
 |
 |
 |
 |
 |
Artigo
| Chemical constituents from the leaves of Eriodictyon californicum (Hook. & Arn.) Torr. (Boraginaceae) |
|
Brenda K. M. SousaI; Talita L. N. GonçalvesI; Lívia M. DutraI; Victória L. A. SantosII I. Núcleo de Estudos e Pesquisas de Plantas Medicinais (NEPLAME), Universidade Federal do Vale do São Francisco, 56304-205 Petrolina - PE, Brasil Received: 02/27/2025 *e-mail: jackson.guedes@univasf.edu.br Eriodictyon californicum is a species belonging to Boraginaceae family, popularly known as "yerba santa" and used in folk medicine to treat rheumatism, headaches, inflammation, and respiratory complications. In the present work, we performed phytochemical analyses of extracts from the leaves of E. californicum in order to isolate its chemical constituents. According to thin-layer chromatography (TLC) analyses, preliminary phytochemical screening indicated the presence of anthocyanins derivatives, flavonoids, condensed and hydrolysable tannins. Furthermore, classical chromatographic techniques (column chromatography, analytical and preparative thin-layer chromatography) led to the isolation of secondary metabolites. The compounds were elucidated by 1D and 2D nuclear magnetic resonance (NMR) (correlation spectroscopy (COSY), heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC)) analysis, Fourier transform infrared (FTIR) spectroscopy, mass spectrometry (MS) analysis, and comparison with literature data. In this study, seven metabolites were identified: sterubin (1), chrysoeriol (2), apigenin (3), hydroxygenkwanin (4), 5,4'-dihydroxy-7,3'-dimethoxyflavanone (5), ethyl cinnamate (6) and 5-hydroxy-7methoxychromone (7). The compounds 1-5 were previously identified in this species, and the compounds 6 and 7 are being reported for the first time in the species E. californicum, as well as in Boraginaceae family. The 1H and 13C NMR data for some compounds have been reviewed. These results revealed that E. californicum is an important source of secondary metabolites, mainly flavonoids with chemophenetic relationships with Boraginaceae family. INTRODUCTION The Boraginaceae family comprises approximately 2000 species, which can be found in more than 100 genera.1 Medicinally, the biological activities of species of this family are related to the presence of bioactive compounds, mainly flavonoids, and polyphenols.1,2 Among the genera of this family, the genus Eriodictyon comprises about 10 species restricted to the southwestern United States and northern Baja California, Mexico.3,4 Eriodictyon californicum, a typical medicinal plant of the Boraginaceae family and employed in the treatment of skin wounds is popularly known as "yerba santa". Its leaves are used by indigenous American tribes in the cure of several diseases, some directly associated with aging, including rheumatism, headaches, and inflammation, in addition to respiratory complications such as asthma, cough, and pulmonary infections.4,5 Nowadays, this species is available for use as a dietary supplement and homeopathic medicine for the supportive treatment of asthma and bronchial diseases.6 Chemically, E. californicum is recognized by the presence of flavonoids as main secondary metabolites. Previous phytochemical investigation using high-performance liquid chromatography (HPLC) describes that "yerba santa" is especially rich in polyphenols and flavonoids.7 Flavonoids are naturally occurring phenolic compounds abundantly present in plants synthesized as secondary metabolites and possess various functions, including antimicrobial and antioxidant agents, visual attractors, photoreceptors, feeding repellent, and light screening.8,9 Furthermore, flavonoids have wide therapeutic potential, and are found in several natural sources, such as leaves, fruits, bulbs, barks, stems, and roots.10,11 Some of the flavonoids identified in the literature12-14 exhibit pharmacological activities, such as antioxidant, anticancer, antimicrobial, anti-inflammatory, antihypertensive and neuroprotective actions. A research showed that extracts and compounds isolated from E. californicum have pharmacological properties, such as anti-inflammatory,4,15,16 antioxidant,6,15 potential cancer chemopreventive,15 antibacterial17 and neuroprotective activities.3,4,16,18 Also, in the past, this species was used to mask the taste of quinine and other bitter medicines, thus, studies have evaluated taste modulating properties of extracts or isolated compounds in the species.7,19-21 In this work, moving forward in the study of the chemical and pharmacological significance of Eriodictyon californicum, we report the isolation and identification of phenolic compounds from the leaves of E. californicum, metabolites with chemophenetic importance to the Boraginaceae family. The 1H and 13C nuclear magnetic resonance (NMR) data for some compounds have been reviewed.
