
 |
 |
 |
 |
 |
Artigo
| Synthesis, molecular docking, dynamics analysis studies, and cytotoxicityactivity evaluation of novel berberine derivative bearing oxadiazole and o-diaminobenzene moieties |
|
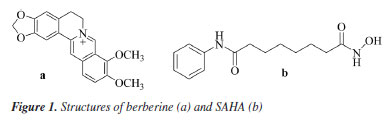
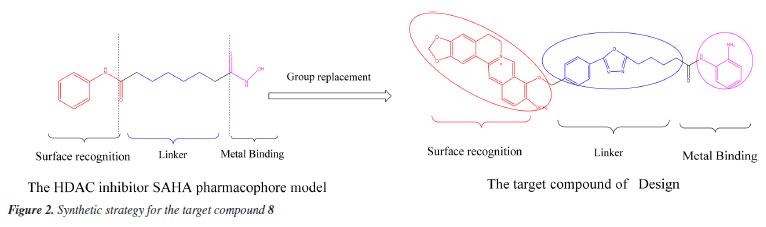
Xiaofei JiangI,II I. School of Science, Kaili University, 556011 Kaili, China Received: 08/02/2024 *e-mail: 407432204@qq.com, wangdaoping@gmc.edu.cn Berberine represents a prominent compound with commendable antitumor activity, a novel berberine derivative with substituted o-diaminobenzene moiety linked at the 9-position through oxadiazole and alkyl chain is designed, synthesized by reference to the histone deacetylase (HDAC) inhibitor structure mode through selective demethylation, substitution, hydrazolysis, acylation, cyclization, elimination, condensation reaction. The structures of these synthetic derivatives were characterized and confirmed by 1H NMR (nuclear magnetic resonance), 13C NMR, and MS (mass spectrometry) spectral data and finally evaluated for antitumor activities against MHCC97H, HCC827, HELA, and HEL cell in vitro by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) method. The results demonstrated that target compound 8 exhibited better antiproliferative activity against the MHCC97H cell line, with a half maximal inhibitory concentration (IC50) = 5.4 ± 0.45 μM L-1 superior to berberine, and the activity against MHCC97H was nearly close to the reference drug doxorubicin. Through the dynamics of molecular docking and simulation, further analysis has demonstrated that the target compound 8 can form effective interactions with the HDAC6 target protein, and the results of binding free energy also revealed a strong affinity between the compound 8 and the HDAC6 protein. The results of this study suggest that compound 8 may be a potential inhibitor of HDAC6, which could have promising applications in the treatment of certain cancers. INTRODUCTION The focus in the scientific community has shifted towards exploring potential antitumor properties of natural products.1 Berberine, depicted in Figure 1, belongs to the class of quaternary protoberberine alkaloids (QPA) and is recognized as a key component of Coptis chinensis.2 Various studies indicate that berberine is extensively employed for the treatment of cancers,3 and diabetes,4 as well as for the treatment of bacterial infection.5 Promising anticancer effects of berberine have been reported6-8 in various types of tumor cell lines, such as breast, hepatoma, lungs, and so on. Previous reports9 showed that the variety of berberine derivatives have better biological activity and bioavailability than berberine. Histone deacetylase (HDAC) has become a hot target for anti-tumor drug research due to its key roles in the metabolism regulation of tumor cells.10 Plenty of studies11 have shown that HDAC inhibitors could exert synergistic anti-tumor effects in combination with other anti-cancer agents. A previous study12 on pharmacological modeling of HDAC inhibitors demonstrated that the head (surface recognition domain) of HDAC inhibitors must be a hydrophobic moiety and is highly compatible with HDAC inhibition effect. Among the heterocyclic compounds, 1,3,4-oxadiazole has become a key building block for the development of innovative medicinal molecules. Heterocycles containing 1,3,4-oxadiazole are essential for molecular planning due to their valuable structure,13,14 which has considerable biological potential. Many pharmaceuticals on the market use medicinal compounds derived from the oxadiazole scaffold, such as the well-known antihypertensive nesapidil, the potent peptide deformylase (PDF) inhibitor furamizole, the human immunodeficiency virus (HIV) integrase inhibitor raltegravir, and the anti-cancer drug zibotentan.15 Furthermore, it has been found that certain oxadiazoles have high inhibitory capability against monoamine oxidase and are effective at the nanomolar level against lipoxygenase.16 The thick cyclic aromatic hydrocarbon structure of berberine indicates that it has strong hydrophobicity. Vorinostat (SAHA) (Figure 1b) is one of the earlier HDAC inhibitors that were approved by the US Food and Drug Administration (FDA) (Figure 2) in 2006.17 On this basis, a series of berberine derivatives were designed and synthesised. These derivatives contained oxadiazole and o-phenylenediamine. The derivatives were synthesized by a series of reactions, including selective demethylation, substitution, hydrazinylation, cyclization, hydrolysis and condensation. These reactions were carried out using a molecular hybridization strategy. The derivatives were then evaluated for their biological activities as anti-tumor drugs. The hope was that this would yield new compounds with significant anticancer activities.
