Chem. Commun. 2009, 1470. [Crossref] 46. Miura, T.; Tanaka, T.; Yada, A.; Murakami, M.; Chem. Lett. 2013, 42, 1308 [Crossref]; Boyer, A.; Org. Lett. 2014, 16, 1660 [Crossref]; Boyer, A.; Org. Lett. 2014, 16, 5878 [Crossref]; Yadagiri, D.; Anbarasan, P.; Chem. - Eur. J. 2013, 19, 15115. [Crossref] 47. Medina, F.; Besnard, C.; Lacour, J.; Org. Lett. 2014, 16, 3232 [Crossref]; Martin, M. L.; Boyer, A.; Eur. J. Org. Chem. 2021, 2021, 5857. [Crossref] 48. Zibinsky, M.; Fokin, V. V.; Angew. Chem. 2013, 125, 1547 [Crossref]; Zhang, W.-B.; Xiu, S.-D.; Li, C.-Y.; Org. Chem. Front. 2015, 2, 47 [Crossref]; Lee, D. J.; Han, H. S.; Shin, J.; Yoo, E. J.; J. Am. Chem. Soc. 2014, 136, 11606 [Crossref]; He, J.; Shi, Y.; Cheng, W.; Man, Z.; Yang, D.; Li, C.-Y.; Angew. Chem., Int. Ed. 2016, 55, 4557 [Crossref]; Yu, Y.; Zhu, L.; Liao, Y.; Mao, Z.; Huang, X.; Adv. Synth. Catal. 2016, 358, 1059. [Crossref] 49. Ran, R.-Q.; Xiu, S.-D.; Li, C.-Y.; Org. Lett. 2014, 16, 6394 [Crossref]; Yang, Y.; Zhou, M.-B.; Ouyang, X.-H.; Pi, R.; Song, R.-J.; Li, J.-H.; Angew. Chem. 2015, 127, 6695 [Crossref]; Lu, X.-L.; Liu, Y.-T.; Wang, Q.-X.; Shen, M.-H.; Xu, H.-D.; Org. Chem. Front. 2016, 3, 725. [Crossref] 50. Schultz, E. E.; Sarpong, R.; J. Am. Chem. Soc. 2013, 135, 4696. [Crossref] 51. Rajasekar, S.; Anbarasan, P.; J. Org. Chem. 2014, 79, 8428. [Crossref] 52. Arafeh, K. M.; Ullah, N.; Nat. Prod. Commun. 2009, 4, 925. [Crossref] 53. Fu, J.; Shen, H.; Chang, Y.; Li, C.; Gong, J.; Yang, Z.; Chem. - Eur. J. 2014, 20, 12881. [Crossref] 54. Zhao, Y.-Z.; Yang, H.-B.; Tang, X.-Y.; Shi, M.; Chem. - Eur. J. 2015, 21, 3562. [Crossref] 55. Pandey, P. S.; Rao, T. S.; Bioorg. Med. Chem. Lett. 2004, 14, 129. [Crossref] 56. Shang, H.; Tian, Y.; Luo, J.; Li, L.; Tang, Y.; Zou, Z.; RSC Adv. 2016, 6, 30835. [Crossref] 57. Choshi, T.; Matsuya, Y.; Okita, M.; Inada, K.; Sugino, E.; Hibino, S.; Tetrahedron Lett. 1998, 39, 2341 [Crossref]; Choshi, T.; Kuwada, T.; Fukui, M.; Matsuya, Y.; Sugino, E.; Hibino, S.; Chem. Pharm. Bull. 2000, 48, 108. [Crossref] 58. Miura, T.; Tanaka, T.; Zhao, Q.; Stewart, S. G.; Murakami, M.; Helv. Chim. Acta 2017, 100, e1600320. [Crossref] 59. Chang, M.-Y.; Kung, Y.-H.; Chen, S.-T.; Tetrahedron 2006, 62, 10843. [Crossref] 60. Yuan, H.; Guo, Z.; Luo, T.; Org. Lett. 2017, 19, 624. [Crossref] 61. Lu, J.-T.; Shi, Z.-F.; Cao, X.-P.; J. Org. Chem. 2017, 82, 7774. [Crossref] 62. Jones, K. D.; Nutt, M. J.; Comninos, E.; Sobolev, A. N.; Moggach, S. A.; Miura, T.; Murakami, M.; Stewart, S. G.; Org. Lett. 2020, 22, 3490. [Crossref] 63. Li, J.; Zhu, S.-R.; Xu, Y.; Lu, X.-C.; Wang, Z.-B.; Liu, L.; Xu, D.-F.; RSC Adv. 2020, 10, 24795. [Crossref] 64. Duan, S.; Xue, B.; Meng, H.; Ye, Z.; Xu, Z.-F.; Li, C.-Y.; Chin. J. Chem. 2021, 39, 1145. [Crossref] |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access