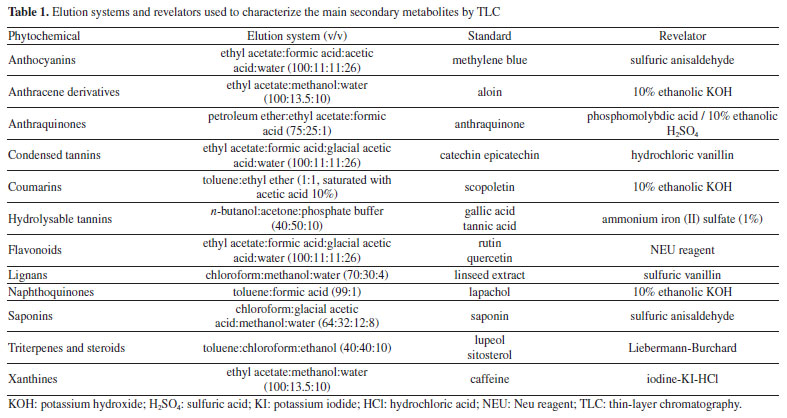
EXPERIMENTAL General experimental procedures The Fourier-transform infrared (FTIR) spectra were acquired in KBr pellets on a Shimadzu spectrometer (IRTracer-100) in mid-IR, equipped with a Universal ATR (attenuated total reflectance) sampling device containing diamond/ZnSe crystal. Spectra were acquired and then processed in the OriginPro 8 (OriginLab Corporation, Massachusetts, United States). The spectra were scanned at room temperature in transmission mode over the wavenumber range of 4000-400 cm-1, with 100 accumulations at a resolution of 4 cm-1. A background spectrum was scanned under the same instrumental conditions before each series of measurements. One-dimensional (1D) and two-dimensional (2D) NMR experiments were performed in DMSO-d6 (dimethyl sulfoxide) or CDCl3 at 298 K on a Bruker™ ASCEND III 400 NMR operating at 9.4 T, observing 1H and 13C at 400 and 100 MHz, respectively. Onebond 1H-13C (heteronuclear single quantum correlation (HSQC)) and long-range (heteronuclear multiple bond correlation (HMBC)) NMR correlation experiments were optimized for an average coupling constant of 1J(C,H) and LRJ(C,H) of 140 and 8 Hz, respectively. All 1H and 13C NMR chemical shifts (d) are expressed in ppm related to the tetramethylsilane (TMS) signal at 0.00 ppm as an internal reference, and the coupling constants (J) in Hz. Mass spectrometric (MS) analysis used a TSQ Quantum Access spectrometer, equipped with an atmospheric pressure chemical ionization (APCI) source and operating in positive and negative acquisition mode. Samples (1 mg mL-1) were prepared in methanol. MS/MS spectra were obtained from the application of energy from 25 to 35 eV. To obtain the mass spectra by direct injection, an Ion Trap-amaZonX spectrometer (Bruker) was used for electrospray ionization-mass spectrometry (ESI-MS, low resolution) and a micrOTOF II (Bruker) for high-resolution electrospray ionization-mass spectrometry (HRESIMS, high resolution) operating with a capillary voltage of 3.5 kV, ESI in positive mode, end plate offset of 500 V, nebulizer 8.0 psi, dry gas (N2) with a flow rate of 5.0 L h-1 and a temperature of 200 ºC. The spectra (m/z 50-1000) were recorded every 2 s. Melting point (m.p.) of the isolated compounds were measured on a melting point apparatus (QUIMIS, model Q340S23), with temperature ranging from 0-310 ºC. Plant material Leaves of E. californicum were collected at Embrapa Semiárido on Bebedouro Experimental Field, in Petrolina (coordinates: 09º23'35" S, 40º30'27" W), state of Pernambuco, Brazil, in February 2020. The identity of the plant was confirmed by Prof. Dr. Lúcia Helena Piedade Kiill, a botanist of the Embrapa Semiárido, based on comparison with a voucher specimen (#HTSA 7850) deposited in the Herbário do Trópico do Semiárido (HTSA). All procedures for access to genetic patrimony and associated traditional knowledge were carried out, and the project was registered in SisGen (Register #A7FE154). Extraction The dried and powdered leaves of E. californicum (611.6 g) were subjected to maceration at room temperature using 95% ethanol as solvent. Three extractions were performed, renewing the solvent every 72 h, until the drug was completely depleted. The extractive solutions were filtered and concentrated in a rotatory evaporator at reduced pressure (40-50 ºC), in order to afford 39.9 g of crude ethanolic extract. Preliminary phytochemical analysis The qualitative presence of different secondary metabolites of crude ethanolic extract was evaluated by thin-layer chromatography (TLC). An aliquot of the extract solutions was applied in plates of silica gel 60 F254 in aluminum supports (Merck, Darmstadt, Germany), eluted in different solvent systems, and applied in specific revelators for each secondary metabolite class, following protocols (Table 1).22 The plates were visualized in a UV camera at 254 and 365 nm. The evaluation of the phytochemical presence in the extracts was based on the spot profiles and comparison with reference standards.