EXPERIMENTAL Materials and methods Preparation of berberine One kilogram of Coptis chinensis was crushed and then soaked in 3% sulfuric acid (H2SO4) with stirring for 24 h, which was sourced from ShiZhu County, Chong Qing, China. After filtration, the resulting filtrate was then neutralized to pH 7 using a saturated calcium hydroxide solution, followed by allowing it to settle without disturbance for 30 min. The mixture was filtered again, and the filtrate was adjusted to pH 2-3 by adding 1 M hydrochloric acid (HCl). Then, 4-5% sodium chloride (NaCl), in proportion to the volume of the filtrate, was added to this adjusted filtrate, and stewing for 30 min. The collected precipitate was recrystallized using methanol, repeating this process 2 to 3 times. The purity of the final product of berberine was verified using thin-layer chromatography (TLC) analysis with a mobile phase composed of benzene (C6H6), ethyl acetate (EtOAc), methanol (MeOH), isopropanol (i-C3H7OH), and ammonia water (NH3·H2O) in a ratio of 6:3:2:2:0.5. Preparation of target compound 8 Preparation of compound 3: berberrubine (compound 2) was prepared according to the first step of Scheme 1, and by the literature.18 Berberine (21.0 g, 10 mmol) in dimethylformamide (DMF) 150 mL was refluxed in a microwave heater for 20 min at 190 ºC, the color changed from yellow to red, indicating product formation. After the completion, upon cooling and precipitation in cold distilled water, the solid was filtered under reduced pressure and recrystallized by CH3OH to yield a pure red solid with 65% yield. Similarly, compound 3, an already reported compound in the previous literature,19 was prepared as follows: dried berberrubine (5.9 g, 18.3 mmol) and potassium carbonate (2.76 g, 20 mmol) in acetonitrile (100 mL) were dissolved and added methyl 4-(bromomethyl) benzoate (8.4 g, 36.6 mmol). The mixture was refluxed for 12 h, after the reaction's completion (monitored by TLC) using solvent system C6H6:EtOAc:MeOH:i-C3H7OH:NH3.H2O (6:3:1.5:1:0.5). The solvent was removed by reducing pressure and concentration. Subsequently, the crude products were purified by 400-mesh Al2O3 column chromatography and eluted with CHCl3:CH3OH (10:1, v:v) to afford compound 3 as a yellow solid. Mp: 210-211 ºC, MS (m/z) 470.2 [M - Cl]+.
9-((4-(Hydrazinecarbonyl)benzyl)oxy)-10-methoxy-5,6-dihydro- [1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium (compound 4) Compound 4 was prepared according to the third step of Scheme 1 and the reported literature.20 A solution of compound 3 (9.0 g, 19.15 mmol) was dissolved in a mixture of ethanol and N2H4.H2O (1:1) (50 mL), the reactant mixture was stirred at 25 ºC for 24 h, after completion of the reaction (monitored by TLC), ethanol was evaporated through reduced pressure at 25 ºC. Then, water was added (50 mL) and the mixture was extracted with chloroform (150 mL × 3). Anhydrous Na2SO4 was added to the solvent to remove water drops and filtered, the solvent was evaporated through reduced pressure and concentrated, the products were obtained and purified through column chromatography using 400-mesh Al2O3 eluted with CH2Cl2/CH3OH (10:1), yielding compound 4, yellow solid with 37% yield. Mp: 160-162 ºC, MS (m/z) 470.4 [M - Cl]+; 1H NMR (600 MHz, DMSO-d6) d 9.90 (s, 1H, -CO-NH-), 9.80 (s, 1H, Ph), 8.95 (s, 1H, Ph), 8.20 (d, J 9.1 Hz, 1H, Ph), 8.02 (d, J 9.1 Hz, 1H, Ph), 7.87 (d, J 7.9 Hz, 2H, Ph), 7.78 (s, 1H, Ph), 7.68 (d, J 7.9 Hz, 2H, Ph), 7.09 (s, 1H, Ph), 6.17 (s, 2H, -O-CH2-O-), 5.40 (s, 2H, -CH2-O-Ph), 4.95 (t, J 6.4 Hz, 4H, -CH2, -NH2), 4.08 (s, 3H, -OCH3), 3.20 (t, J 6.4 Hz, 2H, -CH2); 13C NMR (150 MHz, DMSO-d6) d 165.90, 151.10, 150.30, 148.14, 145.78, 142.32, 139.96, 137.87, 133.54, 133.39, 131.13, 128.87, 127.51, 126.95, 124.36, 122.17, 120.86, 108.89, 105.92, 102.56, 75.22, 57.53, 55.78, 26.80. 