Isolation of compounds The crude ethanolic extract from the leaves of E. californicum (38.2 g) was submitted to vacuum liquid chromatography (VLC) with hexane (Hex, 900 mL), chloroform (CHCl3, 900 mL), ethyl acetate (EtOAc, 900 mL), and methanol (MeOH, 900 mL) as the mobile phases, and silica gel 60 (70-230 mesh) as the stationary phase, giving hexanic (0.1 g), chloroformic (4.9 g), ethyl acetate (18.5 g), and methanolic (3.5 g) fractions, respectively. First, a precipitate on EtOAc fraction was successively washed with methanol and filtered, yielding a white powder. Qualitative TLC analyses confirm purity, giving compound 1 (2.0 g). The EtOAc fraction (7.7 g) was subjected to column chromatography (CC) using silica gel 60 as the stationary phase and as eluents hexane, CHCl3, EtOAc and MeOH, individually or in binary mixtures in ascending order of polarity, affording 108 fractions (100 mL each), that after collected were concentrated in a rotary evaporator. The fractions were evaluated and pooled according to TLC analysis, using the solvent system Hex:EtOAc (3:7), yielding 14 groups (G1 to G14). After the precipitation of G7, the precipitate was washed with methanol, giving a mixture of 1, 2, and 3 (40.4 mg). Similarly, the precipitate of group G8 was submitted to the same procedure as the before fraction, yielding a mixture of 2 and 4 (61.3 mg). The CHCl3 fraction (4.9 g) was subjected to CC in the same conditions as described above, yielding 123 fractions, that were pooled in 26 groups (G1 to G26) according to TLC analysis. The precipitate of group G21 (12.1 mg) was successively washed with methanol, resulting in a white amorphous solid, affording 5 (12.1 mg), and qualitative TLC analysis confirmed purity. Groups G10 (2.8 mg) and G17 (7.8 mg), giving compounds 6 (2.8 mg) and 7 (7.8 mg), respectively. Spectral data Sterubin (1) White amorphous powder; m.p.: 220-223 ºC; ESI-MS: [M + H]+ m/z: 303.0863 (calcd. m/z 303.0863, error: 0.1 ppm), molecular formula C16H14O6; FTIR (KBr) νmax / cm-1 3190 (OH), 2943, 2850 (C-H, aliphatic), 1639 (C=O, enolic), 1303, 1118 (C-O), 1604, 1454, 860, 732 (C=C, aromatic); 1H NMR (400 MHz, DMSO-d6) d 6.89 (br s, 1H, H-2'), 6.76 (br s, 2H, H-5' and H-6'), 6.09 (d, 1H, J 2.4 Hz, H-8), 6.07 (d, 1H, J 2.2 Hz, H-6), 5.42 (dd, 1H, J 12.5 and 3.0 Hz, H-2), 3.78 (s, 3H, OCH3-7), 3.24 (dd, 1H, J 17.2 and 12.5 Hz, H-3α), 2.72 (dd, 1H, J 17.2 and 3.0 Hz, H-3β); 13C NMR (100 MHz, DMSO-d6) d 197.39 (C-4), 167.87 (C-7), 163.66 (C-5), 163.29 (C-9), 146.24 (C-4'), 145.68 (C-3'), 129.74 (C-1'), 118.45 (C-6'), 115.82 (C-5'), 114.85 (C-2'), 103.08 (C-10), 95.04 (C-6), 94.24 (C-8), 79.12 (C-2), 56.33 (OCH3-7), 42.59 (C-3). Chrysoeriol (2) ESI-MS: [M + H]+ m/z: 301.0704 (calcd. m/z 301.0707, error: 1.0 ppm), molecular formula C16H12O6; 1H NMR (400 MHz, DMSO-d6) d 12.99 (s, 1H, OH-5), 7.55 (m, 2H, H-2' and H-6'), 6.96 (m, 1H, H-5'), 6.88 (s, 1H, H-3), 6.51 (d, 1H, J 2.0 Hz, H-8), 6.22 (d, 1H, J 2.0 Hz, H-6), 3.92 (s, 3H, OCH3-3'); 13C NMR (100 MHz, DMSO-d6) d 182.24 (C-4), 164.56 (C-7), 164.08 (C-2), 161.91 (C-5), 157.78 (C-9), 151.15 (C-4'), 148.45 (C-3'), 122.02 (C-1'), 120.77 (C-6'), 116.21 (C-5'), 110.56 (C-2'), 104.20 (C-10), 103.65 (C-3), 99.28 (C-6), 94.48 (C-8), 56.36 (OCH3-3'). Apigenin (3) ESI-MS: [M + H]+ m/z: 271.0602 (calcd. m/z 271.0601, error: -0.3 ppm), molecular formula C15H10O5; 1H NMR (400 MHz, DMSO-d6) d 12.98 (s, 1H, OH-5), 7.91 (d, 2H, J 8.7 Hz, H-2' and H-6'), 6.95 (d, 2H, J 8.8 Hz, H-3' and H-5'), 6.75 (s, 1H, H-3), 6.48 (d, 1H, J 2.2 Hz, H-8), 6.22 (d, 1H, J 2.0 Hz, H-6); 13C NMR (100 MHz, DMSO-d6) d 182.18 (C-4), 164.16 (C-7), 163.96 (C-2), 161.93 (C-5), 161.60 (C4'), 157.76 (C-9), 128.87 (C-2' and C-6'), 121.67 (C-1'), 116.41 (C-3' and C-5'), 103.27 (C-3), 103.94 (C-10), 99.33 (C-6), 94.41 (C-8). Hydroxygenkwanin (4) ESI-MS: [M + H]+ m/z: 301.0702 (calcd. m/z 301.0707, error: 1.6 ppm), molecular formula C16H12O6; 1H NMR (400 MHz, DMSO-d6) d 12.98 (s, 1H, OH-5), 7.45 (m, 2H, H-2' and H-6'), 6.93 (m, 1H, H-5'), 6.70 (s, 1H, H-3), 6.67 (d, 1H, J 2.0 Hz, H-8), 6.35 (d, 1H, J 2.0 Hz, H-6), 3.87 (s, 3H, OCH3-3'); 13C NMR (100 MHz, DMSO-d6) d 182.22 (C-4), 165.49 (C-7), 164.64 (C-2), 161.66 (C-5), 157.60 (C-9), 150.25 (C-4'), 146.21 (C-3'), 121.90 (C-1'), 119.50 (C-6'), 116.41 (C-5'), 113.95 (C-2'), 105.09 (C-10), 103.48 (C-3), 98.31 (C-6), 92.91 (C-8), 56.38 (OCH3-3'). 5,4'-Dihydroxy-7,3'-dimethoxyflavanone (5) White amorphous solid; FTIR (KBr) νmax / cm-1 3414 (OH), 2981, 2850 (C-H, aliphatic), 1639 (C=O, enolic), 1296, 1157 (C-O), 1573, 1400, 840, 744 (C=C, aromatic); 1H NMR (400 MHz, CDCl3) d 12.02 (s, 1H, OH-5), 6.97 (m, 1H, H-2'), 6.95 (m, 2H, H-5' and H-6'), 6.08 (d, 1H, J 2.3 Hz, H-6), 6.05 (d, 1H, J 2.3 Hz, H-8), 5.34 (dd, 1H, J 13.1 and 3.0 Hz, H-2), 3.93 (s, 3H, OCH3-3'), 3.81 (s, 3H, OCH3-7), 3.10 (dd, 1H, J 17.2 and 13.1 Hz, H-3α), 2.79 (dd, 1H, J 17.2 and 3.0 Hz, H-3β); 13C NMR (100 MHz, CDCl3) d 195.99 (C-4), 167.98 (C-7), 164.17 (C-5), 162.84 (C-9), 146.78 (C-3'), 146.24 (C-4'), 130.22 (C-1'), 119.62 (C-6'), 114.54 (C-5'), 108.77 (C-2'), 103.13 (C-10), 95.13 (C-6), 94.28 (C-8), 79.36 (C-2), 56.02 (OCH3-3'), 55.70 (OCH3-7), 43.41 (C-3). Ethyl cinnamate (6) 1H NMR (400 MHz, CDCl3) d 7.69 (d, 1H, J 16.1 Hz, H-7), 7.52 (m, 2H, H-2 and H-6), 7.38 (m, 2H, H-3 and H-5), 7.38 (m, 1H, H-4), 6.44 (d, 1H, J 16.1 Hz, H-8), 4.27 (q, 2H, J 7.2 Hz, H-10), 1.34 (t, 3H, J 7.2 Hz, H-11); 13C NMR (100 MHz, CDCl3) d 166.8 (C-9), 144.60 (C-7), 134.30 (C-1), 130.22 (C-4), 128.88 (C-3 and C-5), 128.1 (C-2 and C-3), 118.30 (C-8), 60.52 (C-10), 14.13 (C-11). 5-Hydroxy-7-methoxychromone (7) 1H NMR (400 MHz, CDCl3) d 12.56 (s, 1H, OH-5), 7.74 (d, 1H, J 6.0 Hz, H-2), 6.39 (d, 1H, J 2.3 Hz, H-8), 6.37 (d, 1H, J 2.3 Hz, H-6), 6.22 (d, 1H, J 6.0 Hz, H-3), 3.86 (s, 3H, OCH3-7); 13C NMR (100 MHz, CDCl3) d 183.30 (C-4), 165.20 (C-7), 162.70 (C-5), 158.20 (C-9), 155.62 (C-2), 111.41 (C-3), 106.70 (C-10), 98.25 (C-6), 92.75 (C-8), 56.06 (OCH3-7).
RESULTS AND DISCUSSION The phytochemical analysis of plants presents an idea about the chemical nature of compounds that confer host plants with certain effective biological activity.23 This analysis is a qualitative method carried out with the aim of identifying the possible chemical constituents present in the vegetal materials from revealers that indicate certain classes of secondary metabolites. Thus, secondary metabolites not only play a vital role in plant defense against environmental stresses but also serve as therapeutic components for humans.24 The results of the phytochemical analysis from leaves of E. californicum are shown in Table 2.
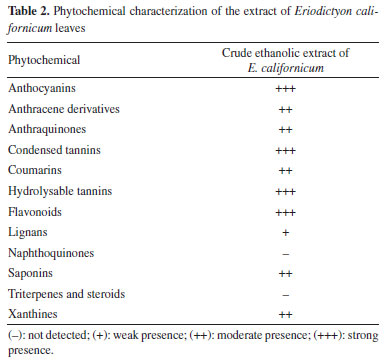
The analysis indicated positive reaction to the presence of anthocyanins, flavonoids, condensed and hydrolysable tannins. Some factors regulate the production of leaf phytochemicals, such as soil nutrients, water or light availability, as well can influence plant phytochemistry by modulating the costs associated with their production and deployment of phytochemicals.25 The phytochemical investigation of the ethyl acetate and chloroform fractions from the leaves of E. californicum affording seven metabolites, including five flavonoids named sterubin (1),26 chrysoeriol (2),27 apigenin (3),28 hydroxygenkwanin (4)29 and 5,4'-dihydroxy-7,3'-dimethoxyflavanone (5),30 in addition to other two uncommon metabolites, one phenylpropanoid known as ethyl cinnamate (6)31 and one chromone identified as 5-hydroxy-7methoxychromone (7).32 Compounds 1-5 were previously found in this species, whereas compounds 6 and 7 are reported for the first time in E. californicum and in the Boraginaceae family. The complete 1H and 13C NMR data for these compounds were reviewed according to 1D and 2D NMR experiments (COSY, HSQC and HMBC), when possible, in combination with FTIR and ESI-MS, as well as by comparison with literature data.26-32 The chemical structures of the compounds are presented in Figure 1. Although the structures of the compounds 6 and 7 have already been described a long time ago, their 1H and 13C NMR data are incomplete or scalar coupling constants values have not been assigned. In this work, the complete and unequivocal 1H and 13C NMR data were reviewed according to 1D and 2D NMR experiments (see Tables 1S-7S in the Supplementary Material).