10-Methoxy-9-((4-(2-(6-methoxy-6-oxohexanoyl)hydrazine-1-carbonyl)benzyl)oxy)-5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium (compound 5) Compound 5 was prepared according to the fourth step of Scheme 1. A solution of compound 4 (1.87 g, 3.98 mmol), Et3N, HOBt (1.07 g, 7.96 mmol), EDCI (1.52 g, 7.96 mmol) and diacid monomethyl (1.44 g, 9.0 mmol) was dissolved in DMF solvent. The mixture was stirred at 20-25 ºC overnight until TLC confirmed complete consumption of the starting material. Subsequently, the reaction was quenched with water and extracted with CHCl3 (150 mL × 3), and concentrated to yield crude product compound 5, which was purified by 400 mesh Al2O3 column chromatography and was eluted with CHCl3:CH3OH (30:1), yielding compound 5, yellow solid with 72% yield. Mp: 210-212 ºC, MS [m/z] 612.2 [M - Cl]+; 1H NMR (600 MHz, DMSO-d6) d 10.32 (s, 1H, -NH-NH), 9.86 (s, 1H, Ph), 9.79 (s, 1H, Ph), 8.93 (s, 1H, Ph), 8.22 (d, J 9.2 Hz, 1H, Ph), 8.01 (d, J 9.1 Hz, 1H, Ph), 7.91 (d, J 8.2 Hz, 2H, Ph), 7.79 (s, 1H, Ph), 7.71 (d, J 8.2 Hz, 2H, Ph), 7.09 (s, 1H, Ph), 6.18 (s, 2H, -O-CH2-O-), 5.43 (s, 2H, -CH2-O-Ph), 4.93 (t, J 6.4 Hz, 2H, -CH2-N), 4.09 (s, 3H, -OCH3), 3.60 (s, 3H, -OCH3), 3.20 (s, 2H, Ph, -CH2-), 2.34 (s, 2H, CO-CH2-), 2.20 (s, 2H, -CO-CH2-), 1.58 (m, 4H, -(CH2)2-). 10-Methoxy-9-((4-(5-(5-methoxy-5-oxopentyl)-1,3,4-oxadiazol-2-yl)benzyl)oxy)-5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium (compound 6) Compound 6 was prepared according to the fifth step of Scheme 1 and the reported reference.21 To a stirred solution of compound 5 (0.77 g, 1.358 mmol), Et3N (1.1 mL, 3.0 mmol) in THF (80 mL) was added p-sultoluene sulfonyl chloride (0. 73 g, 3.78 mmol) to the RB flask, then the mixture was refluxed for 8 h. After completion of the reaction, the solvent was evaporated under reduced pressure and the residue was purified using 400 mesh Al2O3 column chromatography and was eluted with CH2Cl2:CH3OH (30:1) to give compound 6. Yellow solid with 78% yield, Mp: 197-198 ºC, MS [m/z] [M - Cl]+ 594.2; 1H NMR (600 MHz, DMSO-d6) d 9.83 (s, 1H, Ph), 8.93 (s, 1H, Ph), 8.23 (d, J 9.2 Hz, 1H, Ph), 8.02 (t, J 8.0 Hz, 3H, Ph), 7.82 (d, J 8.1 Hz, 2H, Ph), 7.79 (s, 1H, Ph), 7.09 (s, 1H, Ph), 6.18 (s, 2H, 2H, -O-CH2-O-), 5.45 (s, 2H, -CH2-O-Ph), 4.94 (s, J 6.4 Hz, 2H, CH2-N), 4.09 (s, 3H, -OCH3), 3.59 (s, 3H, -OCH3), 3.21 (s, 2H, Ph-CH2-), 2.96 (t, J 7.4 Hz, 2H, -CH2-triazole), 2.39 (t, J 7.4 Hz, 2H, -CH2-CO-), 1.79 (m, 2H, -CH2-), 1.65 (m, 2H, -CH2-). 9-((4-(5-(4-Carboxymethyl)-1,3,4-oxadiazol-2-yl)benzyl)oxy)-10-methoxy-5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium (compound 7) Compound 7 was prepared according to the sixth step of Scheme 1. A suspension of compound 6 (0.63 g, 1.08 mmol) in THF (10 mL) was treated with methanol (5 mL) and NaOH (1.18 g, 30 mmol) in water (10 mL). The mixture was stirred at room temperature overnight, after removal of THF solvents, the residue was diluted with water, the pH was adjusted to 1-2 using 10% aqueous HCl, and the precipitate of the product was filtered, collected, and dried to give compound 7. Yellow solid with 69% yields. MP 220-221 ºC, MS [m/z] 580.3; 1H NMR (600 MHz, DMSO-d6) d 9.61 (s, 1H, Ph), 8.95 (s, 1H, Ph), 8.22 (d, J 7.1 Hz, 1H, Ph), 8.02 (s, 3H, Ph), 7.80 (s, 3H, Ph), 7.1 (s, 1H, Ph), 6.17 (s, 2H, -O-CH2-O-), 5.45 (s, 2H, -CH2-O-Ph), 4.96 (t, J 6.4 Hz, 2H, CH2-N), 4.08 (s, 3H, -OCH3), 3.21 (t, J 7.0 Hz, 2H, Ph-CH2-), 2.92 (t, J 7.5 Hz, 2H, -CH2-triazole), 2.19 (t, J 7.4 Hz, 2H, -CH2-CO-), 1.78 (m, 2H, -CH2-), 1.61 (m, 2H, -CH2-); 13C NMR (150 MHz, DMSO-d6) d 175.05, 167.31, 164.10, 151.02, 150.32, 148.15, 145.76, 142.25, 140.77, 137.97, 133.41, 131.17, 129.69, 126.98, 126.92, 124.39, 123.77, 122.14, 120.86, 120.71, 108.90, 105.89, 102.56, 75.06, 57.52, 55.82, 34.58, 26.80, 25.95 (overlap), 24.88 (overlap), 24.70. 