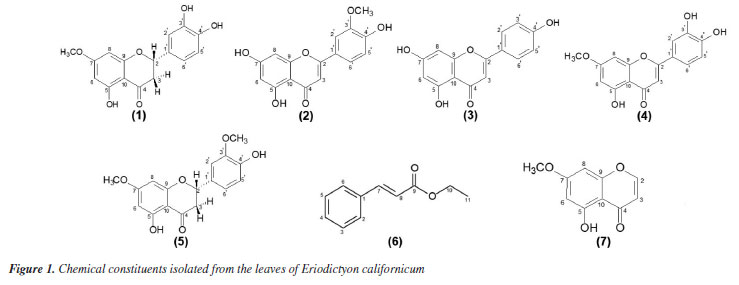
Compound 1 was obtained as a white amorphous powder with the molecular formula C16H14O6 determined by ESI-MS (observed m/z 303.0863 [M + H]+) and NMR analyses. Infrared (IR) spectrum showed absorption bands at 3190, 2943-2850 and 1639 cm-1 typical of hydroxyl, C-H sp3, and enolic carbonyl groups. In addition, absorption bands at 1303-1118 cm-1 were related to C-O of phenols or aromatic ethers, while deformations bands at 1604-1454 and 860732 cm-1 were indicative of the presence of an aromatic ring. The atmospheric-pressure chemical ionization (APCI)-MS spectrum of 1 displayed fragments in positive and negative modes, highlighting the retro-Diels-Alder (RDA) reaction on C-ring of the structure, followed by rearrangement. The main fragment pathway of flavonoids within aglycone is the RDA reaction coupled to losses of small neutral molecules and fragments.33 MS/MS displayed fragmentation pattern similar to the literature.4 The 1H NMR spectrum exhibited characteristic signals of methylene and methine hydrogens at dH 2.72 (dd, 1H, J 17.2 and 3.0 Hz, H-3β), dH 3.24 (dd, 1H, J 17.2 and 12.5 Hz, H-3α) and dH 5.42 (dd, 1H, J 12.5 and 3.0 Hz, H-2), characteristic of flavanones. Furthermore, it was observed two doublets at dH 6.07 (d, 1H, J 2.2 Hz, H-6) and dH 6.09 (d, 1H, J 2.2 Hz, H-8), coupled in meta, representative of aromatic hydrogens of the flavanone A-ring and suggesting the existence of a 6,8-disubstituted ring. Two broad singlets at dH 6.76 (br s, 2H, H-5' and H-6') and dH 6.89 (br s, 1H, H-2'), indicated the presence of a trisubstituted B ring flavonoid in the structure. The singlet at dH 3.78 (s, 3H, OCH3-7) revealed the presence of a methoxyl group. 13C DEPTQ (distortionless enhancement by polarization transfer with quaternary carbon detection) NMR spectrum, as well as one-bond and long-range 1H-13C correlations from HSQC and HMBC NMR experiments, indicated a total of 16 atoms of carbon, which comprised typical signals of aromatic carbons at dC 94.24, 95.04, 103.08, 114.85, 115.82, 118.45, 129.74, 145.68, 146.24, 163.29, 163.66 and 167.87, one methine carbon at dC 79.12 assigned to a saturated oxygenated, one methylene carbon at dC 42.59, one carbonyl carbon at dC 197.39, and finally one carbon at dC 56.33, confirming the presence of a methoxyl group. The hydrogen at dH 5.42 (dd, 1H, J 12.5 and 3.0 Hz, H-2) showed direct 1H-13C HSQC correlation map with the carbon at dC 79.12 and long-range 1H-13C correlations with the carbons at dC 114.85 (C-2'), 118.45 (C-6'), 129.74 (C-1'), and 197.39, which supports the substitution pattern proposed with the carbonyl group located at C-4, characteristic of C-ring of the flavanone. On the other hand, the methylene hydrogens showed one bond 1H-13C HSQC correlation map with the carbon at dC 42.6 and long-range 1H-13C correlations with the carbons at dC 79.12 (C-2), 103.08 (C-10), 129.74 (C-1'), and 197.39, confirming the methylene group at C-3. The location of the methoxyl group at C-7 was established due to the singlet at dH 3.78 with long-range 1H-13C correlation map from HMBC NMR experiment with the carbon at dC 167.87. Moreover, the doublets at dH 6.09 (H-8) and 6.07 (H-6) revealed long-range 1H-13C correlation map from HMBC NMR experiment with the carbon at dC 167.9 (C-7). Also, the hydroxyl group at position C-5 was observed due to the long-range 1H-13C correlation map from HMBC NMR experiment with the carbons at dC 95.04 (C-6), 103.08 (C-10), and 163.66 (C-5). In contrast, the presence of other hydroxyl groups in the molecule in the B ring at C-3' and C-4' were established on the basis of long-range 1H-13C correlation map from HMBC NMR experiment of the hydrogens at dH 6.76 (H-5 and H-6') and 6.89 (H-2') with the carbon dC 145.68 (C-3') and 146.24 (C-4'), respectively, which are typical of the aromatic B-ring. Thus, according to the IR, MS and 1H and 13C NMR 1D/2D data, and comparison with literature data, compound 1 was identified as a flavanone sterubin, a chemophenetic marker of the genus Eriodyction. This compound possesses neuroprotective activity, mainly against Alzheimer disease.3,18 1H and 13C NMR data analyses of G7 in comparison with literature data, resulted in the identification of a mixture of metabolites named sterubin (1),26 chrysoeriol (2)27 and apigenin (3).28 On the other hand, 1H and 13C NMR data of G8, resulted the mixture of flavones known as chrysoeriol (2) and hydroxygenkwanin (4).29 The G21 resulted in the identification of 5,4'-dihydroxy-7,3'dimethoxyflavanone (5).30 All these flavonoids have been previously reported in E. californicum. While sterubin (1) is a chemophenetic marker of the genus Eriodyction,4 chrysoeriol (2) has been identified in the species E. angustifolium,20,34,35 E. californicum15,20,35-37 and E. trichocalyx,38 apigenin (3) was isolated from E. angustifolium,E. californicum,E. tomentosum,34 E. trichocalyx38 and E. senssifolium,39 the compound hydroxygenkwanin (4) was found in E. angustifolium and E. californicum,35 and 5,4'-dihydroxy-7,3'dimethoxyflavanone (5) was identified only in E. californicum.37 The 1H NMR spectrum of compound 6 revealed the presence of signals at dH 1.28 (t, 3H, J 7.2 Hz, CH3) and 4.27 (q, 2H, J 7.2 Hz, OCH2), assigned to an ethoxyl group (OCH2CH3). Additionally, the signals at dH 7.39 (m, 3H, H-3, H-5, and H-4) and 7.52 (m, 2H, H-2 and H-6) were attributed to the chemically equivalent aromatic hydrogens, suggesting the presence of a monosubstituted aromatic ring. The signals at dH 6.44 (d, 1H, J 16.1 Hz, H-8) and 7.69 (d,J 16.1 Hz, 1H, H-8) indicate a relative trans configuration between vicinal olefinic protons. The 13C NMR spectrum showed 7 signals referring to 9 carbons. The signals at dC 14.14 and 60.51 were attributed to terminal methyl of the ethoxy group and oxymethylene carbon, respectively. Also, it was observed the presence of the signals at dC 118.30 (C-8) and 144.61 (C-7) for the olefinic carbons, and characteristic signals at dC 128.07 (C-2 and C-6), 128.89 (C-3 and C-5) and 130.23 (C-4) in the aromatic region. Non-hydrogenated carbons were determined through the long-range 1H-13C correlation map from HMBC NMR experiment. The 1H-1H COSY correlation map of 6 showed coupling of signals at dH 1.28 (H-11)/dH 4.27 (H-10), correlations between aromatic hydrogens dH 7.39 (H-3 and H-5; H-4)/dH 7.52 (H-2 and H-6), and also of vicinal olefinic protons at dH 6.44 (H-8)/dH 7.69 (H-7). The direct 1H-13C correlation map from HSQC NMR experiment map allowed establishing the one-bond correlation between the signals at dH 7.52 (m, 2H,)/dC 128.1 (C-2 and C-6), dH 7.39 (m, 2H)/dC 128.9 (C-3 and C-5) and dH 7.39 (m, 1H)/dC 130.2 (C-4), associated to the aromatic system. The long-range 1H-13C HMBC correlation map showed the correlations of the olefinic hydrogens at dH 7.69 (H-7)/dC 166.80 (C9, J3), characteristic to an ester carbonyl, and at dH 6.44 (H-8) with the non-hydrogenated aromatic carbon at dC 134.30 (C1, J3) and 166.80 (C-9, J2). The correlation map also showed the signal at dH 4.26 (H-10)/dC 14.14 (C-11, J2) and dC 166.80 (C-9, J2), confirming that the ethoxy group is attached to the carbonyl carbon. These and another key correlation were illustrated in Figure 2. Comparison with the literature data31 allowed to identify compound 6 as the phenylpropanoid ethyl (E)-3-phenylprop-2-enoate, popularly known as ethyl cinnamate.