9-((4-(5-(5-((2-Aminophenyl)amino)-5-oxopentyl)-1,3,4-oxadiazol-2-yl)benzyl)oxy)-10-methoxy-5,6-dihydro-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinolin-7-ium (target compound 8) Compound 8 was prepared according to the seventh step of Scheme 1. Compound 7 (0.32 mmol, 0.186 g) was dissolved in DMF (10 mL), was added EDCI (0.1 mmol, 19.3 mg, HOBT), o-diaminobenzene (0.11 mmol, 11.7 mg), NMM (4-methylmorpholine) (234 mg, 2 mmol), the mixture was stirred at 20-25 ºC overnight until TLC indicated complete consumption of the starting material. Then, it was quenched with water and extracted with CHCl3. The organic layer was washed with brine. After drying over anhydrous Na2SO4, concentrated to give the crude product compound 8, which was purified by 400 mesh Al2O3 column chromatography and was eluted with CH2Cl2:CH3OH (30:1), yellow solid with 42% yield. Mp: 225.6-226.3 ºC, MS [m/z] [M - Cl]+ 670.3; 1H NMR (600 MHz, DMSO-d6) d 9.83 (s, 1H, Ph), 9.14 (s, 1H, Ph), 8.93 (s, 1H, Ph), 8.23 (d, J 9.2 Hz, 1H, Ph), 8.03 (dd, J 7.2, 8.8 Hz, 3H, Ph), 7.27-7.87 (m, 2H, Ph), 7.15 (dd, J 7.9, 1.5 Hz, 1H, Ph), 7.09 (s, 1H, Ph), 6.90 (td, J 7.6, 1.6 Hz, 1H, Ph), 6.72 (dd, J 8.0, 1.5 Hz, 1H, Ph), 6.53 (td, J 7.6, 1.5 Hz, 1H, Ph), 6.18 (s, 2H, -O-CH2-O-), 5.45 (s, 2H, -CH2-O-Ph), 4.94 (t, J 6.4 Hz, 2H, CH2-N), 4.83 (s, 2H, Ph-NH2), 4.09 (s, 3H, -OCH3), 3.20 (t, J 6.4 Hz, 2H, Ph-CH2-), 2.99 (t, J 7.4 Hz, 2H, CH2-triazole), 2.40 (t, J 7.3 Hz, 2H, -CH2-CO-), 1.84 (m, 2H), 1.71 (m, 2H); 13C NMR (150 MHz, DMSO-d6) d 171.31, 167.30, 164.14, 151.03, 150.34, 148.17, 145.75, 142.39, 142.27, 140.81, 138.00, 133.42, 131.18, 129.68, 127.00 (overlap), 126.93 (overlap), 126.23, 125.79, 124.38, 123.91, 123.78, 122.14, 120.87, 120.72, 116.63, 116.35, 108.91, 105.89, 102.57, 75.07, 57.53, 55.81, 35.63, 26.79, 25.89, 25.05, 24.88. Cytotoxicity activity The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) method, as indicated in the referenced text,22 was used to determine the cytotoxicity of berberine and compound 8. Liver cancer cells (MHCC97H), non-small cell lung cancer cells (HCC827), cervical cancer cells (HELA), and erythroid leukemia cells (HEL): cells were routinely cultured in DMEM (Dulbecco's modified eagle medium) supplemented with 10% heat-inactivated FBS (fetal bovine serum), were plated at a density of 1 × 104 cells per well in 96-well microplates. The plates were then incubated for 48 h at 37 ºC with 5% CO2. Each tumor cell line was treated with each test compound 8 and berberine at various concentrations in triplicate for incubation for 48 h. Following incubation, 10 μL of MTT reagent (5 mg mL-1) was added and the plates incubated at 37 ºC for 4 h once more. Subsequently, the MTT reagent was discarded and 150 μL of DMSO dissolved the formazan crystals. The absorbance at 490 nm was measured using an enzyme immunoassay instrument. The half-maximum inhibitory concentration (IC50) values were determined by SPSS 16.0 software (IBM, Armonk, USA, 2009) based on the reduction in absorbance observed in the negative control assay. The entire assay was performed in triplicate and the data were reported as mean ± standard deviation (SD). Molecular docking and dynamic analysis studies Molecular docking The molecular docking were performed using the Glide module within the Schrödinger Maestro (v 11.1) software.23 Protein preparation was carried out using the Protein Preparation Wizard. The HDAC6 (PDB ID: 6PYE) was optimized using the Protein Preparation Wizard adding bond orders and hydrogen atoms to the crystal structure using the OPLS3 force field. Prime was used to fix missing residues or atoms in the protein and to remove co-crystallized water molecules. PROPKA was used to check for the protonation state of ionizable protein groups (pH = 7.0). The hydrogen bonds were optimized through the reorientation of hydroxyl bonds, thiol groups, and amide groups. In the end, the system was minimized with an RMSD (root mean square deviation) convergence threshold of 0.3 Å.24,25 All compounds were prepared following the default settings of the LigPrep module. The force field adopted was OPLS3 and Epik 3.9 was selected as an ionization tool at pH 7.2 ± 0.2. Tautomers generation was unflagged and the maximum number of conformers generated was set at 32.26,27 The native ligand of the HDAC6 protein was selected as the centroid for a 12 Å × 12 Å × 12 Å box (x = -15.4, y = 4.76, z = 37.17), employing the Standard Precision (SP) docking mode. The final score was determined using an energy-minimized pose, expressed as the Glide score. Molecular dynamics simulation To further investigate the molecular mechanism of binding between the compound 8 and HDAC6 protein, molecular dynamics simulations