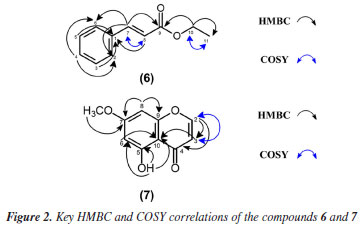
The 1H NMR spectrum of compound 7 showed the signals at dH 7.74 (d, 1H, J 6.0 Hz, H-2) and 6.22 (d, 1H, J 6.0 Hz, H-3) for unsaturated methine hydrogens with vicinal coupling, and two doublets in dH 6.37 (d, 1H, J 2.3 Hz, H-6) and 6.39 (d, 1H, J 2.3 Hz, H-8), typical of the aromatic hydrogens on ring A. The signal at dH 3.86 (s, 3H, OCH3-7), suggested the presence of the methoxyl group and the signal at dH 12.56 (s, 1H, OH-5), was assigned to a hydroxyl group. The 13C NMR spectrum (100 MHz, CDCl3) revealed the presence of 5 carbons signals, among which, the signal in dC 56.34 indicated a free methoxyl. The signals at dC 92.77 (C-8) and 98.26 (C-6) are characteristic of the aromatic carbons and the signals at dC 111.43 (C-3) and 155.65 (C-2) were assigned to unsaturated carbons of the cis configuration of the molecule. Further signals were determined based on two-dimensional analysis. Through 1H-1H COSY correlation map was observed couplings between the vicinal olefinic hydrogens at dH 6.22 (H-3)/dH 7.74 (H-2), confirming the structure of the chromone skeleton. The long-range 1H-13C HMBC correlation map allowed to observe long distance between dH 3.81 (OCH3-7)/dC 165.2 (C-7, J3), confirming the location of the methoxyl on ring A. The signal at dH 6.39 (d, 1H, J 2.3 Hz, H-8) correlated with the non-hydrogenated carbon at dC 106.7 (C-10, J2), and with the carbons at dC 165.2 (C-7, J3) and 158.2 (C-9, J3), characteristic of oxygenated aromatic carbons. Besides that, revealed the correlations of the signal at dH 7.74 (H-2)/dC 183.3 (C-4, J3), coupling with a carboxylic carbon. These and another key correlation were illustrated in Figure 2. The comparison of the NMR data corroborate the identification of these constituent as the chromone 5-hydroxy-7-methoxychromone (7).32 This compound has been identified in another vegetal species, but is being reported for the first time in Eriodictyon californicum and in the Boraginaceae family.
CONCLUSIONS The phytochemical study of E. californicum leaves resulted in the identification of seven compounds, including five flavonoids. These compounds were previously identified in this species. Besides these, it was identified cinnamate derivative and a chromone, reported for the first time in E. californicum and in Boraginaceae family. Thus, the work contributed to the expansion of the phytochemical knowledge of the genus Eriodictyon and of the Boraginaceae family through the isolation of a new compound from the species E. californicum.
SUPPLEMENTARY MATERIAL Supplementary data associated with this article (Figures 1S-84S and Tables 1S-7S) can be found in the online version at http://quimicanova.sbq.org.br as PDF file, with free access.
ACKNOWLEDGMENTS The authors are grateful to Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA), Rede de Pesquisa em Biodiversidade e Sustentabilidade (process CNPq 406399/2022-0) and Rede Baiana de Bioprospecção e Desenvolvimento de Fármacos - BioproFar-BA (process PIE0001/2024) for financial support.
REFERENCES 1. Fayed, M. A. A.; Phytomedicine Plus 2021, 1, 100036. [Crossref] 2. Shahbaz, A.; Abbasi, B. A.; Iqbal, J.; Fatima, I.; Zahra, S. A.; Kanwal, S.; Devkota, H. P.; Capasso, R.; Ahmad, A.; Mahmood, T.; Saudi J. Biol. Sci. 2021, 28, 6086. [Crossref] 3. Kazmi, I.; Al-Abbasi, F. A.; Afzal, M.; Nadeem, M. S.; Altayb, H. N.; Saudi J. Biol. Sci. 2023, 30, 103560. [Crossref] 4. Maher, P.; Fischer, W.; Liang, Z.; Soriano-Castell, D.; Pinto, A. F. M.; Rebman, J.; Currais, A.; Front. Pharmacol. 2020, 11, 208. [Crossref] 5. Albuquerque, P. B. S.; de Oliveira, W. F.; Silva, P. M. S.; Correia, M. T. S.; Kennedy, J. F.; Coelho, L. C. B. B.; Carbohydr. Polym. 2022, 277, 118824. [Crossref] 6. Mödinger, Y.; Schön, C.; Wilhelm, M.; Pickel, C.; Grothe, T.; J. Med. Food 2021, 24, 1235. [Crossref] 7. Ley, J. P.; Krammer, G.; Reinders, G.; Gatfield, I. L.; Bertram, H.-J.; J. Agric. Food Chem. 2005, 53, 6061. [Crossref] 8. Rakha, A.; Umar, N.; Rabail, R.; Butt, M. S.; Kieliszek, M.; Hassoun, A.; Aadil, R. M.; Biomed. Pharmacother. 2022, 156, 113945. [Crossref] 9. Mutha, R. E.; Tatiya, A. U.; Surana, S. J.; Future Journal of Pharmaceutical Sciences 2021, 7, 25. [Crossref] 10. Alizadeh, S. R.; Ebrahimzadeh, M. A.; Eur. J. Med. Chem. 2022, 229, 114068. [Crossref] 11. Jia, Y.; Fu, Y.; Man, H.; Yan, X.; Huang, Y.; Sun, S.; Qi, B.; Li, Y.; Food Res. Int. 2022, 161, 111784. [Crossref] 12. Patel, D. K.; Pharmacological Research - Modern Chinese Medicine 2022, 4, 100155. [Crossref] 13. Slika, H.; Mansour, H.; Wehbe, N.; Nasser, S.; Iratni, R.; Nasrallah, G.; Shaito, A.; Ghaddar, T.; Kobeissy, F.; Eid, A.; Biomed. Pharmacother. 2022, 146, 112442. [Crossref] 14. Beigh, S.; Rehman, M. U.; Khan, A.; Patil, B. R.; Makeen, H. A.; Rasool, S.; Rashid, S.; Arafah, A.; Kamal, M. A.; Phytomedicine Plus 2022, 2, 100221. [Crossref] 15. Liu, Y.-L.; Ho, D. K.; Cassady, J. M.; Cook, V. M.; Baird, W. M.; J. Nat. Prod. 1992, 55, 357. [Crossref] 16. Fischer, W.; Currais, A.; Liang, Z.; Pinto, A.; Maher, P.; Redox Biol. 2019, 21, 101089. [Crossref] 17. Salle, A. J.; Jann, G. J.; Wayne, L. G.; Arch. Biochem. Biophys. 1951, 32, 121. [Crossref] 18. Hofmann, J.; Fayez, S.; Scheiner, M.; Hoffmann, M.; Oerter, S.; AppeltMenzel, A.; Maher, P.; Maurice, T.; Bringmann, G.; Decker, M.; Chem. - Eur. J. 2020, 26, 7299. [Crossref] 19. Ley, J. P.; Krammer, G.; Kindel, G.; Gatfield, I. L.; Bertram, H.-J.; Developments in Food Science, vol. 43; Bredie, W. L. P.; Petersen, M. A., eds.; Elsevier, 2006, p. 173. [Crossref] 20. Reichelt, K. V.; Hartmann, B.; Weber, B.; Ley, J. P.; Krammer, G. E.; Engel K.-H.; J. Agric. Food Chem. 2010, 58, 1850. [Crossref] 21. Fletcher, J. N.; Kinghorn, A. D.; Slack, J. P.; McCluskey, T. S.; Odley, A.; Jia, Z.; J. Agric. Food Chem. 2011, 59, 13117. [Crossref] 22. Wagner, H.; Bladt, S.; Plant Drug Analysis: A Thin Layer Chromatography Atlas, 2nIV ed.; Springer: Berlin, Germany, 1996. [Crossref] 23. Afzal, T.; Bibi, Y.; Ishaque, M.; Masood, S.; Qayyum, A.; Nisa, S.; Shah, Z. H.; Alsamadany, H.; Chung, G.; Saudi J. Biol. Sci. 2022, 29, 1185. [Crossref] 24. Zhang, Y.-W.; Shi, Y.-C.; Zhang, S.-B.; Plant Diversity 2023, 45, 326. [Crossref] 25. Martins-Noguerol, R.; Matías, L.; Pérez-Ramos, I. M.; Moreira, X.; Francisco, M.; Pedroche, J.; DeAndrés-Gil, C.; Gutiérrez, E.; Salas, J. J.; Moreno-Pérez, A. J.; Davy, A. J.; Muñoz-Vallés, S.; Figueroa, M. E.; Cambrollé, J.; Sci. Total Environ. 2023, 869, 161806. [Crossref] 26. Alqasoumi, S. I.; Basudan, O. A.; Alam, P.; Abdel-Kader, M.; Pak. J. Pharm. Sci. 2016, 29, 97. [Link] accessed in April 2025 27. Erenler, R.; Sen, O.; Yıldız, I.; Aydın, A.; Rec. Nat. Prod. 2016, 10, 766. [Link] accessed in April 2025 28. Fajriah, S.; Megawati, M.; Darmawan, A.; Journal of Tropical Life Science 2016, 6, 7. [Crossref] 29. Park, S. H.; Cui, X.; Ahn, D.; Lee, E. B.; Cha, D. S.; Jeon, H.; Zee, O. P.; Kim, Y. C.; Kim, D. K.; Nat. Prod. Sci. 2014, 20, 80. [Link] accessed in April 2025 30. Ibrahim, A.-R. S.; Galal, A. M.; Ahmed, M. S.; Mossa, G. S.; Chem. Pharm. Bull. 2003, 51, 203. [Crossref] 31. Pathiranage, A. L.; Martin, L. J.; Osborne, M.; Meaker, K.; J. Lab. Chem. Educ. 2018, 6, 156. [Link] accessed in April 2025 32. Vasconcelos, J. M. J.; Silva, A. M. S.; Cavaleiro, J. A. S.; Phytochemistry 1998, 49, 1421. [Crossref] 33. Śliwka-Kaszyńska, M.; Anusiewicz, I.; Skurski, P.; Molecules 2022, 27, 1032. [Crossref] 34. Bacon, J. D.; Hannan, G. L.; Fang, N.; Mabry, T. J.; Biochem. Syst. Ecol. 1986, 14, 591. [Crossref] 35. Taguchi, N.; Hata, T.; Kamiya, E.; Homma, T.; Kobayashi, A.; Aoki, H.; Kunisada, T.; Int. J. Cosmet. Sci. 2020, 42, 336. [Crossref] 36. Tutin, F.; Clewer, H. W. B.; J. Chem. Soc., Trans. 1909, 95, 81. [Crossref] 37. Johnson, N. D.; Biochem. Syst. Ecol. 1983, 11, 211. [Crossref] 38. Bohm, B. A.; Constant, H.; Biochem. Syst. Ecol. 1990, 18, 491. [Crossref] 39. Arriaga-Giner, F. J.; Weber, E.; Schober, I.; Yatskievych, G.; Z. Naturforsch., C 1988, 43, 337. [Crossref]
Guest Editor handled this article: Antonio E. M. Crotti |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access