of the HDAC6-8 complexes were performed using the GROMACS 2020 software package (University of Groningen, Groningen, Netherlands, 2020). The protein utilized the AMBER99SB-ILDN force field parameters, while the compound 8 employed the gaff2 universal force field parameters. Small molecule topologies were constructed using the acpype program, and charges were fitted using the RESP method.28,29 The TIP3P explicit water model was chosen, with atoms in the protein at a minimum distance of 1.0 nm from the edge of the water box. Based on docking results, sodium or chloride ions were used to neutralize the system charge.30 The molecular dynamics simulation workflow included four steps: energy minimization, heating, equilibration, and production dynamics simulation. Initially, the heavy atoms of the protein (and small molecules) were constrained, and water molecules underwent 10,000 steps of energy minimization (including 5,000 steps of steepest descent and 5,000 steps of conjugate gradient method). Constraints were then released, and the entire system underwent 10,000 steps of energy minimization. After energy optimization, the system was gradually heated to 300 K over 50 ps; following heating, the system was equilibrated under the NPT ensemble for 50 ps. Finally, the system underwent 200 ns of molecular dynamics simulation under the NPT ensemble. Trajectory data were saved every 20 ps and analyzed using the trjconv module. The binding free energy of the compound 8 with HDAC6 protein was calculated using the MMPBSA method implemented in GROMACS 2020 program.31
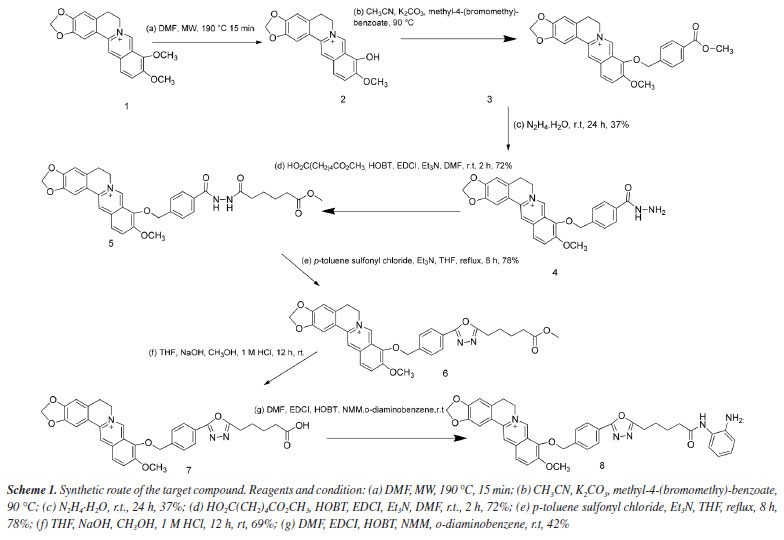
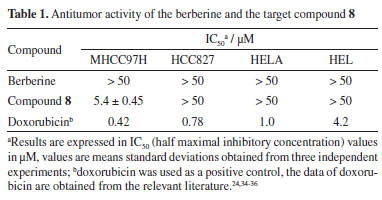
RESULTS AND DISCUSSION In the Experimental section, the NMR and MS spectra of the target compound are displayed. The synthetic route for preparing the target compound 8 is shown in Scheme 1. In the initial stage of the process, berberine underwent selective demethylation in situ under microwave action with DMF at 160 ºC as a reaction solvent. This process yielded the berberine base, which was then subjected to substitution with methyl 4-(bromomethyl)benzoate, resulting in the formation of compound 3. Compound 3 with hydrazine hydrate produced compound 4. The acylation of compound 4 provided the amide product 5. Cyclization of product 5 led to the key intermediate compound 6. Compound 7 was obtained by the treatment of ethanol sodium and tetrahydrofuran. Finally, the target compound 8 was prepared after a condensation reaction in good yield. The structures of the intermediate are not reported in the literature and target compound 8 was characterized and confirmed by 1H NMR, 13C NMR, and MS spectral data. In the past decades, a variety of berberine derivatives have been designed and synthesized to obtain compounds with stronger anti-tumor activity. Iwasa et al.32 found that the alkyl chain substitution at positions 8 and 13 of berberine can improve the cytotoxic activity significantly. Furthermore, Orfila et al.33 found a hydroxyl associated at the position 8 of berberine have great impact on the cytotoxic activity (IC50 < 0.03 µM). Liao et al.22 also reported a kind of conjugate structures based on the honokiol and berberine, and the results revealed the inhibitory effects (IC50 = 7.858 mmol L-1) against HepG2 cell in vitro. All of the results expressed significantly the modification in the molecular structure of berberine can yield better cytotoxic activity than that of berberine. To evaluate the activities of compound 8 against human liver cancer cells (MHCC97H), human non-small cell lung cancer cells (HCC827), human cervical cancer cell line (HELA), and human erythroid leukemia cells (HEL) in vitro, the cytotoxicity of berberine and their derivative were compared through MTT assay in four human cancer cell lines and their IC50 values (Table 1). Our results showed that compound 8 exhibited better inhibitory activities with IC50 values of 5.4 ± 0.45 µM against MHCC97, and compound 8 demonstrated selectivity inhibitory activities against specific cell lines MHCC97H. Moreover, compound 8 did not have good activity against other cell lines.
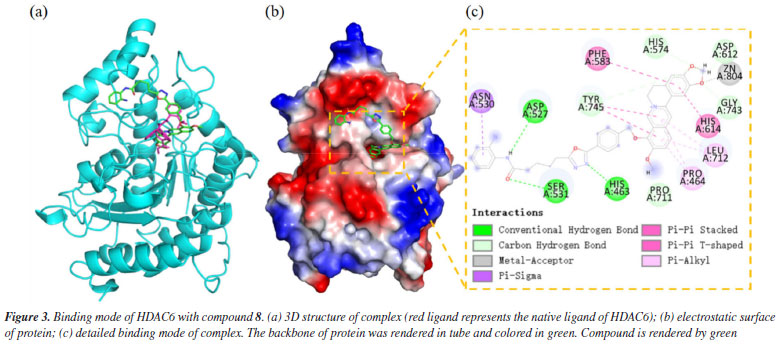
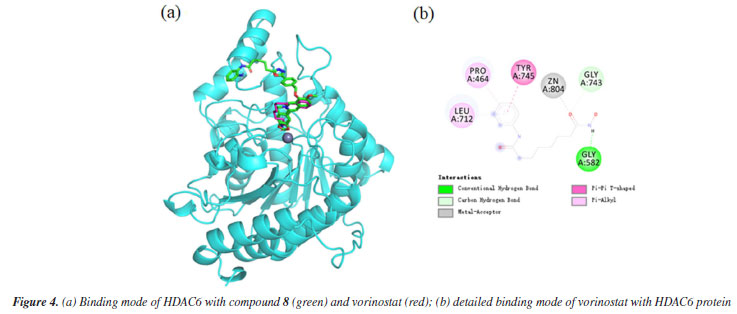
The docking results demonstrated a favorable binding affinity (docking score: -7.24 kcal mol-1) between compound 8 and the HDAC6 target protein, with a binding energy lower than that of the native ligand inhibitor. This indicates that the target protein HDAC6 exhibits a strong affinity for the small molecule. Visualization of the interactions was performed using PyMOL 2.1 (Schrödinger, LLC., USA, 2018), which elucidated the binding pattern between the compound and the protein. The analysis distinctly highlighted the amino acid residues within the protein pocket that interact with the compound. Notably, compound 8 showed a high degree of overlap with the binding mode of the native ligand inhibitor in the HDAC6 protein's active site, further suggesting its potential as an effective inhibitor (Figure 3a). Specifically, compound 8 was observed to form significant hydrogen bonds with the active site residues ASP-527, SER-531, and HIS-463, which substantially contribute to the stabilization of the molecule within the protein's cavity. Additionally, the compound exhibited strong hydrophobic interactions with residues PHE-583, HIS-614, PRO-464, LEU-712, and ASN-530. Moreover, π-π stacking interactions between the benzene rings of the compound and the phenyl rings of PHE-583 and HIS-614 were observed, playing a crucial role in the molecule's stabilization. To differentiate the binding modes of SAHA and compound 8, we analyzed the binding mode of SAHA with HDAC6 (Figure 4). The results showed that SAHA forms hydrogen bonds with GLY-582 in HDAC6, engages in π-π conjugation with TYR-745, and establishes hydrophobic interactions with PRO-464 and LEU-712. Additionally, the compound's carbonyl group coordinates with Zn2+, promoting stable binding between the small molecule and the protein, thereby facilitating its activity. We also found that SAHA and compound 8 exhibit highly similar binding modes within the HDAC6 active cavity, sharing analogous pharmacophores, which could be key to compound 8 activity.
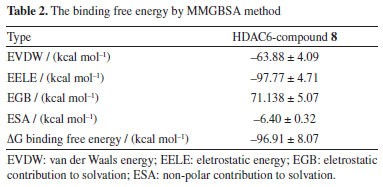
To explore the molecular mechanism underlying the interaction between compound 8 and the HDAC6 protein, we utilized the MMPBSA tool to compute the binding free energy from a 200 ns molecular dynamics simulation. Binding free energy serves as a fundamental metric for analyzing changes in the binding mode of compound 8 by assessing its thermodynamic properties. Negative binding free energy values (ΔG binding energy) indicate system stability, while positive values suggest instability (see Table 2). According to Table 2, van der Waals forces significantly contribute to stabilizing the small molecule, followed by electrostatic interactions. The binding free energy between compound 8 and the HDAC6 protein was -96.91 ± 8.07 kcal mol-1, with electrostatic contributions being predominant (-97.77 ± 4.71 kcal mol-1). This suggests that the compound can stably reside within the protein's binding pocket, forming strong hydrogen bonds with surrounding residues. Additionally, the compound's good fit within the protein cavity results in substantial van der Waals interactions, further stabilizing the small molecule (-63.88 ± 4.09 kcal mol-1). Overall, compound 8 demonstrates a strong affinity for the HDAC6 protein, facilitating the formation of a stable complex and thereby exerting its biological activity.
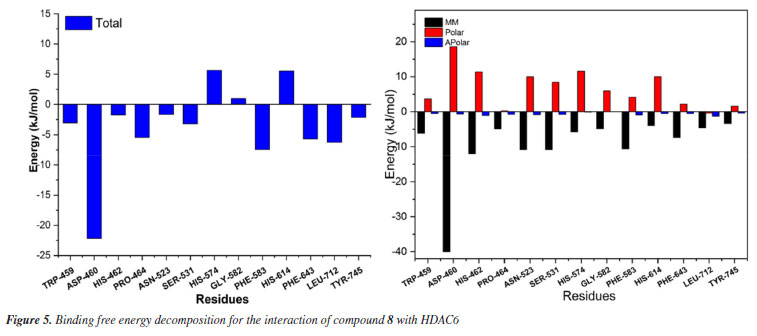
To gain a deeper understanding of each residue's contribution to the small molecule's binding, an energy decomposition analysis was performed for each residue using MMPBSA. As shown in Figure 5, the interactions between compound 8 and the protein are primarily categorized into polar, apolar, and MM (molecular mechanics) energies, with MM energy playing a dominant role. Higher solvation energy can be detrimental to binding, while hydrophobic interactions enhance stability and binding affinity. Detailed analysis reveals that amino acids such as TRP-459, ASP-460, HIS-462, PRO-464, ASN-523, SER-531, HIS-574, GLY-582, PHE-583, HIS-614, PHE-643, LEU-712, and TYR-745 play a significant role in stabilizing the small molecule, particularly ASP-460, with an energy contribution of -5.30 kcal mol-1. Notably, residues HIS-574, GLY-582, PHE-583, and HIS-614 contribute positively, indicating potential barriers to the small molecule's binding. These insights provide critical guidance for the further optimization and modification of compound design.
CONCUSIONS In summary, through the seven-step reaction process, we synthesized a novel berberine derivative, compound 8, which integrates the pharmacophores of HDAC inhibitors for potential anti-tumor activity. The anti-tumor efficacy of compound 8 was evaluated in vitro against four cell lines. The compound exhibited modest anti-tumor effects against HCC827, HeLa, and HEL cells, but demonstrated superior inhibitory activity against MHCC97H cells, with an IC50 value of 5.4 ± 0.45 µM, compared to berberine. Molecular docking and dynamics analysis studies not only illustrate the effective use of computational tools to predict and analyze molecular interactions but also highlight the potential of compound 8 as a promising HDAC6 inhibitor. The findings from this research provide valuable insights for future experimental validations, potentially guiding the design of novel therapeutic agents targeting HDAC6. Consequently, compound 8 may serve as a lead compound in pharmaceutical chemistry.
SUPPLEMENTARY MATERIAL 1H NMR, 13C NMR, and MS spectra of compounds prepared and the Molecular Docking, Dynamics Analysis Studies section are available as Supplementary Material at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
ACKNOWLEDGMENTS This work was supported by the PhD program of Kaili University (grant number BS20240220), Guizhou province education commission natural Science Foundations (grant number [2021]074) and Natural Science Foundation of Guizhou Province (grant number QKHJC-ZK[2021]YB052) for financial support of this study.
REFERENCES 1. Wang, Y.; Li, Y.; Liu, X.; Cho, W. C. S.; Curr. Cancer Drug Targets 2013, 13, 506. [Crossref] 2. Hu, Y.; Davies, G. E.; Fitoterapia 2010, 81, 358. [Crossref] 3. Pandey, M. K.; Sung, B.; Kunnumakkara, A. B.; Sethi, G.; Chaturvedi, M. M.; Aggarwal, B. B.; Cancer Res. 2008, 68, 5370. [Crossref] 4. Dong, H.; Wang, N.; Zhao, L.; Lu, F.; Evidence-Based Complementary and Alternative Medicine 2012, 2012, 591654. [Crossref] 5. Yu, Y.; Yi, Z. B.; Liang, Y. Z.; FEBS Lett. 2007, 581, 4179. [Crossref] 6. Kim, S.; Han, J.; Kim, N.-Y.; Lee, S. K.; Cho, D. H.; Choi, M.-Y.; Kim, J. S.; Kim, J.-H.; Choe, J.-H.; Nam, S. J.; Lee, J. E.; Oncol. Rep. 2012, 27, 210. [Crossref] 7. Wang, J.; Liu, Q.; Yang, Q.; Int. J. Mol. Med. 2012, 30, 1166. [Crossref] 8. Wang, L.; Yang, X.; Li, X.; Stoika, R.; Wang, X.; Lin, H.; Ma, Y.; Wang, R.; Liu, K.; New J. Chem. 2020, 44, 14024. [Crossref] 9. Ding, Y. P.; Ye, X. L.; Zhou, L.; Luo, S.; Li, X. G.; Chin. J. Org. Chem. 2012, 32, 677. [Crossref] 10. Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C.; Cell Res. 2007, 17, 195. [Crossref] 11. Ropero, S.; Esteller, M.; Mol. Oncol. 2007, 1, 19. [Crossref] 12. Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T.; J. Med. Chem. 2010, 53, 8546. [Crossref] 13. Pangal, A.; Shaikh, J.; Res. J. Chem. Sci. 2013, 3, 79. [Link] accessed in March 2025 14. Khan, M.; Fazal, Z.; Alam, A.; Ibrahim, M.; Ali, T.; Ali, M.; Khan, H. D.; Lett. Drug Des. Discovery 2023, 20, 2018. [Crossref] 15. Begum, F.; Yousaf, M.; Iqbal, S.; Ullah, N.; Hussain, A.; Khan, M.; Khalid, A.; Algarni, A. S.; Abdalla, A. N.; Khan, A.; Lodhi, M. A.; Al-Harrasi, A.; ACS Omega 2023, 8, 46816. [Crossref] 16. Jha, A.; Sen, A.; Malla, R. R.; Russ. J. Bioorg. Chem. 2021, 47, 670. [Crossref] 17. Frew, A. J.; Johnstone, R. W.; Bolden, J. E.; Cancer Lett. 2009, 280, 125. [Crossref] 18. Han, X.; Shao, K.; Hu, W.; Chem. Res. Chin. Univ. 2018, 34, 571. [Crossref] 19. Chen, J.-X.; Lin, W.-E.; Chen, M.-Z.; Zhou, C.-Q.; Lin, Y.-L.; Chen, M.; Jiang, Z.-H.; Chen, W.-H.; Jiang, Z.-H.; Chen, W.-H.; Bioorg. Med. Chem. Lett. 2012, 22, 7056. [Crossref] 20. Li, Z. F.; Zhang, M.; Cui, L. Y.; Lu, D. L.; Zhang, J.; Chin. J. Org. Chem. 2008, 28, 479. [Crossref] 21. He, S. P.; Dong, G. Q.; Wang, Z. B.; Chen, W.; Huang, Y. H.; Li, Z. G.; Jiang, Y.; Liu, N.; Yao, J. Z.; Miao, Z. Y.; Zhang, W. N.; Sheng, C. Q.; ACS Med. Chem. Lett. 2015, 6, 239. [Crossref] 22. Liao, S. L.; Zhang, G. G.; Cao, H.; Chen, B. Y.; Li, W. M.; Chin. J. Org. Chem. 2018, 38, 1549. [Crossref] 23. Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Pollard, W. T.; Banks, J. L.; J. Med. Chem. 2004, 47, 1750. [Crossref] 24. Kusharyanti, I.; Larasati, L.; Susidarti, R. A.; Meiyanto, E.; Indonesian Journal of Cancer Chemoprevention 2011, 2, 267. [Crossref] 25. Rajeswari, M.; Santhi, N.; Bhuvaneswari, V.; Bioinformation 2014, 10, 157. [Crossref] 26. Mahbub, N. I.; Hasan, M. I.; Rahman, M. H.; Naznin, F.; Islam, M. Z.; Moni, M. A.; Informatics in Medicine Unlocked 2022, 30, 100888. [Crossref] 27. Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J. Y.; Wang, L.; Lupyan, D.; Dahlgren, M. K.; Knight, J. L.; Kaus, J. W.; Cerutti, D. S.; Krilov, G.; Jorgensen, W. L.; Abel, R.; Friesner, R. A.; J. Chem. Theory Comput. 2016, 12, 281. [Crossref] 28. Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J. L.; Dror, R. O.; Shaw, D. E.; Proteins 2010, 78, 1950. [Crossref] 29. Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Berendsen, H. J. C.; J. Comput. Chem. 2005, 26, 1701. [Crossref] 30. Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L.; J. Chem. Phys. 1983, 79, 926. [Crossref] 31. Miller III, B. R.; McGee Jr., T. D.; Swails, J. M.; Homeyer, N.; Gohlke, H.; Roitberg, A. E.; J. Chem. Theory Comput. 2012, 8, 3314. [Crossref] 32. Iwasa, K.; Moriyasu, M.; Yamori, T.; Turuo, T.; Lee, D.-U.; Wiegrebe, W.; J. Nat. Prod. 2001, 64, 896. [Crossref] 33. Orfila, L.; Rodríguez, M.; Colman, T.; Hasegawa, M.; Merentes, E.; Arvelo, F.; J. Ethnopharmacol. 2000, 71, 449. [Crossref] 34. Yang, Z.; Ya, P.; Yang, H.; Journal of Guiyang Medical College 2012, 37, 235 (in Chinese). [Link] accessed in March 2025 35. Jiang, X. F.; Liu, Y.; Cao, J. F.; Lat. Am. J. Pharm. 2017, 36, 2105. [Link] accessed in March 2025 36. Racané, L.; Cindrić, M.; Zlatar, I.; Kezele, T.; Milić, A.; Brajša, K.; Hranjec, M.; J. Enzym. Inhib. Med. Chem. 2021, 36, 163. [Crossref]
Editor handled this article: Nelson H. Morgon |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access